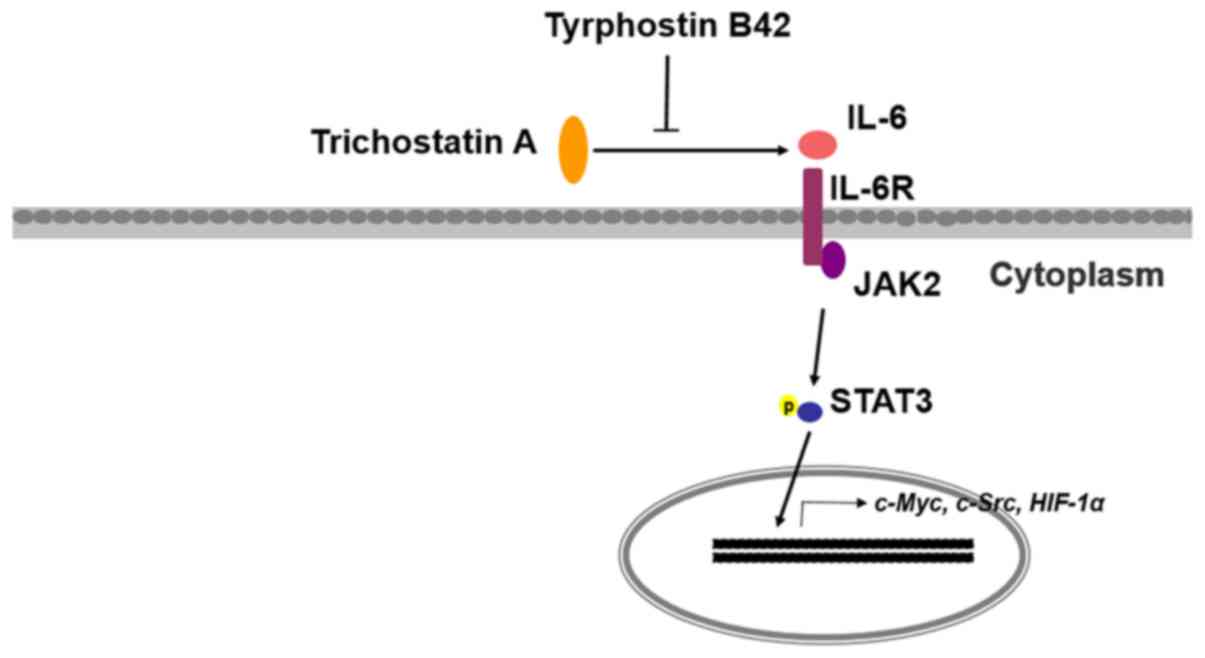
Introduction
Pancreatic cancer is a type of aggressive malignant
tumor with drug-resistance, poor prognosis and high mortality. The
etiology of pancreatic cancer remains unclear, and the factors
including genetics, diet and chronic pancreatitis contribute to the
occurrence and development of pancreatic cancer. Due to the changes
in lifestyle habits, dietary structure and an increase in
environmental pollution, the incidence of pancreatic cancer has
increased gradually (1,2). Clinically, surgery is regarded as the
main treatment method for pancreatic cancer. However, only a small
number of patients with pancreatic cancer can undergo surgery.
Patients who are unable to undergo surgery presently are treated
with 5-fluorouracil, cisplatin, gemcitabine, or a combination of
radiation therapy and chemotherapy (3–5). These
auxiliary treatments can to some extent inhibit the further
deterioration of patients with pancreatic cancer, but whether they
can extend the life of patients warrants further clinical
observations. Thus, it is urgent to develop new treatments and
effective drugs for pancreatic cancer.
In recent years, the importance of epigenetic
alterations has been confirmed in cancer development, including the
role of aberrant DNA methylation and histone acetylation on
aberrant silencing of multiple tumor-suppressor genes in a
diversity of human cancers (6).
HDAC inhibitors, which interfere with the function of histone
deacetylase (HDAC), are emerging as potent anticancer agents as a
result of their effective anti-proliferative activity in a wide
variety of tumors, mediated by mitotic defects through the aberrant
acetylation of histone and non-histone proteins (3,7).
However, our previous study revealed that pancreatic cancer cells
(PCCs) were resistant to HDAC inhibitor trichostatin A (TSA)
(8). Over-proliferative activity
and inhibition of apoptosis indicated that some
proliferation-related signaling pathways may be involved in the
process of TSA-mediated resistance in PCCs (8,9).
According to previous studies, IL-6 is a
well-studied proinflammatory cytokine with numerous links to a
variety of malignancies (10). IL-6
has been noted to be overexpressed in patients with pancreatic
cancer (11). In vivo, IL-6
can induce the phosphorylation of Janus kinase (JAK) via the
activation of cytokine receptors, which then creates receptor
docking sites for recruitment of cytoplasmic signal transducers and
activators of transcription-3 (STAT) proteins. Once in the nucleus,
STAT molecules bind specific promoter DNA sequences that result in
the transcription of genes that regulate the proliferation,
differentiation, and apoptosis of cancer cells (12). STAT3 belongs to a family of
transcription factors that relay cytokine receptor generated
signals into the nucleus (13,14).
Once in the nucleus, STAT molecules bind specific promoter DNA
sequences that result in the transcription of genes that regulate
cell proliferation, differentiation, and apoptosis (e.g., Bcl-2,
CCND1 and c-Myc) (11,12,15).
The apoptosis-associated proteins Bcl-2 and Bax play an important
role in regulating cell survival and are key transcriptional
targets of STAT3 (16–18). Evidence has revealed that
hypoxia-inducible factor 1α (HIF-1α) plays pivotal roles in tumor
invasion, metastasis, and drug resistance. It has been well
documented that hypoxia is one of the fundamental biological
phenomena, which is related to the development and aggressiveness
of pancreatic cancer (19,20). Furthermore, inhibition of the
STAT3/cyclinD1 pathway increased the sensitivity of cancer cells to
chemotherapy (21).
In the present study, we first investigated the
activity of IL-6/JAK2/STAT3 signaling in patients with pancreatic
cancer and in PCCs with (PANC-1-TSA) or without (PANC-1) TSA
resistance. Secondly, tyrphostin B42 had been reported to possess
inhibitory activity on the proliferation of various tumor cells
(22,23), and to be associated with
IL-6/JAK2/STAT3 signaling (24).
Therefore, in the present study the effects of tyrphostin B42 on
TSA-induced over-proliferation and resistance of PCCs were also
evaluated. Our results revealed the crucial role of IL-6/JAK2/STAT3
signaling in TSA-resistance of PCCs, resulting in the inhibition of
apoptosis and induction of proliferation. Treatment with tyrphostin
B42 attenuated TSA-mediated resistance by antagonizing
IL-6/JAK2/STAT3 signaling.
Materials and methods
Patients with pancreatic cancer
In the present study, a total of 96 patients with
pancreatic cancer were enrolled from the First Affiliated Hospital
of Wenzhou Medical University (Wenzhou, China) between March 2015
and August 2016. All pancreatic cancer cases, which were confirmed
by pathological examination, enrolled in the current study had not
received any radiation or chemotherapy before surgery. The
demographic and clinical characteristics of the subjects are shown
in Table I, including age, sex,
smoking, tumor stage, TNM classification, lymph node metastasis,
and vascular infiltration. Based on the TNM classification system
promulgated by the American Joint Committee on Cancer (AJCC), the
pathologic stage was divided into localized and aggressive cancer,
as determined by transrectal ultrasound, magnetic resonance imaging
(MRI) and emission computed tomography (ECT) (25).
 | Table I.Demographic and characteristics of
patients with pancreatic cancer. |
Table I.
