Introduction
Colorectal cancer remains one of the most prevalent
malignancies. While primary tumors are often resectable leading to
a better prognosis, the presence of metastatic lesions represent
the worst scenario medicine has to cope with. Colorectal cancer
metastasizes mainly to the liver and almost 70% of patients with
this cancer present distant invasion at the time of diagnosis. The
mechanisms leading to metastatic formation and growth remain poorly
characterized, especially those linked with stromal cells, known to
facilitate tumor cell escape from the immune system and support
tumor growth (1,2). Therefore, a better understanding of
the tumor microenvironment is required for a wider comprehension of
metastatic disease. One of the receptors expressed in stromal
cells, along with tumor cells, is CXCR4, one of the two known
receptors for CXCL12, also known as stromal cell-derived factor 1
(SDF-1) (3).
CXCR4 has been described to be upregulated in human
melanoma metastasis or colorectal cancer lymph node and liver
metastases and has been linked with poor survival (4–6). This
receptor has been shown to mediate different pro-metastatic events
in tumor cells in vitro, such as invasion and migration and
cancer cell extravasation in vivo (7–9).
Interestingly, this receptor is not only expressed by tumor cells
but also has been found in hepatic stellate cells (HSCs), liver
resident cells. Under inflammatory conditions, these cells become
activated and trans differentiate into myofibroblast-like cells,
supporting fibrosis, liver cancer and liver metastasis (10–12).
In the liver, HSCs are the main population of cancer-associated
fibroblasts (CAFs), which are the main component of the tumor
microenvironment (13) and arise as
a modulator of the tumor response. Intriguingly, tumor/fibroblast
interaction has been postulated as a relevant event during the
progression of cancer with the CXCR4/CXCL12 chemokine axis one of
the main catalysts of malignancy (14). Furthermore, HSCs, along with liver
sinusoidal endothelial cells (LSECs), are one of the main sources
of CXCL12 in the liver where they mediate not only the recruitment
of CXCR4-expressing tumor cells, but also CXCR4-expressing immune
cells.
During cancer development and metastasis formation,
the cytotoxicity against cancer cells is compromised due to immune
suppression. This pathological process is in part orchestrated by
myeloid-derived suppressor cells (MDSCs), CXCR4-expressing immature
myeloid cells able to suppress the cytotoxic capacity of T
lymphocytes (15). During liver
metastasis, these cells are recruited into the organ (16) from the circulation leading to
uncontrolled tumor growth and disease progression. In fact, this
chemokine and its CXCR4 receptor seem to be relevant for tumor
progression, as reported through the reduction of CXCL12
availability and CXCR4 blockage within the tumor microenvironment,
reducing liver metastasis in nude mice (17). Moreover, CXCR4 blockage has been
confirmed to be effective for reducing in vivo lung cancer
metastasis (18), pointing out the
interaction of CXCR4 with its lig and CXCL12 as a mediator in this
process. However, the cellular and molecular mechanisms by which
CXCR4 interaction with CXCL12 drives disease progression remain
poorly understood, especially when it comes to stromal cells.
Therefore, the present study aimed to investigate
the efficacy of treatment with the CXCR4 antagonist AMD3100 in an
orthotopic model of colorectal cancer liver metastasis and the
cellular mechanisms involved in the processes disrupted by this
antagonist.
Materials and methods
Animals
Six-week-old male Balb/c mice were obtained from
Janvier Labs (France). Housing, care, and experimental conditions
were carried out in conformity with institutional guidelines and
national and international laws for experimental animal care. The
animals were fed a standard chow and had access to water ad
libitum. All the proceedings were approved by the Basque Country
University Ethics Committee (CEID) in accordance with
institutional, national and international guidelines regarding the
protection and care of animals used for scientific purposes.
Cell lines
All in vitro and in vivo experiments
were conducted using the murine C26 colon adenocarcinoma (C26) cell
line (also known as MCA-26, CT-26) syngeneic with Balb/c mice and
purchased from ATCC (LGC Standards S.L.U. Barcelona, Spain). Cells
were cultured in RPMI-1640 medium supplemented with 10%
heat-inactivated fetal bovine serum (FBS), penicillin (10,000
U/ml), streptomycin (10,000 µg/ml) and amphotericin B (25 µg/ml)
all purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Primary murine hepatic stellate cell
isolation and culture
HSCs were isolated as previously described with some
modifications (19,20). Briefly, the liver was perfused with
collagenase from Clostridium histolyticum (Sigma-Aldrich, St.
Louis, MO, USA) and the resulting cell suspension was subjected to
isopycnic centrifugation through a Percoll gradient (GE Healthcare,
Chicago, IL, USA). The fraction enriched in HSCs was cultured in
culture medium without serum supplemented with antibiotics and
antimycotics incubated at 37°C in 5% CO2 overnight
before the experimental procedures.
Preparation of conditioned culture
medium
Tumor cells were seeded at a concentration of
5×104 cells/cm2 and cultured overnight in
RPMI-1640 supplemented with 10% FBS and antibiotics. Then, the
culture medium was substituted with fresh medium supplemented with
1% FCS. After 24 h, the cell culture supernatant was collected and
used as C26-conditioned medium (C26-cm) for further studies. HSC
conditioned medium was also prepared. To do so, HSCs were cultured
at a concentration of 7,5×104 cells/cm2 as
previously described (10). Then,
culture medium was replaced with fresh medium supplemented with 1%
FBS and HSCs were cultured for an additional 24 h. The supernatant
was collected and used as HSC-conditioned medium for further
studies. In some cases, HSCs were exposed to C26-cm diluted 2-fold
with fresh medium for 24 h. Then, the supernatant was collected and
used as tumor-activated HSC-cm (taHSC-cm).
Western blot analyses
CXCR4 expression was analyzed by western blotting in
C26 cells and HSCs under several conditions. To do so, the tumor
cells were either untreated or treated with HSC-cm or taHSC-cm, and
HSCs were either untreated or treated with C26-cm. After 48 h of
treatment, the cultured medium was removed and cell lysates were
obtained. To obtain cell lysates for protein extraction, the cells
were washed twice with cold 1X phosphate-buffered saline (PBS)
before adding 100 µl/106 cells of 1X Laemmli sample
buffer (Bio-Rad, USA) with 1% β-mercaptoethanol and 1 M NaCl.
Sample electrophoresis was performed using 10% SDS-PAGE gels and
run for 1.5 hat 120 mV on ice. Proteins were transferred onto a
nitrocellulose membrane in wet conditions at 380 mA for 3.5 hon
ice. Then, the membranes were incubated with rabbit anti-mouse
CXCR4 (1:1,000; cat. no. PA3-305; Thermo Fisher Scientific)
overnight at 4°C in 5% milk containing TBS-T (2% 1 M Tris-HCL pH,
7.5 10% 5 M NaCl, 0.1% Tween-20), followed by a 1-h incubation with
specific goat anti-rabbit biotinylated secondary antibodies
(1:2,000; cat. no. 65-6140). Streptavidine-HRP conjugated (1:500)
was used for detection. Antibodies and detection systems were
purchased from Thermo Fisher Scientific. Finally, the bands were
visualized using Luminata™ Crescendo Western HRP Substrate
(Millipore, Billerica, MA, USA). SnapGene software (Syngene,
Frederick, MD, USA) was used to measure the protein expression
(Each experiment was performed three times).
