Introduction
Chronic myeloid leukemia (CML) is one of the most
prevalent types of myeloproliferative neoplasm, and its multidrug
resistance (MDR) is usually associated with a poor clinical outcome
(1,2). MDR1 and MDR-associated protein 1
(MRP1) belong to the protein family of ATP-binding cassette
transporters, which use the energy released by ATP hydrolysis to
bind drugs and export them from the cell (3,4).
Adriamycin-resistant CML K562 (K562/ADM) cells reportedly
overexpress MDR1 and MRP1 (5),
meaning that the resistance of this cell line is associated with
abnormalities in drug efflux. In the present study, we found that
the proliferation of K562/ADM cells was significantly inhibited
upon knockdown of the Tribbles homolog 2 (TRIB2) gene, compared
with that noted in the untreated cells. The reason for this growth
inhibition in the resistant cell line may be associated with cell
drug-resistance reversal. It is known that TRIB2 is expressed in
mammals. TRIB protein family members encode pseudo-kinase proteins
that are highly conserved in evolution, and act as adaptors in
signaling pathways for important cellular processes (6,7).
Previous studies have focused on the pathological role of TRIB2 in
various diseases, including CML, and metabolic and neurological
diseases, in which it has been identified as a critical signaling
modulator and mediator (8,9). Related reports suggest that TRIB2
overexpression can indeed promote tumor resistance by activating
relevant cell pathways (10).
However, little is known concerning the effects of TRIB2 gene
knockdown on drug-resistant proteins.
Previous studies have found that the downregulation
of drug-resistance proteins may partly depend on inhibition of the
ERK pathway in cancer (11–13). In the present study, we explored
changes in the ERK signaling pathway after knockdown of the TRIB2
gene in K562/ADM cells. The aim of the present study was to explore
the effect of the downregulation of TRIB2 expression on the
chemotherapy resistance and proliferation of K562/ADM cells. The
results provide a novel basis for the treatment of clinical drug
resistance mechanisms, and potential routes for therapeutic
strategies in CML.
Materials and methods
Cell lines and cell culture
Human CML K562 cells and the MDR sub-cell line
K562/ADM cells were obtained from the Institute of Medical
Molecular Genetics of Binzhou Medical University (Yantai, China).
The cells were maintained in RPMI-1640 basic medium (1X)
supplemented with 10% fetal bovine serum (FBS; both from Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in a
humidified atmosphere containing 5% CO2. K562/ADM cells
were maintained in the presence of 7 µM ADM (Wanle, Shenzhen,
China). Prior to the experiment, the cells were cultured in
drug-free medium for 1 week. In addition, K562/ADM and
K562/ADM-TRIB2 cells were pretreated for 24 h with 10 µM U0126
(Shanghai Selleck Chemicals Co., Ltd., Shanghai, China) in order to
study the ERK pathway.
Cell transfection
A pair of vector sequences targeting TRIB2 were
generated and named pGPU6/GFP/Neo-shNC, pGPU6/GFP/Neo-TRIB2 and
pGPU6/GFP/Neo-TRIB2-1. Firstly, cells were seeded at a density of
0.9–4×105/well into a 6-well plate with 1.6 ml 10%
FBS-containing RPMI-1640 medium the day before transfection. After
incubation for 24 h, cell confluence was 40–80% on the day of
transfection. DNA (0.4 µg) was dissolved in TE buffer and added to
3.2 µl Enhancer; 10 µl Effectene Transfection reagent (Qiagen China
Co., Ltd., Shanghai, China) was added after 5 min of incubation.
After incubating for a further 6–18 h, the medium was removed and
replaced with fresh medium. Transfection efficiency was determined
using fluorescence microscopy and the non-transfected K562/ADM
cells were used as the control. After 48 h of transfection, G418
(500 ng/ml; Thermo Fisher Scientific, Inc.) was added to the
medium. Stable positive clones were obtained after 4 weeks of
selection. Administration of G418 was halted at 2 weeks prior to
the experiment.
Cell proliferation assay
K562/ADM cells (8×103) with or without
knockdown of TRIB2 were divided into 0, 24, 48 and 72 h
experimental groups, seeded into 96-well plates and incubated with
medium containing 10% FBS. Each group consisted of five parallel
wells, with parental K562/ADM cells serving as the control. Then,
10 µl CCK-8 (Dojindo Molecular Technologies, Inc., Shanghai, China)
solution was added to each well and incubated for a further 4 h.
Then, the absorbance at 450 nm was measured using a fluorescence
spectrophotometer (F-7000; Hitachi, Ltd., Tokyo, Japan). The
absorbance values were collected for processing using GraphPad
Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA).
CCK-8 analysis of the half maximal
inhibitory concentration (IC50)
The CCK-8 was used to determine the inhibition ratio
of cells incubated with various concentrations of ADM (0–16 µM for
the K562 cells; 15–120 µM for the K562/ADM cells, K562/ADM-Con
cells or K562/ADM-TRIB2 cells). After dilution in RPMI-1640 medium
for 24 h, 10 µl CCK-8 solution was added to each well and incubated
for 4 h. The absorbance was then measured at 450 nm with a
microplate reader. A blank well containing only medium and ADM was
used as a control. The concentration of ADM that resulted in the
IC50 was calculated. Resistant fold=IC50
K562/ADM/IC50 K562. Reversal fold=IC50
K562/ADM-TRIB2 group/IC50 control.
Flow cytometry
K562, K562/ADM, and cells transfected with
pGPU6/GFP/Neo-TRIB2 and pGPU6/GFP/Neo-shNC were seeded into a
6-well plate at a density of 5×105 cells/well. The
non-transfected group was defined as the blank control. A total of
5 µM ADM was applied to the wells. After incubation for 1 h, the
cells were harvested via centrifugation and washed twice with
ice-cold PBS. The cell-associated mean fluorescence intensity (MFI)
of ADM was detected using a FACSCalibur flow cytometer (FACS FC500;
Beckman Coulter, Inc., Brea, CA, USA), with excitation/emission
wavelengths of 485/580 nm.
Reverse transcription-polymerase chain
reaction (RT-PCR) and RT-quantitative PCR (RT-qPCR)
According to the manufacturer's instructions, total
RNA was isolated using TRIzol reagent (Thermo Fisher Scientific,
Inc.) and assessed at a ratio of A260/A280 by spectrophotometry
(Nano Drop 2000; Nano Drop Technologies, Inc., Wilmington, DE,
USA). RNA (1–2 µg) was used to synthesize the first-strand cDNA.
