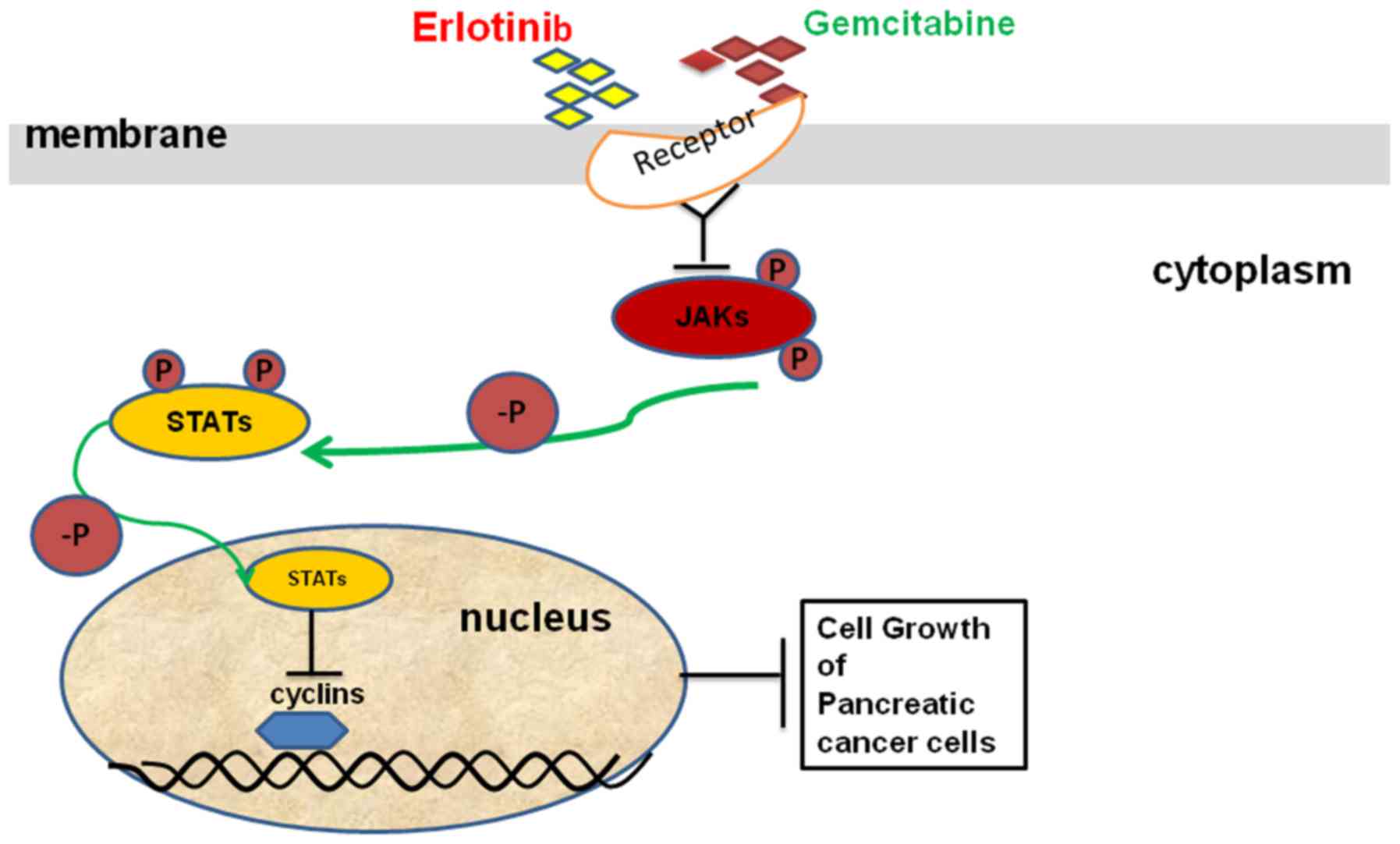
Introduction
Pancreatic cancer (PC) is the 10th most commonly
diagnosed cancer in the world. In 2013, it was the fourth leading
cause of deaths in the United States (1). The 5-year overall survival rate in
patients with PC is roughly 5%. Currently, surgery is considered to
be the most effective treatment for these patients (2). However, only 20% of patients diagnosed
with pancreatic cancer are eligible to undergo the surgery
(3). For most patients with PC,
long-term survival rates cannot be improved even after undergoing
radical resection. Moreover, chemotherapeutic drugs, such as
gemcitabine or fluorouracil/folinic acid have been demonstrated to
be effective against PC. Single-agent gemcitabine is a first-line
drug for treating patients with advanced pancreatic cancer or
patients after R0 or R1 resection (4,5).
However, this treatment approach produces only modest improvements
(6).
Gemcitabine is a deoxycytidine analog and its
cytotoxic activity is based on several activities of DNA synthesis
(4,7). To date, gemcitabine remains the
cornerstone of neo-adjuvant, adjuvant and palliative therapy for PC
(7). Along with gemcitabine,
erlotinib is the first additional drug used to improve the overall
survival of patients with advanced pancreatic cancer (4). Erlotinib is an oral small-molecule
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor
(TKI), which can block the signal transduction pathway essential
for cancer differentiation, proliferation, apoptosis, angiogenesis,
invasion, and metastasis (8). The
combination of gemcitabine and erlotinib has been reasonably
effective in treating patients with locally advanced or metastatic
pancreatic cancer (4,9). However, the mechanism of action of
gemcitabine in combination with erlotinib in pancreatic cancer
remains unknown and requires further investigation.
The Janus kinase/signal transducers and activators
of transcription (JAK-STAT) pathway is the principal signaling
mechanism for a wide array of cytokines and growth factors. JAK
activation stimulates cell proliferation, differentiation,
migration and apoptosis in mammals (10). The activated JAKs subsequently
phosphorylate additional targets, including both receptors and the
major substrates, STATs. STATs are latent transcription factors
that reside in the cytoplasm until activated. The seven mammalian
STATs bear a conserved tyrosine residue near the C-terminus that is
phosphorylated by JAKs. This phosphotyrosine permits the
dimerization of STATs through interaction with a conserved SH2
domain (10), which then enter the
nucleus. Once in the nucleus, dimerized STATs bind specific
regulatory sequences to activate or suppress transcription of
target genes. Furthermore, the activation of the JAK-STAT pathway
was determined to be associated with human pancreatic cancer
(11).
The present study investigated the effectiveness of
gemcitabine in combination with erlotinib on recurrent pancreatic
cancer growth in vivo. In this study, a nude mice model of
orthotopic xenotransplantation after tumor resection was
established using pancreatic cancer cell lines, BxPC-3 and PANC-1.
Consequently, mice were divided in different experimental groups
and treated for 3 weeks, after which the recurrent tumor tissues
were dissected and analyzed ex vivo.
Materials and methods
Cell lines
The human pancreatic cancer cell lines BxPC-3 and
PANC-1 were purchased from the Chinese Academy of Sciences
(Shanghai, China) and the American Type Culture Collection (ATCC,
Manassas, VA, USA), respectively. Human pancreatic adenocarcinoma
cell lines BxPC-3, and PANC-1 were both cultured in DMEM (Gibco,
Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented
with 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific,
Inc.) in a humidified atmosphere containing 5% CO2/95%
air at 37°C. For the animal experiments, the cells were
trypsinized, resuspended in Matrigel (BD Biosciences, Franklin
Lakes, NJ, USA) at a concentration of 1×107 cells/30 µl,
and stored on ice until injection.