Flow cytometric analyses
Flow cytometric analyses were carried out for the
quantification of CXCR4 expression in C26 cells and quantification
of MDSCs in the circulation of tumor-bearing mice. To do so, C26
cells and PMNCs (for collection of PMNCs see later on) were fixed
in 70% ethanol for 10 min before the unspecific background was
blocked by a 30-min incubation in PBS containing 5% FBS. The cells
were incubated for 2 h with either rat anti-mouse CXCR4 monoclonal
antibody (1:500; cat. no. MAB21651; R&D Systems Inc.,
Minneapolis, MN, USA), FITC-conjugated rat anti-mouse CD11b (1:500;
cat. no. ab24874; Abcam, Cambridge, UK) and rat anti-mouse-Ly6G
primary antibody (1:500; cat. no. NBP1-28168; Novus Biologicals,
Littleton, CO, USA). Then, cells were incubated with Alexa-488
(excitation 488, emission 525) and Alexa-594 (excitation 594,
emission 617) conjugated-secondary antibody for CXCR4 and MDSC
detection, respectively. Immunolabelled cells were analyzed by flow
cytometry (Gallios; Beckman Coulter, Brea, CA, USA). Flow
cytometric analyses were performed three times for tumor cells and
a pool of 5 mice were used for MDSC quantification.
Cell viability assay
C26 viability was analyzed using PrestoBlue™ Cell
Viability reagent (Life Technologies Inc.). C26 tumor cells were
seeded at a concentration of 5×103 cells/cm2
and let expand for 3 h. Afterwards, the tumor cells were incubated
with increasing concentrations (0.1, 1 and 10 nM) of CXCL12
(OriGene, Rockville, MD, USA) with or without 10 µg/ml AMD3100
(from 2 h prior to the addition of CXCL12), for 48 h (AMD3100 was
previously diluted in PBS for stock solution). Then, cell viability
was quantified by adding PrestoBlue™ reagent for 120 min. The
reaction was measured with the Ascent Multiskan (Labsystems). Cell
viability was represented as percentage of the increase respect to
cell viability at time 0 (Cell viability assay was carried out
three times).
Gelatin zymography
MMP-2 and MMP-9 activity was measured by gelatin
zymography in C26 cells or HSC supernatants. The cells were treated
with either 1 nM CXCL12, or AMD3100 (10 µg/ml from 2 h prior to the
addition of CXCL12) or the combination of both for 24 h. Cell
culture supernatants were collected and run on 1% gelatin
containing electrophoresis gel. After 2 washes in 2.5% TritonX-100,
the gels were incubated overnight in a developing buffer containing
divalent cations for the proper activity of MMPs. After wards, the
gel was stained with Coomassie Blue to visualize degradation bands.
Images were captured through Quantity One program (Bio-Rad
Laboratories, Hercules, CA, USA) (MMP-2 and MMP-9 secretion was
measured three times).
Migration assay
For the quantification of the migratory potential of
C26 cells and HSCs, modified Boyden chambers were utilized.
Briefly, cells were seeded onto type I collagen-coated 8-µm
diameter pore membrane inserts (Greiner Bio-One GmbH,
Frickenhausen, Germany). Cells were incubated in RPMI-1640
supplemented with 1% FBS and antibiotics and allowed to adhere and
expand for 3 h before the addition of AMD3100 (10 µg/ml) in both
upper and lower chambers. After incubation of the cells with
AMD3100 for 2 h, the medium was replaced and the cells were treated
with either AMD3100 (10 µg/ml), 1 nM CXCL12 or a combination of
both for 18 h supplemented with 1% FBS. Then, the non-migrated
cells were gently removed from the upper side of the insert and the
remaining cells on the external side of the insert were subjected
to 70% ethanol fixation and DAPI mounting medium for visualization
of the nuclei. The number of migrated tumor cells and HSCs was
quantified in 10 fields at ×10 magnification under Axioscope
fluorescence microscope (Carl Zeiss, Oberkochen, Germany). The
migratory potential is represented as the percentage of migrated
treated cells in respect to the untreated cells (migration studies
were performed three times).
Experimental development of liver
metastasis
C26 tumor cells were in trasplenically injected at a
concentration of 2×105 cells in 100 µl PBS. Briefly, a
thin cut was made in the left flank of each anesthetized mouse and
the spleen was exposed. Then, the tumor cell suspension of the
grafted C26 colorectal cancer cells was slowly injected in the
spleen distal pole. Afterwards, the spleen was relocated and the
wound was closed. Mice were treated daily with 5 mg/kg AMD3100 or
vehicle solution starting 24 h after tumor cell injection. All
animals were sacrificed 14 days after tumor cell injection and
livers were frozen and kept at −80°C for immunehistochemical
analyses or paraffin embedded for hematoxylin and eosin staining
and quantification of liver tissue area occupied by the tumor.
In vivo metastasis assay was performed in duplicate with 5
animals in each group/experiment (n=10 each group).
Immunohistochemical analysis
Frozen liver sections (10-µm thick) were analyzed
for the quantification of different immune cell populations. Liver
sections were stained with specific primary antibodies:
Ratanti-mouse CD11b as neutrophil marker (Abcam), ratanti-mouseLy6G
as granulocytic marker (Novus Biologicals) and Cy3-conjugated mouse
anti-human αSMA (1:500; cat. no. C6198; Sigma-Aldrich) as HSC
activation marker. After blocking and incubation with primary
antibodies, surface molecules were detected by the use of secondary
antibodies conjugated either with Alexa-488 or Alexa-594. For αSMA
staining, samples were incubated with rabbit anti-mouse IgG F (ab')
(cat. no. 31192; Thermo Fisher Scientific) diluted in PSB for the
blockage of endogenous mouse IgGs for 1 h prior to the primary
antibody incubation (0.15 mg/ml). The number of
CD11b+Ly6G+ cells along with in tratumoral
αSMA expression was analyzed in the metastatic livers and
quantified by image analyses with ImageJ software (NIH, Bethesda,
MD, USA).
Isolation of blood PMNCs
For isolation of PMNCs, mice were anesthetized prior
to their sacrifice and the abdominal cavity was opened exposing the
cava vein. Sterile PBS-EDTA was inoculated into the cava vein right
before extraction of blood using the same syringe. Erythrocytes
were discarded using Lympholyte M (Cederlane, Canada) density
gradient and PMNCs and lymphocyte fraction collected for flow
cytometry analyses as described above. Results represent the pool
of 5 mice/group.
Statistical analyses
Data are expressed as mean ± standard deviation (SD)
of three independent experiments. Statistical analysis was
performed using SPSS version 13.0 (SPSS, Inc. Chicago, IL, USA).
Individual comparisons were performed using two-tailed, unpaired
Student's t-test. Differences were considered to be significant for
P<0.05.