The primers used in this experiment were designed and synthesized
by Shanghai GenePharma, Co., Ltd. (Shanghai, China) and are
presented in Table I. Prime Script™
RT reagent kit with gDNA Eraser (Takara Bio, Inc., Otsu, Japan) was
used to perform the RT reaction. Then, Premix Taq™ (Takara Bio,
Inc.) was used to perform PCR amplification on the Eppendorf
Mastercycler Personal system (Eppendorf China Ltd., Hong Kong,
China). The reaction system contained forward primer, reverse
primer, Premix Taq and template cDNA. The PCR products were
separated on 1% agarose gels (Takara Bio, Inc.), stained with G-Red
nucleic acid dyes (1:10,000; BioTeke Corporation, Beijing, China).
The images were captured with a Tanon gel imaging system (Tanon,
Shanghai, China). The parental non-transfected K562/ADM cells were
used as the blank control. GAPDH served as an internal standard for
quality control and quantification of target genes.
 | Table I.Primers used in reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers used in reverse
transcription-quantitative polymerase chain reaction.
| Gene | Primer
sequence | Product length
(bp) |
|---|
| MDR1 | Forward:
5′-GGAGCCTACTTGGTGGCACATAA-3′ | 121 |
|
| Reverse:
5′-TGGCATAGTCAGGAGCAAATGAAC-3′ |
|
| MRP1 | Forward:
5′-CAGCCCTTCCTGACAAGCTA-3′ | 133 |
|
| Reverse:
5′-GTGGCCTCATCCAACACAAG-3′ |
|
| TRIB2 | Forward:
5′-CTCCGAACTTGTCGCATTG-3′ | 233 |
|
| Reverse:
5′-CACATAGGCTTTGGTCTCAC-3′ |
|
| GAPDH | Forward:
5′-CAGCCCTTCCTGACAAGCTA-3′ | 133 |
|
| Reverse:
5′-GTGGCCTCATCCAACACAAG-3′ |
|
RT-qPCR was performed with a Step One™ Real-Time PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.) and
SYBR Premix Ex Taq™ (Takara Bio, Inc.). The reaction system of PCR
contained SYBR Premix Ex Taq™, the forward primer, the reverse
primer, template cDNA and nuclease-free distilled water. The
results were calculated using the 2−ΔΔCq value.
Western blot analysis
Cells cultured in 6-well plates were harvested and
washed with cold PBS three times. A total of 120 µl RIPA lysis
buffer (Beyotime Institute of Biotechnology, Shanghai, China) was
added to extract the proteins. Protein samples were separated via
10 or 6% SDS-PAGE (Beyotime Institute of Biotechnology) with a
constant voltage of 80 V for 0.5 h, which was then switched to 120
V for a further 1 h. Proteins were then transferred onto
polyvinylidine difluoride membranes (EMD Millipore, Billerica, MA,
USA). The membranes were blocked with 5% skimmed dry milk in 1X
Tris-buffered saline with Tween-20 (pH 8.0) at room temperature for
2 h, and then incubated overnight at 4°C with six styles of primary
antibodies respectively. The primary antibodies were rabbit
polyclonal anti-TRIB2 (1:500; cat. no. 204119; Abcam, Cambridge,
UK), anti-MDR1 (1:500; cat no. 0563R; BIOSS, Beijing, China),
anti-MRP1 (1:500; cat. no. 0657R; BIOSS), anti-STAT3 (1:500; cat.
no. 1141R; Bioworld Technology, Inc., Nanjing, China), anti-p-ERK
(1:500, cat. no. 5016; Bioworld Technology, Inc.) and anti-GAPDH
(1:1,000; cat. no. AP0063; Bioworld Technology, Inc.). Following
this, the membranes were incubated with a horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G (1:5,000;
cat no. 13278; Bioworld Technology, Inc.) for 2 h. Finally, images
were captured using a FluorChem FC2 gel imaging system (Protein
Simple, San Jose, CA, USA). The intensity of each band was
normalized to GAPDH in the respective lane, and the K562/ADM cells
were as control.
Data analysis
Statistical analyses were performed using SPSS 21.0
software (IBM Corp., Armonk, NY, USA). Independent two-sample
t-tests were used to analyze differences between two groups.
One-way analysis of variance (ANOVA) was used to analyze
differences among three or more groups. A post-hoc test of ANOVA
was conducted by performing a Tukey's test. Data are expressed as
the mean ± standard deviation. Statistical significance was
accepted at P<0.05.
Results
Calculation of the level of ADM
drug-resistance in the K562 and K562/ADM cell groups
The IC50 was calculated by performing a
CCK-8 spectrophotometric assay. The data from the K562/ADM cells
were markedly higher compared with K562 cells, with a resistance
ratio of 26.22 for K562/ADM to K562 cells (Table II). There was a significant
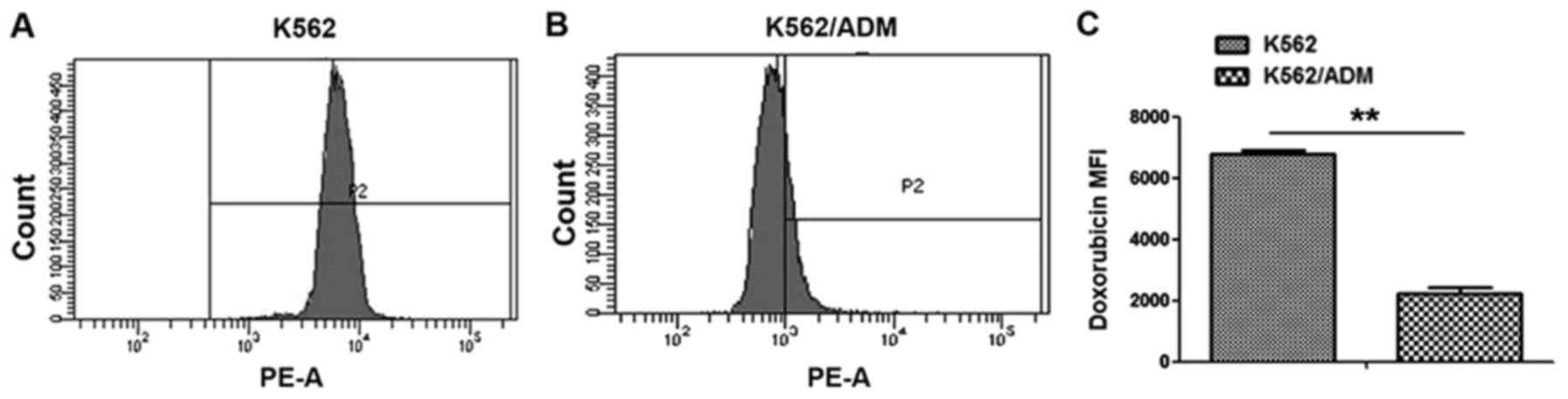
difference in intracellular ADM accumulation between the K562/ADM
group and the K562 group (Fig. 1).