Drugs
Erlotinib (Tarceva) was purchased from Roche
Pharmaceutical Ltd. (Basel, Switzerland) as a fine powder, and was
consequently dissolved in distilled water containing 6% (w/v)
Captisol (Aoke Biotechnology, Ltd., Qingdao, China). Gemcitabine
(Gemzar) was purchased from Eli Lilly and Company (Neuilly sur
Seine, France) and dissolved in 0.9% saline solution.
Orthotopic xenotransplantation of
human pancreatic cancer cells and tumor resection
Four-week-old female nude mice weighing 18–20 g were
obtained from Beijing HuaFuKang Bioscience Co., Ltd. (Beijing,
China). All the animals were housed (acclimatized for 10 days in a
sterile environment, in which bedding, food, and water were
autoclaved) in an environment with a temperature of 22±1°C,
relative humidity of 50±1% and a light/dark cycle of 12/12 h. All
animal studies (including the mice euthanasia procedure) were
performed in compliance with the regulations and guidelines of
Guizhou Medical University Institutional Αnimal Care and conducted
according to the AAALAC and the IACUC guidelines.
Mice were divided in four groups (each
with n=12)
After establishment of orthotopic
xenotransplantation of human pancreatic cancer cells after tumor
resection (12) they were treated
as follows: the control group received a placebo (0.9% saline,
every three days) via intraperitoneal injection (i.p.); the
administration of gemcitabine was performed via i.p. in the
gemcitabine group (group G) (50 mg/kg i.p., twice a week) and
erlotinib was administered by oral gavage in the erlotinib group
(group E) (50 mg/kg oral gavage, once every three days). The mode
of drug administration in the gemcitabine-erlotinib combination
group (E+G group) was performed by oral gavage of erlotinib (50
mg/kg) on the first day and i.p. of gemcitabine (50 mg/kg) was
administered the next day. On the third day, the mice were left to
rest and then administration of the drugs continued in the next
three days. The treatments lasted for 21 days, after which all mice
were sacrificed and the tumors were examined ex vivo
according to a previously described method (24). A recurrent tumor was defined as a
new tumor mass found during relaparotomy which was performed four
weeks after the first surgery, removing all the tumor mass that was
in the area where we inoculated.
Physical measurements of tumor
volume
At necropsy, the tumors were dissected away from the
normal pancreas and their measured tumor volume was calculated as:
volume (mm3) = (length × width2)/2 (13).
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) assay
Paraffin blocks were cut into slices with 4-µm
thickness. A TUNEL assay was conducted using a TUNEL apoptosis
detection kit (Nanjing, KeyGen Biotech, Co., Ltd., Jiangsu, China)
following the manufacturer's protocol. Paraffin sections were
incubated with proteinase K at 37°C for 30 min. Endogenous
peroxidase activity was blocked by incubation with 3% hydrogen
peroxide in methanol. Positive control sections were treated with
DNase I 50 U/µl, while the negative control sections were incubated
with label solution (without the terminal deoxynucleotidyl
transferase enzyme). All the other sections were incubated with
TUNEL reaction mixture at 37°C for 1 h in a humidity chamber. The
conjugated horseradish peroxidase was visualized with
diaminobenzidine. Finally, the sections were counterstained with
hematoxylin and the images were captured in a positive position
under the microscope BX51 (Olympus Corp., Tokyo, Japan). The
results were expressed as the percentage of TUNEL-positive cells
per ×40 magnification. A total of ten ×40 fields were examined from
three tumors in each of the treatment groups.
Western blot analysis
The tumor tissue was first laced in a homogenizer,
and then on ice for homogenization with RIPA buffer containing a
protease inhibitor cocktail and a phosphatase inhibitor cocktail
(Beijing Solarbio Science & Technology, Co., Ltd., Beijing,
China). The cell lysates were centrifuged and boiled with SDS
loading buffer. Then the protein samples were subjected to
SDS-polyacrylamide gel electrophoresis (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) and electrophoretically transferred to 0.45-µm
polyvinylidene difluoride membranes (Millipore, Billerica, MA,
USA). After incubation with 5% non-fat dry milk in TBS containing
0.1% Tween-20 (TBST) for 1 h, the membranes were washed once with
TBST and probed with the indicated primary antibodies overnight at
4°C. The following primary antibodies were used: anti-JAK1 (rabbit
polyclonal; cat. no. AF5012), phospho-JAK1 (Tyr1022)
(rabbit polyclonal; cat. no. AF2012), anti-JAK2 (rabbit polyclonal;
cat. no. AF6022), phospho-JAK2 (Tyr221) (rabbit
polyclonal; cat. no. AF3023), anti-JAK3 (mouse monoclonal; cat. no.
BF0256), phospho-JAK3 (Tyr981) (rabbit polyclonal; cat.
no. AF8160), phospho-STAT1 (Tyr701) (rabbit polyclonal;
cat. no. AF3300) and anti-STAT1 (rabbit polyclonal; cat. no.
AF6300) were all purchased from Affinity Biosciences (Cambridge,
UK); anti-STAT3 (rabbit monoclonal; cat. no. ab76315), and
anti-pSTAT3 Try705 (rabbit monoclonal; cat. no. ab68153)
were obtained from Abcam (Cambridge, MA, USA); anti-HIF-1α (rabbit
polyclonal; cat. no. BS3514) and anti-cyclin D1 (rabbit polyclonal;
cat. no. BS6532) were purchased from Bioworld Technology Co., Ltd.
(Nanjing, China); anti-p53 (rabbit, monoclonal; cat. no. 2527) was
acquired from Cell Signaling Technology, Inc. (Danvers, MA, USA);
while GAPDH was purchased from EarthOx Life Sciences (Millbrae, CA,
USA). All the primary antibodies were diluted 1:500-1:1,000 in
primary antibody dilution buffer (Beyotime Institute of
Biotechnology, Haimen, China). The membranes were washed three
times with TBST and incubated with a 1:1,000 dilution of
horseradish peroxidase-conjugated anti-rabbit antibodies polyclonal
antibodies (cat. no. SP-9001; ZSGB-BIO, Beijing, China) for 1 h.
After being washed three times with TBST, the membranes were
incubated with enhanced chemiluminescence substrate (ECL;
Millipore) for 5 min. Chemiluminescent signals were visualized by
exposing the membrane to ChemiDoc™™ XRS+ (Bio-Rad Laboratories,
Inc.). Immunoblot analyses were performed at least three times for
all the antibodies. The immune complexes were analyzed with ImageJ
software (NIH, Bethesda, MD, USA) which assessed the band intensity
and relative protein abundance.
Immunohistochemical staining
The recurrent tumor tissues were fixed in 10%
formalin at 4°C for 24 h and embedded in paraffin for
immunohistochemistry analyses. The sections covered with tumor
tissues were deparaffinized and rehydrated using xylene and an
ethanol gradient. The following primary antibodies were used:
anti-caspase-9 (rabbit polyclonal; bs-0049R) and anti-caspase-3
(rabbit polyclonal; bs-0081R), both purchased from BIOSS (Beijing,
China). The primary antibodies were diluted 1:200 in PBS. Sections
were incubated with active-caspase-9- and active-caspase-3
polyclonal antibodies (pAb), as well as a 1:3,000 dilution of
horseradish peroxidase-conjugated anti-rabbit polyclonal antibodies
(cat. no. 111-035-003; Jackson ImmunoResearch Laboratories, Inc.