Results
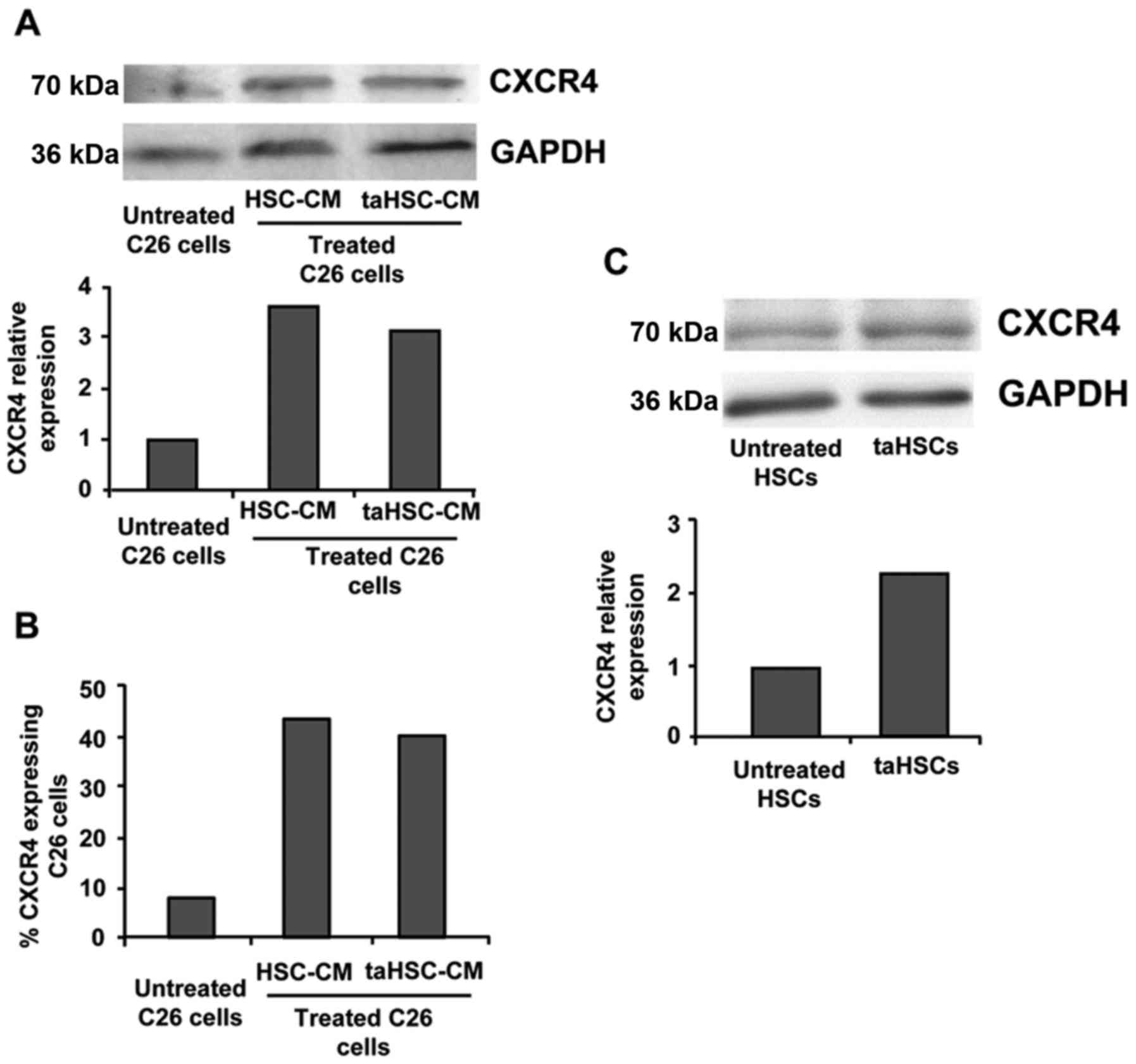
Tumor/stroma crosstalk stimulates
CXCR4 expression in C26 cells and HSCs
Tumor/stroma interaction is known to modulate a wide
variety of proteins in both cancer and stromal cells (10). As mentioned above, CXCR4 expression
is localized in HSCs in the liver and tumor cells. We treated C26
cells with soluble factors derived from either quiescent HSCs or
taHSCs, which resulted in a 4-fold increase in the CXCR4 expression
in C26 tumor cells compared to untreated C26 cells when analyzed by
western blotting (Fig. 1A) and FACS
(Fig. 1B). In the same way, HSCs
treated with soluble factors derived from C26 cells showed a 2-fold
increased expression of CXCR4 when compared to that of control HSCs
(Fig. 1C). Thus, CXCR4 expression
was increased in the HSCs and tumor cells after their paracrine
interaction.
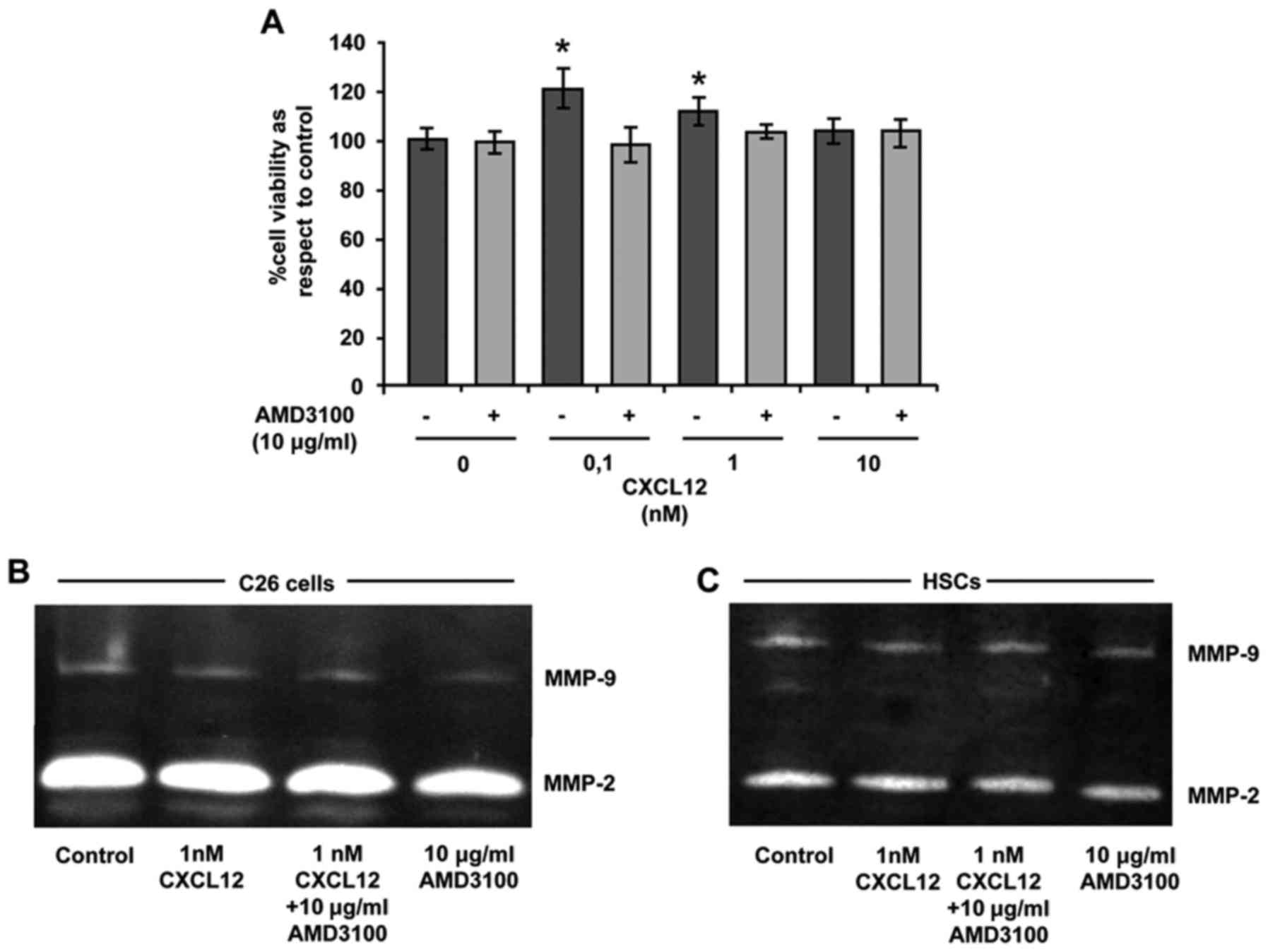
CXCL12 stimulates the proliferation of
C26 cancer cells through CXCR4
Since CXCL12 is known to induce proliferation in
CXCR4-expressing cells the effect of CXCL12 on the viability of C26
cells after treatment with increasing concentrations of CXCL12 was
quantified. As shown in Fig. 2A,
0.1 and 1 nM CXCL12 significantly enhanced C26 viability after a
24-hincubation, while 10 nM did not alter tumor cell proliferation.