All values were statistically significant (P<0.01).
| Table II.IC50 values for K562/ADM
cells and K562 cells toward ADM by CCK-8 assay (means ± SD;
n=5). |
Table II.
IC50 values for K562/ADM
cells and K562 cells toward ADM by CCK-8 assay (means ± SD;
n=5).
|
| IC50 ±
SD (µM) |
|
|---|
|
|
|
|
|---|
| Treatment | K562 | K562/ADM | RF |
|---|
| Adriamycin | 3.219± 0.921 |
84.801±0.0183a | 26.22 |
Determination of relative protein
expression in K562 and K562/ADM cells
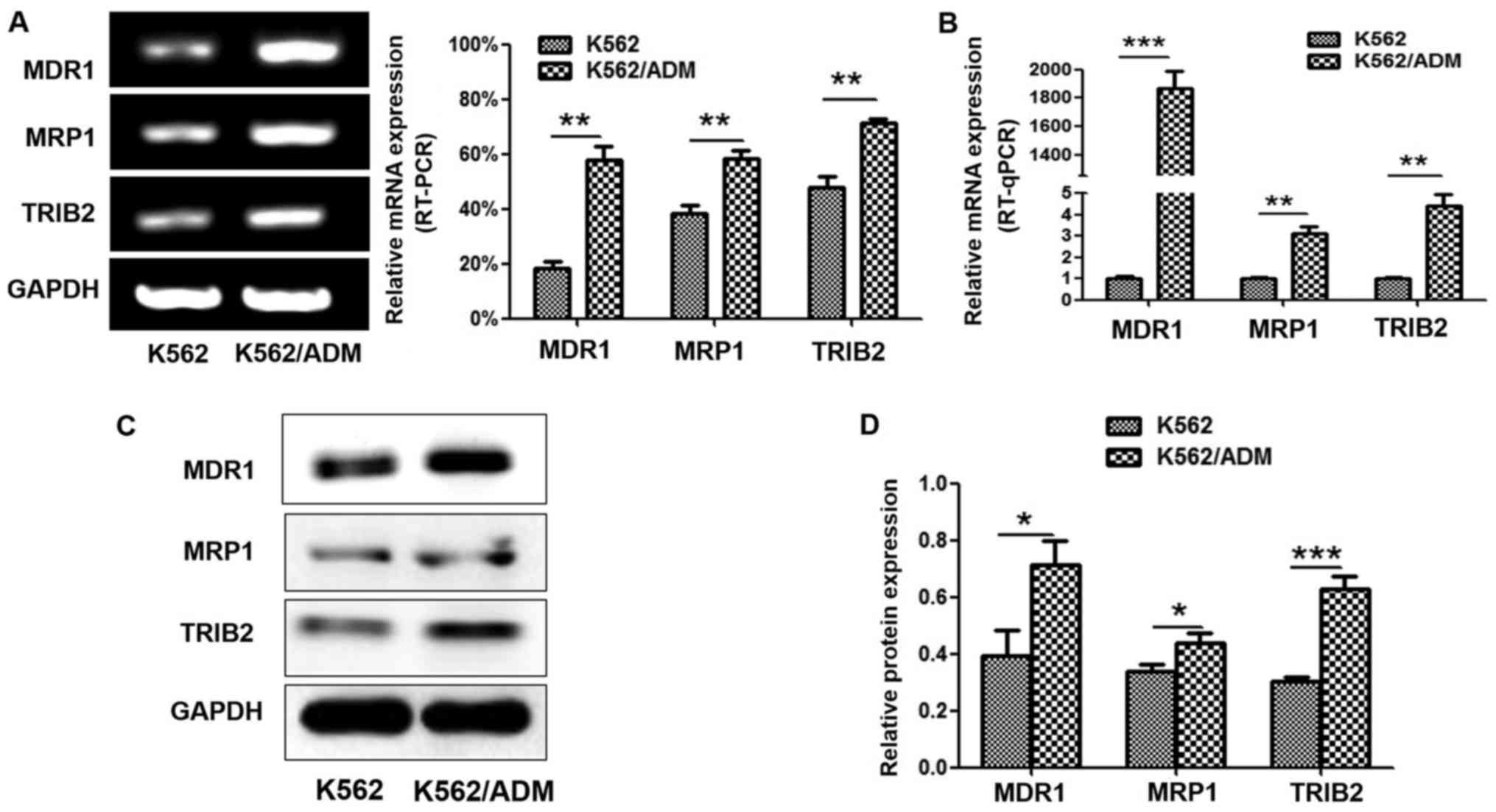
We evaluated MDR1, MRP1 and TRIB2 expression in the
non-resistant K562 cells and the ADM-resistant K562/ADM cells. The
results indicated that the K562 and K562/ADM cells both showed high
expression levels of all three proteins. The K562/ADM cells
exhibited higher mRNA expression of TRIB2, MDR1 and MRP1, compared
with the K562 cells (Fig. 2A and
B). Western blot analyses revealed the same expression trends
at the protein level (Fig. 2C and
D).
