Introduction
Colorectal cancer (CRC) is one of the most commonly
diagnosed cancer in both men and women and is the second leading
cause of cancer-associated death worldwide (1). Currently, the treatment options for
CRC include surgery, chemotherapy, radiotherapy and targeted
therapies, which depend on stage of the disease and physical health
of the patient. The chemotherapeutic agents commonly used in CRC
treatments are 5-fluorouracil, oxaliplatin and irinotecan (2). Although combination therapy of these
drugs has improved response rate and progression-free survival, its
application is often limited due to acquired drug resistance which
can occur in nearly all patients with CRC (3). Therefore, novel therapeutic compounds
and strategies to overcome drug resistance are urgently needed.
Tumor suppressor p53 plays an important role in the
regulation of DNA repair, cell cycle arrest, and apoptosis in the
presence of cellular stress. Mutations in p53, the most common
genetic change in human cancer, have been detected in ~40–50% of
sporadic CRC (4). Mutant p53 has
been shown to be involved in proliferation, invasion, migration,
angiogenesis and cell survival. It is also responsible for drug
resistance of cancer cells (5).
Mutant p53 induced chemoresistance through several mechanisms,
including induction of drug efflux, disruption of cell cycle
regulation, evasion of apoptosis and upregulation of DNA repair
(6). It has been reported that
mutated p53 in CRC is associated with resistance to commonly used
chemotherapeutic agents, including, 5-FU, doxorubicin, oxaliplatin
and irinotecan (7–9). Therefore, finding a novel anticancer
compound that remains effective without p53 chemoresistance may
offer a potential strategy for the treatment of chemoresistant
cancer cells.
Cepharanthine (CEP), a biscoclaurine alkaloid
extracted from Stephania cepharantha Hayata, possesses
various biological activities such as anti-inflammatory,
anti-malarial, anti-HIV-1, anti-oxidant, and anti-allergic
(10). Notably, CEP has been found
to exert anticancer activities against several types of cancer
cells such as leukemia, oral squamous cell carcinoma,
hepatocellular carcinoma, myeloma, cholangiocarcinoma,
osteosarcoma, cervical adenocarcinoma, nasopharyngeal carcinoma and
non-small cell lung cancer both in vitro and in vivo
(11–21). It has been reported that CEP
inhibited tumor growth through numerous mechanisms, including
induction of host immune responses, inhibition of NF-κB and STAT3
signaling pathways and reduction of angiogenesis by inhibiting
expression of two major pro-angiogenic factors, vascular
endothelial growth factor (VEGF) and interleukin-8 (IL-8) (16,18,22–24).
Mechanistic studies revealed that CEP induced apoptosis via
activating caspase-3 and −9, stimulating pro-apoptotic signaling
pathways such as JNK, ERK and p38 MAPK pathways, inhibiting
anti-apoptotic gene Bcl-xl expression and inducing oxidative stress
(12,14–16,18,24,25).
Moreover, CEP was found to induce cell cycle arrest by
downregulating the expression of cyclin D1, CDK-6 and c-Myc
(15,18). Remarkably, CEP has been found to
induce cell cycle arrest of adenosquamous cell carcinoma carrying
p53 mutation (25).
To the best of our knowledge, there were only a few
reports of antitumor effect of CEP in CRC cells (26). More importantly, the mechanisms
underlying anticancer effect of CEP in CRC have never been
investigated. In the present study, we therefore evaluated the
anticancer activity of CEP using a p53 wild-type human colorectal
cancer cell line, COLO-205, and a p53 mutant human colorectal
cancer cell line, HT-29, and determined its underlying mechanisms
of action.
Materials and methods
Chemical reagents
Cepharanthine was from Abcam (Cambridge, UK). Bovine
serum albumin (BSA), dimethyl sulfoxide (DMSO), hydrogen peroxide
(H2O2), N-acetylcysteine (NAC), oxaliplatin,
SN-38, trypan blue, 5-fluorouracil,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were from Sigma-Aldrich (St. Louis, MO, USA). Propidium iodide (PI)
was from Santa Cruz Biotechnology (Dallas, TX, USA). Chloroform,
ethanol and 2-propanolol were from Merck (Kenilworth, NJ, USA).
Nuclease-free water was from Qiagen (Hilden, Germany).
Cell lines and culture
Human colorectal cancer cell lines HT-29 and
COLO-205 were from American Type Culture Collection (Manassas, VA,
USA) whereas SW-620 and HCT-116 were kindly provided by Dr Amornpun
Sereemaspun (Chulalongkorn University, Thailand). HT-29 and SW-620
were grown in Dulbecco's modified Eagle's minimal essential medium
(DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco), 100 U/ml penicillin and 100 µg/ml
streptomycin (Gibco). COLO-205 and HCT-116 were cultured in
RPMI-1640 medium containing 10% FBS, 100 U/ml penicillin and 100
µg/ml streptomycin. The cells were maintained at 37°C in a
humidified 5% CO2 atmosphere.
Cell viability assay
Cell viability was determined by using the MTT
reduction assay. Briefly, cells were seeded in 96-well plates at a
density of 5×103 cells/well. After overnight incubation,
the cells were treated with 5-FU, oxaliplatin, SN-38 or CEP at
0.01, 0.1, 1, 10 and 100 µM for 48 h. Thereafter, MTT solution was
added and incubated for additional 4 h. The medium was removed and
200 µl of DMSO was added to each well. The absorbance of the
converted dye was measured at a wavelength of 570 nm using
LabSystems Multiskan MS microplate reader (Thermo Scientific,
Vantaa, Finland).
Cell cycle analysis
HT-29 and COLO-205 cells were seeded in 6-well
plates at a density of 3×105 cells/well and incubated
overnight. HT-29 cells were treated with 5, 10, and 20 µM of CEP or
0.2% DMSO (solvent control) whereas COLO-205 cells were treated
with 10, 20 and 40 µM of CEP or 0.2% DMSO. After 12, 24 or 36 h of
incubation, the cells were washed with phosphate-buffered saline
(PBS), harvested by trypsinization and centrifuged at 1,500 rpm for
5 min. The cell pellets were washed with cold PBS and fixed with
70% ethanol for 20 min at −20°C. The cells were then washed with
cold PBS and incubated with 500 µl of assay buffer containing 5 µl
of 4 mg/ml RNase A at room temperature. After 30-min incubation,
the cells were stained with 5 µl of 0.05 µg/ml PI for another 30
min at room temperature in the dark. DNA content of the stained
cells was analyzed using BD LSR II flow cytometer.
Real-time RT-PCR
Total RNA was isolated using TRIzol reagent (Life
Technologies) according to the manufacturer's instructions. RNA
concentration and purity was determined using NanoDrop ND-2000
(Thermo Scientific, Rockford, IL, USA). RNA (500 ng) was
reverse-transcribed into cDNA using Improm II™ Reverse
Transcription system (Promega, Madison, WI, USA). Amplification of
target genes was carried out on StepOnePlus™ Real-Time PCR system
(Applied Biosystems, Waltham, MA, USA) using SYBR GreenER™ qPCR
Supermix (Life Technologies) and the primers listed in Table I. Expression of the gene of interest
was normalized to the glyceroldehyde-3-phosphate dehydrogenase
(GAPDH) and relative expression was calculated using the ∆∆CT
method.
 | Table ISequences of primers used for
real-time RT-PCR analysis. |
Table I
Sequences of primers used for
real-time RT-PCR analysis.
| Target genes | Primer
sequences |
|---|
| Bcl-2 | F: 5′-TCA TGT GTG
TGG AGA GCG TCA A-3′ |
|
| R: 5′-CTA CTG CTT
TAG TGA ACC TTT TGC-3′ |
| Bcl-xl | F: 5′-TTG GAC AAT
GGA CTG GTT GA-3′ |
|
| R: 5′-GTA GAG TGG
ATG GTC AGT G-3′ |
| Bax | F: 5′-GAC GAA CTG
GAC AGT AAC ATG-3′ |
|
| R: 5′-AGG AAG TCC
AAT GTC CAG CC-3′ |
| Bak | F: 5′-ATG GTC ACC
TTA CCT CTG CAA-3′ |
|
| R: 5′-TCA TAG CGT
CGG TTG ATG TCG-3′ |
| Cyclin A | F: 5′-CTG CTG CTA
TGC TGT TAG CC-3′ |
|
| R: 5′-TGT TGG AGC
AGC TAA GTC AAA A-3′ |
| Cyclin D | F: 5′-TTG TTG AAG
TTG CAA AGT CCT GG-3′ |
|
| R: 5′-ATG GTT TCC
ACT TCG CAG CA-3′ |
| Cyclin E | F: 5′-TCC TGG ATG
TTG ACT GCC TT-3′ |
|
| R: 5′-CAC CAC TGA
TAC CCT GAA ACC T-3′ |
| p21 | F: 5′-CCT GTC ACT
GTC TTG TAC CCT-3′ |
|
| R: 5′-GCG TTT GGA
GTG GTA GAA ATC T-3′ |
| GAPDH | F: 5′-AAG GTC GGA
GTC AAC GGA TTT GGT-3′ |
|
| R: 5′-ATG GCA TGG
ACT GTG GTC ATG AGT-3′ |
Western blotting
Cell lysates were prepared by homogenizing cells in
RIPA lysis buffer (Thermo Fisher Scientific) containing protease
inhibitors (Sigma). Protein concentration of supernatants was
determined by a microplate assay with the Bio-Rad DC Protein assay
reagents (Hercules, CA, USA) using bovine serum albumin as a
standard. Twenty micrograms of protein lysate were separated on an
8% sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and then transferred to a PVDF membrane. The membrane
was blocked in 3% non-fat dry milk (NFDM) and then incubated with
antibodies against p21Waf1/Cip1 (dilution 1:1,000, 2947;
Cell Signaling Technology, Danvers, MA, USA), cyclin A2 (dilution
1:2,000, 4656; Cell Signaling Technology), cyclin D1 (dilution
1:1,000, 2922; Cell Signaling Technology) or β-actin (dilution
1:1,000, 4970; Cell Signaling, Technology) overnight at 4°C. The
membrane was then washed and incubated with HRP-linked secondary
antibody (Cell Signaling Technology) for 1 h at room temperature.