Blocking of CXCR4 by addition of AMD3100 (10 µg/ml) before CXCL12
addition was able to revert the effect of the chemokine in regards
to C26 cell viability (Fig. 2A).
Therefore, CXCR4 mediated the proliferation of CXCL12-stimulated
C26 cells.
Effect of CXCL12 on MMP-2 and MMP-9
expression in C26 tumor cells and HSCs
The remodeling of the ECM represents a key event
during organ colonization and tumor growth. Tumor and stromal cells
contribute to this process by secreting metalloproteases (MMPs),
mainly gelatinocytic MMP-2 and MMP-9. Thus, the presence of these
MMPs in the conditioned medium derived from C26 cells and HSCs was
analyzed after the treatment with either CXCL12 (1 nM), AMD3100 (10
µg/ml) or a combination of both. While CXCL12 was unable to
increase the levels of any MMP over basal levels, treatment with
AMD3100 slightly decreased the MMP-2 and MMP-9 secretion in tumor
and stromal cells compared to those present in the supernatants of
the control and CXCL12-treated cell cultures (Fig. 2B and C).
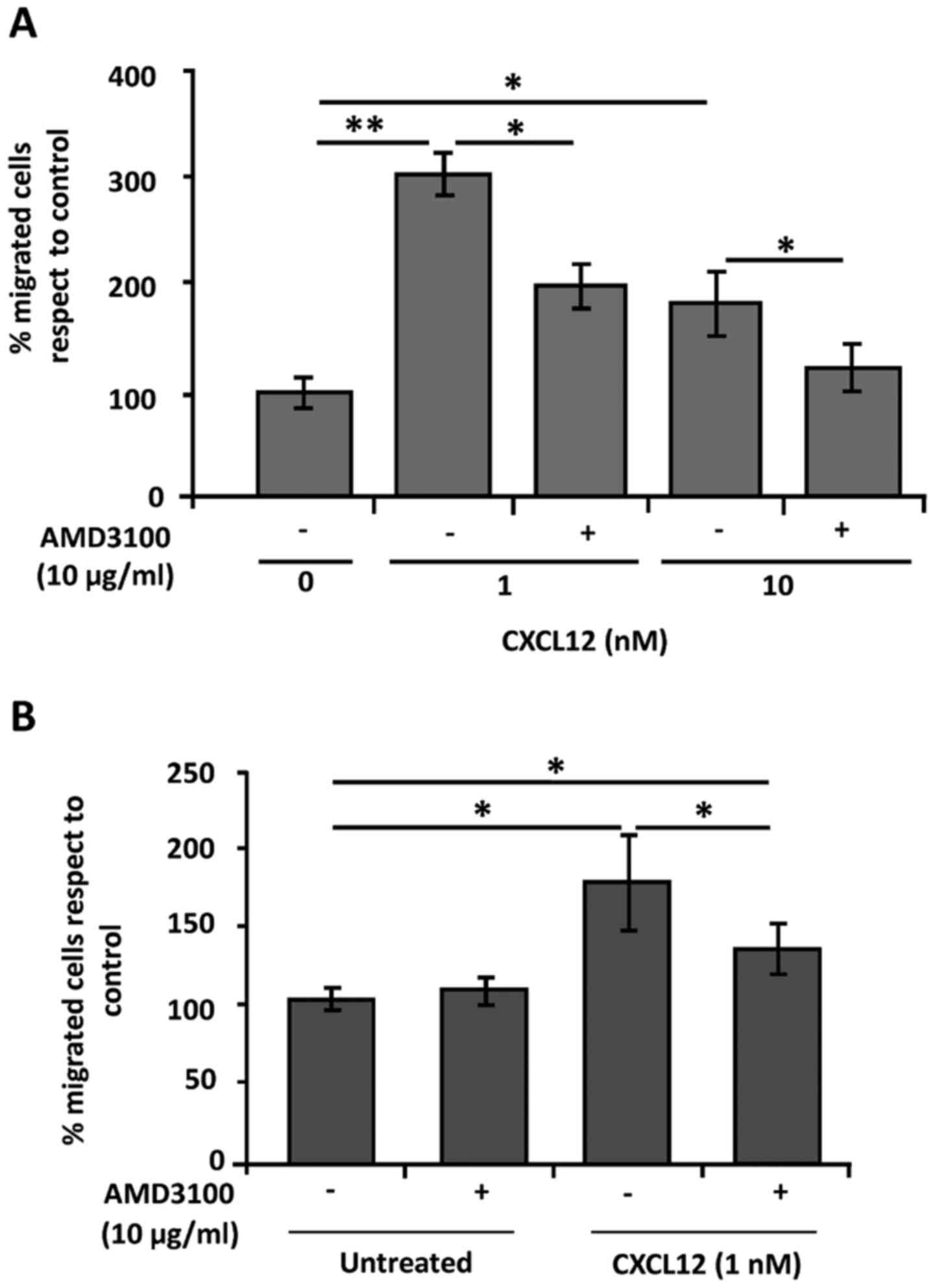
CXCR4 mediates the migration of C26
and HSCs towards CXCL12
CXCL12 has been pointed out as a potent chemokine
for CXCR4-expressing cells promoting their migration. Thus, we
investigated the role of the CXCR4 receptor in the migration of C26
cells and primary HSCs after activation with CXCL12. As shown in
Fig. 3A, CXCL12 induced a 4-fold
increase in the migratory potential of C26 cells compared to that
of the untreated cells. Blockage of CXCR4 by AMD3100 treatment
inhibited the effect of CXCL12 in decreasing the percentage of
migrated C26 cells (Fig. 3A).