Construction of a stable TRIB2
transfection cell system
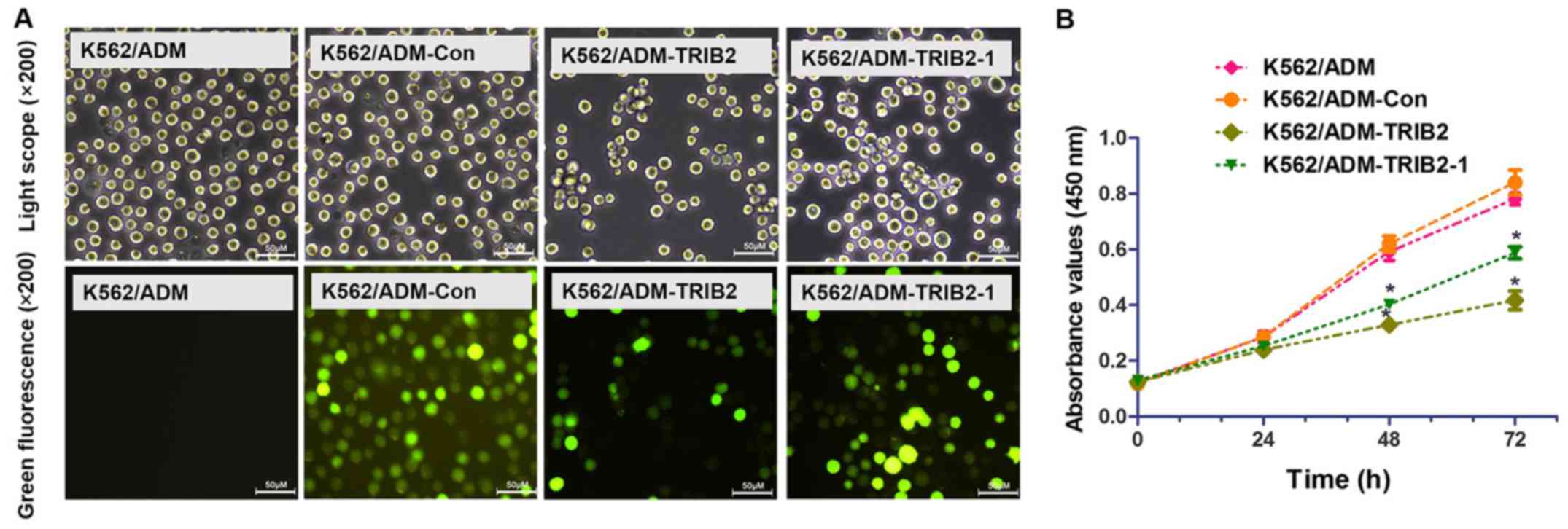
To evaluate the functional changes in the K562/ADM
cells following silencing of TRIB2, we transfected
pGPU6/GFP/Neo-shNC, pGPU6/GFP/Neo-TRIB2 and pGPU6/GFP/Neo-TRIB2-1
(Table III) into K562/ADM cells,
with untreated K562/ADM cells serving as the control. Green
fluorescence is only detected in cells successfully transfected
with GFP. Therefore, cells with green fluorescence indicate a high
efficiency of transfection. After 4 weeks of G418 selection, we
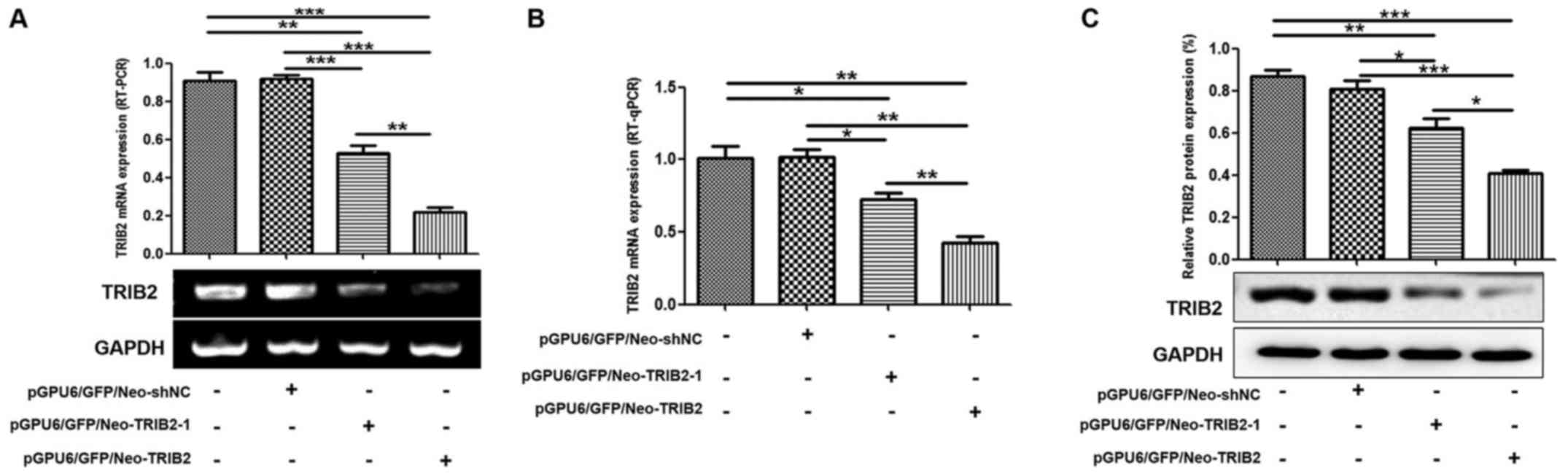
successfully obtained stable positive clones (Fig. 3A). RT-PCR, RT-qPCR and western blot
analyses revealed that TRIB2 expression was markedly decreased in
the K562/ADM-TRIB2 group and the K562/ADM-TRIB2-1 group at the
transcription and translation levels, compared with that in the
K562/ADM group. TRIB2 expression in the K562/ADM-Con group was not
significantly different from the negative control group (Fig. 4).
| Table III.Construction of the expression vector
and its interference sequence (5′-3′). |
Table III.
Construction of the expression vector
and its interference sequence (5′-3′).
| Vector
construction | Interference
sequence |
|---|
|
pGPU6/GFP/Neo-shNC |
GTTCTCCGAACGTGTCACGT |
|
pGPU6/GFP/Neo-TRIB2 |
TAGCGAGATATGGGAGATC |
|
pGPU6/GFP/Neo-TRIB2-1 |
CTTGTCGCATTGCGTTTCTTG |
TRIB2 knockdown inhibits cell
proliferation
A CCK-8 assay was performed to evaluate cell
proliferation (Fig. 3B). According
to the growth curve, we found that the proliferation of cells
treated with pGPU6/GFP/Neo-TRIB2 and pGPU6/GFP/Neo-TRIB2-1 was
markedly inhibited, while cells transfected with pGPU6/GFP/Neo-shNC
exhibited no significant difference. The results also showed that
pGPU6/GFP/Neo-TRIB2 was more effective than pGPU6/GFP/Neo-TRIB2-1.
Then, pGPU6/GFP/Neo-TRIB2 was used to explore the effect of the
downregulation of TRIB2 expression on the chemotherapy resistance
and proliferation of K562/ADM cells.