Protein bands were detected using Luminata™ Cescendo Western HRP
substrate (Millipore, Billerica, MA, USA) and analyzed using Image
Studio software (LI-COR, Lincoln, NE, USA). β-actin was used as the
loading control for protein normalization.
Immunofluorescence staining
After growing HT-29 cells on 8-well Lab-Tek chamber
slides (Thermo Fisher Scientific) overnight, the cells were treated
with CEP at various concentrations for 24 h. The cells were then
fixed in 4% paraformaldehyde for 15 min at room temperature. After
washing with PBS, the cells were permeabilized with 0.25% Triton
X-100 for 10 min at room temperature. Thereafter, the cells were
washed and incubated with blocking solution containing 2% normal
goat serum (Abcam) and 1% bovine serum albumin for 20 min at room
temperature. The cells were then washed with PBS and incubated with
a p21Waf1/Cip1 rabbit antibody (dilution 1:800, 2947;
Cell Signaling Technology) overnight at 4°C followed by incubation
with Alexa Fluor 488 conjugated goat anti-rabbit secondary antibody
(Invitrogen) for 30 min at room temperature in the dark. The cells
were washed, mounted using fluoroshield mounting medium with DAPI
(Abcam) and observed with an LSM 800 Confocal Laser Scanning
Microscope (Zeiss, Oberkochen, Germany).
Measurement of ROS generation
Intracellular ROS was quantified using
membrane-permeable fluorescent probe dichlorodihydrofluorescein
diacetate (DCFH-DA). Cells were seeded in 96-well plates at a
density of 5×103 cells. After overnight incubation, the
culture medium was replaced with 100 µl of Hank's buffered salt
solution (HBSS) containing 10 µM DCFH-DA and incubated for 30 min
at 37°C in the dark. The cells were washed with PBS and incubated
with various concentrations of CEP for 1 h. The cells were then
washed with cold PBS and suspended in 200 µl of 1% Triton X-100.
The fluorescence intensity was measured using a fluorescent
microplate reader (Thermo Scientific) at an excitation wavelength
of 485 nm and an emission wavelength of 535 nm.
Statistical analysis
Data are presented as mean ± standard error of mean
(SEM) from at least three independent experiments. Statistical
analysis was carried out by one-way analysis of variance (ANOVA)
followed by a Bonferroni/Dunn post hoc comparison test. P-values
<0.05 were considered statistically significant.
Results
Cepharanthine exhibits a potent
anticancer activity in p53-mutated colorectal cancer cells
Initially, we evaluated the cytotoxicity of
cepharanthine (CEP) compared to the most common chemotherapeutic
agents for CRC, including 5-fluorouracil, oxaliplatin, and SN-38
(an active metabolite of irinotecan), in a p53 mutant HT-29 cells
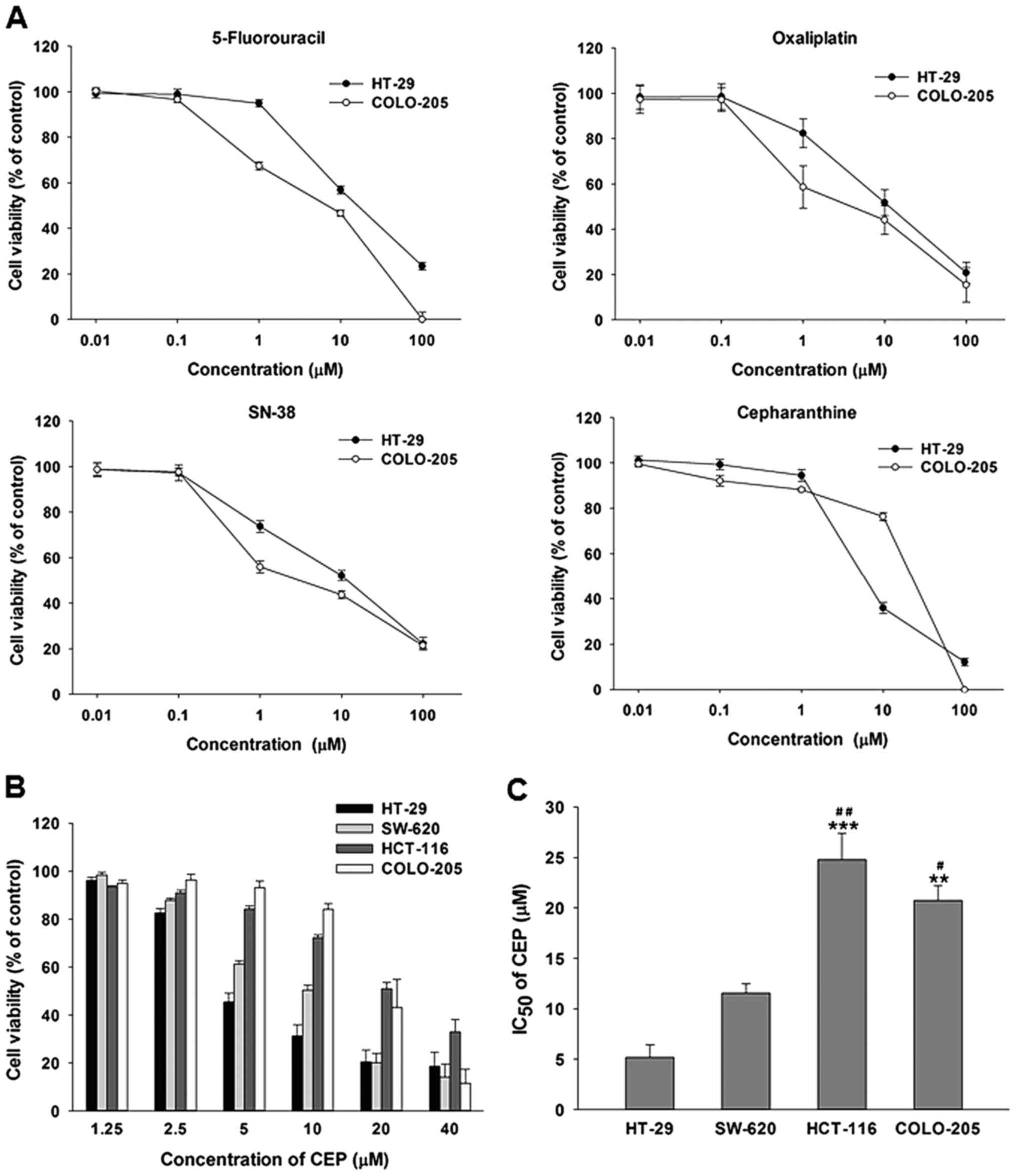
and a p53 wild-type COLO-205 cells. As shown in Fig. 1A, all four compounds decreased cell
viability of both cell lines in a concentration-dependent manner.
However, the IC50 values illustrated that the COLO-205
cells were more susceptible to the three FDA-approved anticancer
drugs than the HT-29 cells (Table
II). Interestingly, CEP was more toxic to the HT-29 cells than
the COLO-205 cells as evidenced by a ~4-fold lower IC50
value.
| Table II.IC50 of anticancer agents
and cepharanthine (CEP) in HT-29 and COLO-205 cells. |
Table II.
IC50 of anticancer agents
and cepharanthine (CEP) in HT-29 and COLO-205 cells.
|
| IC50
(µM) |
|---|
|
|
|
|---|
| Drug | HT-29 | COLO-205 |
|---|
| 5-Fluorouracil | 12.64±2.61 |
2.67±1.20 |
| Oxaliplatin |
2.83±0.32 |
1.23±0.45 |
| SN-38 |
2.40±0.37 |
1.20±0.14 |
| CEP |
5.18±1.23 | 20.74±1.47 |
To further confirm the anticancer activity of CEP in
p53 mutant colorectal cancer cells, we determined the cell
viability of 4 human colorectal cancer cell lines treated with CEP
at 2.5, 5, 10, 20 and 40 µM for 48 h. These cell lines were p53
mutant CRC cells; HT-29 and SW-620 and p53 wild-type CRC cells;
HCT-116 and COLO-205. As shown in Fig.