Similarly, CXCL12 activation of primary HSCs also enhanced their
migratory potential by 2-fold (Fig.
3B). The effect of this chemokine was partially compromised by
using AMD3100, which points to CXCR4 as one of the mediators of
CXCL12-induced HSC recruitment into metastatic lesions (Fig. 3B). These results uncover the role of
CXCL12/CXCR4 crosstalk during tumor cell and HSC migration.
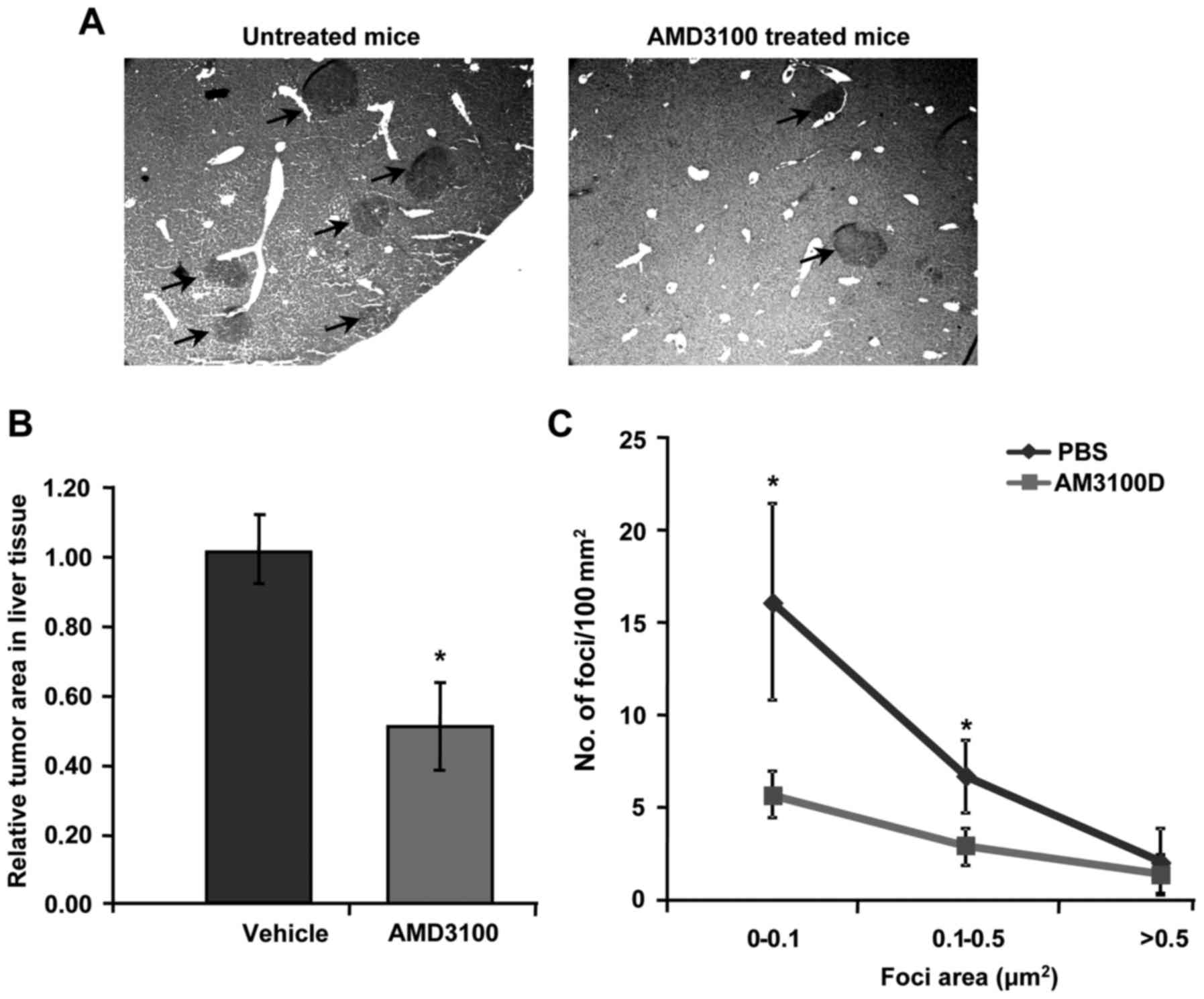
CXCR4 blockage via AMD3100
administration reduces the number and size of metastatic foci in
the liver
To further investigate the significance of CXCR4
during metastatic spread, in vivo liver metastasis assay was
carried out using colorectal cancer C26 cell line. Mice were
administered with a daily dose of AMD3100 (5 mg/kg) starting the
day after tumor cell injection. After 14 days, mice were sacrificed
and the livers were collected for tumor burden analysis by
quantifying the liver tissue area occupied by metastatic foci.
Daily treatment with AMD3100 antagonist resulted in a significant
reduction in the tumor burden in the liver of tumor-bearing mice
compared to those treated with vehicle solution (Fig. 4A). In fact, AMD3100-treated
tumor-bearing mice showed a 2-fold decrease in the metastatic area
occupied in the liver along with a reduced number of foci of every
size, and a significant reduction of small foci counts (Fig. 4B and C). Therefore, blocking CXCR4
is linked with reduced liver metastatic growth of colorectal C26
cancer cells in mice.