(West Grove, PA, USA). Finally, the sections were counterstained
with hematoxylin and images were captured in a positive position
under the microscope BX51 (Olympus Corp.). The staining intensity
and percentage of positive cells were calculated and scored in
sections. The staining intensity of the active-caspase-9 and
active-caspase-3-positive cells was divided into four grades:
‘negative (−)’-, ‘+’, ‘++’ and ‘+++’ and scored as 0, 1, 2 and 3,
respectively. The positive cell percentages were divided into six
categories: ‘negative (−)’, ‘1-20%’, ‘21-40%’, ‘41-60%’, ‘61-80%’
and ‘81-100%’ and scored as 0, 1, 2, 3, 4, 5 and 6, respectively.
Total scores of every visual field were determined using the
formula: staining intensity scores of positive cells × percentage
scores of positive cells = total scores of each visual field.
Statistical analysis
Data were presented as the mean ± SD. Comparisons
among groups were performed by one-way ANOVA followed by the least
significant difference (LSD) test. Comparisons between two groups
were analyzed using Student's t-tests. P<0.05 was considered to
indicate a statistically significant result. All statistical
analyses were performed using SPSS statistical software package,
version 17.0 (SPSS, Inc., Chicago, IL, USA).
Results
The combination of gemcitabine and
erlotinib inhibit recurrent tumor growth in mice
To investigate the effect of the combination of
gemcitabine and erlotinib on recurrent tumor in vivo, an
orthotopic tumor model was established and validated using BxPC-3
and PANC-1 cell lines (12).
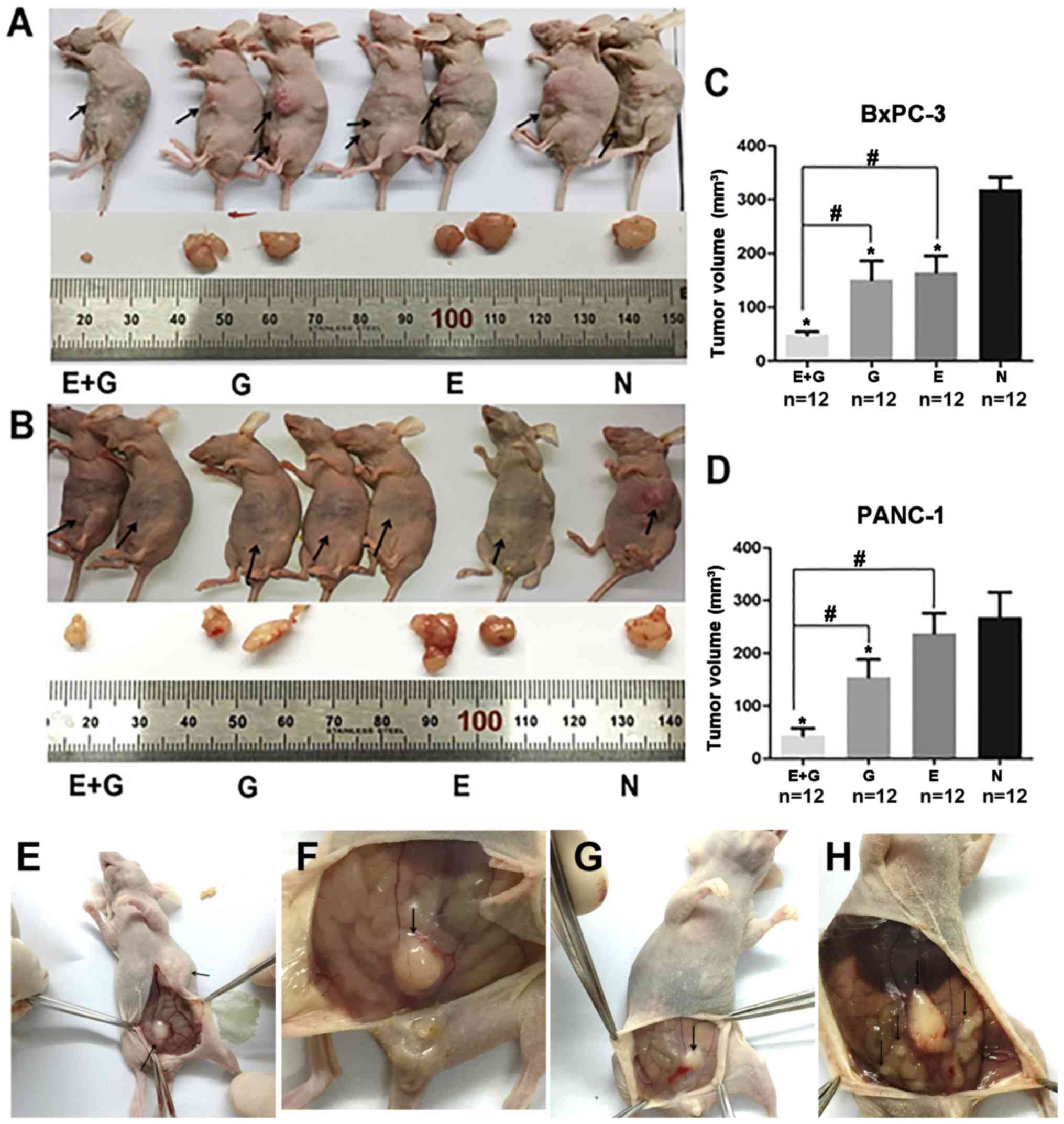
Briefly, recurrent and metastatic tumors were observed three weeks
after tumor resection (Fig. 1). A
significantly smaller tumor volume was observed in the group of
mice treated with the combination of gemcitabine and erlotinib
compared to the other groups (P<0.05, for both cell lines)
(Fig. 1). Hence, our data indicated
that the combination of gemcitabine with erlotinib inhibited
recurrent tumor growth more effectively than all the three other
groups.