1B, CEP decreased the viability of HT-29, SW-620, HCT-116 and
COLO-205 cells in a concentration-dependent manner. However, HT-29
and SW-620 with p53 mutations were more susceptible to the
cytotoxic effect of CEP than HCT-116 and COLO-205 cells with
wild-type p53 (Fig. 1C). These
results suggested that CEP was very effective in controlling the
growth of cancer cells harboring p53 mutations.
Cepharanthine induces cell cycle
arrest and apoptosis in both p53-mutant and p53 wild-type
colorectal cancer cells
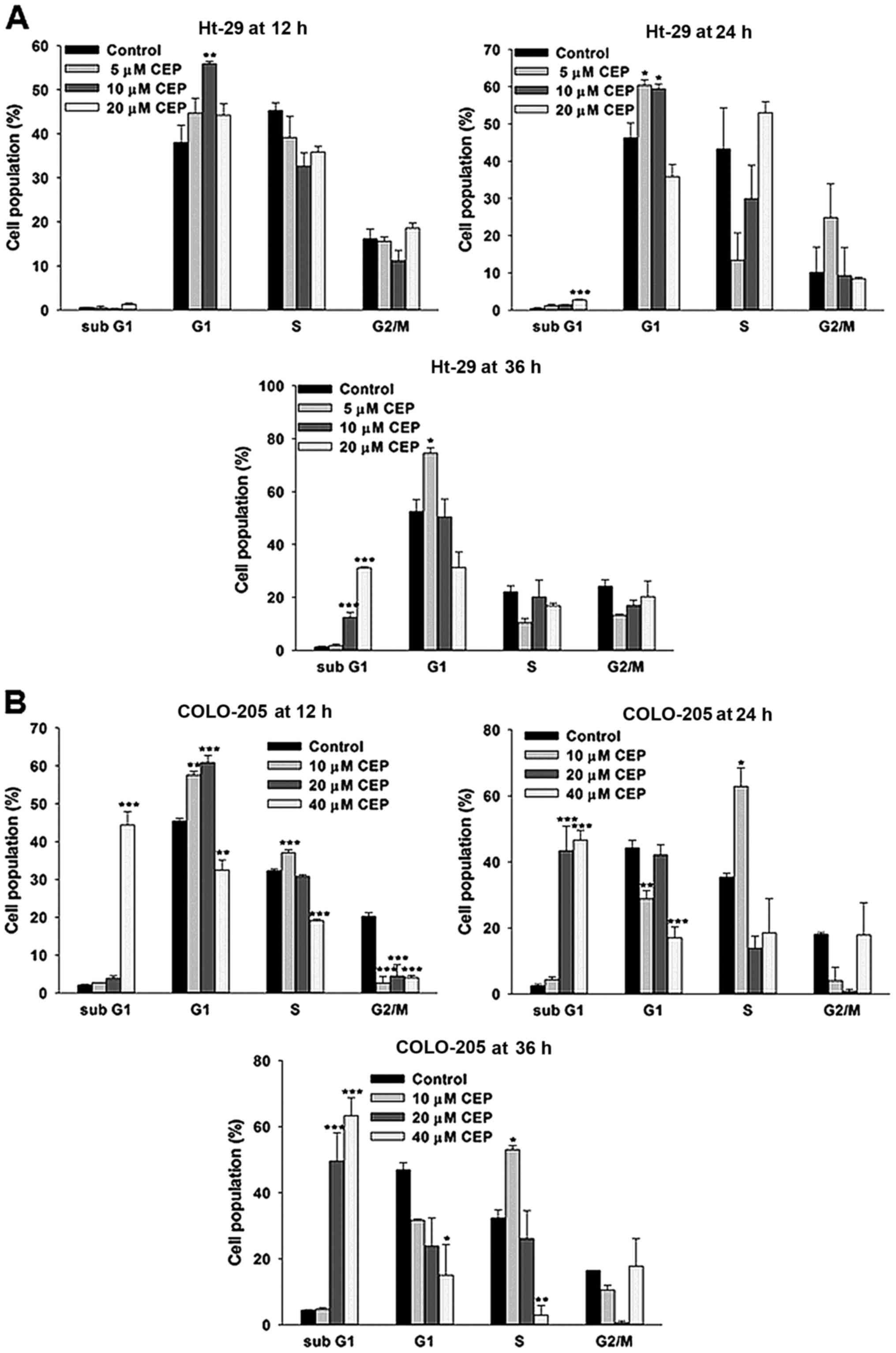
We then determined whether the growth-inhibitory
effect of CEP is related to cell cycle arrest. HT-29 and COLO-205
cells were treated CEP and cell cycle was analyzed by PI staining
followed by flow cytometry. As shown in Fig. 2A, the percentages of HT-29 cells in
the G1 phase were significantly increased following treatment with
10 µM CEP for 12 h. Similar results were detected in HT-29 cells
treated with 5 and 10 µM CEP for 24 h. However, it should be noted
that 20 µM of CEP significantly increased accumulation of the
sub-G1 fraction, an indicator of apoptotic cell death. At 36 h of
incubation, treatment with CEP only at 5 µM caused cell
accumulation at the G1 phase whereas 10 and 20 µM of CEP
significantly induced a sub-G1 accumulation 10- and 24-fold above
control, respectively. We also observed a significant accumulation
of COLO-205 cells in the G1/S phases following treatment with CEP
at 10 and 20 µM whereas CEP at 40 µM induced apoptotic cell death
as early as 12 h of incubation (Fig.
2B). Interestingly, for the longer incubation time-point, CEP
at 10 µM resulted in accumulation of the COLO-205 cells in S phase
while apoptosis was significantly detected in the cells treated
with CEP at 20 and 40 µM. These results indicated that CEP at low
concentrations effectively disturbed cell cycle progression while
CEP at higher concentrations clearly induced apoptosis in both
HT-29 and COLO-205 cells.
Cepharanthine induces changes in cell
cycle-regulated gene expression in colorectal cancer cells
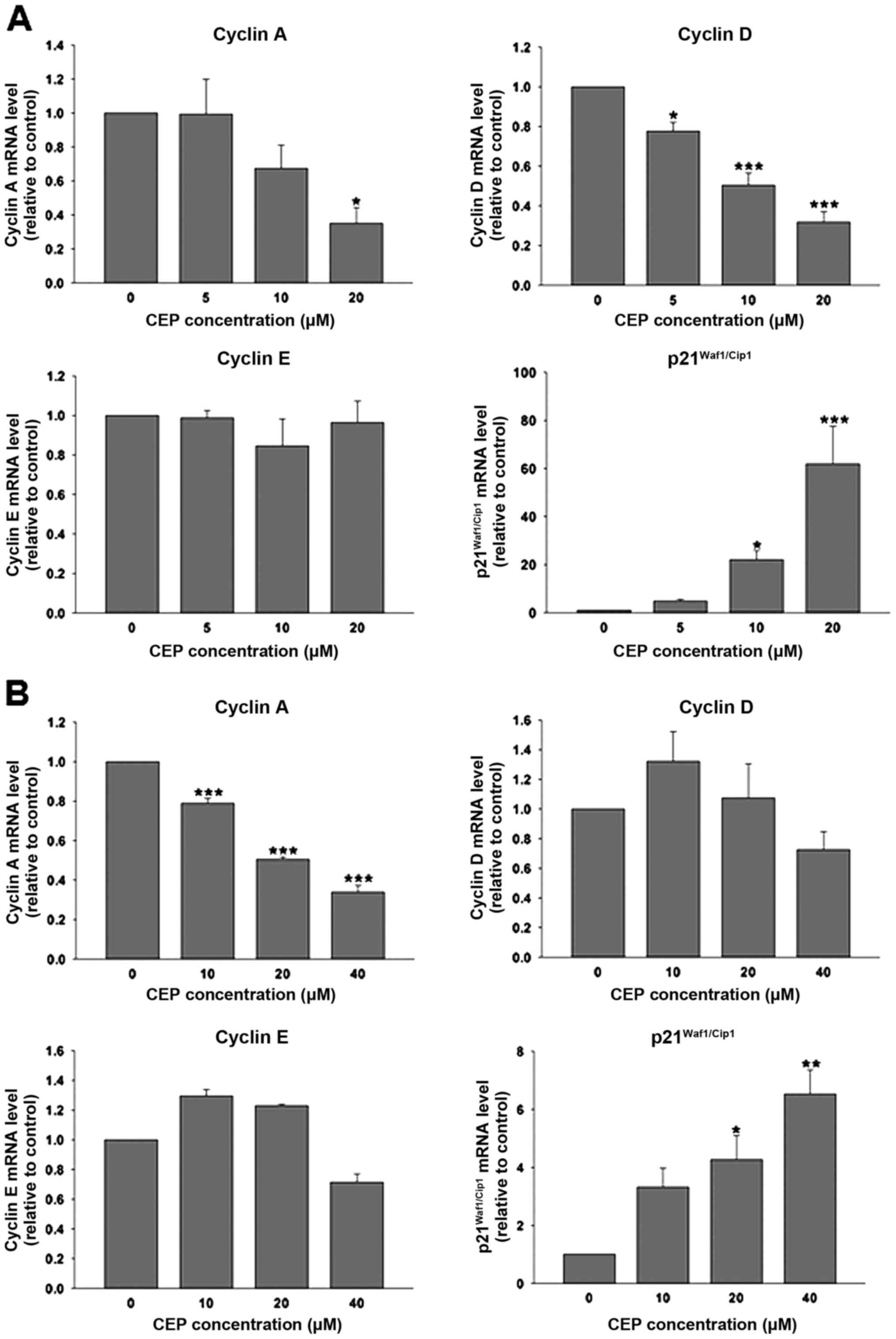
To address the mechanism responsible for the effect
of CEP on cell cycle arrest, we measured the mRNA levels of cell
cycle regulators: cyclins A, D and E; and p21, a CDK inhibitor
using real-time reverse transcription polymerase chain reaction. As
shown in Fig. 3A, CEP at 20 µM
significantly decreased cyclin A mRNA level in HT-29 cells by ~65%.