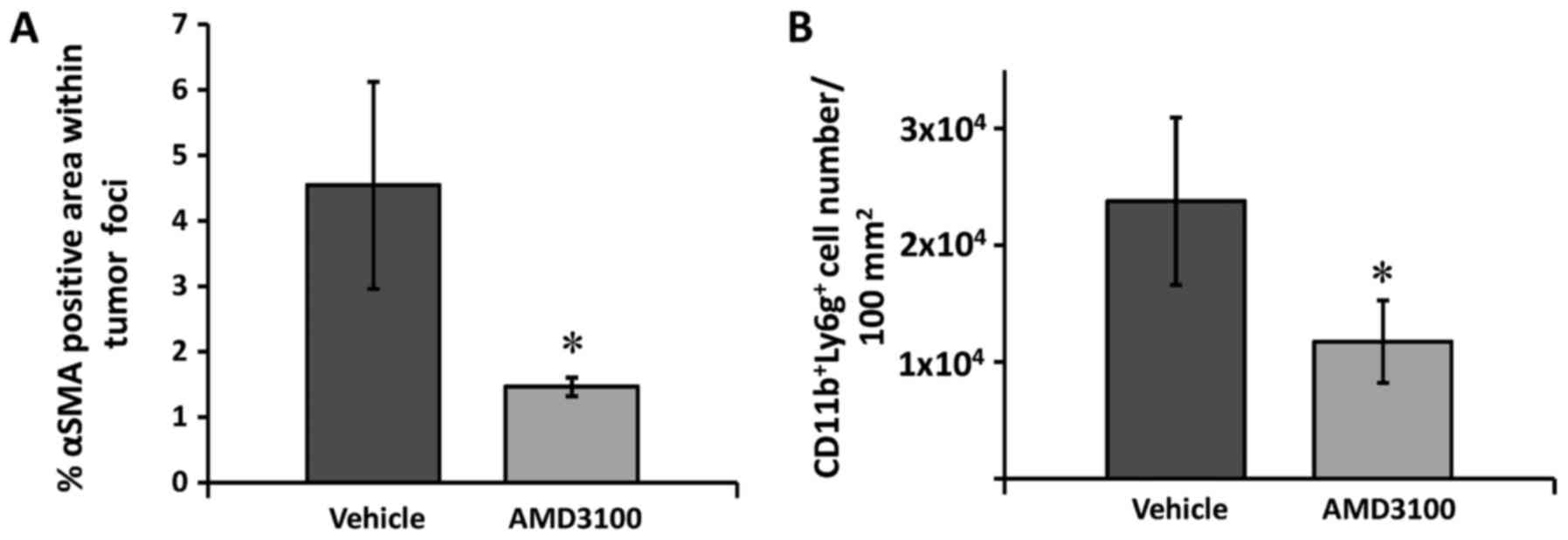
αSMA-expressing cell recruitment is
decreased within liver foci in the AMD3100-treated mice
Activated HSCs express αSMA and help support
angiogenesis (10). To confirm the
obtained in vitro results, we analyzed the infiltration of
αSMA-expressing cells in the tumor foci of the vehicle and
AMD3100-treated mice 14 days after tumor cell injection. The
immunohistochemical analyses revealed a significant 2-fold decrease
in the area positively stained for αSMA within the tumor foci in
the liver of the AMD3100-treated mice compared to that of the
vehicle-treated mice (Fig. 5A),
which corroborates the in vitro results showing reduced
migration of HSCs uponantagonist treatment. Hence, HSC expression
of CXCR4 appears to be involved in chemotaxis leading to the
recruitment of HSCs into tumor areas.
AMD3100 treatment is related to
reduced CD11b+Ly6G+ MDSC recruitment into the
metastatic liver
HSCs are known to promote immune suppression via
different pathways (11). In fact,
evidence supports their role in the induction of MDSCs (21), which may drive immune suppression in
the tumor microenvironment. These immune cells express CXCR4, and
their chemotactic migration towards CXCL12 has been reported.
Therefore, we investigated the effect of AMD3100 on the recruitment
of CD11b+Ly6G+ MDSCs into the metastatic
liver. The quantification of CD11b+Ly6G+
MDSCs in tumor-bearing mice revealed a marked 2-fold decrease in
the number of positive cells in the liver of the AMD3100-treated
mice, compared to that of the vehicle-treated mice (Fig. 5B), which is in line with the
decrease in αSMA-positive cells in the tumor foci in the
AMD3100-treated tumor-bearing mice. This observation lets us
conclude that the infiltration of MDSCs into the metastatic liver
may be mediated by expression of CXCR4.
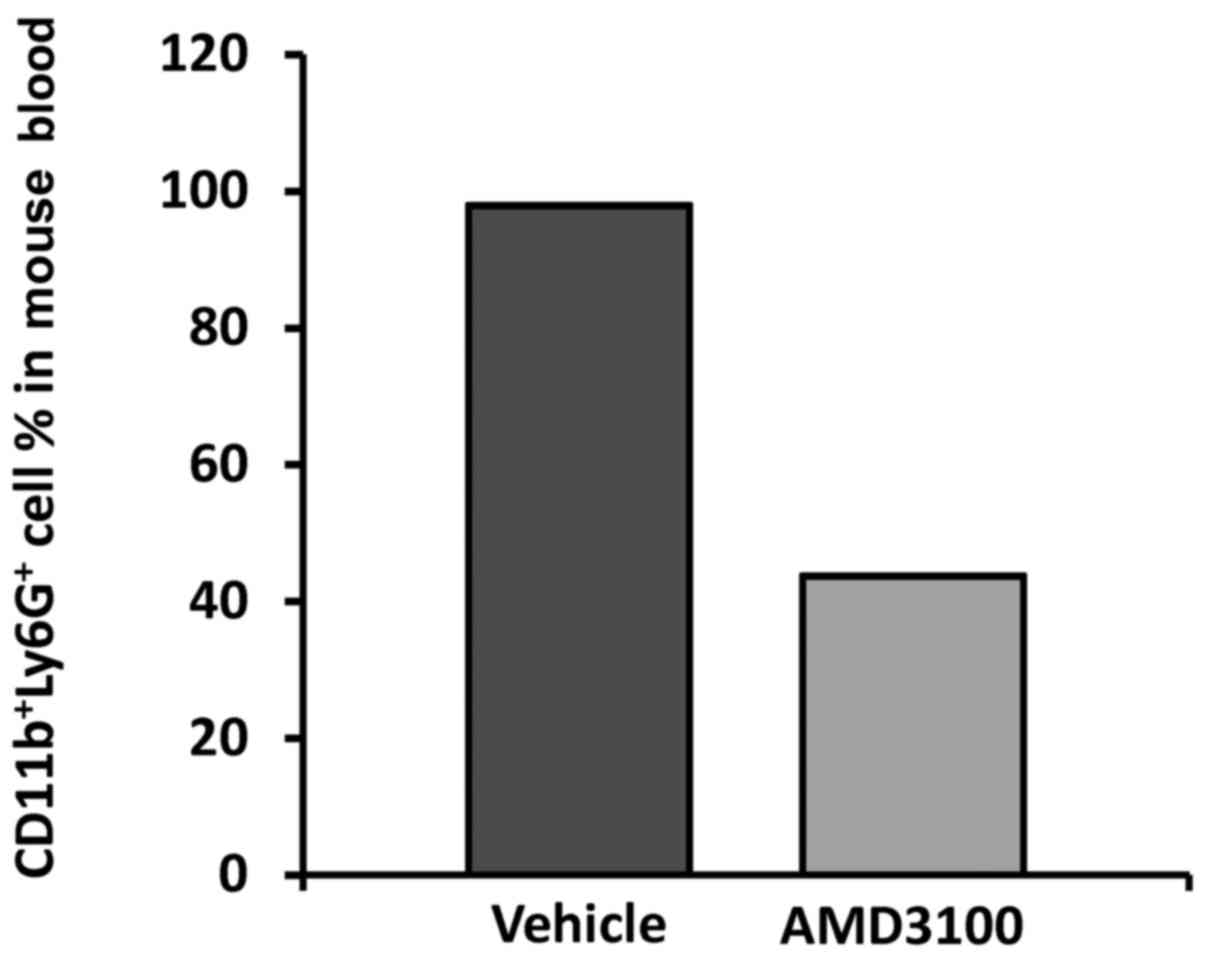
Circulating
CD11b+Ly6G+ number is reduced in the
AMD3100-treated mice
The increase in MDSCs in the liver could represent
an induction in the target organ but could also reflect an increase
in the circulating number of MDSCs. Giving the influence of
HSC-derived factors in these cells, the differentiation of the
circulating pool could be compromised due to attenuated HSC
activation and infiltration into the metastatic lesions. In order
to assess the rate of blood born MDSCs, tumor-bearing mice treated
with AMD3100 or vehicle solution were sacrificed after 14 days of
tumor cell inoculation and blood was collected from 4 animals per
experimental group for PMNC isolation. Then, the presence of
double-positive CD11b+Ly6G+ cell percentage
was calculated by flow cytometry. The results revealed a
significant 2-fold decrease in CD11b+Ly6G+
cell population in the AMD3100-treated tumor-bearing mice, compared
to the mice treated with vehicle where almost 98% of
Ly6G+ cells were positive for the monocytic marker
CD11b. In contrast, only 43.8% of Ly6G+ cells exhibited
a double-positive phenotype in the AMD3100-treated tumo-bearing
mice (Fig. 6). Consequently,
blockage of CXCR4 and the subsequent response related to CXCL12
ligation promoted a reduced MDSC percentage in the blood of the
AMD3100-treated mice.