TRIB2 knockdown decreases
IC50 and increases intracellular ADM accumulation
The IC50 value was calculated by
performing a CCK-8 spectrophotometric assay. The IC50 of
the K562/ADM-Con group was not observed to be significantly
different compared with the K562/ADM cells, while a reduction in
the IC50 value was obvious in the K562/ADM-TRIB2 group,
with a reversal fold of 12.12 (Table
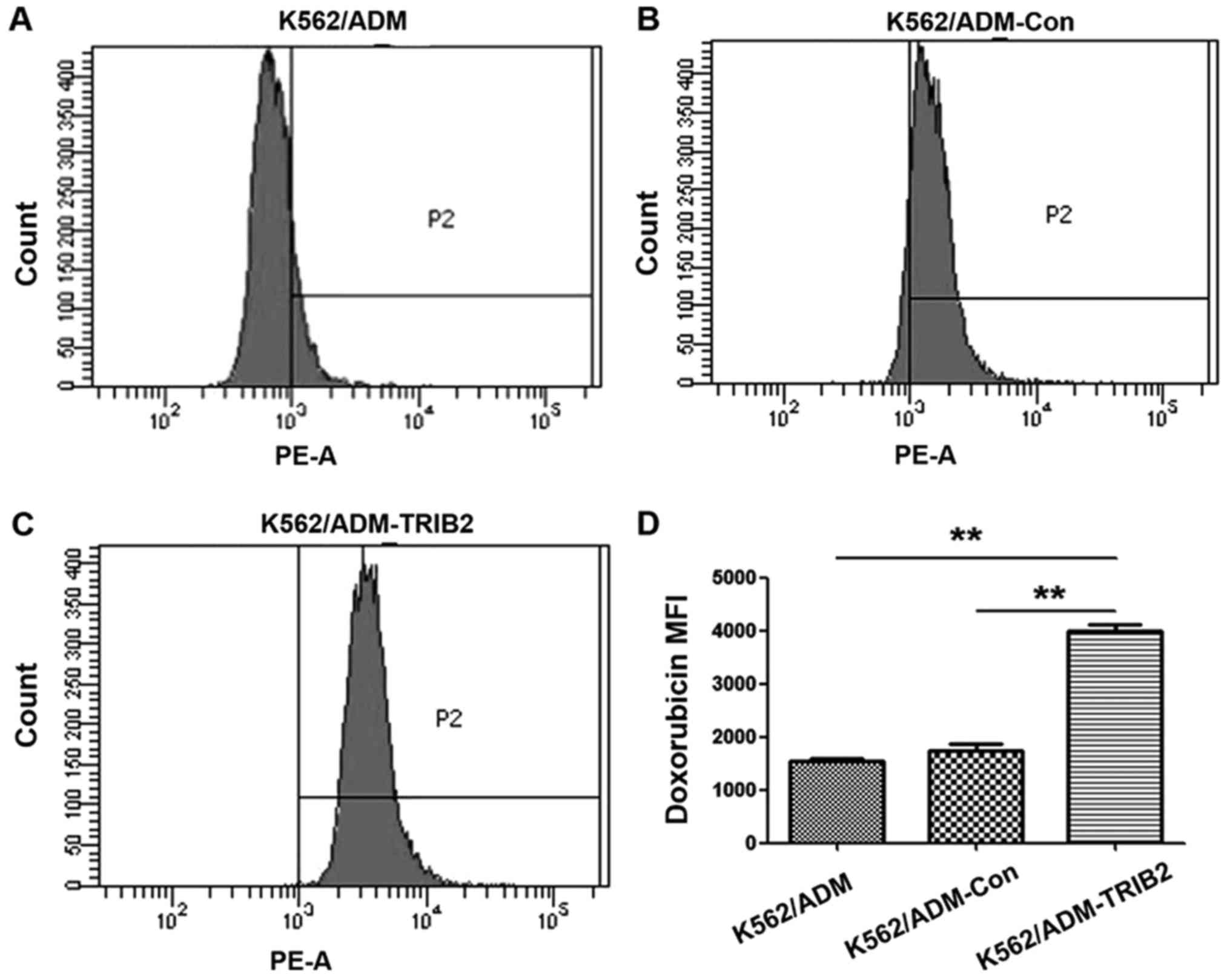
IV). The intracellular ADM accumulation in K562/ADM-TRIB2 cells
was considerably higher than that in the K562/ADM cells and
K562/ADM-Con cells (Fig. 5). All
values were statistically significant (P<0.01).
| Table IV.IC50 values of K562/ADM
cells, K562/ADM-Con and K562/ADM-TRIB2 cells toward Adriamycin by
CCK-8 assay (means ± SD of triplicate experiments). |
Table IV.
IC50 values of K562/ADM
cells, K562/ADM-Con and K562/ADM-TRIB2 cells toward Adriamycin by
CCK-8 assay (means ± SD of triplicate experiments).
|
| IC50 ±
SD (µM) |
|
|---|
|
|
|
|
|---|
| Treatment | K562/ADM | K562/ADM-Con | K562/ADM-TRIB2 | RF |
|---|
| Adriamycin |
84.012±0.037 |
75.946±0.134 |
39.041±0.321a | 12.12 |
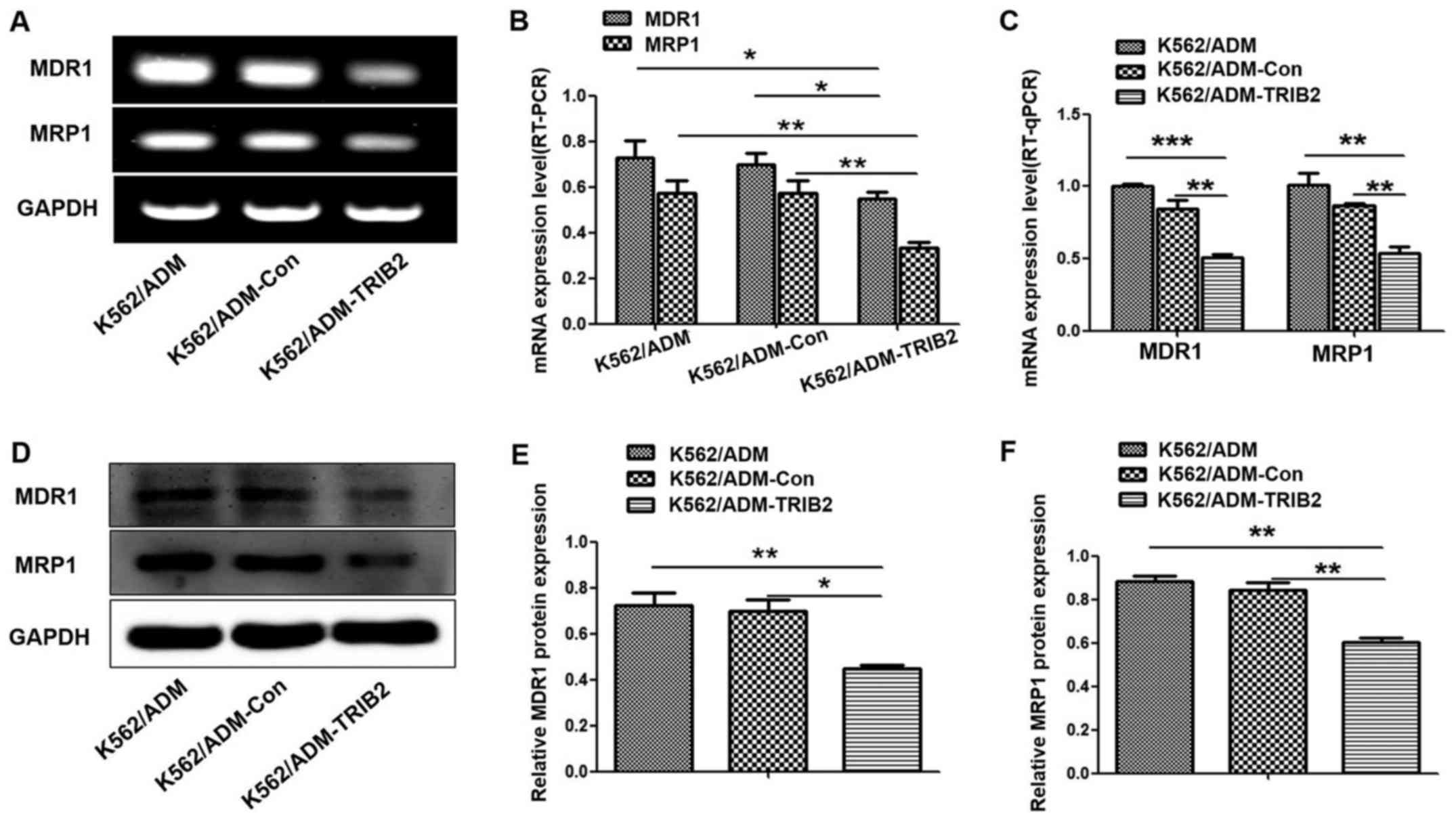
Decreased expression of MDR1 and MRP1
by TRIB2 knockdown
MDR1 and MRP1 are ABC transporters overexpressed in
many drug-resistant tumor cells, which contribute to the
development of MDR. Therefore, we assessed whether TRIB2 knockdown
could influence the expression of MDR1 and MRP1. Notably, western
blotting, RT-PCR and RT-qPCR analyses (Fig. 6) illustrated that the expression
levels of MDR1 and MRP1 were lower in the K562/ADM-TRIB2 cells,
compared with levels in the control cells. This indicated that
TRIB2 may be involved in key steps of MDR development in CML.