Demographic and characteristics of
patients with pancreatic cancer.
|
|
| IL-6
expression |
|
|
|---|
|
|
|
|
|
|
|---|
|
Characteristics | Number (%) | Negative (%) | Positive (%) | P-value | Odds ratio (95%
CI) |
|---|
| Total |
| 96 | 19 (19.8) | 77 (80.2) |
|
| Sex |
|
|
|
|
|
|
Female | 25 (26) | 5
(5.2) | 20 (20.8) | 0.976 | 0.982
(0.314–3.075) |
|
Male | 71 (74) | 14 (14.6) | 57 (59.3) |
|
|
| Smoking |
|
|
|
|
|
|
Yes | 44 (45.8) | 9
(9.4) | 35 (36.5) | 0.881 | 0.926
(0.339–2.532) |
| No | 52 (54.2) | 10 (10.4) | 42 (43.8) |
|
|
| Age (years) |
|
|
|
|
|
|
≤60 | 43 (44.8) | 8
(8.3) | 35 (39.6) | 0.793 | 1.146
(0.415–3.162) |
|
>60 | 53 (55.2) | 11 (11.5) | 42 (43.8) |
|
|
| Tumor stage |
|
|
|
|
|
|
T1+T2 | 8 (8.3) | 3
(3.1) | 5
(5.2) | 0.197 | 2.100
(0.446–9.891)a |
|
T3+T4 | 63 (65.6) | 14 (14.6) | 49 (51.0) |
|
|
|
Unknown | 25 (26.0) | 2
(2.1) | 23 (24.0) |
|
|
| Lymph node
metastasis |
|
|
|
|
|
|
Yes | 38 (39.6) | 3
(3.1) | 35 (36.5) | 0.027 | 4.321
(1.082–17.252)b |
| No | 37 (38.5) | 10 (10.4) | 27 (28.1) |
|
|
|
Unknown | 21 (21.9) | 4
(4.2) | 17 (17.7) |
|
|
| Tumor
differentiation |
|
|
|
|
|
|
Mildly | 19 (19.8) | 9
(9.4) | 10 (10.4) | 0.003 | 6.120
(1.999–18.734)c |
|
Moderately | 50 (52.1) | 7
(7.3) | 43 (44.8) |
|
|
|
Poorly | 27 (28.1) | 3
(3.1) | 25 (26.0) |
|
|
| Vascular
invasion |
|
|
|
|
|
|
Perineural | 26 (16.7) | 2
(2.1) | 24 (25.0) | 0.049 | 4.653
(1.001–21.633)d |
| Adipose
tissue | 68 (51.0) | 19 (19.9) | 49 (51.0) |
|
|
All subjects were informed about the contents of the
study and provided their informed consent. This study was approved
by the Ethics Committee of Wenzhou Medical University.
Immunohistochemical analysis
Sections, 4-µm-thick, from pancreatic cancer tissues
in patients were dewaxed with xylene and hydrated using sequential
ethanol (100, 95, 85, and 75%) and distilled water washes. The
endogenous peroxidase was blocked with 3% hydrogen peroxide.
Antigen retrieval was performed by heating the sections in 0.1%
sodium citrate buffer (pH 6.0). The expression of IL-6 (CAS No:
ab6672, 1:400; Abcam, Cambridge, MA, USA) was determined using the
immunochemical streptavidin-peroxidase method. All samples were
semi-quantitatively or quantitatively assessed by two independent
investigators in a blinded manner. Slides were examined and images
were captured using a DM4000 B LED microscope system (Leica
Microsystems GmbH, Hannheim, Germany) and a DFC 420 C 5M digital
microscope camera (Leica Microsystems GmbH). For H-score
assessment, 10 fields were chosen at random at an ×400
magnification and the staining intensity in the malignant cell
nuclei was scored as 0, 1, 2, or 3 corresponding to the presence of
negative, weak, intermediate, and strong brown staining,
respectively. The total number of cells in each field and the
number of cells stained at each intensity were counted. The average
percentage of positive cells was calculated and the following
formula was applied:
IHA H-score=(% of cells stained at intensity
category 1×1)+(% of cells stained at intensity category 2×2)+(% of
cells stained at intensity category 3×3) (26).
Cell culture and drug treatment
Human PCCs (PANC-1) were obtained from the American
Type Culture Collection (ATCC® CRL-1469™; Manassas, VA,
USA). Trichostatin A (TSA; CAS No: 58880-19-6; Selleckchem,
Houston, TX, USA)-induced resistance of PCCs (PANC-1-TSA) were
generated in our laboratory. PANC-1 and PANC-1-TSA cells were
maintained in DMEM (Gibco; Invitrogen; Thermo Fisher Scientific,
Inc., Grand Island, NY, USA) and supplemented with 10% fetal bovine
serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin (all
from Invitrogen; Thermo Fisher Scientific, Inc.) were treated with
5% tyrphostin B42 (CAS No: 133550-30-8; Selleckchem) or recombinant
IL-6 (CAS No: 200-06; PeproTech, Inc., Rocky Hill, NJ, USA) for 24
h. Tyrphostin B42a and IL-6 were dissolved in 100% dimethyl
sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) and diluted
with the culture medium for experiments. The final concentration of
DMSO for all treatments was maintained at 0.1%.
Establishment of TSA-resistant PCCs
(PANC-1-TSA)
PANC-1-TSA was obtained by culture of PCCs (PANC-1)
in vitro with intermittent increase of the concentration of
TSA in the culture for 30 weeks. After cultivating PANC-1 cells
with different concentrations of TSA for 1 week, we ascertained the
cell death conditions and chose the concentration (0.05 µmol/l) of
median lethal dose (LD80) as the initial concentration
to cultivate the resistant cell line. When cells grew stably and
entered the logarithmic growth phase, the concentration gradually
increased at 0.01 µmol/l every 2 weeks. After 15 concentration
gradients and 30 weeks of cultivation, the final concentration was
stable at 0.2 µmol/l.
CCK-8 assay
Cell suspension (100 µl; 5.0×103
cells/well) was dispensed in a 96-well plate with the outer wells
left empty for the addition of water. The plate was pre-incubated
for 24 h in humidified tyrphostin B42 and DMSO was used as a
control. A CCK-8 solution (10 µl) was added (Dojindo Molecular
Technologies, Inc., Tokyo, Japan) to each well of the plate. The
plate was then incubated for 4 h. The absorbance at 450 nm was
measured using a microplate reader (Thermo Fisher Scientific,
Inc.). Six parallel wells were set up in each experiment, and all
of the measurements were repeated at least three times.