This compound also downregulated the expression of cyclin D in a
concentration-dependent manner; however, expression of cyclin E was
unaffected by CEP. Notably, the expression of
p21Waf1/Cip1 mRNA in HT-29 cells was dramatically
upregulated in response to CEP treatment. CEP at 5, 10 and 20 µM
increased p21Waf1/Cip1 mRNA levels ~5, 20 and 60 times
above control, respectively. In COLO-205 cells, CEP significantly
downregulated the cyclin A mRNA expression levels in a
concentration-dependent manner (Fig.
3B) whereas the expression of cyclin D and E were unaltered by
CEP. Similar to HT-29, CEP at 20 and 40 µM significantly induced
the expression of p21Waf1/Cip1 mRNA in COLO-205 cells,
although to a lesser extent.
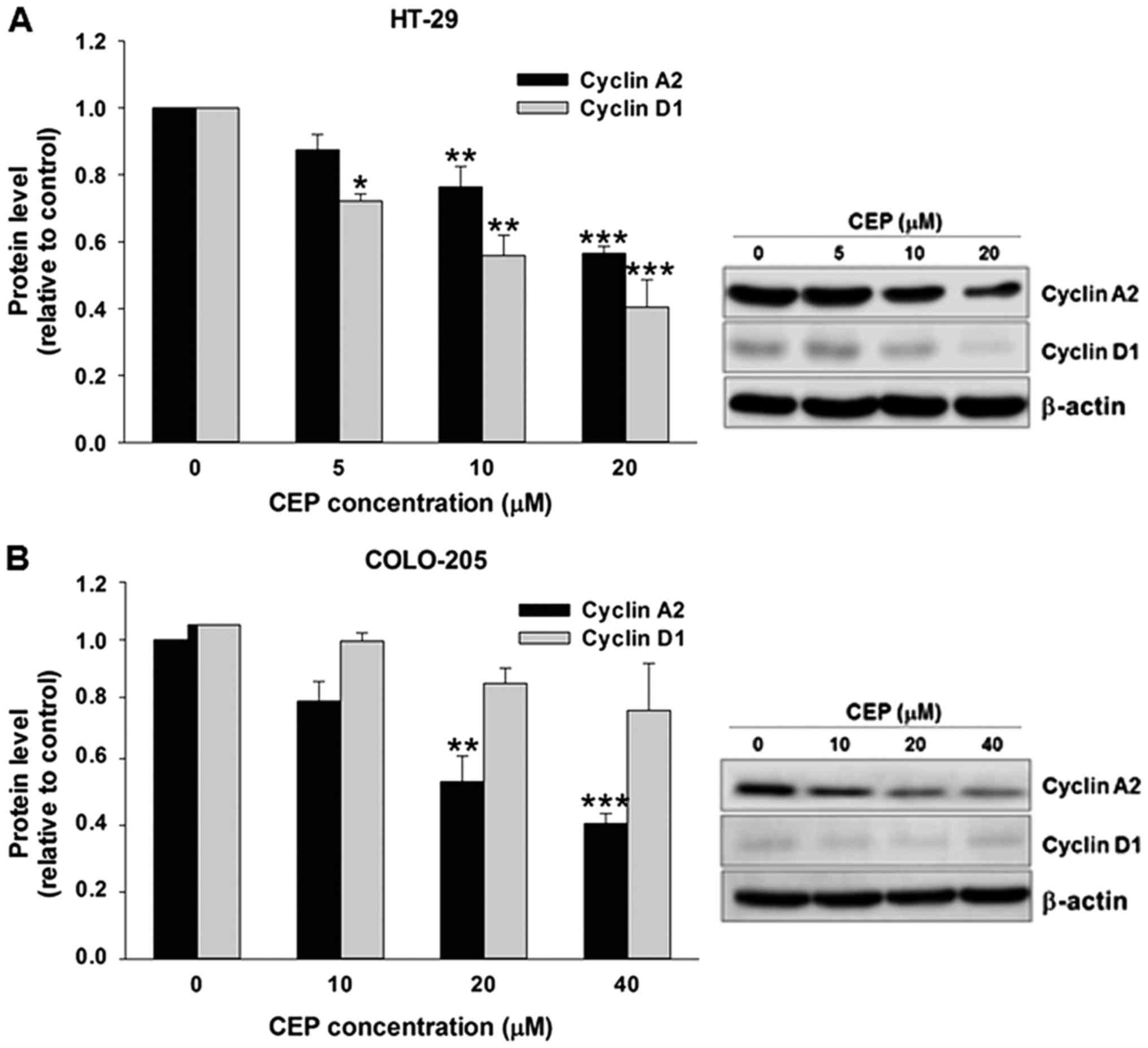
Western blot analysis also showed that CEP
suppressed expression of cyclin A2 and cyclin D1 proteins in HT-29
cells (Fig. 4A). Similarly,
treatment with CEP resulted in a decrease in cyclin A2 protein
level in COLO-205 cells in a concentration-dependent manner
(Fig. 4B). The increased
expressions of p21Waf1/Cip1 in HT-29 cells were further
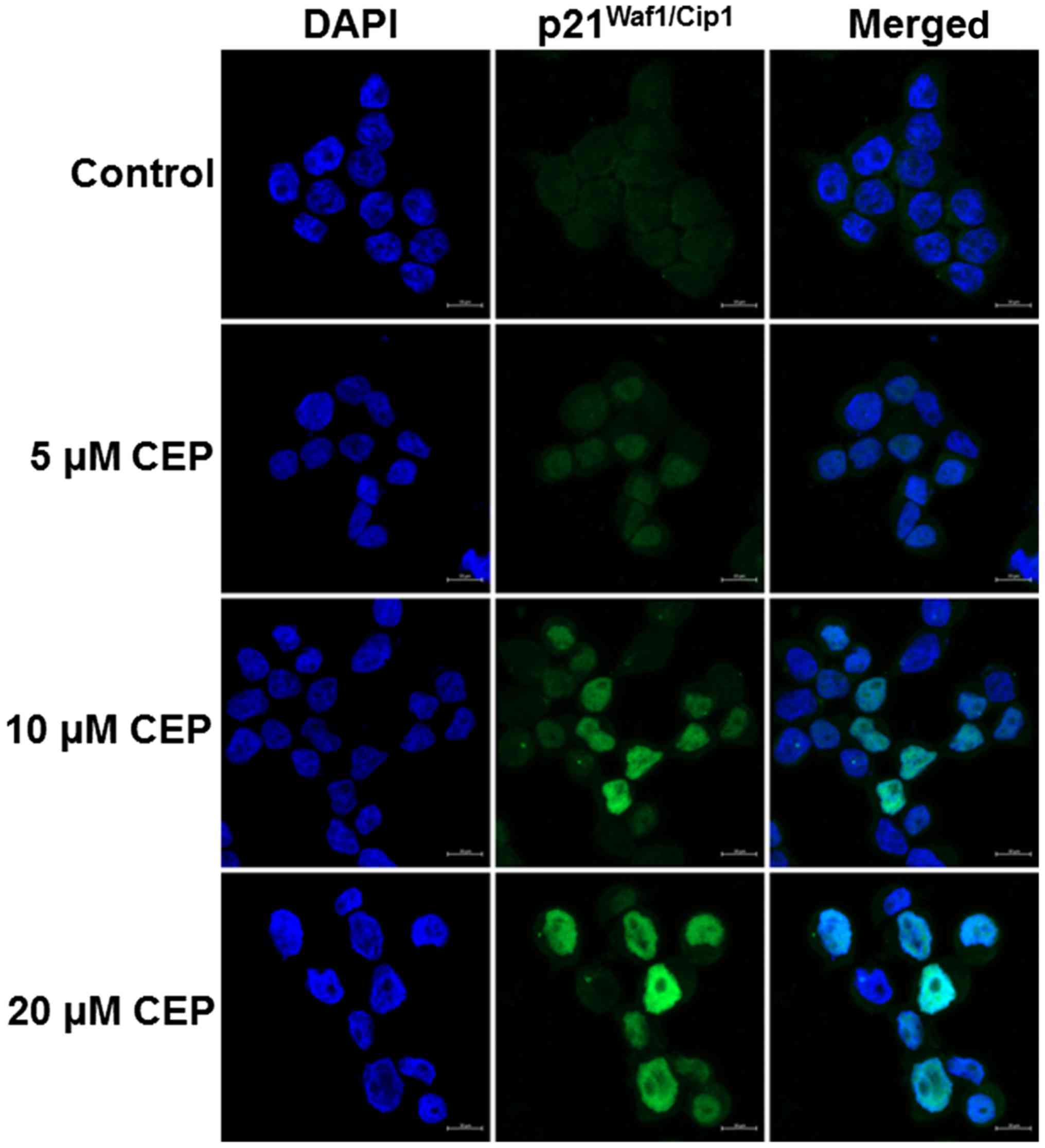
confirmed by immunofluorescence staining. HT-29 cells were treated
with 5, 10 and 20 µM of CEP for 24 h then immunolabeled for the
presence of p21Waf1/Cip1 and stained with DAPI. In
agreement with mRNA and protein expression results, CEP increased
p21Waf1/Cip1 fluorescence intensity in the nucleus of
the HT-29 cells in a concentration-dependent manner (Fig. 5).