The combination of gemcitabine and
erlotinib induces tumor apoptosis in mice
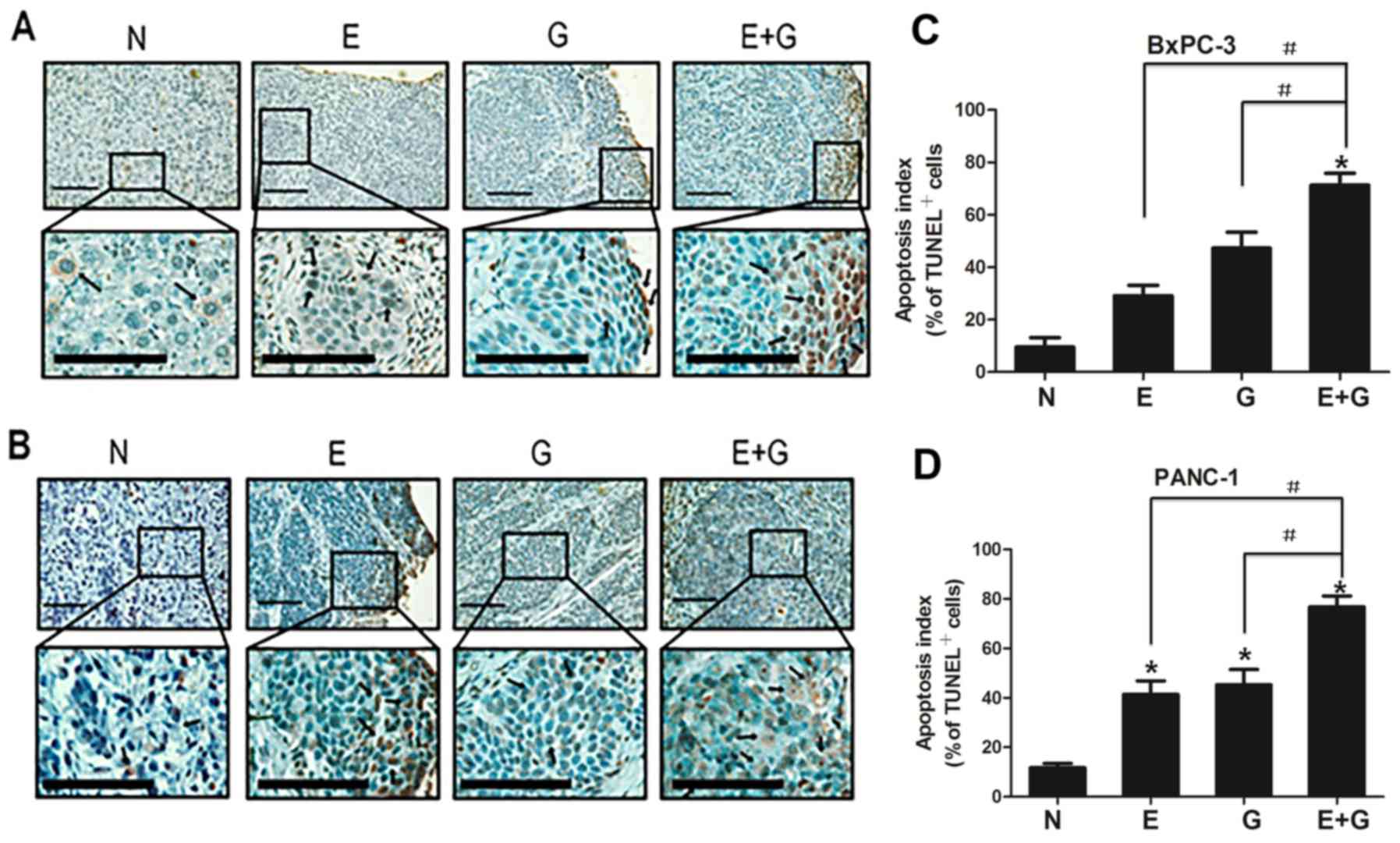
To assess the apoptotic feature in recurrent tumors
among groups, apoptotic cells were counted after TUNEL staining.
Positive apoptotic cells were significantly increased in the mice
treated with gemcitabine (group G) and erlotinib (group E) compared
with the control group (group N) (P<0.05, for both cell lines)
(Fig. 2). In addition, positive
apoptotic cells were significantly increased in the group of mice
treated with a combination of gemcitabine-erlotinib (group E+G)
compared with the monotherapy groups (group G and group E)
(P<0.05, for both cell lines). The apoptotic index in the BxPC-3
and PANC-1 cells in group N was 9.5±3.63 and 11.7±1.77,
respectively. The ones in group E were 29.1±4.01 and 41.4±5.50,
respectively. The ones in group G were 47.3±6.11 and 45.3±6.15),
respectively. The ones in group E+G were 71.34±4.55 and 77.6±4.65),
respectively (Table I).
 | Table I.Apoptotic index in recurrent tumors
of both BxPC-3 and PANC-1 cells. |
Table I.
Apoptotic index in recurrent tumors
of both BxPC-3 and PANC-1 cells.
| Cell line | N group (mean ±
SD) | G group (mean ±
SD) | E group (mean ±
SD) | E+G group (mean ±
SD) | P-value |
|---|
| BxPC-3 |
9.5±3.63 |
47.3±6.11 |
29.1±4.01 |
71.34±4.55 |
P<0.05a |
| PANC-1 |
11.7±1.77 |
45.3±6.15 |
41.4±5.50 |
77.6±4.65 |
P<0.05a |
The combination of gemcitabine and
erlotinib suppresses the JAK-STAT signaling cascade
Since STAT3 activation is quite common in pancreatic
cancer, and the phosphorylation of tyrosine residue 705 in the
STAT3 protein is a crucial event for its activation (14), we examined the effects of
gemcitabine combination with erlotinib on the phosphorylation of
STAT3. As shown in Fig. 3A-D the
phosphorylation of the tyrosine 705 residue in the STAT3 protein
was significantly lower in group E and group E+G compared to group
N (P<0.05). Phosphorylation levels of the STAT3 protein were
decreased in group E+G compared with group E and group G,
respectively (Fig. 3A-D). In
addition, the difference in the phosphorylation of STAT3 between
groups N and group G was not significant in both cell lines
(Fig. 3A-D).
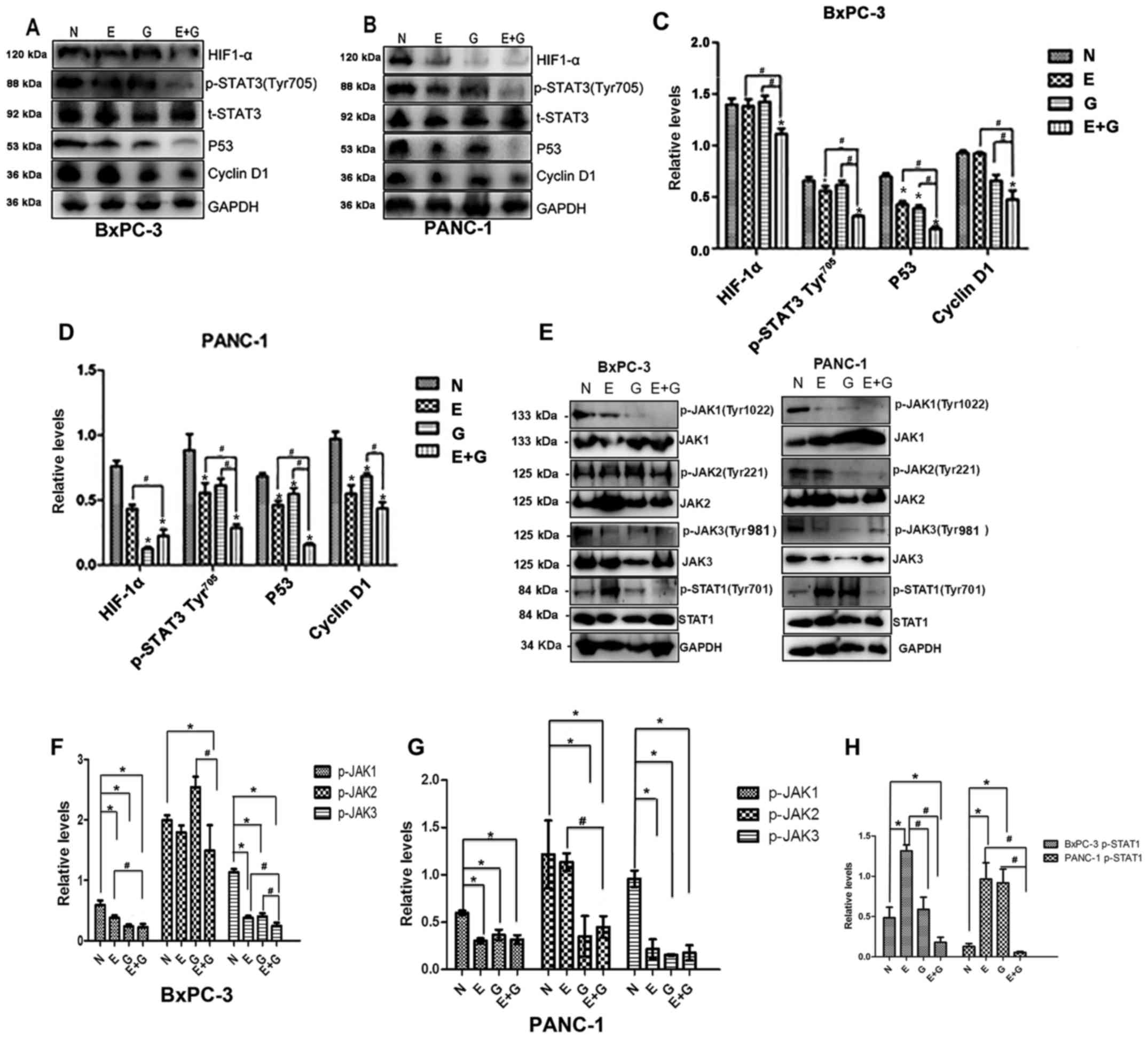 | Figure 3.The gemcitabine-erlotinib (E+G)
combination group inhibits the activity of the JAK-STAT pathway, as
well as the expression of downstream HIF-1α, cyclin D1 and p53. (A
and B) The protein levels of phosphorylated STAT3 Tyr705
(p-STAT3 Tyr705), HIF-1α, p53, cyclin D1 in BxPC-3 and
PANC-1 cells were analyzed by western blotting, respectively. (C
and D) The protein expression of p-STAT3 Tyr705, HIF-1α,
p53, cyclin D1 in BxPC-3 and PANC-1 cells was calculated using
one-way ANOVA. *P<0.05 denotes the controls vs. the treatment
groups. #P<0.05 denotes the monotherapy (E or G) vs.