Quantitative real-time polymerase
chain reaction (qRT-PCR) analysis
Total RNA was isolated using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.). The concentration and purity of
RNA were determined using a spectrophotometer. cDNA was synthesized
with reverse transcriptase (Thermo Fisher Scientific, Inc.).
qRT-PCR assays were carried out using FastStart Universal SYBR
Green Master Mix (Roche Diagnostics GmbH, Mannheim, Germany). PCR
parameters were as follows: 95°C for 10 min, then 95°C for 15 sec,
and 60°C for 30 sec for 40 cycles. The PCR assays were performed in
triplicate and were conducted using a Real-Time PCR Detection
System (ABI 7500; Applied Biosystems; Thermo Fisher Scientific,
Inc., Carlsbad, CA, USA). The expression of mRNA was calculated
using the 2−ΔΔCt method as previously described
(27). Three replicate reactions
per sample and endogenous control were used to ensure statistical
significance. All the primers that were used in this study are
listed in Table II.
| Table II.qRT-PCR primers in this study. |
Table II.
qRT-PCR primers in this study.
| Gene | Forward
(5′→3′) | Reverse
(5′→3′) |
|---|
| HIF-1α |
GCAGCAACGACACAGAAACT |
AGCGGTGGGTAATGGAGAC |
| CCND1 |
CCTGTCCTACTACCGCCTCA |
TCCTCCTCTTCCTCCTCCTC |
| c-Src |
CGAGAAAGTGAGACCACGAA |
TGCGGGAGGTGATGTAGAA |
| c-Myc |
CCTCCACTCGGAAGGACTATC |
TTCGCCTCTTGACATTCTCC |
| BCL-2 |
CAACACAGACCCACCCAGA |
TGGCTTCATACCACAGGTTTC |
| Bax |
TTTCTGACGGCAACTTCAACTG |
CGGAGGAAGTCCAATGTCCAG |
| GAPDH |
TCCCATCACCATCTTCCAGG |
GATGACCCTTTTGGCTCCC |
Western blot analysis
Whole-cell protein extracts and nuclear protein
extracts from PCCs were prepared with RIPA Lysis Buffer [50 mM
Tris-HCl, pH 8.0, with 150 mM sodium chloride, 1.0% Igepal CA-630
[(NP-40]), 0.5% sodium deoxycholate, and 0.1% sodium dodecyl
sulfate]) and a Nuclear Extract kit (Active Motif, Carlsbad, CA,
USA), respectively, according to the manufacturer's instructions.
Protein concentrations were determined using an assay kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Lysates containing 100 µg
of protein were mixed with loading buffer containing 5%
β-mercaptoethanol and heated for 5 min at 100°C. The samples were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred onto nitrocellulose membranes by
blotting. The membranes were probed with antibodies JAK2 (D2E12)
(1:1,000; rabbit monoclonal; cat. no. 3230), STAT3 (124H6)
(1:1,000; mouse monoclonal; cat. no. 9139), and p-STAT3 (D3A7)
(1:1,000; rabbit monoclonal; cat. no. 9145; all from Cell Signaling
Technolgy, Inc., Danvers, MA, USA) using an enhanced
chemiluminescence kit (cat. no. 34095; Thermo Fisher Scientific),
and GAPDH (1:5,000; cat. no. AP0063; Bioworld Technology, St. Louis
Park, MN, USA) was used as a control.
Statistical analysis
All in vitro experiments were repeated at
least three times to confirm the results. All the statistical
calculations were performed using Prism 6.0 (GraphPad Software,
Inc., San Diego, CA, USA) and the data were expressed as the mean ±
SEM. Statistical significance was determined with the Student's
t-test (two-tailed) when comparing two groups of data set. P-values
<0.05 were considered to indicate statistical significance.
Asterisks shown in the figures indicate significant differences of
the experimental groups in comparison with the corresponding
control groups.
Results
IL-6/JAK2/STAT3 signaling is activated
in pancreatic cancer and exacerbated by TSA
We first evaluated the activity of IL-6, an upstream
inducer of JAK2/STAT3 signaling, in patients with pancreatic
cancer. As shown in Fig. 1A and
Table I, the expression levels of
IL-6 in pancreatic cancer tissues were increased, and positively
associated with lymph node metastasis (P=0.027), tumor
differentiation (P=0.003) and vascular infiltration (P=0.049) of
pancreatic cancer. In pancreatic cancer patients, enhanced IL-6
expression had a high risk of lymph node metastasis [Odds ratio
(OR)=4.321; 95% CI: 1.082–17.252; P=0.027; metastasis vs.
non-metastasis), poorly differentiated (OR=6.120; 95% CI:
1.999–18.734; P=0.003; (poorly + moderately) vs. mildly)], and
perineural vascular infiltration (OR=4.653, 95% CI: 1.001–21.633;
P=0.035; perineural vs. adipose), suggesting that IL-6 levels and
downstream JAK2/STAT3 signaling may be essential for the
development of pancreatic cancer.
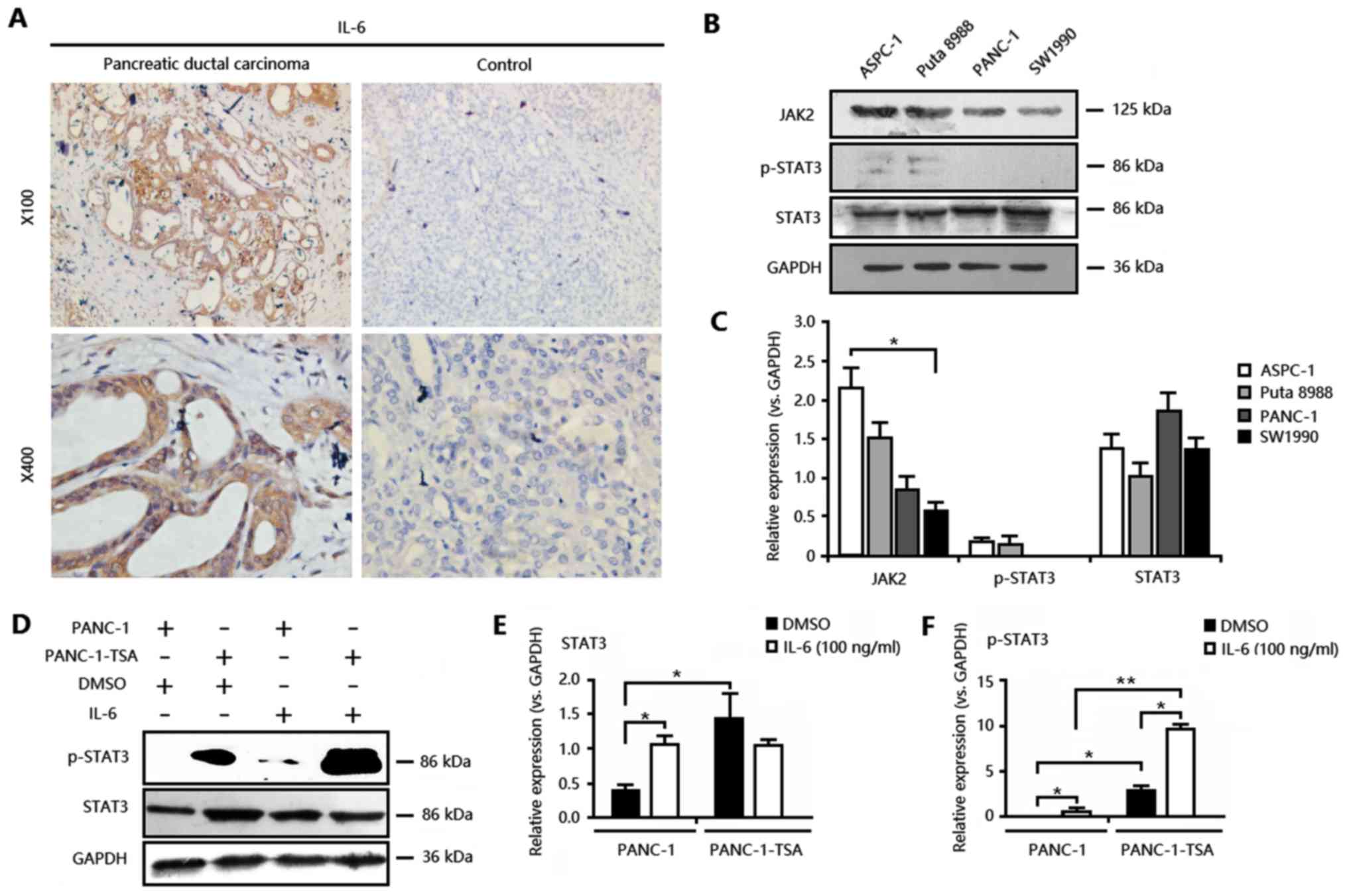 | Figure 1.IL-6/JAK2/STAT3 signaling is
activated in pancreatic cancer and exacerbated by TSA. (A) Enhanced
expression of IL-6 in human pancreatic cancer tissues. Bar, 200 µm.