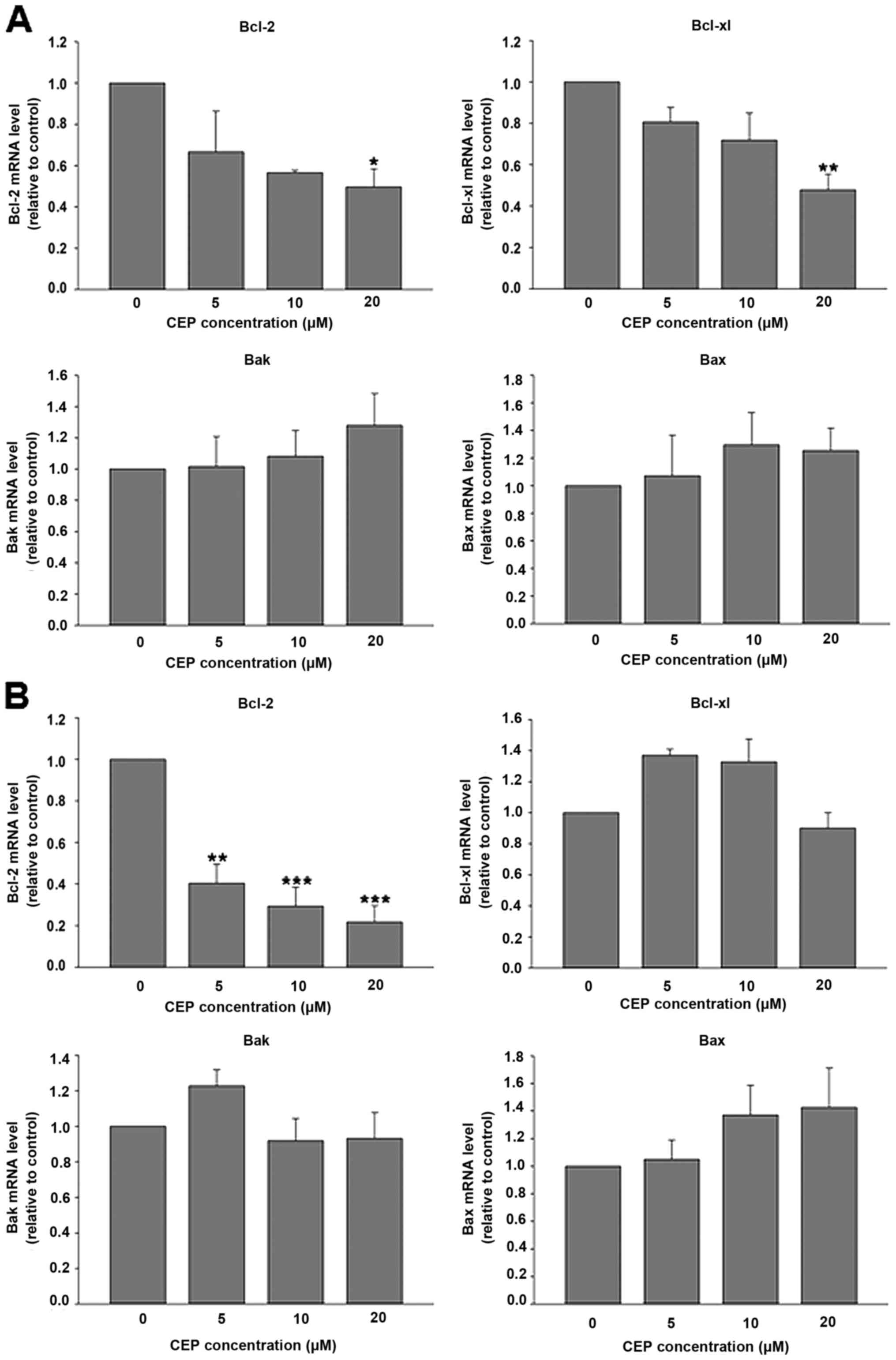
Cepharanthine decreases Bcl-2
expression in colorectal cancer cells
The intrinsic pathway of apoptosis is mainly
involved in anticancer activity of several chemotherapeutic agents.
This pathway is regulated by members of the Bcl-2 family proteins
(27). Therefore the mRNA levels of
Bcl-2 family, such as anti-apoptotic proteins, including Bcl-2 and
Bcl-xl; and pro-apoptotic proteins, including Bax and Bak were
evaluated after CEP treatment. Real-time RT-PCR analysis revealed
that CEP at 20 µM significantly downregulated the mRNA expression
of anti-apoptotic proteins, Bcl-2 and Bcl-xl in HT-29 cells
(Fig. 6A). In contrast, CEP did not
alter mRNA expression of pro-apoptotic proteins, Bak and Bax. As
illustrated in Fig. 6B, CEP
significantly decreased Bcl-2 mRNA expression in COLO-205 cells in
a concentration-dependent manner. However, mRNA levels of Bcl-xl,
Bak and Bax were unaffected following CEP treatment. These results
indicated that downregulation of Bcl-2 mRNA expression was likely
responsible for CEP-induced apoptosis in colorectal cancer
cells.
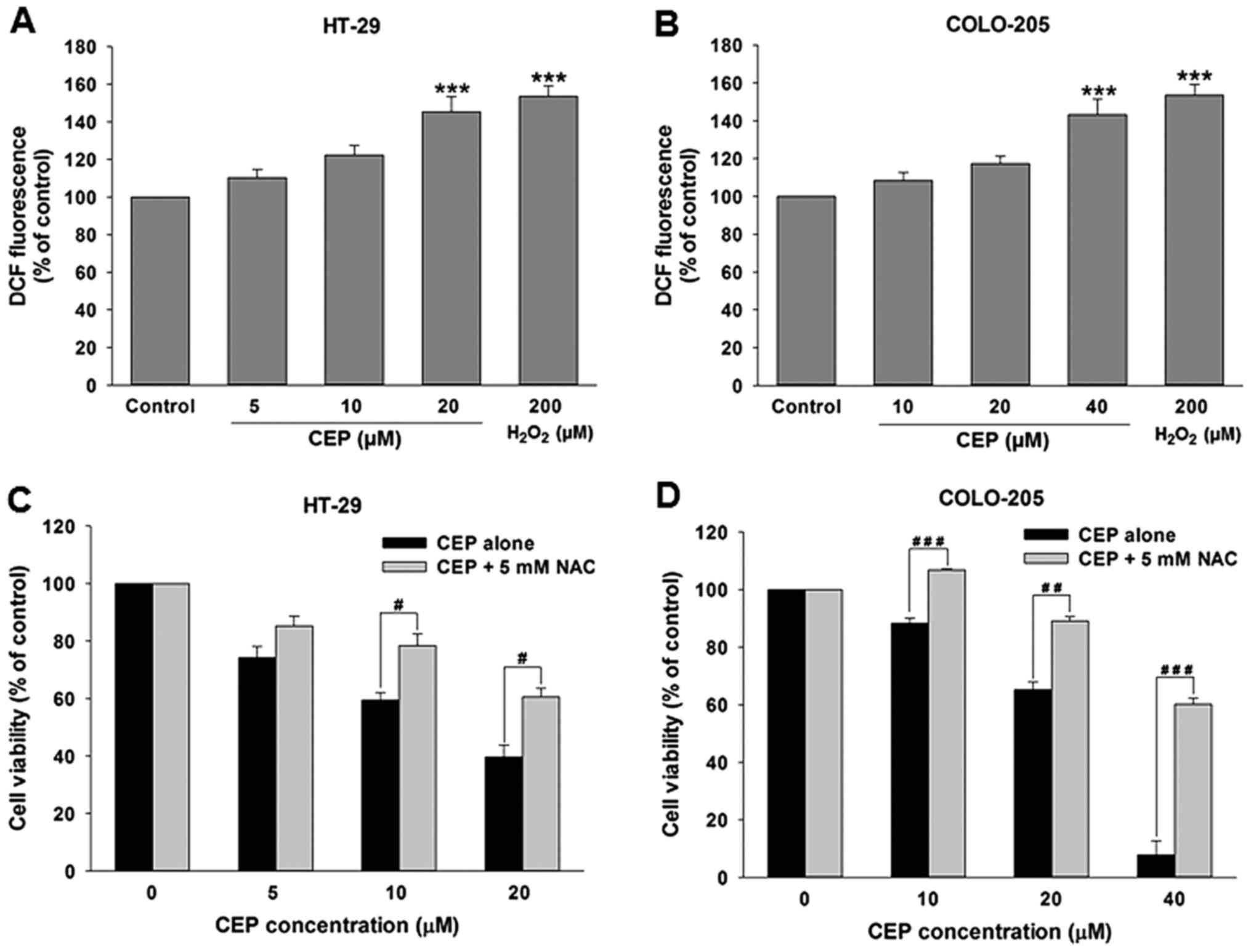
Cepharanthine induces ROS production
in colorectal cancer cells
Overproduction of reactive oxygen species (ROS) can
lead to oxidative stress causing cell death. It has been shown that
various anticancer drugs such as cisplatin, vinblastin, doxorubicin
and camptothecin induce cell death through generation of ROS in
several cancer cells (28). We
therefore determined whether CEP is capable of triggering ROS
production in colorectal cancer cells. HT-29 cells were treated
with CEP at concentrations of 5, 10 and 20 µM and COLO-205 cells
were treated with CEP at concentrations of 10, 20 and 40 µM. One
hour after treatment ROS level was determined by measuring the
dichlorodihydrofluorescein (DCF) fluorescence intensity with
dichlorodihydrofluorescein diacetate (DCFH-DA) assay. As
illustrated in Fig. 7A, treatment
of HT-29 cells with 10 and 20 µM of CEP significantly induced ROS
production. Similarly, ROS level was significantly elevated in
COLO-205 cells after treatment with 40 µM of CEP (Fig. 7B).