the combination (E+G) groups. (E) Phosphorylation levels of JAK1
(Tyr1022), JAK2 (Tyr221),
JAK3(Tyr981) and STAT3 (Tyr701) in BxPC-3 and
PANC-1 cells were analyzed by western blotting, respectively. (F
and G) The protein intensity of phospho-JAK1 (p-JAK1), p-JAK2 and
p-JAK3 in BxPC-3 and PANC-1 cells was calculated using one-way
ANOVA. #P<0.01; *P<0.05. (H) The protein intensity
of p-STAT1 in BxPC-3 and PANC-1 cells was assessed using one-way
ANOVA. #P<0.01, *P<0.05. E, erlotinib; G,
gemcitabine. |
Next, we evaluated the expression levels of p53, a
molecule strongly associated with tumor growth and the
phosphorylation of STAT3 and STAT1. Upstream of STAT3, the
expression levels of JAK1, JAK2, JAK3 were also evaluated (Fig. 3). The expression level of p53 was
significantly lower in group E+G compared with group G or group E,
respectively, and it was significantly lower in group G, group E
and group E+G when they were compared with group N in both cell
lines. The phosphorylation level of JAK1 was lower in group G,
group E and group E+G when they were compared with group N in both
cell lines, and it was also lower when group E+G was compared with
group E. The phosphorylation level of JAK2 was significantly lower
in group E+G when it was compared with group N or group G in
recurrent tumors of the BxPC-3 cell line. While in recurrent tumors
of the PANC-1 cell line, the phosphorylation level of JAK2 was
significantly lower in group E+G when it was compared with group N
or group E, and it was also lower in group G when it was compared
with group N. As for the phosphorylation level of JAK3, it was
lower in group E, group G or group E+G when they were compared with
group N in both cell lines, respectively. In recurrent tumors of
the BxPC-3 cell line, the phosphorylation level of JAK3 was lower
in group E+G when compared with groups E, G and N respectively. The
phosphorylation level of JAK3 was decreased in group E+G when
compared with groups E and G, respectively. In recurrent tumors of
PANC-1 cell line, the phosphorylation level of JAK3 in group N was
higher than that of the other groups. However, no significant
difference in the phosphorylation level of JAK3 was found between
group E+G and group E or group G, respectively. The phosphorylation
level of STAT1 was significantly lower in group E+G when it was
compared with group N, group E or group G, respectively. In
addition, it was higher in group E when it was compared with group
N, group G and group E+G in recurrent tumors of the BxPC-3 cell
line, respectively. In the PANC-1 cell line recurrent tumors, the
phosphorylation level of STAT1 was significantly lower in group N
when it was compared with group E and group G, respectively.
Furthermore, it was also significantly lower in group E+G when it
was compared with group E and group G in recurrent tumors of the
PANC-1 cell line, respectively.
Furthermore, we evaluated the expression level of