(B) The expression of JAK2, STAT3 and phosphorylated STAT3
(p-STAT3) determined by western blotting was upregulated in
different cell types, including PANC-1, ASPC-1, Puta 8988, and
SW1990. (C) Relative expression levels of JAK2, STAT3 and p-STAT3
according to the results obtained with western blotting,
*P<0.05, SW1990 vs. ASPC-1. (D) The expression and
phosphorylation of STAT3 in PANC-1 and PANC-1-TSA cells treated
with DMSO or IL-6 (100 ng/ml) for 24 h. (E) Relative expression
levels of STAT3 according to the results obtained with western
blotting. *P<0.05, vs. PANC-1 cells treated with DMSO. (F) The
phosphorylation of STAT3 according to the results obtained with
western blotting. *P<0.05, vs. PANC-1 cells treated with DMSO;
**P<0.01, PANC-1 cells with IL-6 (100 ng/ml) vs. PANC-1-TSA
cells with IL-6 (100 ng/ml). PANC-1, non-resistant pancreatic
cancer cells; PANC-1-TSA, TSA-resistant pancreatic cancer
cells. |
We next investigated the activity of IL-6/JAK2/STAT3
signaling in different PCC cell lines. We found overexpression of
JAK2 and STAT3, but not the phosphorylation of STAT3, in PCC cell
lines including ASPC-1, SW1990, Puta 8988, and PANC-1 (Fig. 1B and C). Among them, PANC-1 cells
exhibited the highest expression of the STAT3 protein. In addition,
in PANC-1 cells, IL-6 activated JAK2/STAT3 signaling by inducing
the expression and phosphorylation of STAT3 (Fig. 1D-F). Similarly, in PANC-1-TSA cells,
IL-6 also induced the phosphorylation of STAT3, although the
changes of STAT3 expression did not exhibit significance (Fig. 1D-F). These results identified again
the crucial role of IL-6/JAK2/STAT3 signaling in pancreatic cancer.
Notably, the activity of IL-6/JAK2/STAT3 signaling in PANC-1-TSA
cells was higher when compared with those in PANC-1 cells.
Moreover, IL-6 treatment exacerbated this difference, as indicated
by the phosphorylation, but not the expression, of IL-6/JAK2/STAT3
signaling in PANC-1-TSA cells. Thus, over-activation of
IL-6/JAK2/STAT3 signaling in PCC cells may be one explanation for
TSA-induced resistance.
Expression levels of downstream target
genes of IL-6/JAK2/STAT3 signaling in PCCs are upregulated by
TSA
Given that TSA triggers the resistance in PCCs by
inducing the activation of IL-6/JAK2/STAT3 signaling, we
investigated whether TSA exerted similar effects on
STAT3-downstream target genes. Thus, the mRNA expression levels of
c-Myc, c-Src, HIF-α, and CCND1 in PANC-1-TSA cells were quantified
by the qRT-PCR. Enhanced levels of c-Myc and c-Src are required for
the proliferation of PCCs, which are regulated by cyclin D1
(encoded by the CCND1 gene), a key molecule involved in regulating
cell cycle progression (28–30).
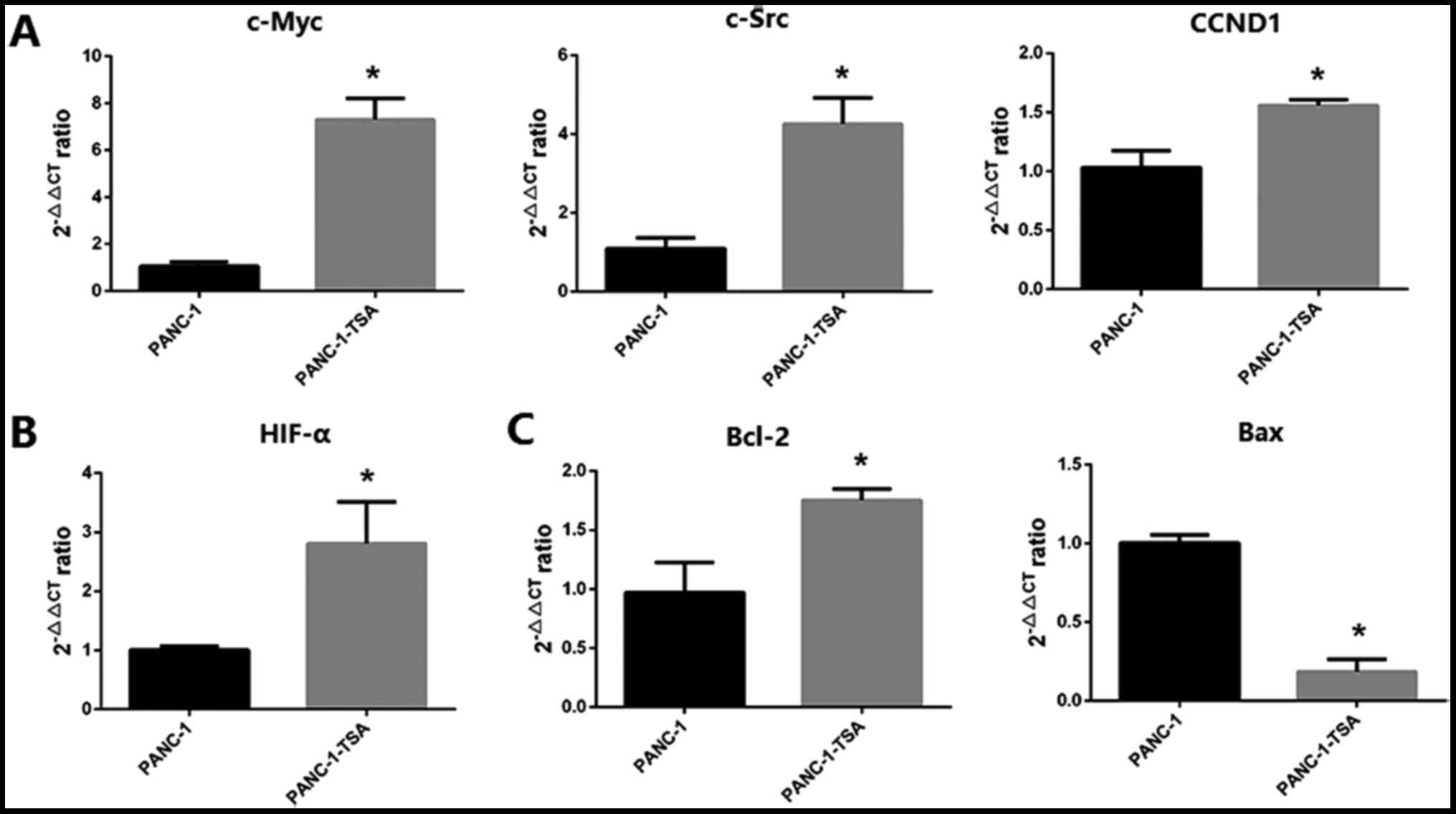
As shown in Fig. 2A, the mRNA
expression levels of c-Myc, c-Src and CCND1 were markedly increased
in PANC-1-TSA cells compared with those in PANC-1 cells
(P<0.05), indicating that PANC-1-TSA cells may have a more
proliferative activity than PANC-1 cells. In addition,
overexpression of HIF-α mRNA was also observed in PANC-1-TSA cells
(Fig. 2B). In response to a hypoxic
tumor microenvironment, TSA-induced HIF-α expression plays an
important role in differentiation, tumor growth and angiogenesis in
pancreatic cancer (31).