To confirm whether ROS production is involved in
CEP-mediated cytotoxicity, both HT-29 and COLO-205 cells were
pre-incubated with 5 mM of N-acetyl cysteine (NAC), a ROS
inhibitor, for 30 min before treatment with CEP. As shown in
Fig. 7C and D, the cytotoxicity of
CEP to both HT-29 and COLO-205 cells was significantly reduced by
pretreatment with NAC, suggesting that ROS production is partly
responsible for CEP-induced colorectal cancer cell death.
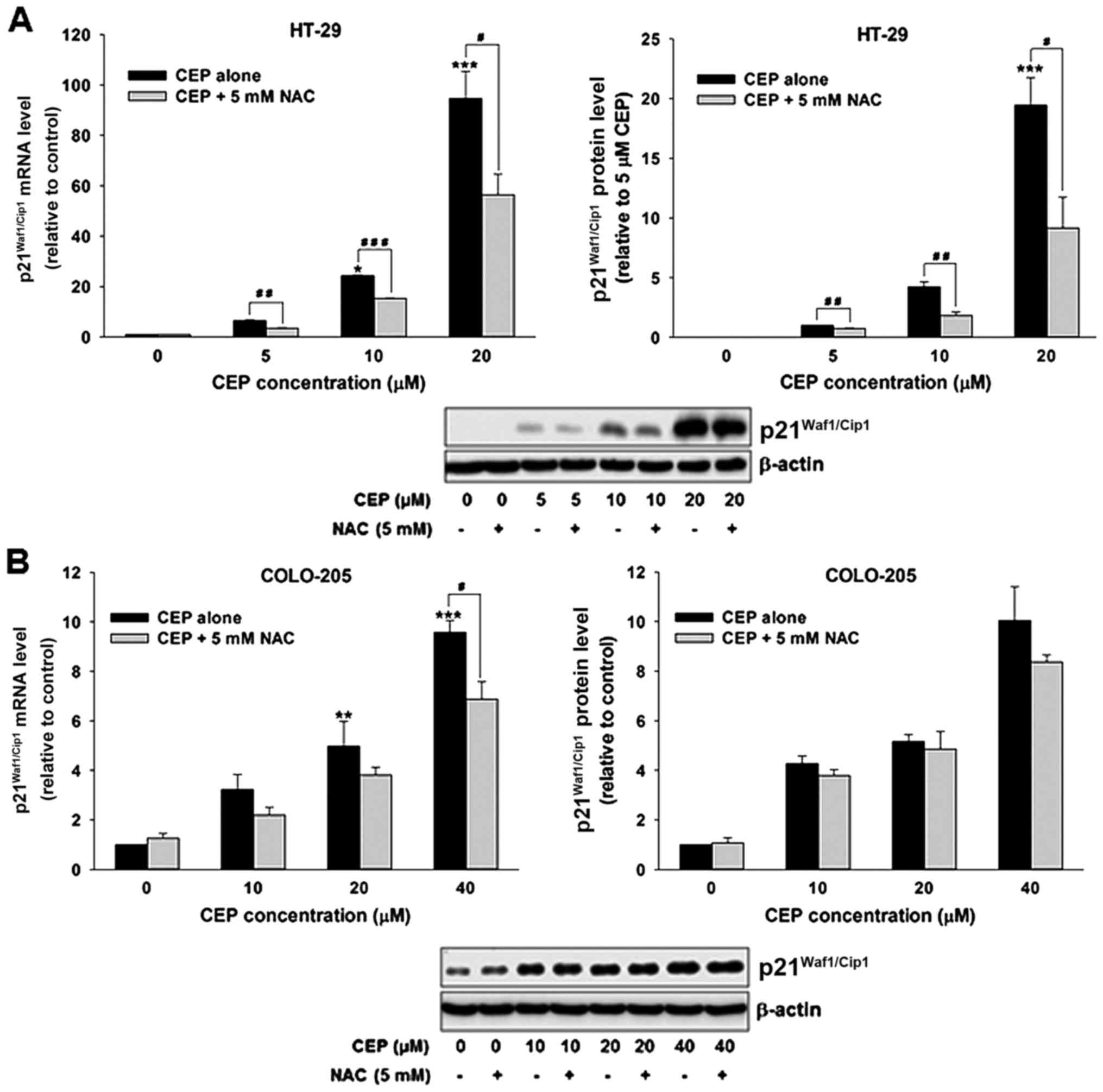
Many studies have reported links between ROS and
p21Waf1/Cip1, we therefore determined whether ROS is
involved in the induction of p21Waf1/Cip1 following CEP
treatment in colorectal cancer cells. As shown in Fig. 8A, CEP increased
p21Waf1/Cip1 expression on the mRNA and protein levels
in HT-29 cells. Remarkably, co-incubation with NAC significantly
attenuated the p21Waf1/Cip1 induction in CEP-treated
cells. Although treatment with CEP resulted in upregulation of
p21Waf1/Cip1 mRNA and protein levels in COLO-205, the
significant difference in p21Waf1/Cip1 mRNA level
between cells treated with CEP alone and in combination with NAC
was only detected after treatment with 40 µM CEP (Fig. 8B). Taken together, these results
suggest that ROS is critically required for p21Waf1/Cip1
induction in response to CEP treatment in p53 mutant colorectal
cancer cells.
Discussion
Many lines of evidence have demonstrated that p53
mutations, the most common genetic change identified in human
cancer, are associated with resistance to several types of
chemotherapeutic agents, including alkylating agents, topoisomerase
I/II inhibitors, and antimetabolites (29–31).
This highlights the need for a novel anticancer agent that remains
effective in cancer cells harboring mutant p53. Cepharanthine
(CEP), a biscoclaurine alkaloid isolated from Stephania
cepharantha Hayata, was shown to possess anticancer activities
in many types of cancer cells (11–21).
Interestingly, it was reported that CEP could inhibit the growth of
human adenosquamous cell carcinoma containing p53 mutation
(25). In the present study, we
investigated the anticancer activity of CEP against colorectal
cancer cells using a p53 wild-type human colorectal cancer cell
line, COLO-205, and a p53 mutant human colorectal cancer cell line,
HT-29. We have found that CEP induced colorectal cancer cell death
in vitro in a time- and concentration-dependent manner.
Interestingly, CEP was more cytotoxic to the p53 mutant HT-29 cells
than the p53 wild-type COLO-205 cells. The IC50 value of
the HT-29 cells treated with CEP was ~4 times lower than that in
the COLO-205 cells. In contrast, the commonly used chemotherapeutic
agents, 5-FU, oxaliplatin and SN-38, an active metabolite of
irinotecan were more cytotoxic to the COLO-205 cells than the HT-29
cells. In addition to approved drugs, promising chemotherapeutic
agents such as 3′-hydroxypterostilbene, 3,5,4′-trimethoxystilbene,
and myricetin (32–34) were shown to be more toxic to the
COLO-205 cells than the HT-29 cells.
To further confirm the anticancer activity of CEP
against p53 mutant colorectal cancer cells, SW-620, another p53
mutant colorectal cancer cell line and HCT-116, a p53 wild-type
colorectal cancer cell were treated with CEP. The MTT assays
revealed that CEP was more cytotoxic to the two p53 mutant
colorectal cancer cell lines, HT-29 and SW-620, than the two p53
wild-type colorectal cancer cell lines, HCT-116 and COLO-205,
suggesting that CEP has potential to be used as a novel
chemotherapeutic agent for colorectal cancer cells harboring p53
mutation.
Many chemotherapeutic agents exert their anticancer
activity via induction of cell cycle arrest and apoptosis. Previous
studies demonstrated that CEP could induce cell cycle arrest at G1
and S phase in various types of cancer (13,15,18).