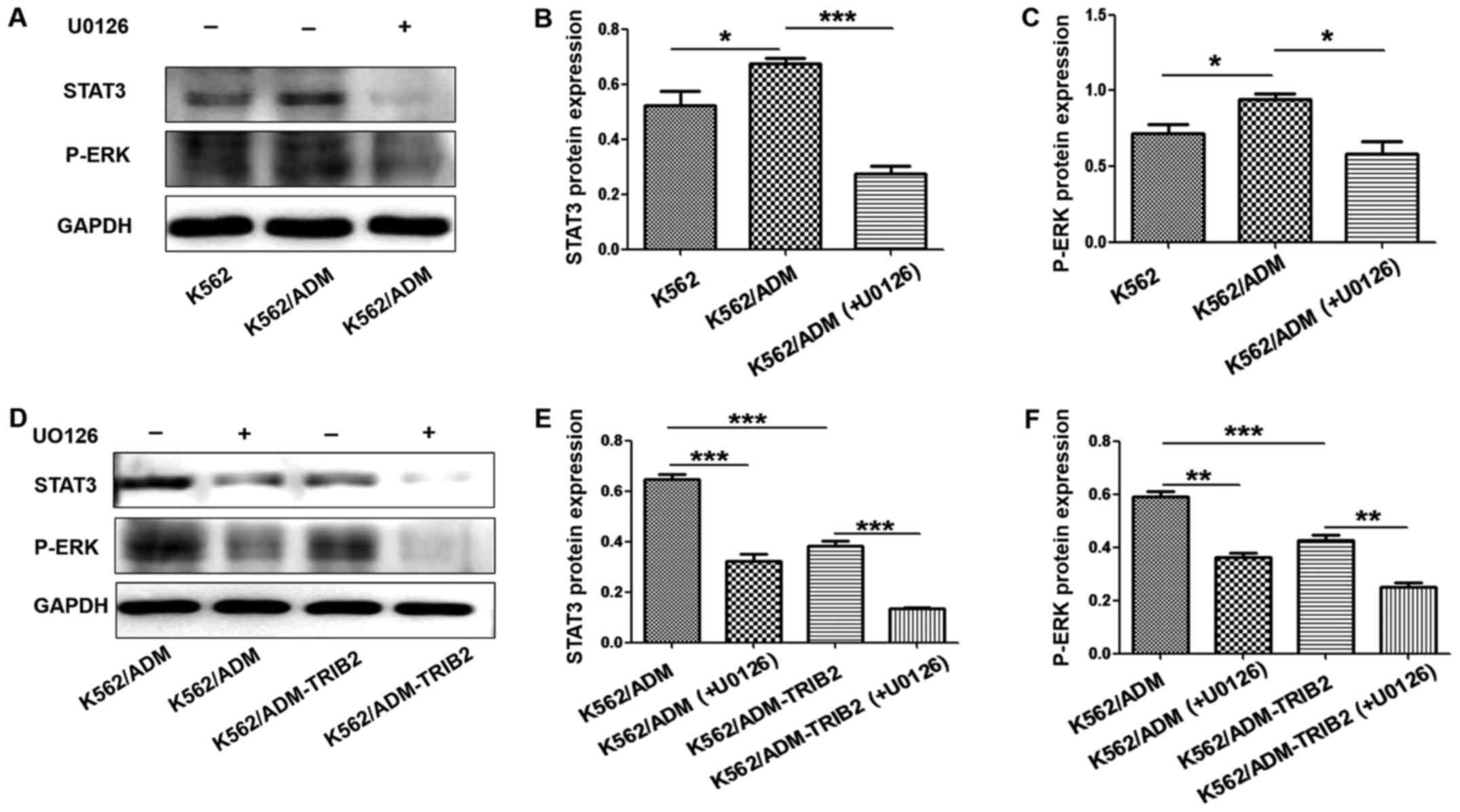
Inhibition of the ERK pathway in
K562/ADM-TRIB2 cells
The expression of p-ERK and STAT3 in K562/ADM cells
was higher compared with that noted in the K562 cells. However,
after treatment with U0126, p-ERK and STAT3 expression was
significantly decreased (Fig.
7A-C). These results suggested that expression of the ERK
pathway was active in CML K562/ADM cells and U0126 could
specifically block this pathway. Furthermore, the expression of
p-ERK and STAT3 was clearly downregulated in the K562/ADM-TRIB2
cells. After treatment with U0126, the expression of p-ERK and
STAT3 in K562/ADM-TRIB2 cells was significantly decreased,
indicating that TRIB2 knockdown may act by blocking ERK pathway
activity (Fig. 7D-F). These results
suggest that downregulation of TRIB2 affects cell resistance by
altering the expression of p-ERK and STAT3 in CML K562/ADM
cells.