cyclin D1 and HIF-1α, downstream proteins of STAT3. We found that
the expression level of cyclin D1 was significantly lower in group
E+G than that in the monotherapy groups in recurrent tumors of the
BxPC-3 cell line. The expression level of cyclin D1 was higher in
group N when it was compared with the monotherapy groups. Cyclin D1
was lower in group E+G when it was compared with group G in
recurrent tumors of the PANC-1 cell line. The expression level of
HIF-1α was found to be significantly lower in group E+G than in any
of the other three groups and the expression level of HIF-1α was
found to be in recurrent tumors of the BxPC-3 cell line,
significantly lower in group G than in group N (P<0.05) in
recurrent tumors of the PANC-1 cell line (Fig. 3A-D).
The combination of gemcitabine with
erlotinib induces apoptosis of pancreatic cancer cells via
caspase-dependent pathway
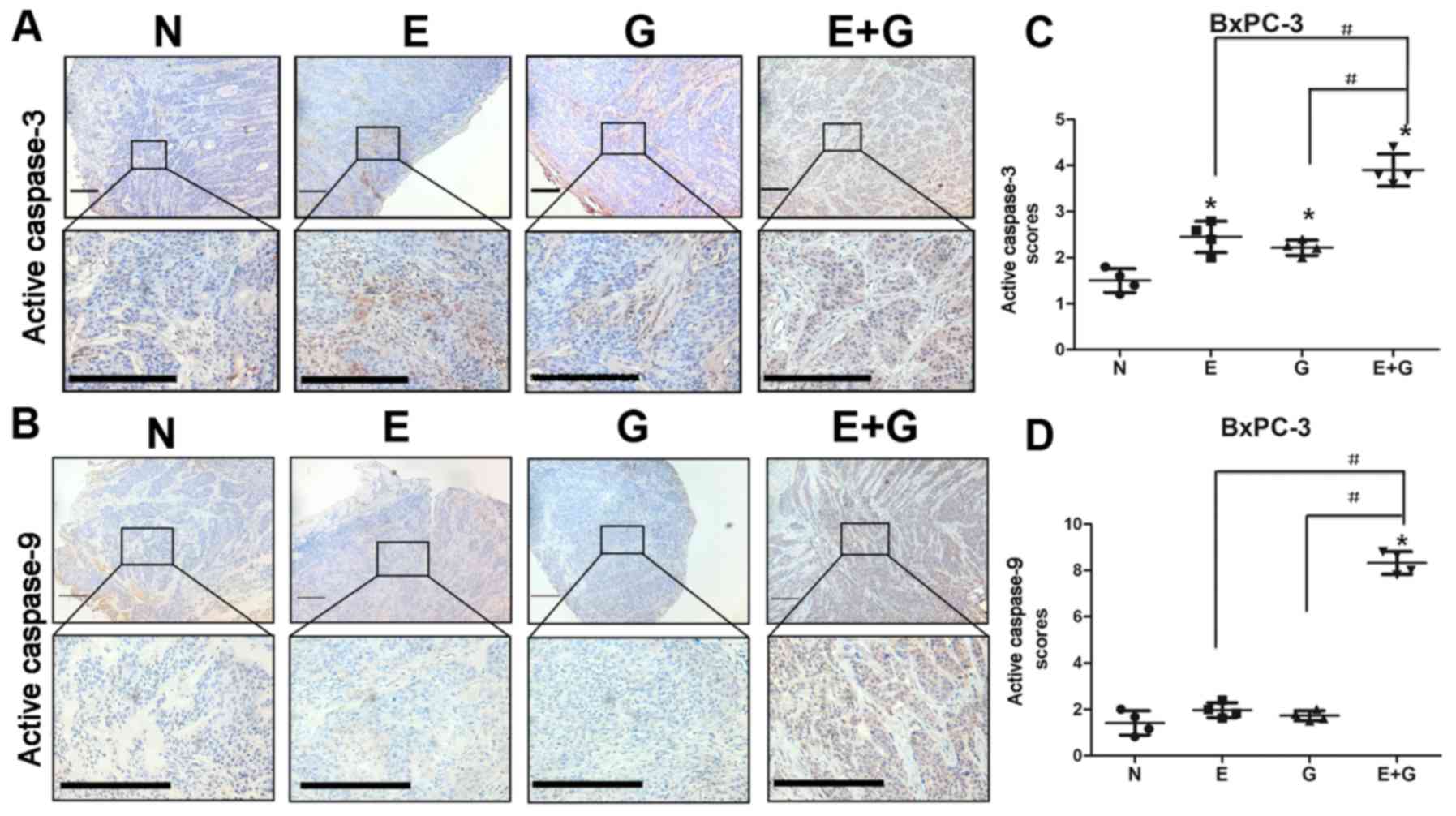
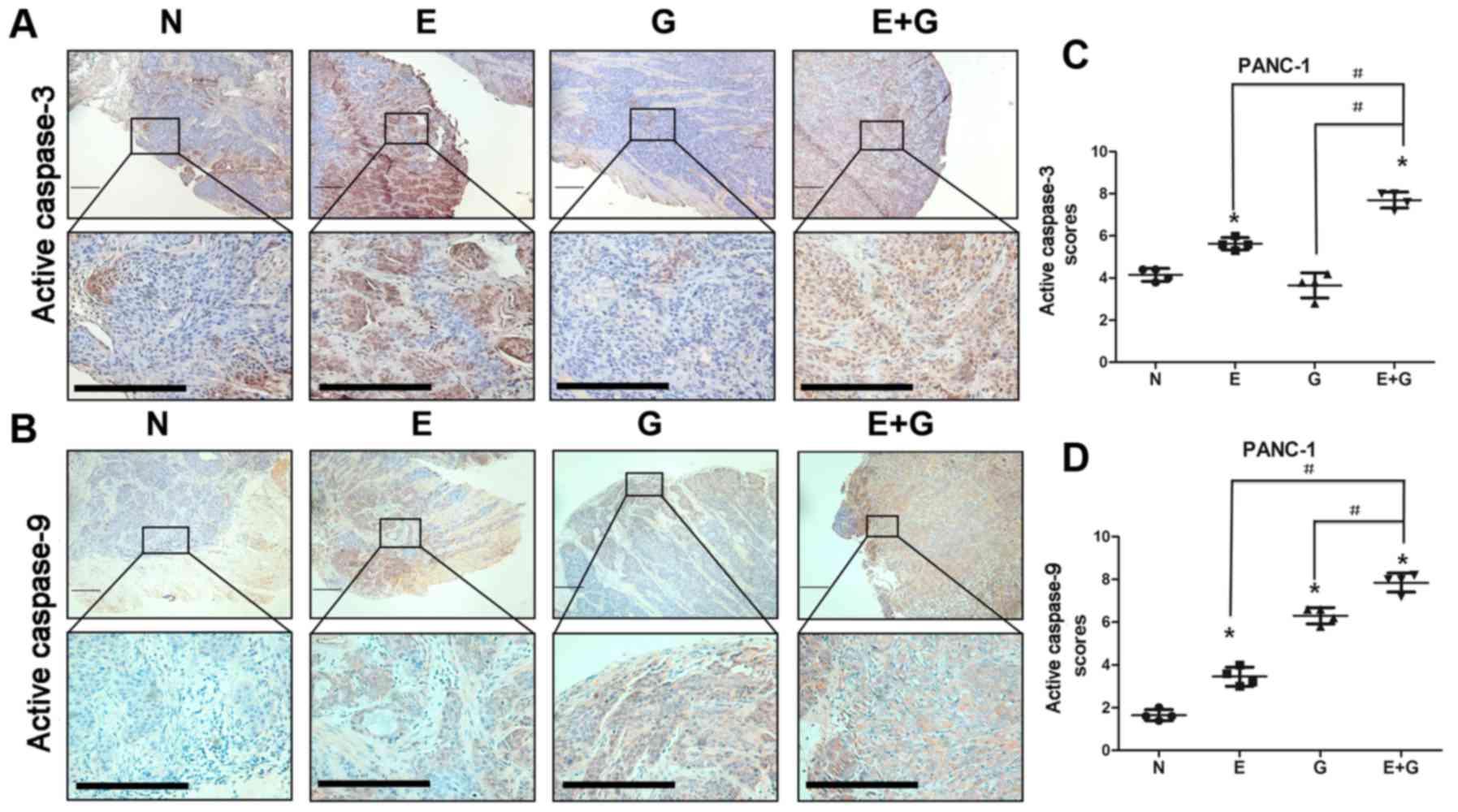
It was reported that reduction in STAT3
phosphorylation at tyrosine 705 residue could cause activation of
caspase-9 and caspase-3 (15). In
the present study, we investigated the state of caspase-9 and
caspase-3 by immunohistochemistry in all groups. Our data indicated
that caspase-9 and caspase-3 were activated in mice bearing BxPC-3
and PANC-1 xenografts and treated with gemcitabine and erlotinib
compared to the other groups (Figs.
4 and 5). These results
indicated that the combination of gemcitabine and erlotinib induced
apoptosis of pancreatic cancer cells via caspase-dependent
pathway.
Discussion
In patients with pancreatic cancer following R0 or
R1 resection, benefits from chemotherapy after surgery are usually
very modest. Compared to single-agent gemcitabine treatment,
gemcitabine in combination with erlotinib has been demonstrated to
be effective for advanced pancreatic cancer. Nonetheless, its
effectiveness in patients with PC after R0 or R1 resection has not
yet been reported. Poor penetration of drugs into the dense and
under vascularized tumor stroma in pancreatic cancer patients leads
to poor response to chemotherapy. It has been reported that
gemcitabine in combination with erlotinib is additive in the
KRAS–mutated pancreatic cancer cells PANC-1, whereas it is
antagonistic in the KRAS wild-type pancreatic cancer cell line
BxPC-3 (16). However, the effect
of gemcitabine in combination with erlotinib on recurrent tumor
remains unknown. We hypothesized that gemcitabine in combination
with erlotinib may also be effective for patients after resection
due to better penetration of drugs into the tumor stroma. Herein,
we explored its effect on recurrent tumors in a nude mouse model
after resection. We found that gemcitabine in combination with
erlotinib could inhibit tumor growth and induce apoptosis more
effectively than gemcitabine or erlotinib alone, or the control
group in vivo. The underlying mechanism may be JAK-STAT
pathway inhibition induced by gemcitabine in combination with
erlotinib.