Similar to previous studies, we have found that CEP caused
accumulation of HT-29 and COLO-205 cells at the G1 and the G1 and S
phase of the cell cycle, respectively. Remarkably, cell cycle
arrest effect was detected following CEP treatment at the lowest
concentration while its apoptotic-induction effect was noted at the
highest concentration in both HT-29 and COLO-205 cells. The time
course studies revealed that CEP at the intermediate concentration
(10 µM) significantly caused accumulation of the HT-29 cells at the
G1 phase after 24-h treatment and induced apoptosis at 36 h whereas
treatment of COLO-205 cells with CEP at the intermediate
concentration (20 µM) significantly disrupted progression of cell
cycle as early as 12 h while significant apoptotic cell death was
observed at 24 h after treatment. These results suggested that CEP
was able to induce both cell cycle arrest and apoptosis and its
anticancer activity likely depended on concentration and duration
of the treatment. Accumulating evidence has demonstrated that
mutations of p53 are capable of not only disrupting the
antiproliferative properties but also promoting various oncogenic
responses to cell growth, metastasis, invasion, chemoresistance,
and apoptosis resistance (5).
Moreover, it was reported that mutant p53 could delay cell growth
arrest in fibroblasts (35). Thus,
it is likely that mutation of p53 is responsible for the delayed
cell cycle arrest in HT-29 cells relative to COLO-205 cells.
Bcl-2 family proteins, including anti-apoptotic
proteins such as Bcl-2 and Bcl-xl and pro-apoptotic proteins such
as Bax and Bak are regulators of mitochondria or intrinsic
apoptotic pathway (36). It has
been reported that CEP induced apoptosis through upregulation of
Bax and downregulation of Bcl-2 and Bcl-xl in various cancer cells
(18,19,21).
In agreement with these observations, this study revealed that CEP
decreased level of Bcl-2 mRNA in COLO-205 cells in a
concentration-dependent manner. Notably, significant reductions of
mRNA levels of the two key anti-apoptotic proteins, Bcl-2 and
Bcl-xl were observed in HT-29 cells following CEP treatment at high
concentration only. Taken together, these results suggested that
apoptotic-inducing effect of CEP was mediated through
downregulation of Bcl-2 and Bcl-xl in colorectal cancer cells.
Previously, several studies demonstrated that CEP reduced
expression of Bcl-2 and Bcl-xl through inhibition of STAT3
signaling pathway in cervical adenocarcinoma and osteosarcoma
(18,19). Therefore, apoptosis-inducing effects
of CEP on human colorectal cancer cells may be mediated through
inhibition of STAT3 resulting in suppression of downstream gene
products, including Bcl-2 and Bcl-xl. However, the effects of CEP
on the STAT3 signaling pathway in colorectal cancer cells require
further elucidation.
The cell cycle is regulated at the checkpoints by
cyclin/cyclin-dependent kinase (CDK) complexes, which are
inactivated by CDK inhibitors (CKIs) (37). In the present study, we have
documented that CEP downregulated the expression of cyclin A and
both cyclin A and D in COLO-205 and HT-29 cells, respectively.
Numerous previous studies demonstrated that CEP downregulated the
expression of cyclins D1 and E in several cancer cells including
osteosarcoma, oral squamous cell carcinoma and myeloma (13,15,18).
However, one of the most surprising finding of this study was that
CEP was able to decrease the expression of cyclin A in both
COLO-205 and HT-29 cells, suggesting that cyclin A may be a
therapeutic target of CEP in human colorectal cancer cells.
Oxidative stress is an overproduction of reactive
oxygen species (ROS) beyond the cellular antioxidant capacity. It
was reported that oxidative stress can trigger p53-dependent and
p53-independent apoptotic cell death pathway in cancer cells
(38). Numerous natural agents with
potent anticancer activity such as epigallocatechin-3-gallate
(EGCG), resveratrol, and curcumin were shown to induce apoptosis in
cancer cells via a ROS-dependent pathway (39). Previous studies demonstrated that
CEP triggered apoptosis in human hepatocellular carcinoma cells and
non-small cell lung cancer cells through ROS (14,21).
Similarly, the results of this study showed that CEP significantly
increased ROS level in HT-29 and COLO-205 cells. Moreover, the
cytotoxicity of CEP was significantly abolished by N-acetylcysteine
(NAC), a specific ROS inhibitor, indicating that cytotoxic effect
of CEP is partly mediated by ROS.
Although p53 is a known transcriptional regulator of
p21Waf1/Cip1, several different compounds such as
phenoxodiol, ascofuranone, and MLN4924, an inhibitor of NEDD8
activating enzyme, have been demonstrated to induce
p21Waf1/Cip1 independently of p53 status
(40–42). Similarly, CEP was shown to induce
cell cycle arrest at G1 phase by activating p21Waf1/Cip1
in a human adenosquamous cell carcinoma cell line harboring mutant
p53 (25). Our results also showed
that CEP dramatically increased mRNA and protein levels of
p21Waf1/Cip1 in HT-29 cells. Remarkably, NAC was able to
attenuate CEP-induced p21Waf1/Cip1 expression in HT-29
cells. Together, these findings suggest that upregulation of
p21Waf1/Cip1by ROS mainly contribute to anticancer
activity of CEP in p53 mutant colorectal cancer cells. Although
several studies demonstrated that upregulation of
p21Waf1/Cip1 led to increased production of ROS in
various cancer cells such as prostate, bladder and head and neck
(43,44), our results raised the possibility
that CEP induced production of ROS, leading to transcription of
p21Waf1/Cip1, independently of p53. The high levels of
p21Waf1/Cip1, in turn, activated ROS production. A
recent study demonstrated that cells undergoing
p21Waf1/Cip1-dependent cell death had higher sensitivity
to oxidants (43), thus it is
likely that upregulation of p21Waf1/Cip1 makes HT-29
cells susceptible to cytotoxicity of CEP. In the present study, we
also uncovered that CEP increased p21Waf1/Cip1 in
COLO-205 cells but less extensive than in HT-29 cells and NAC had
no significant effect on CEP-induced p21Waf1/Cip1
expression. Since p53 regulates growth arrest and apoptosis mainly
through the activation or suppression of various target genes,
therefore expression of p21Waf1/Cip1 might not play an
important role in anticancer activity of CEP in p53 wild-type
colorectal cancer cells. In fact, we observed that CEP dramatically
downregulated Bcl-2 expression in COLO-205 cells.
In conclusion, these results clearly illustrated
that CEP has a remarkable anticancer activity against both p53
wild-type and p53 mutant colorectal cancer cells. Mechanistic
studies revealed that the anticancer effect of CEP in colorectal
cancer cells involves induction of cell cycle arrest and apoptosis
which may be associated with upregulation of
p21Waf1/Cip1, downregulation of cyclin A and Bcl-2 and
induction of ROS. It should be noted, however, that this study was
only performed on two cell lines. Further elucidation and
verification of these observations both in vitro and in
vivo are warranted. In addition, toxicity of CEP should be
thoroughly tested for clinical application.
Acknowledgements
We thank Dr Amornpun Sereemaspun at the Department
of Anatomy, Faculty of Medicine, Chulalongkorn University for
kindly providing SW-620 and COLO-205 cell lines and Mr. Noppadol
Saardlam at the Immunology Laboratory, Faculty of Dentistry,
Chulalongkorn University for flow cytometric analysis. This study
was supported by the 90th Anniversary of Chulalongkorn University
Fund (Ratchadaphiseksompot Endowment Fund), grant no.
GCUGR11255725066M (A.R.), Thailand Research Fund, grants no.
TGR5880214 (A.M.) and TRG5880242 (P.W.), Rathchadaphiseksomphot
Endowment Fund for Development of New Faculty Staff, grant no. GDNS
59-007-30-001 (P.W.), the Chulalongkorn Academic Advancement into
its 2nd Century (CUAASC) Project and Special Task Force for
Activating Research (STAR) Ratchadaphiseksomphot Endowment Fund,
grant no. GSTAR 59-005-30-001.
References
|
1
|
O'Keefe SJ: Diet, microorganisms and their
metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol.
13:691–706. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kotelevets L, Chastre E, Desmaële D and
Couvreur P: Nanotechnologies for the treatment of colon cancer:
From old drugs to new hope. Int J Pharm. 514:24–40. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hu T, Li Z, Gao CY and Cho CH: Mechanisms
of drug resistance in colon cancer and its therapeutic strategies.
World J Gastroenterol. 22:6876–6889. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takayama T, Miyanishi K, Hayashi T, Sato Y
and Niitsu Y: Colorectal cancer: Genetics of development and
metastasis. J Gastroenterol. 41:185–192. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Muller PA and Vousden KH: p53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
He C, Li L, Guan X, Xiong L and Miao X:
Mutant p53 gain of function and chemoresistance: The role of mutant
p53 in response to clinical chemotherapy. Chemotherapy. 62:43–53.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dart DA, Picksley SM, Cooper PA, Double JA
and Bibby MC: The role of p53 in the chemotherapeutic responses to
cisplatin, doxorubicin and 5-fluorouracil treatment. Int J Oncol.