Discussion
CXCR4 has been reported in several human and mouse
cancer cell lines (9,22,23).
However, modulation of the expression of this receptor in the tumor
microenvironment remains poorly characterized. CAFs have been shown
to promote the expression of a wide variety of cell receptors and
adhesion molecules.
Herein, we showed that soluble factors derived from
control or tumor-activated HSCs increased the expression of CXCR4
in tumor cells. This increase may be mediated by the production of
IL-1 and IL-6 by HSCs, which has been shown to promote CXCR4
upregulation in different cell types (24,25).
Moreover, both human and mouse HSCs express CXCR4. In fact, the
increased expression of this receptor has been linked to
progressive HSC activation under culture conditions. Furthermore,
the upregulation of CXCR4 has also been reported in fibrotic
livers, primarily related to activated HSCs (26). Interestingly, HSC activation has
also been observed during metastatic colonization of the liver
(10). Consistent with these
findings, we reported upregulated CXCR4 expression in
tumor-activated HSCs in vitro.
The metastatic process is orchestrated by
circulating tumor cells in concert with cells of the target organ.
Tumor cells migrate towards soluble stimulus, which attract them to
the colonizing organ. Among others, CXCL12, the main lig and for
CXCR4, has been reported to drive tumor cell migration and has been
shown to be upregulated in the liver under pathological conditions,
including cancer (27). In line
with several reports, our results showed that C26 migration was
increased after CXCL12 stimulation (25) whereas C26 treatment with CXCR4
antagonist AMD3100 led to attenuated response to this chemokine.
This phenomenon could be in part mediated by Rho and Rac GTPases,
involved in metastatic colonization, as observed in colon cancer
and hepatoma cells after CXCR4-mediated CXCL12 stimulation
(28), therefore, promoting cancer
cell extravasation. Indeed, the formation of pseudopodia was
described in CXCL12-treated multiple myeloma cells (29), which may also account for the
facilitation of tumor migration by CXCL12-treated C26 cells.
Once in a target organ, cancer cells begin to
proliferate in an uncontrolled manner driving tumor growth. Target
organ cells support the tumor growth by the secretion of a wide
variety of growth factors, cytokines and chemokines. In the liver,
CXCL12 is produced by HSCs and LSECs in addition to malignant cells
(30). Activated LSECs and HSCs
secrete CXCL12, which may have an effect on CXCR4-expressing
metastasizing colorectal cancer cells. Our results showed, however,
that CXCL12 slightly increased the proliferation of C26 cells, a
response that could be more pronounced in vivo, since
stromal cells stimulate CXCR4 expression in C26 cells. The
activation of the MAPK pathway could be responsible for this
proliferation enhancement, as previously reported in other cell
types (31). Moreover, this
chemokine switches on the PI3K/AKT pathway, driving tumor growth
(32), which could account for the
increased C26 cancer cell viability.
Tumor growth is usually accompanied by ECM
remodeling, a required step for disease progression. ECM remodeling
is carried out by both tumor and stromal cells, such as HSCs in the
liver. Intriguingly, the migration of HSCs was also fostered after
treatment with this chemokine, which was partially abrogated after
blocking CXCR4 using AMD3100. After interaction of C26 cells with
liver sinusoidal endothelial cells (LSECs), Valcarcel et al
observed an increase in VEGF, known to be an upstream inductor of
CXCL12 in the liver (33), along
with the initiation of an inflammatory response (20) that may account for HSC recruitment
in tumor foci. This observation agrees with previous reports
linking stimulated migration ability of human primary HSCs treated
with CXCL12, along with CXCL12-mediated contraction mechanism in
mouse primary HSC line, which may play a significant role in this
process (34,35). Surprisingly, we observed no changes
in MMP-2 or MMP-9 activation in both C26 and HSCs treated with
CXCL12, in line with other groups reporting no effect in
CXCL12-treated cells (36). This
result could be explained by the fact that HSCs do secrete CXCL12,
which could activate in an autocrine manner the secretion of MMPs
(37). In concordance with these
results, we also observed a decrease in the expression of MMP-2 in
these cells after AMD31000 treatment, which avoids the interaction
of CXCL12 with its receptor CXCR4.
The production of CXCL12 in the liver is elevated
(38) and this organ becomes a
perfect collecting site for CXCR4-expressing circulating cells.
Thus, this might be one of the reasons why the liver represents one
of the main target organs for metastatic colorectal cancer cells.
In fact, the administration of CXCR4 antagonist AMD3100 in
colorectal liver metastasis model reduced by 2-fold the metastatic
area in the liver. Several reports have shown similar results by
hijacking the circulating CXCL12 or administering AMD3100 into nude
mice, therefore, impairing the metastatic colonization of the liver
by tumor cells. The CXCR4-CXCL12 interaction mediated proliferative
response observed in colorectal cancer cells (39,40)
can be blocked by AMD3100 administration in the
tumor-microenvironment, contributing to the reported decrease in
the metastatic area of AMD3100-treated tumor-bearing mice. This
observation may be further mediated by reduced αSMA-expressing
cells into metastatic foci, which impairs the angiogenic response
and, consequently, the blood-mediated oxygen and element supply to
the tumor mass, a required step for disease development. In
addition, CXCR4-CXCL12 interplay has been shown to be responsible
for elevated VEGF availability in tumor areas, driving new vessel
formation (41). Interestingly,
this group reported that HSCs mediated VEGF upregulation during
liver metastasis, which may facilitate LSEC recruitment and
transition from avascular to vascular stage of metastatic foci
(10). Therefore, AMD3100
administration might interfere in the VEGF secretion by
CXCL12-activated HSCs, leading to impaired neovascularization due
to deficient LSEC recruitment.
These activated myofibroblast-like cells secrete a
wide array of cytokines, such as, IL-1, IL-6 or IL-10 (42). IL-1 cytokine, along with VEGF,
promote the differentiation and generation of MDSCs from the bone
marrow, a highly relevant immunosuppressive cell population,
profoundly linked with cancer progression and immune tolerance.
Chou et al showed not only that HSCs can promote generation
of MDSCs but also that these MDSCs possess a potent immune
inhibitory activity (43). Our
results showed that the proportion of circulating
CD11b+Ly6G+ cells was elevated in untreated
tumor-bearing mice, compared to those mice treated with AMD3100.