As we aforementioned, the JAK-STAT pathway is very
important in tumor proliferation. In the present study, we
determined that the phosphorylation levels of JAK1, JAK2, JAK3 and
their downstream STAT1 and STAT3 were significantly decreased in
the E+G group. STAT1, a member of the family of STAT, has been
reported to be expressed in 88% of primary pancreatic cancer
patients (17). Reduced
phospho-STAT1 has been reported to attenuate tumor growth and
metastasis through downregulation of MUC4 mucin in pancreatic
cancer (18). In our study, we
revealed that gemcitabine in combination with erlotinib could lower
the phosphorylation level of STAT1. These results revealed that
gemcitabine in combination with erlotinib could also inhibit
recurrent pancreatic cancer tumor growth by decreasing the
phosphorylation levels of STAT1.
The phosphorylation of the tyrosine 705 residue in
the STAT3 protein is a crucial event for its activation, that leads
to formation of STAT3 homodimers and translocation into the nuclei
(14). Nuclear localized STAT3
dimers bind to the promoters of various target genes, which are
involved in cancer cell proliferation, survival and invasion, and
regulate their transcription. (19). Nagaraj et al reported that
gemcitabine in combination with erlotinib could not inhibit the
phosphorylation of the serine 727 residue in STAT3 in vitro
(20); while we found that
gemcitabine in combination with erlotinib could inhibit the
phosphorylation of tyrosine residue 705 in STAT3. Phosphorylation
of tyrosine residue 705 or serine residue 727 in STAT3 may function
differently, some genes may be preferentially regulated by only one
or both phosphorylation of tyrosine residue 705 or serine residue
727 in STAT3 (21). The different
roles that phosphorylation of tyrosine residue 705 or serine
residue 727 in STAT3 may play in pancreatic cancer still remain
unclear. We also determined that the expression of STAT3 downstream
proteins, cyclin D1 and HIF-1α were downregulated. Reduction in
STAT3 phosphorylation at tyrosine residue 705 could cause
activation of caspase-9 and caspase-3, while apoptotic signaling
could be mediated via activation of the cell death initiator
procaspase-9, whereas activated caspase-9 in turn cleaves
executioner caspase-3 (15,20). The stroma of pancreatic cancer is
often hypoxic, as indicated by the increased expression of HIF-1α
in stromal cells from human tumor sections, and HIF-1α expression
was revealed to be correlated with tumor size, worse prognosis,
advanced stage, presence of lymph node metastasis, and a higher
incidence of hepatic metastasis in pancreatic cancer (22). In addition, during a study that
monitored HIF-1α activity in vivo during tumor progression
by using a human pancreatic cancer line engineered to express a
reporter of HIF-1α activity, the authors observed that the activity
of HIF-1α was accompanied by local invasion, peritoneal
dissemination, and liver metastasis of pancreatic cells (23). Thus, a decreased expression level of
HIF-1α by gemcitabine in combination with erlotinib may also
inhibit recurrent tumor growth.
The p53 protein is an important tumor-suppressor
gene, which can arrest cell cycle progression at several points and
induce apoptosis for cells undergoing uncontrolled growth (24). However, p53 mutants possess
tumor-promoting functions (25).
Loss or mutation of p53 occurs in 50% of human pancreatic cancers
(26), including BxPC-3 and PANC-1
cells (27). It has been determined
that STAT3 binds to the p53 promoter in vitro and in
vivo, while blocking STAT3 in cancer cells upregulating the
expression of p53, thus leading to p53-mediated tumor cell
apoptosis (28). In the present
study, we determined that reduced phosphorylation of STAT3 did not
increase the expression of p53. Conversely, the expression of p53
was decreased in the gemcitabine in combination with erlotinib
group. This may indicate that gemcitabine in combination with
erlotinib may also inhibit tumor growth via downregulation of
p53.
Cyclin D1 is an important regulator of
cyclin-dependent kinase 4 (CDK4) and CDK6. It regulates cell cycle
transition from the G1 to the S phase, where overexpression of
cyclin D1 results in dysregulated CDK activity, rapid cell growth
under restricted mitogenic signaling conditions, bypass of key
cellular checkpoints, and ultimately, neoplastic growth (29). Hypoxia is most commonly present in
the solid tumor microenvironment, and especially in pancreatic
cancer. Hypoxia-inducible factor-1 (HIF-1; consists of highly
regulated HIF-1a and a constitutively expressed HIF-1b) is the most
important transcription factor resulting from intratumoral hypoxia,
which can mediate many adaptive physiological responses (22,30).
In pancreatic cancer, HIF-1a expression levels are associated with
tumor progression, fibrotic focus, angiogenesis, cell migration,
and hepatic metastasis (22,30).
Cyclin D1 and HIF-1α are both downstream proteins of STAT3.
Decrease in the phosphorylation of STAT3 led to downregulation of
cyclin D1 and HIF-1α, thus inhibiting tumor growth.
Our results indicated that gemcitabine in
combination with erlotinib decreased the expression of p53 and the
phosphorylation of tyrosine residue 705 in STAT3, which led to
downregulation of cyclin D1 and HIF-1α and activation of both
caspase-9 and caspase-3. Downregulation of cyclin D1 and HIF-1α
contributed to the inhibition of G1/S cell cycle transition and
angiogenesis. Activation of caspase-9 and caspase-3 resulted in
apoptosis of pancreatic cancer cells. Decreased expression of p53
could also decrease its tumor-promoting ability.
The heterogeneity of genetic alterations associated
with pancreatic cancer emphasizes the need to identify whether the
aforementioned treatment could function for pancreatic cancer cells
of different genetic background. In the present study, we used two
pancreatic cancer cell lines; BxPC-3 with wild-type KRAS, and
PANC-1 with mutated type KRAS. KRAS mutation exists in almost 90%
of pancreatic cancer cells, which leads to constitutive activation
of Ras signaling and resistance to TKI (31,32).
Notably, we revealed that gemcitabine in combination with erlotinib
could inhibit phosphorylation of tyrosine 705 residue in STAT3 in
recurrent tumors of both cell lines. This suggests that inhibition
of the phosphorylation of the 705 tyrosine residue in STAT3 by
gemcitabine in combination with erlotinib does not depend on the
state of KRAS.
Our results clearly revealed that the combination of
gemcitabine and erlotinib could inhibit recurrent pancreatic tumor
growth via downregulation of the phosphorylation levels of JAKs and
STATs and as a result to induce apoptosis of pancreatic cancer
cells (Fig. 6). Collectively, our
findings provide compelling evidence for the use of gemcitabine in
combination with erlotinib in the treatment of pancreatic cancer
after R0 or R1 resection, and this combination may be used as a
more effective treatment for pancreatic cancer after R0 or R1
resection than gemcitabine alone.
Acknowledgements
The present study was supported by Guizhou Province
Science and Technology Projects [(2014)2015 and (2016)7232]. We
would like to thank the Central Laboratory at Yongchuan Hospital,
Chongqing Medical University and Clinical Research Center,
Affiliated Hospital of Guizhou Medical University for housing all
the experiments.
Glossary
Abbreviations
Abbreviations:
|
i.p.
|
intraperitoneal injection
|
|
PC
|
pancreatic cancer
|
|
EGFR
|
epidermal growth factor receptor
|
|
TKI
|
tyrosine kinase inhibitor
|
|
JAK
|
Janus kinase
|
|
STAT
|
signal transducers and activators of
transcription
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling
|
|
TBST
|
TBS containing 0.1% Tween-20
|
|
CDK4
|
cyclin-dependent kinase 4
|
|
HIF-1
|
hypoxia-inducible factor-1
|
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hartwig W, Werner J, Jäger D, Debus J and
Büchler MW: Improvement of surgical results for pancreatic cancer.