In addition to the induction of proliferation, TSA
also exerted an inhibitory effect on the apoptosis of PCCs. As
shown in Fig. 2C, the expression of
Bcl-2 mRNA was upregulated in PANC-1-TSA cells whereas the
expression of Bax was downregulated compared with that in PANC-1
cells (P<0.05), suggesting that TSA inhibited the apoptosis of
PCCs through the mitochondrial pathway.
Tyrphostin B42 inhibits TSA-mediated
over-proliferation of PCCs through IL-6/JAK2/STAT3 signaling
As aforementioned, PANC-1-TSA cells may possess more
proliferative activity than PANC-1 cells. In this case whether
tyrphostin B42 improves TSA-mediated resistance in PCCs through the
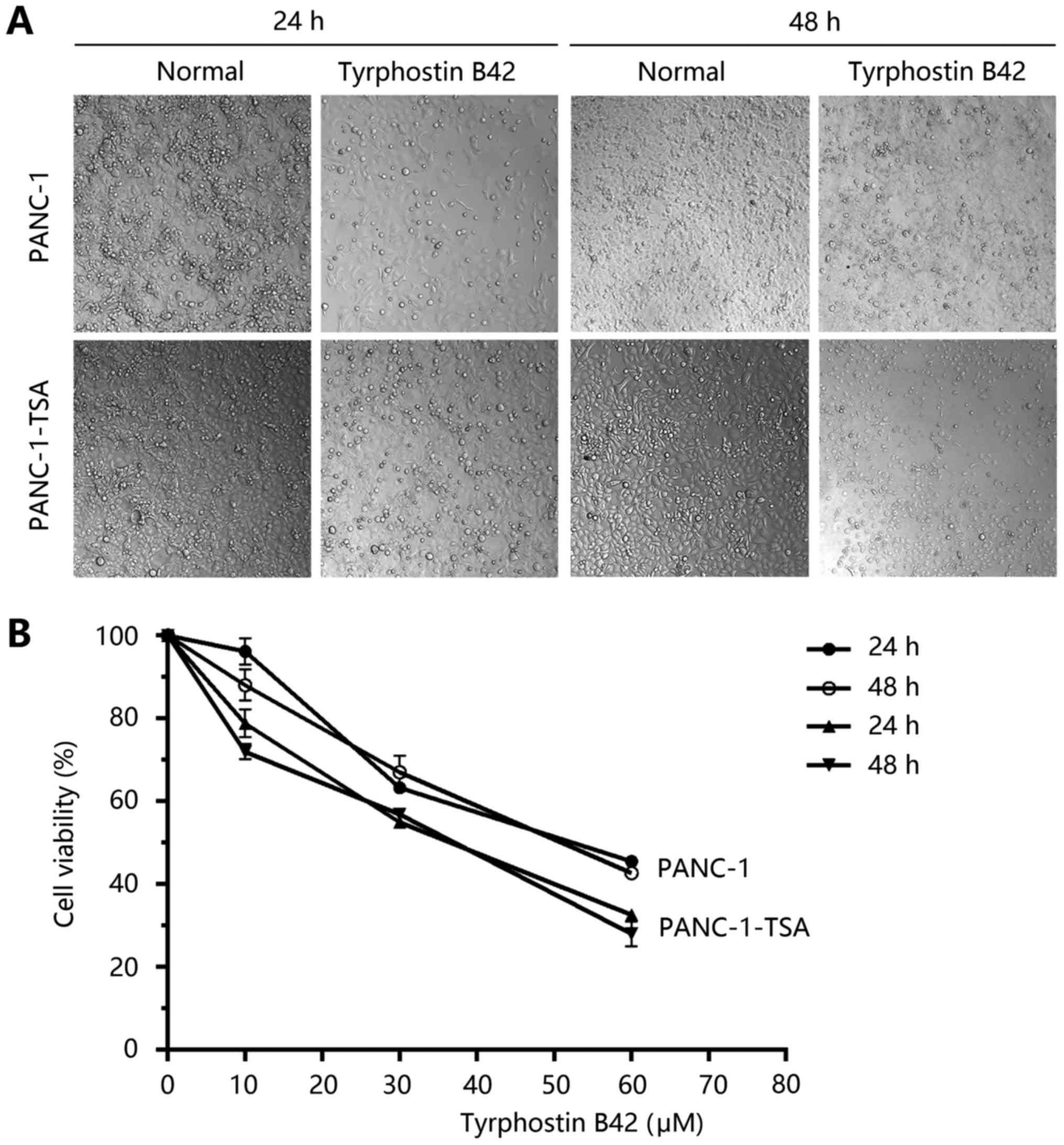
inhibition of proliferation remains unknown. As shown in Fig. 3A, we determined that tyrphostin B42
(30 µM) significantly decreased the number of proliferated PCCs,
especially in PANC-1-TSA cells in a time-dependent manner. The
results from the CCK-8 assay indicated that the viabilities of
PANC-1 and PANC-1-TSA cells were decreased with the treatment of
tyrphostin B42 (Fig. 3B,
P<0.05). In addition, high-doses of tyrphostin B42 (≥30 µM) had
a stronger inhibitory effect on the proliferation of PANC-1-TSA
cells compared with PANC-1 cells (Fig.
3B, P<0.05). These results revealed that tyrphostin B42
inhibited the over-proliferative activity of PCCs and improved
TSA-mediated resistance.