24:115–125. 2004.PubMed/NCBI
|
|
8
|
Ravi R, Jain AJ, Schulick RD, Pham V,
Prouser TS, Allen H, Mayer EG, Yu H, Pardoll DM, Ashkenazi A, et
al: Elimination of hepatic metastases of colon cancer cells via
p53-independent cross-talk between irinotecan and Apo2
ligand/TRAIL. Cancer Res. 64:9105–9114. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arango D, Wilson AJ, Shi Q, Corner GA,
Arañes MJ, Nicholas C, Lesser M, Mariadason JM and Augenlicht LH:
Molecular mechanisms of action and prediction of response to
oxaliplatin in colorectal cancer cells. Br J Cancer. 91:1931–1946.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rogosnitzky M and Danks R: Therapeutic
potential of the biscoclaurine alkaloid, cepharanthine, for a range
of clinical conditions. Pharmacol Rep. 63:337–347. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu J, Suzuki H, Zhou YW, Liu W, Yoshihara
M, Kato M, Akhand AA, Hayakawa A, Takeuchi K, Hossain K, et al:
Cepharanthine activates caspases and induces apoptosis in Jurkat
and K562 human leukemia cell lines. J Cell Biochem. 82:200–214.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu J, Suzuki H, Akhand AA, Zhou YW,
Hossain K and Nakashima I: Modes of activation of mitogen-activated
protein kinases and their roles in cepharanthine-induced apoptosis
in human leukemia cells. Cell Signal. 14:509–515. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harada K, Supriatno, Yamamoto S, Kawaguchi
S, Yoshida H and Sato M: Cepharanthine exerts antitumor activity on
oral squamous cell carcinoma cell lines by induction of p27Kip1.
Anticancer Res. 23B:1441–1448. 2003.
|
|
14
|
Biswas KK, Tancharoen S, Sarker KP,
Kawahara K, Hashiguchi T and Maruyama I: Cepharanthine triggers
apoptosis in a human hepatocellular carcinoma cell line (HuH-7)
through the activation of JNK1/2 and the downregulation of Akt.
FEBS Lett. 580:703–710. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kikukawa Y, Okuno Y, Tatetsu H, Nakamura
M, Harada N, Ueno S, Kamizaki Y, Mitsuya H and Hata H: Induction of
cell cycle arrest and apoptosis in myeloma cells by cepharanthine,
a biscoclaurine alkaloid. Int J Oncol. 33:807–814. 2008.PubMed/NCBI
|
|
16
|
Seubwai W, Vaeteewoottacharn K, Hiyoshi M,
Suzu S, Puapairoj A, Wongkham C, Okada S and Wongkham S:
Cepharanthine exerts antitumor activity on cholangiocarcinoma by
inhibiting NF-kappaB. Cancer Sci. 101:1590–1595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Uthaisar K, Seubwai W, Srikoon P,
Vaeteewoottacharn K, Sawanyawisuth K, Okada S and Wongkham S:
Cepharanthine suppresses metastatic potential of human
cholangiocarcinoma cell lines. Asian Pac J Cancer Prev. 13
Suppl:149–154. 2012.PubMed/NCBI
|
|
18
|
Chen Z, Huang C, Yang YL, Ding Y, Ou-Yang
HQ, Zhang YY and Xu M: Inhibition of the STAT3 signaling pathway is
involved in the antitumor activity of cepharanthine in SaOS2 cells.
Acta Pharmacol Sin. 33:101–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fang ZH, Li YJ, Chen Z, Wang JJ and Zhu
LH: Inhibition of signal transducer and activator of transcription
3 and cyclooxygenase-2 is involved in radiosensitization of
cepharanthine in HeLa cells. Int J Gynecol Cancer. 23:608–614.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu G, Wu D, Liang X, Yue H and Cui Y:
Mechanisms and in vitro effects of cepharanthine hydrochloride:
Classification analysis of the drug-induced
differentially-expressed genes of human nasopharyngeal carcinoma
cells. Oncol Rep. 34:2002–2010. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hua P, Sun M, Zhang G, Zhang Y, Tian X, Li
X, Cui R and Zhang X: Cepharanthine induces apoptosis through
reactive oxygen species and mitochondrial dysfunction in human
non-small-cell lung cancer cells. Biochem Biophys Res Commun.
460:136–142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ebina T, Ishikawa K and Murata K:
Antitumor effect of Cepharanthin in the double grafted tumor
system. Gan To Kagaku Ryoho. 17:1165–1171. 1990.(In Japanese).
PubMed/NCBI
|
|
23
|
Takahashi-Makise N, Suzu S, Hiyoshi M,
Ohsugi T, Katano H, Umezawa K and Okada S: Biscoclaurine alkaloid
cepharanthine inhibits the growth of primary effusion lymphoma in
vitro and in vivo and induces apoptosis via suppression of the
NF-kappaB pathway. Int J Cancer. 125:1464–1472. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Harada K, Ferdous T, Itashiki Y, Takii M,
Mano T, Mori Y and Ueyama Y: Cepharanthine inhibits angiogenesis
and tumorigenicity of human oral squamous cell carcinoma cells by
suppressing expression of vascular endothelial growth factor and
interleukin-8. Int J Oncol. 35:1025–1035. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Harada K, Bando T, Yoshida H and Sato M:
Characteristics of antitumour activity of cepharanthin against a
human adenosquamous cell carcinoma cell line. Oral Oncol.
37:643–651. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bun SS, Laget M, Chea A, Bun H, Ollivier E
and Elias R: Cytotoxic activity of alkaloids isolated from
Stephania rotunda [corrected]. Phytother Res. 23:587–590. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brunelle JK and Letai A: Control of
mitochondrial apoptosis by the Bcl-2 family. J Cell Sci.
122:437–441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fang J, Nakamura H and Iyer AK:
Tumor-targeted induction of oxystress for cancer therapy. J Drug
Target. 15:475–486. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Giaccone G and Pinedo HM: Drug Resistance.
Oncologist. 1:82–87. 1996.PubMed/NCBI
|
|
30
|
Vazquez A, Bond EE, Levine AJ and Bond GL:
The genetics of the p53 pathway, apoptosis and cancer therapy. Nat
Rev Drug Discov. 7:979–987. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hientz K, Mohr A, Bhakta-Guha D and
Efferth T: The role of p53 in cancer drug resistance and targeted
chemotherapy. Oncotarget. 8:8921–8946. 2017.PubMed/NCBI
|
|
32
|
Pan MH, Gao JH, Lai CS, Wang YJ, Chen WM,
Lo CY, Wang M, Dushenkov S and Ho CT: Antitumor activity of
3,5,4′-trimethoxystilbene in COLO 205 cells and xenografts in SCID
mice. Mol Carcinog. 47:184–196. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheng TC, Lai CS, Chung MC, Kalyanam N,
Majeed M, Ho CT, Ho YS and Pan MH: Potent anti-cancer effect of
3′-hydroxypterostilbene in human colon xenograft tumors. PLoS One.
9:e1118142014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ko CH, Shen SC, Lee TJ and Chen YC:
Myricetin inhibits matrix metalloproteinase 2 protein expression
and enzyme activity in colorectal carcinoma cells. Mol Cancer Ther.
4:281–290. 2005.PubMed/NCBI
|
|
35
|
Wyllie F, Haughton M, Bartek J, Rowson J
and Wynford-Thomas D: Mutant p53 can delay growth arrest and loss
of CDK2 activity in senescing human fibroblasts without reducing
p21(WAF1) expression. Exp Cell Res. 285:236–242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shin SY, Ahn S, Koh D and Lim Y:
p53-dependent and -independent mechanisms are involved in
(E)-1-(2-hydroxyphenyl)-3-(2-methoxynaphthalen-1-yl)prop-2-en-1-one
(HMP)-induced apoptosis in HCT116 colon cancer cells. Biochem
Biophys Res Commun. 479:913–919. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mukhtar E, Adhami VM, Khan N and Mukhtar
H: Apoptosis and autophagy induction as mechanism of cancer
prevention by naturally occurring dietary agents. Curr Drug
Targets. 13:1831–1841. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jeong JH, Kang SS, Park KK, Chang HW,
Magae J and Chang YC: p53-independent induction of G1 arrest and
p21Waf1/Cip1 expression by ascofuranone, an isoprenoid antibiotic,
through downregulation of c-Myc. Mol Cancer Ther. 9:2102–2113.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Aguero MF, Venero M, Brown DM, Smulson ME
and Espinoza LA: Phenoxodiol inhibits growth of metastatic prostate
cancer cells. Prostate. 70:1211–1221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jia L, Li H and Sun Y: Induction of
p21-dependent senescence by an NAE inhibitor, MLN4924, as a
mechanism of growth suppression. Neoplasia. 13:561–569. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Masgras I, Carrera S, de Verdier PJ,
Brennan P, Majid A, Makhtar W, Tulchinsky E, Jones GD, Roninson IB
and Macip S: Reactive oxygen species and mitochondrial sensitivity
to oxidative stress determine induction of cancer cell death by
p21. J Biol Chem. 287:9845–9854. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fitzgerald AL, Osman AA, Xie TX, Patel A,
Skinner H, Sandulache V and Myers JN: Reactive oxygen species and
p21Waf1/Cip1 are both essential for p53-mediated senescence of head
and neck cancer cells. Cell Death Dis. 6:e16782015. View Article : Google Scholar : PubMed/NCBI
|