Discussion
MDR is a major clinical obstacle for effective tumor
chemotherapy, and its impact on chemotherapy in the clinic is
worsening. Therefore, novel targeted therapeutic approaches are
being explored in order to increase the efficacy of chemotherapy
against blood cancers and diseases (14). We observed that TRIB2, MDR1 and MRP1
expression levels were higher in K562/ADM cells, compared with
levels in the K562 cells, at both the protein and mRNA levels. This
indicated that TRIB2 was involved in the development of MDR in
K562/ADM cells.
Numerous studies have shown that the HOX gene family
plays an important role in tumor resistance. Knockdown of HOXA5,
HOXA10 or HOXB4 was found to reverse multidrug resistance of human
CML K562/ADM cells (2,15–17).
Related studies have also shown that miR-3142 and miR-146a are
overexpressed in K562/ADM cells compared with that noted in K562
cells, which promotes normal cell proliferation and enhanced
resistance to ADM in vitro (18,19).
Similarly, knockdown of miR-224 and let-7i was shown to reverse the
MDR of human CML K562/ADM cells (20). It has been reported that TRIB2
expression is significantly increased in tumor tissues from
patients, correlating with the increased phosphorylation of AKT,
FOXO3a, MDM2 and C/EBPα (10,21).
We found that the knockdown of TRIB2 could decrease MDR1 and MRP1
activity in human CML K562/ADM cells, and also reverse
intracellular drug accumulation. In the present study, we first
focused on evaluating the association between TRIB2 knockdown and
the expression of resistance proteins. Our results indicated that
the expression levels of drug-resistant proteins were inhibited by
suppressing TRIB2, which reduced the efflux of intracellular ADM
and reversed cell resistance. In summary, TRIB2 repression could
partially reverse the MDR of K562/ADM cells by inhibiting cellular
efflux functions and downregulating the expression levels of MDR1
and MRP1, thus elevating intracellular chemotherapeutic
accumulation.
To further investigate the mechanism underlying the
role of TRIB2 knockdown in reducing cell resistance, we examined
the activity of the ERK pathway. ERK1 and ERK2 constitute the
ERK1/2 signal transduction pathway. This is mainly composed of the
RAS/RAF/MEK/ERK signal transduction cascade, which can be
stimulated by various external stimuli (22,23).
Expression of the ERK signal transduction pathway and drug efflux
proteins in hepatocellular carcinoma, gastric cancer and breast
cancer cells is reported to be significantly higher compared with
the corresponding controls (24–26).
Numerous studies have shown that excessive activation of ERK is
positively correlated with the presence of numerous resistant
tumors, the mechanism of which may be modulated by the
overexpression of resistance-related genes and proteins (27,28).
We detected that the expression levels of p-ERK and STAT3 in
K562/ADM cells were higher when compared with the levels in K562
cells. Meanwhile, knockdown of TRIB2 in normal K562/ADM cells
resulted in the reduced activity of p-ERK and STAT3. To test our
hypothesis, the ERK signal transduction pathway inhibitor U0126 was
used to downregulate ERK phosphorylation. U0126 is an inhibitor of
mitogen-activated protein kinase 1 and 2 (MEK1/2), which blocks
phosphorylation and activation of ERK1/2 (29–31).
In the present study, western blotting showed that p-ERK was
significantly inhibited in K562/ADM cells treated with U0126. This
indicated that the gene encoding ERK was involved in the effects of
TRIB2 knockdown.
Furthermore, this study investigated the ERK
downstream product STAT3, which correlates with cell proliferation
(32). STAT3-targeted therapy can
reverse drug resistance in CML (33). Our results showed that the activity
of STAT3 in K562/ADM cells following TRIB2 knockdown was also lower
compared with that noted in the control group. While these initial
findings are promising, more comprehensive and detailed studies
need to be conducted, including in vivo animal models and
more precise ERK1/2 pathway assays. Nevertheless, we demonstrated
that decreased expression of resistant proteins is associated with
inhibition of the ERK pathway through the knockdown of TRIB2,
thereby reversing cell MDR.
Acknowledgements
The present study was supported by the National
Natural Science Foundation (grant no. 31371321), the Shandong
Science and Technology Committee (grant nos. ZR2014HQ079,
ZR2014HL056 and ZR2013HL003), the Foundation of Shandong
Educational Committee (grant nos. J17KA121 and J13LE11) and Young
Backbone Teacher Development Support Project of Binzhou Medical
University.
Competing interests
The authors declare no competing interests.
Glossary
Abbreviations
Abbreviations:
|
CML
|
chronic myeloid leukemia
|
|
CCK-8
|
Cell Counting Kit-8
|
|
GFP
|
green fluorescent protein
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
SDS-PAGE
|
sodium dodecyl sulphate-polyacrylamide
gel electrophoresis
|
|
TRIB2
|
tribble homologue 2
|
|
MDR1
|
multidrug resistance 1
|
|
MRP1
|
multidrug resistance-associated
protein 1
|
References
|
1
|
Clarke CJ and Holyoake TL: Preclinical
approaches in chronic myeloid leukemia: From cells to systems. Exp
Hematol. 47:13–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cheng Z, Yang N, Liang W, Yan X, Li L and
Pan L: Effect of phosphatase and tensin homology deleted on
chromosome 10 (PTEN) gene transfection on reversal of multidrug
resistance in K562/ADM cells. Leuk Lymphoma. 53:1383–1389. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yin J and Zhang J: Multidrug
resistance-associated protein 1 (MRP1/ABCC1) polymorphism: From
discovery to clinical application. Zhong Nan Da Xue Xue Bao Yi Xue
Ban. 36:927–938. 2011.PubMed/NCBI
|
|
4
|
Reznicek J, Ceckova M, Ptackova Z,
Martinec O, Tupova L, Cerveny L and Staud F: MDR1 and BCRP
transporter-mediated drug-drug interaction between rilpivirine and
abacavir and effect on intestinal absorption. Antimicrob Agents
Chemother. 61:e00837–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen JR, Jia XH, Wang H, Yi YJ, Wang JY
and Li YJ: Timosaponin A-III reverses multi-drug resistance in
human chronic myelogenous leukemia K562/ADM cells via
downregulation of MDR1 and MRP1 expression by inhibiting PI3K/Akt
signaling pathway. Int J Oncol. 48:2063–2070. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liang KL, O'Connor C, Veiga JP, McCarthy
TV and Keeshan K: TRIB2 regulates normal and stress-induced
thymocyte proliferation. Cell Discov. 2:150502016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gilby DC, Sung HY, Winship PR, Goodeve AC,
Reilly JT and Kiss-Toth E: Tribbles-1 and −2 are tumour
suppressors, down-regulated in human acute myeloid leukaemia.