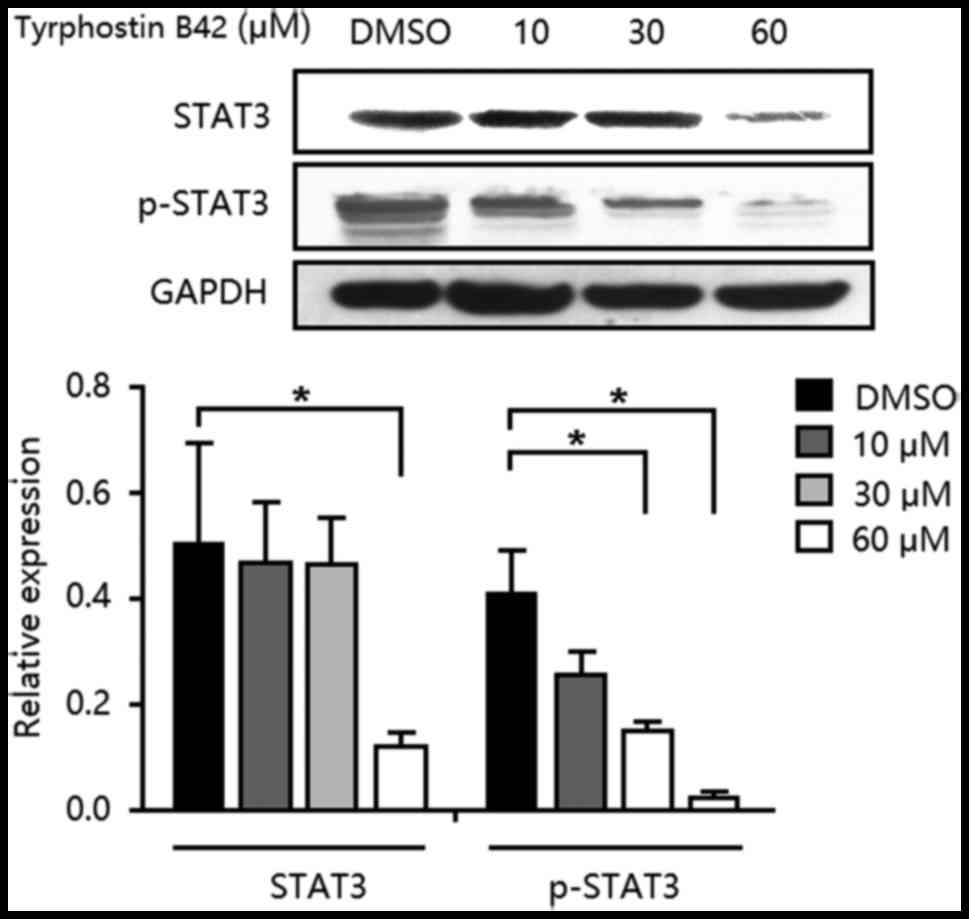
After TSA treatment, IL-6/JAK2/STAT3 signaling was
activated and then induced the over-proliferation of PCCs. In the
present experiment, in PANC-1-TSA cells, tyrphostin B42 reduced the
expression of STAT3 and inhibited the phosphorylation of p-STAT3
(Fig. 4). Moreover, high-doses of
tyrphostin B42 (≥30 µM) exerted stronger inhibitory effects on
IL-6/JAK2/STAT3 signaling. Consequently, tyrphostin B42-mediated
inhibition of PCC proliferation may be induced through
IL-6/JAK2/STAT3 signaling.
Tyrphostin B42 inhibits the expression
of downstream target genes of IL-6/JAK2/STAT3 signaling
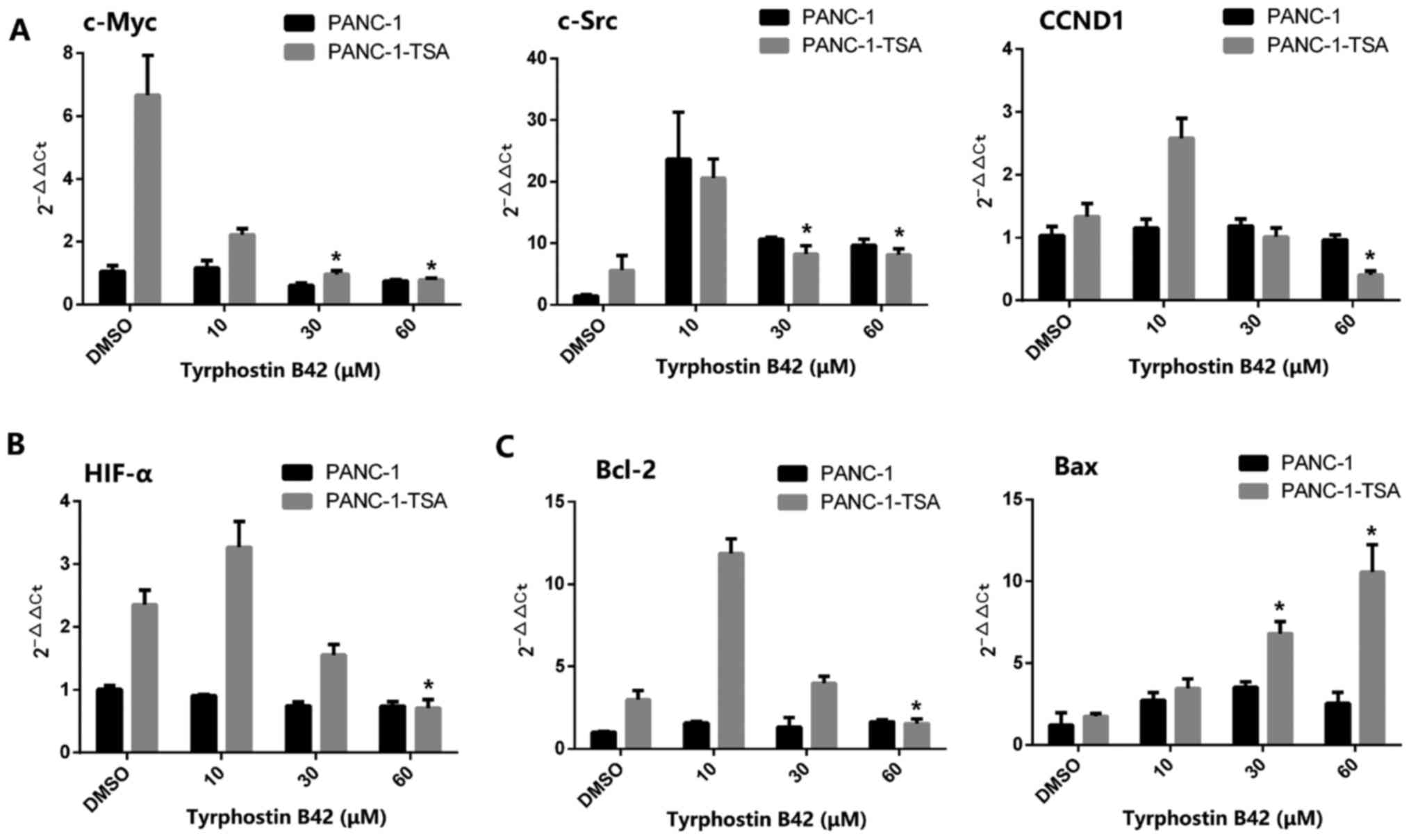
In PANC-1 cells, tyrphostin B42 (0–60 µM) did not
downregulate the expression of downstream target genes of
IL-6/JAK2/STAT3 signaling, including c-Myc, c-Src and CCND1
(Fig. 5A). Similarly, in PANC-1-TSA
cells, a low-dose (<30 µM) of tyrphostin B42 did not inhibit but
promote the expression of these target genes. However, high-doses
(≥30 µM) of tyrphostin B42 downregulated the mRNA expression of
c-Myc, c-Src and CCND1. In addition, high-doses of tyrphostin B42
also reduced low-dose-mediated overexpression of HIF-α mRNA in
PANC-1-TSA cells (Fig. 5B).
Furthermore, decreased expression of Bcl-2 and increased expression
of Bax in PANC-1-TSA cells were inhibited with tyrphostin B42
treatment at low doses (<30 µM) (Fig. 5C). These results indicated that
tyrphostin B42 at higher doses (≥30 µM) exerted an inhibitory
effect on the expression of downstream target genes of
IL-6/JAK2/STAT3 signaling, leading to the inhibition of
proliferation, the induction of apoptosis, and the reduction of
TSA-mediated resistance in PCCs.