Lancet Oncol. 14:e476–e485. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang Q, Zeng L, Chen Y, Lian G, Qian C,
Chen S, Li J and Huang K: Pancreatic cancer epidemiology,
detection, and management. Gastroenterol Res Pract.
2016:89623212016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Y, Hu GF, Zhang QQ, Tang N, Guo J,
Liu LY, Han X, Wang X and Wang ZH: Efficacy and safety of
gemcitabine plus erlotinib for locally advanced or metastatic
pancreatic cancer: A systematic review and meta-analysis. Drug Des
Devel Ther. 10:1961–1972. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ducreux M, Cuhna AS, Caramella C,
Hollebecque A, Burtin P, Goéré D, Seufferlein T, Haustermans K, Van
Laethem JL, Conroy T, et al ESMO Guidelines Committee, : Cancer of
the pancreas: ESMO Clinical Practice Guidelines for diagnosis,
treatment and follow-up. Ann Oncol. 26 Suppl 5:v56–v68. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Binenbaum Y, Na'ara S and Gil Z:
Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug
Resist Updat. 23:55–68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bareschino MA, Schettino C, Troiani T,
Martinelli E, Morgillo F and Ciardiello F: Erlotinib in cancer
treatment. Ann Oncol. 18 Suppl 6:vi35–vi41. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Czarnecka AM, Korzeń P, Nowak-Dement A,
Kukwa W, Korniluk J and Szczylik C: Prolonged complete response
following gemcitabine-erlotinib combined therapy in advanced
pancreatic cancer. Oncol Lett. 11:1101–1104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rawlings JS, Rosler KM and Harrison DA:
The JAK/STAT signaling pathway. J Cell Sci. 117:1281–1283. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mace TA, Shakya R, Elnaggar O, Wilson K,
Komar HM, Yang J, Pitarresi JR, Young GS, Ostrowski MC, Ludwig T,
et al: Single agent BMS-911543 Jak2 inhibitor has distinct
inhibitory effects on STAT5 signaling in genetically engineered
mice with pancreatic cancer. Oncotarget. 6:44509–44522. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Egberts JH, Cloosters V, Noack A,
Schniewind B, Thon L, Klose S, Kettler B, von Forstner C, Kneitz C,
Tepel J, et al: Anti-tumor necrosis factor therapy inhibits
pancreatic tumor growth and metastasis. Cancer Res. 68:1443–1450.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Naito S, von Eschenbach AC, Giavazzi R and
Fidler IJ: Growth and metastasis of tumor cells isolated from a
human renal cell carcinoma implanted into different organs of nude
mice. Cancer Res. 46:4109–4115. 1986.PubMed/NCBI
|
|
14
|
Huang G, Yan H, Ye S, Tong C and Ying QL:
STAT3 phosphorylation at tyrosine 705 and serine 727 differentially
regulates mouse ESC fates. Stem Cells. 32:1149–1160. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Agarwal C, Tyagi A, Kaur M and Agarwal R:
Silibinin inhibits constitutive activation of Stat3, and causes
caspase activation and apoptotic death of human prostate carcinoma
DU145 cells. Carcinogenesis. 28:1463–1470. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Torres C, Linares A, Alejandre MJ,
Palomino-Morales RJ, Delgado JR and Perales S: Interplay between
gemcitabine and erlotinib over pancreatic adenocarcinoma cells.
Pancreas. 45:269–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun Y, Yang S, Sun N and Chen J:
Differential expression of STAT1 and p21 proteins predicts
pancreatic cancer progression and prognosis. Pancreas. 43:619–623.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Seshacharyulu P, Ponnusamy MP, Rachagani
S, Lakshmanan I, Haridas D, Yan Y, Ganti AK and Batra SK: Targeting
EGF-receptor(s) - STAT1 axis attenuates tumor growth and metastasis
through downregulation of MUC4 mucin in human pancreatic cancer.
Oncotarget. 6:5164–5181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye C, Zhao W, Li M, Zhuang J, Yan X, Lu Q,
Chang C, Huang X, Zhou J, Xie B, et al: δ-tocotrienol induces human
bladder cancer cell growth arrest, apoptosis and chemosensitization
through inhibition of STAT3 pathway. PLoS One. 10:e01227122015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nagaraj NS, Washington MK and Merchant NB:
Combined blockade of Src kinase and epidermal growth factor
receptor with gemcitabine overcomes STAT3-mediated resistance of
inhibition of pancreatic tumor growth. Clin Cancer Res. 17:483–493.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ouédraogo ZG, Biau J, Kemeny JL, Morel L,
Verrelle P and Chautard E: Role of STAT3 in genesis and progression
of human malignant gliomas. Mol Neurobiol. 54:5780–5797. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yuen A and Díaz B: The impact of hypoxia
in pancreatic cancer invasion and metastasis. Hypoxia (Auckl).
2:91–106. 2014.PubMed/NCBI
|
|
23
|
Kizaka-Kondoh S, Itasaka S, Zeng L, Tanaka
S, Zhao T, Takahashi Y, Shibuya K, Hirota K, Semenza GL and Hiraoka
M: Selective killing of hypoxia-inducible factor-1-active cells
improves survival in a mouse model of invasive and metastatic
pancreatic cancer. Clin Cancer Res. 15:3433–3441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun C, Zhang M, Shan X, Zhou X, Yang J,
Wang Y, Li-Ling J and Deng Y: Inhibitory effect of cucurbitacin E
on pancreatic cancer cells growth via STAT3 signaling. J Cancer Res
Clin Oncol. 136:603–610. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stojanovic N, Hassan Z, Wirth M, Wenzel P,
Beyer M, Schäfer C, Brand P, Kroemer A, Stauber RH, Schmid RM, et
al: HDAC1 and HDAC2 integrate the expression of p53 mutants in
pancreatic cancer. Oncogene. 36:1804–1815. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rosenfeldt MT, O'Prey J, Morton JP, Nixon
C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al:
p53 status determines the role of autophagy in pancreatic tumour
development. Nature. 504:296–300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Izetti P, Hautefeuille A, Abujamra AL, de
Farias CB, Giacomazzi J, Alemar B, Lenz G, Roesler R, Schwartsmann
G, Osvaldt AB, et al: PRIMA-1, a mutant p53 reactivator, induces
apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic
cancer cell lines. Invest New Drugs. 32:783–794. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Niu G, Wright KL, Ma Y, Wright GM, Huang
M, Irby R, Briggs J, Karras J, Cress WD, Pardoll D, et al: Role of
Stat3 in regulating p53 expression and function. Mol Cell Biol.
25:7432–7440. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qie S and Diehl JA: Cyclin D1, cancer
progression, and opportunities in cancer treatment. J Mol Med
(Berl). 94:1313–1326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao X, Gao S, Ren H, Sun W, Zhang H, Sun
J, Yang S and Hao J: Hypoxia-inducible factor-1 promotes pancreatic
ductal adenocarcinoma invasion and metastasis by activating
transcription of the actin-bundling protein fascin. Cancer Res.
74:2455–2464. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Berry W, Algar E, Kumar B, Desmond C, Swan
M, Jenkins BJ and Croagh D: Endoscopic ultrasound-guided
fine-needle aspirate-derived preclinical pancreatic cancer models
reveal panitumumab sensitivity in KRAS wild-type tumors. Int J
Cancer. 140:2331–2343. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zehir A, Benayed R, Shah RH, Syed A,
Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et
al: Mutational landscape of metastatic cancer revealed from
prospective clinical sequencing of 10,000 patients. Nat Med.
23:703–713. 2017. View Article : Google Scholar : PubMed/NCBI
|