Immunol Lett. 130:115–124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yokoyama T and Nakamura T: Tribbles in
disease: Signaling pathways important for cellular function and
neoplastic transformation. Cancer Sci. 102:1115–1122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hannon MM, Lohan F, Erbilgin Y, Sayitoglu
M, O'Hagan K, Mills K, Ozbek U and Keeshan K: Elevated TRIB2 with
NOTCH1 activation in paediatric/adult T-ALL. Br J Haematol.
158:1–634. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hill R, Madureira PA, Ferreira B, Baptista
I, Machado S, Colaco L, Santos M, Liu NS, Dopazo A, Ugurel S, et
al: TRIB2 confers resistance to anti-cancer therapy by activating
the serine/threonine protein kinase AKT. Nat Commun. 8:146872017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen T, Wang C, Liu Q, Meng Q, Sun H, Huo
X, Sun P, Peng J, Liu Z, Yang X and Liu K: Dasatinib reverses the
multidrug resistance of breast cancer MCF7 cells to doxorubicin by
downregulating P-gp expression via inhibiting the activation of ERK
signaling pathway. Cancer Biol Ther. 16:106–114. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yuan WQ, Zhang RR, Wang J, Ma Y, Li WX,
Jiang RW and Cai SH: Asclepiasterol, a novel C21 steroidal
glycoside derived from asclepias curassavica, reverses tumor
multidrug resistance by down-regulating P-glycoprotein expression.
Oncotarget. 7:31466–31483. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sheng Y, You Y and Chen Y: Dual-targeting
hybrid peptide-conjugated doxorubicin for drug resistance reversal
in breast cancer. Int J Pharm. 512:1–13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jacobs RW, Awan FT, Leslie LA, Usmani SZ
and Ghosh N: The shrinking role of chemotherapy in the treatment of
chronic lymphocytic leukemia. Expert Rev Hematol. 9:1177–1187.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li N, Jia X, Wang J, Li Y and Xie S:
Knockdown of homeobox A5 by small hairpin RNA inhibits
proliferation and enhances cytarabine chemosensitivity of acute
myeloid leukemia cells. Mol Med Rep. 12:6861–6866. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H, Jia XH, Chen JR, Yi YJ, Wang JY,
Li YJ and XIE SY: HOXB4 knockdown reverses multidrug resistance of
human myelogenous leukemia K562/ADM cells by downregulating P-gp,
MRP1 and BCRP expression via PI3K/Akt signaling pathway. Int J
Oncol. 49:2529–2537. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yi YJ, Jia XH, Wang JY, Li YJ, Wang H and
Xie SY: Knockdown of HOXA10 reverses the multidrug resistance of
human chronic mylogenous leukemia K562/ADM cells by downregulating
P-gp and MRP-1. Int J Mol Med. 37:1405–1411. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao L, Shan Y, Liu B, Li Y and Jia L:
Functional screen analysis reveals miR-3142 as central regulator in
chemoresistance and proliferation through activation of the
PTEN-AKT pathway in CML. Cell Death Dis. 8:e28302017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu W, He J, Yang Y, Guo Q and Gao F:
Upregulating miR-146a by physcion reverses multidrug resistance in
human chronic myelogenous leukemia K562/ADM cells. Am J Cancer Res.
6:2547–2560. 2016.PubMed/NCBI
|
|
20
|
Zhou H, Li Y, Liu B, Shan Y, Li Y, Zhao L,
Su Z and Jia L: Downregulation of miR-224 and let-7i contribute to
cell survival and chemoresistance in chronic myeloid leukemia cells
by regulating ST3GAL IV expression. Gene. 626:106–118. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Keeshan K, He Y, Wouters BJ, Shestova O,
Xu L, Sai H, Rodriguez CG, Maillard L, Tobias JW, Valk P, et al:
Tribbles homolog 2 inactivates C/EBPα and causes acute myelogenous
leukemia. Cancer Cell. 10:401–411. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang S and Liu G: Targeting the
Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol Lett.
13:1041–1047. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guo Y, Ding Y, Zhang T and An H: Sinapine
reverses multi-drug resistance in MCF-7/dox cancer cells by
downregulating FGFR4/FRS2alpha-ERK1/2 pathway-mediated NF-kappaB
activation. Phytomedicine. 23:267–273. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Buonato JM and Lazzara MJ: ERK1/2 blockade
prevents epithelial-mesenchymal transition in lung cancer cells and
promotes their sensitivity to EGFR inhibition. Cancer Res.
74:309–319. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao YY, Yu L, Liu BL, He XJ and Zhang BY:
Downregulation of P-gp, Ras and p-ERK1/2 contributes to the arsenic
trioxide-induced reduction in drug resistance towards doxorubicin
in gastric cancer cell lines. Mol Med Rep. 12:7335–7343. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fan DP, Zhang YM, Hu XC, Li JJ and Zhang
W: Activation of AKT/ERK confers non-small cell lung cancer cells
resistance to vinorelbine. Int J Clin Exp Pathol. 7:134–143.
2013.PubMed/NCBI
|
|
28
|
Ochi N, Takigawa N, Harada D, Yasugi M,
Ichihara E, Hotta K, Tabata M, Tanimoto M and Kiura K: Src mediates
ERK reactivation in gefitinib resistance in non-small cell lung
cancer. Exp Cell Res. 322:168–177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Droebner K, Pleschka S, Ludwig S and Planz
O: Antiviral activity of the MEK-inhibitor U0126 against pandemic
H1N1v and highly pathogenic avian influenza virus in vitro and in
vivo. Antiviral Res. 92:195–203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shukla V, Coumoul X, Wang RH, Kim HS and
Deng CX: RNA interference and inhibition of MEK-ERK signaling
prevent abnormal skeletal phenotypes in a mouse model of
craniosynostosis. Nat Genet. 39:1145–1150. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tong Y, Huang H and Pan H: Inhibition of
MEK/ERK activation attenuates autophagy and potentiates
pemetrexed-induced activity against HepG2 hepatocellular carcinoma
cells. Biochem Biophys Res Commun. 456:86–91. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gu FM, Li QL, Gao Q, Jiang JH, Huang XY,
Pan JF, Fan J and Zhou J: Sorafenib inhibits growth and metastasis
of hepatocellular carcinoma by blocking STAT3. World J
Gastroenterol. 17:3922–3932. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gleixner KV, Schneeweiss M, Eisenwort G,
Berger D, Herrmann H, Blatt K, Greiner G, Byrgazov K, Hoermann G,
Konopleva M, et al: Combined targeting of STAT3 and STAT5: A novel
approach to overcome drug resistance in chronic myeloid leukemia.
Haematologica. 102:1519–1529. 2017. View Article : Google Scholar : PubMed/NCBI
|