Discusssion
Pancreatic cancer is considered to be one of the
most lethal solid tumors with early metastasis and high resistance
to chemotherapy (19). Multiple
biochemical and molecular alterations, including genetic
alterations, epigenetic changes, redundancies and crosstalk of cell
signaling pathways, contribute to the development and
drug-resistance of pancreatic cancer (28). In addition, abnormal activation of
some pathways may be involved in resistance to pancreatic cancer
chemotherapy. Accumulating studies provide evidence that the
IL6/JAK2/STAT3 signaling pathway plays an important role in drug
resistance and is highly expressed in different types of
drug-resistant cancers, including pancreatic cancer. IL-6 is a
pleiotropic cytokine produced by a variety of cell types including
macrophages, fibroblasts and cancer cells. IL-6 binds to membrane
receptors (e.g., IL-6R) and then activates non-receptor tyrosine
kinases, including JAK2. These phosphotyrosine residues serve as a
docking site for the recruitment of STAT3 proteins, which act as
cellular mediators of IL-6 (39). STAT3 is an oncogene that is
activated by p-STAT3 in response to extracellular signals and JAK2
pathway activation (30). Once the
tyrosine is phosphorylated, two STAT3 monomers form dimers through
reciprocal phosphotyrosine-SH2 interactions, translocate to the
nucleus, where they bind to STAT3-specific DNA-response elements of
target genes, and induce gene transcription (31). Thus, IL-6 induces the activation of
its downstream cascade the JAK2/STAT3 signaling pathway, resulting
in tumorigenesis by regulating cell cycle progression, angiogenesis
and tumor cell evasion of the immune system (32–34).
Evidence reveals an important role of IL-6/JAK2/STAT3 signaling in
the development of pancreatic cancer (31).
It has been reported that PCCs were resistant to
HDAC inhibitor trichostatin A (TSA) (8). Aberrant activation of Wnt/β-catenin
signaling contributes to TSA resistance through
epithelial-mesenchymal transition. In the present study, we
initially observed increased IL-6 expression in human pancreatic
tissues and identified the crucial role of JAK2/STAT3 signaling in
TSA resistance in PCCs. The elevated levels of IL-6 were positively
associated with lymph node metastasis, tumor differentiation and
vascular infiltration of pancreatic cancer. TSA-resistant cells
exhibited significantly upregulated expression and phosphorylation
of STAT3 along with enhanced expression of c-Myc, c-Src, HIF-α, and
CCND1 as compared to TSA-nonresistant cells. Moreover, in
aggressive malignant pancreatic cancer cell lines, the
significantly elevated expression of IL-6 predicted a more
aggressive cell type and a poorer clinical outcome. On the basis of
these data, JAK2 could be considered as a potential target for
chemotherapy-resistant pancreatic carcinoma. Thus, targeted
inhibition of the over-activation of IL-6/JAK2/STAT3 signaling can
provide a strategy for the treatment of TSA resistance.
Tyrphostin B42 (AG490) is an inhibitor of epidermal
growth factor receptor (EGFR) by competing to binding sites with
receptor tyrosine kinases (RTKs) (35). Tyrphostin B42 has been reported to
have an inhibitory effect on the proliferation of many tumor cells
through IL-6/JAK2/STAT3 signaling (22–24). A
previous study revealed that in vitro AG-490 (60–100 µM)
blocked the constitutive activation of Stat3sm, and
inhibited spontaneous as well as interleukin 2-induced growth of
mycosis fungoides tumor cells (36). In vivo, combined therapy with
AG-490 and IL-12 induced greater antitumor effects than either
agent alone in a murine myeloma tumor model (37). In the present study, we found that
tyrphostin B42 (30–60 µM) inhibited the proliferation of PCCs in a
dose-dependent manner. The inactivation of IL-6/JAK2/STAT3
signaling was responsible for tyrphostin B42-mediated inhibition of
proliferation of PCCs. Inactivated IL-6/JAK2/STAT3 signaling
resulted in the imbalance of proliferation- and
apoptosis-associated gene expression. Furthermore, tyrphostin B42
also inhibited the expression of c-Src mRNA in non-resistant PCCs,
indicating that tyrphostin B42 not only attenuates TSA-mediated
resistance, but also has a therapeutic potential for pancreatic
cancer.
However, an evident limitation in the present study
is that it did not build TSA-resistant tumor animal models to
evaluate the protective effects of tyrphostin B42 in vivo.
In addition, tyrphostin B42 treatment at low concentrations (less
than 10 µM) did not exert an inhibitory effect on the expression of
downstream target genes of IL-6/JAK2/STAT3 signaling, revealing
that other mechanisms appear to be involved in the regulation of
proliferation and apoptosis of TSA-mediated resistance in PCCs, and
thus it is important for tyrphostin B42 to have an appropriate
dose/concentration used in vivo.
In conclusion, our in vitro experiments
indicated that aberrant activation of IL-6/JAK2/STAT3 signaling was
likely one of the main mechanisms triggering the resistance to TSA,
leading to over-proliferation and inhibition of apoptosis of PCCs
(Fig. 6). Tyrphostin B42 at certain
concentrations effectively attenuates TSA-mediated resistance in
PCCs by antagonizing IL-6/JAK2/STAT3 signaling. The present study
helps to better understand the TSA-resistance mechanism in
pancreatic cancer, and provide a theoretical basis for the
screening of antitumor drugs.
Acknowledgements
This study was supported by the Natural Science
Foundation of Zhejiang province, China (LY17H050005), the National
Natural Science Foundation of China (nos. 81570583 and 81572087),
and the Wenzhou Municipal Science and Technology Plan Project
(Y20150037).
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
FITC
|
fluorescein isothiocyanate
|
|
HDAC
|
histone deacetylase
|
|
JAK
|
Janus kinase
|
|
PCCs
|
pancreatic cancer cells
|
|
RT-qPCR
|
reverse-transcriptase quantitative
polymerase chain reaction
|
|
STAT
|
signal transducers and activators of
transcription
|
|
TSA
|
trichostatin A
|
References
|
1
|
Castellanos JA, Merchant NB and
Nagathihalli NS: Emerging targets in pancreatic cancer:
Epithelial-mesenchymal transition and cancer stem cells. Onco
Targets Ther. 6:1261–1267. 2013.PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ouaïssi M, Cabral S, Tavares J, da Silva
AC, Mathieu Daude F, Mas E, Bernard J, Sastre B, Lombardo D and
Ouaissi A: Histone deacetylase (HDAC) encoding gene expression in
pancreatic cancer cell lines and cell sensitivity to HDAC
inhibitors. Cancer Biol Ther. 7:523–531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Donadelli M, Costanzo C, Beghelli S,
Scupoli MT, Dandrea M, Bonora A, Piacentini P, Budillon A, Caraglia
M, Scarpa A and Palmieri M: Synergistic inhibition of pancreatic
adenocarcinoma cell growth by trichostatin A and gemcitabine.
Biochim Biophys Acta. 1773:1095–1106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu W, Li J, Wu S, Li S, Le L, Su X, Qiu
P, Hu H and Yan G: Triptolide cooperates with Cisplatin to induce
apoptosis in gemcitabine-resistant pancreatic cancer. Pancreas.
41:1029–1038. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim DH, Kim M and Kwon HJ: Histone
deacetylase in carcinogenesis and its inhibitors as anti-cancer
agents. J Biochem Mol Biol. 36:110–119. 2003.PubMed/NCBI
|
|
7
|
Zhang X, Jiang SJ, Shang B and Jiang HJ:
Effects of histone deacetylase inhibitor trichostatin A combined
with cisplatin on apoptosis of A549 cell line. Thorac Cancer.
6:202–208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang B, Zou Q, Sun M, Chen J, Wang T, Bai
Y, Chen Z, Chen B and Zhou M: Reversion of trichostatin A
resistance via inhibition of the Wnt signaling pathway in human
pancreatic cancer cells. Oncol Rep. 32:2015–2022. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen Z, Yang Y, Liu B, Wang B, Sun M,
Zhang L, Chen B, You H and Zhou M: Promotion of
metastasis-associated gene expression in survived PANC-1 cells
following Trichostatin a treatment. Anticancer Agents Med Chem.
15:1317–1325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taniguchi K and Karin M: IL-6 and related
cytokines as the critical lynchpins between inflammation and
cancer. Semin Immunol. 26:54–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Okitsu K, Kanda T, Imazeki F, Yonemitsu Y,
Ray RB, Chang C and Yokosuka O: Involvement of interleukin-6 and
androgen receptor signaling in pancreatic cancer. Genes Cancer.
1:859–867. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bharadwaj U, Marin-Muller C, Li M, Chen C
and Yao Q: Mesothelin overexpression promotes autocrine IL-6/sIL-6R
trans-signaling to stimulate pancreatic cancer cell proliferation.
Carcinogenesis. 32:1013–1024. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vera J, Rateitschak K, Lange F, Kossow C,
Wolkenhauer O and Jaster R: Systems biology of JAK-STAT signalling
in human malignancies. Prog Biophys Mol Biol. 106:426–434. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Quintás-Cardama A and Verstovsek S:
Molecular pathways: Jak/STAT pathway: Mutations, inhibitors, and
resistance. Clin Cancer Res. 19:1933–1940. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thoennissen NH, Iwanski GB, Doan NB,
Okamoto R, Lin P, Abbassi S, Song JH, Yin D, Toh M, Xie WD, et al:
Cucurbitacin B induces apoptosis by inhibition of the JAK/STAT
pathway and potentiates antiproliferative effects of gemcitabine on
pancreatic cancer cells. Cancer Res. 69:5876–8584. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gozgit JM, Bebernitz G, Patil P, Ye M,
Parmentier J, Wu J, Su N, Wang T, Ioannidis S, Davies A, et al:
Effects of the JAK2 inhibitor, AZ960, on Pim/BAD/BCL-xL survival
signaling in the human JAK2 V617F cell line SET-2. J Biol Chem.
283:32334–32343. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bai J, Sui J, Demirjian A, Vollmer CM Jr,
Marasco W and Callery MP: Predominant Bcl-XL knockdown disables
antiapoptotic mechanisms: tumor necrosis factor-related
apoptosis-inducing ligand-based triple chemotherapy overcomes
chemoresistance in pancreatic cancer cells in vitro. Cancer Res.
65:2344–2352. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hodge DR, Hurt EM and Farrar WL: The role
of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer.
41:2502–2512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang R, Cheng L, Xia J and Wang Z, Wu Q
and Wang Z: Gemcitabine resistance is associated with
epithelial-mesenchymal transition and induction of HIF-1α in
pancreatic cancer cells. Curr Cancer Drug Targets. 14:407–417.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen H, Guan YL, Yuan G, Zhang Q and Jing
N: A perylene derivative regulates HIF-1 alpha and Stat3 signaling
pathways. Bioorg Med Chem. 22:1496–1505. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qin A, Yu Q, Gao Y, Tan J, Huang H, Qiao Z
and Qian W: Inhibition of STAT3/cyclin D1 pathway promotes
chemotherapeutic sensitivity of colorectal caner. Biochem Biophys
Res Commun. 457:681–687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soto-Cruz I, Rangel-Corona R,
Valle-Mendiola A, Moreno-Morales X, Santiago-Pérez R, Weiss-Steider
B and Cáceres-Cortés JR: The tyrphostin B42 inhibits cell
proliferation and HER-2 autophosphorylation in cervical carcinoma
cell lines. Cancer Invest. 26:136–144. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu Y, Zhang J, Wu J, Zhong S and Li H:
Inhibition of JAK2 Reverses paclitaxel resistance in human ovarian
cancer cells. Int J Gynecol Cancer. 25:1557–1564. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen B, Liu Y, Zhang Y, Li J, Cheng K and
Cheng L: IL-21 is positively associated with intervertebral disc
degeneration by interaction with TNF-α through the JAK-STAT
signaling pathway. Inflammation. 40:612–622. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
American Joint Committee on Cancer (AJCC)
TNM staging system, . 2013.American Cancer Society. http://www.cancer.org/cancer/pancreaticcancer/detailedguide/pancreatic-cancer-stagingApril
5–2016
|
|
26
|
Detre S, Saclani Jotti G and Dowsett MA: A
‘quickscore’ method for immunohistochemical semiquantitation:
Validation for oestrogen receptor in breast carcinomas. J Clin
Pathol. 48:876–878. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Silvestris N, Gnoni A, Brunetti AE,
Vincenti L, Santini D, Tonini G, Merchionne F, Maiello E, Lorusso
V, Nardulli P, et al: Target therapies in pancreatic carcinoma.
Curr Med Chem. 21:948–965. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Roxburgh CS and McMillan DC: Therapeutics
targeting innate immune/inflammatory responses through the
interleukin-6/JAK/STAT signal transduction pathway in patients with
cancer. Transl Res. 167:61–66. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim BH, Yi EH and Ye SK: Signal transducer
and activator of transcription 3 as a therapeutic target for cancer
and the tumor microenvironment. Arch Pharm Res. 39:1085–1099. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bowman T, Garcia R, Turkson J and Jove R:
STATs in oncogenesis. Oncogene. 19:2474–2488. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pop VV, Seicean A, Lupan I, Samasca G and
Burz CC: IL-6 roles-Molecular pathway and clinical implication in
pancreatic cancer-A systemic review. Immunol Lett. 181:45–50. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Miyamoto Y, Hosotani R, Doi R, Wada M, Ida
J, Tsuji S, Kawaguchi M, Nakajima S, Kobayashi H, Masui T, et al:
Interleukin-6 inhibits radiation induced apoptosis in pancreatic
cancer cells. Anticancer Res. 21:2449–2456. 2001.PubMed/NCBI
|
|
34
|
Fofaria NM and Srivastava SK: STAT3
induces anoikis resistance, promotes cell invasion and metastatic
potential in pancreatic cancer cells. Carcinogenesis. 36:142–150.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Caceres-Cortes JR: A potent anti-carcinoma
and anti-acute myeloblastic leukemia agent, AG490. Anticancer
Agents Med Chem. 8:717–722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nielsen M, Kaltoft K, Nordahl M, Röpke C,
Geisler C, Mustelin T, Dobson P, Svejgaard A and Odum N:
Constitutive activation of a slowly migrating isoform of Stat3 in
mycosis fungoides: Tyrphostin AG490 inhibits Stat3 activation and
growth of mycosis fungoides tumor cell lines. Proc Natl Acad Sci
USA. 94:pp. 6764–6769. 1997; View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Burdelya L, Catlett-Falcone R, Levitzki A,
Cheng F, Mora LB, Sotomayor E, Coppola D, Sun J, Sebti S, Dalton
WS, et al: Combination therapy with AG-490 and interleukin 12
achieves greater antitumor effects than either agent alone. Mol
Cancer Ther. 1:893–899. 2002.PubMed/NCBI
|