This decrease in MDSCs may be due to the down regulation of
differentiation stimuli secreted by both tumor cells and HSCs when
blocking CXCR4, hence, leading to lower numbers of MDSCs. In fact,
these CD11b+Ly6G+ cells express CXCR4 and
migrate towards CXCL12 (44),
therefore, accumulating in tumor areas. Our in vivo studies
revealed that blocking CXCR4 drove to an attenuated recruitment of
this suppressive cell subset into metastatic liver, in concordance
with several studies reporting decreased numbers of these cells in
breast carcinoma or subcutaneous lesions of hepatoma and breast
carcinoma cells using CXCR4 antagonists (45,46).
This is also in close relation with the reduced number of HSCs
recruited to the tumor foci after blockage of CXCR4 signaling by
AMD3100. Interestingly, the soluble factors produced after C26
interaction with LSECs recruit HSCs through a COX-2 mediated
pathway (data not shown), and the same pathway is required for MDSC
accumulation driven by recruited activated HSCs (21).
Taken together, our data uncovered the relevance of
tumor and stromal crosstalk driving CXCR4-CXCL12 interplay
stimulation, which in turn favors the recruitment of tumor cells
and MDSCs into the liver. This is in relation to the infiltration
of αSMA-expressing activated HSCs into metastatic foci which
fosters the recruitment and accumulation of differentiated
CD11b+Ly6g+ cell subsets from the circulation
of tumor-bearing mice, driving liver metastasis. All of these
processes are reduced by the administration of AMD3100, implying
CXCR4/CXCL12 interaction as a perfect target for the inactivation
of multiple pro-tumoral processes involving both tumor and host
cells.
Acknowledgements
This study was financially supported in part by a
postdoctoral fellowship from the University of the Basque Country
to A.B. and by funds from the Basque Government-Saiotek to B.A.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MDSCs
|
myeloid-derived suppressor cells
|
|
HSCs
|
hepatic stellate cells
|
|
αSMA
|
alpha smooth muscle actin
|
|
CAFs
|
cancer-associated fibroblasts
|
|
LSECs
|
liver sinusoidal endothelial cells
|
|
ECM
|
extracellular matrix
|
References
|
1
|
Van den Eynden GG, Majeed AW, Illemann M,
Vermeulen PB, Bird NC, Høyer-Hansen G, Eefsen RL, Reynolds AR and
Brodt P: The multifaceted role of the microenvironment in liver
metastasis: Biology and clinical implications. Cancer Res.
73:2031–2043. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Colangelo T, Polcaro G, Muccillo L,
D'Agostino G, Rosato V, Ziccardi P, Lupo A, Mazzoccoli G, Sabatino
L and Colantuoni V: Friend or foe? The tumour microenvironment
dilemma in colorectal cancer. Biochim Biophys Acta. 1867:1–18.
2017.PubMed/NCBI
|
|
3
|
Trautmann F, Cojoc M, Kurth I, Melin N,
Bouchez LC, Dubrovska A and Peitzsch C: CXCR4 as biomarker for
radioresistant cancer stem cells. Int J Radiat Biol. 90:687–699.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brand S, Dambacher J, Beigel F, Olszak T,
Diebold J, Otte JM, Göke B and Eichhorst ST: CXCR4 and CXCL12 are
inversely expressed in colorectal cancer cells and modulate cancer
cell migration, invasion and MMP-9 activation. Exp Cell Res.
310:117–130. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ottaiano A, Franco R, Aiello Talamanca A,
Liguori G, Tatangelo F, Delrio P, Nasti G, Barletta E, Facchini G,
Daniele B, et al: Overexpression of both CXC chemokine receptor 4
and vascular endothelial growth factor proteins predicts early
distant relapse in stage II–III colorectal cancer patients. Clin
Cancer Res. 12:2795–2803. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamada S, Shimada M, Utsunomiya T, Morine
Y, Imura S, Ikemoto T, Mori H, Arakawa Y, Kanamoto M, Iwahashi S
and Saito Y: CXC receptor 4 and stromal cell-derived factor 1 in
primary tumors and liver metastases of colorectal cancer. J Surg
Res. 187:107–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li JK, Yu L, Shen Y, Zhou LS, Wang YC and
Zhang JH: Inhibition of CXCR4 activity with AMD3100 decreases
invasion of human colorectal cancer cells in vitro. World J
Gastroenterol. 14:2308–2313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wendel C, Hemping-Bovenkerk A, Krasnyanska
J, Mees ST, Kochetkova M, Stoeppeler S and Haier J: CXCR4/CXCL12
participate in extravasation of metastasizing breast cancer cells
within the liver in a rat model. PLoS One. 7:e300462012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
O'Boyle G, Swidenbank I, Marshall H,
Barker CE, Armstrong J, White SA, Fricker SP, Plummer R, Wright M
and Lovat PE: Inhibition of CXCR4-CXCL12 chemotaxis in melanoma by
AMD11070. Br J Cancer. 108:1634–1640. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Olaso E, Salado C, Egilegor E, Gutierrez
V, Santisteban A, Sancho-Bru P, Friedman SL and Vidal-Vanaclocha F:
Proangiogenic role of tumor-activated hepatic stellate cells in
experimental melanoma metastasis. Hepatology. 37:674–685. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Puche JE, Saiman Y and Friedman SL:
Hepatic stellate cells and liver fibrosis. Compr Physiol.
3:1473–1492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Song Y, Kim SH, Kim KM, Choi EK, Kim J and
Seo HR: Activated hepatic stellate cells play pivotal roles in
hepatocellular carcinoma cell chemoresistance and migration in
multicellular tumor spheroids. Sci Rep. 6:367502016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cojoc M, Peitzsch C, Trautmann F,
Polishchuk L, Telegeev GD and Dubrovska A: Emerging targets in
cancer management: role of the CXCL12/CXCR4 axis. Onco Targets
Ther. 6:1347–1361. 2013.PubMed/NCBI
|
|
14
|
Mishra P, Banerjee D and Ben-Baruch A:
Chemokines at the crossroads of tumor-fibroblast interactions that
promote malignancy. J Leukoc Biol. 89:31–39. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kusmartsev S and Gabrilovich DI: Effect of
tumor-derived cytokines and growth factors on differentiation and
immune suppressive features of myeloid cells in cancer. Cancer
Metastasis Rev. 25:323–331. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang H, Li Z, Wang L, Tian G, Tian J,
Yang Z, Cao G, Zhou H, Zhao L, Wu Z and Yin Z: Critical role of
myeloid-derived suppressor cells in tumor-induced liver immune
suppression through inhibition of NKT cell function. Front Immunol.
8:1292017.PubMed/NCBI
|
|
17
|
Goodwin TJ, Zhou Y, Musetti SN, Liu R and
Huang L: Local and transient gene expression primes the liver to
resist cancer metastasis. Sci Transl Med. 8:364ra1532016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
D'Alterio C, Barbieri A, Portella L, Palma
G, Polimeno M, Riccio A, Ieranò C, Franco R, Scognamiglio G, Bryce
J, et al: Inhibition of stromal CXCR4 impairs development of lung
metastases. Cancer Immunol Immunother. 61:1713–1720. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Smedsrød B and Pertoft H: Preparation of
pure hepatocytes and reticuloendothelial cells in high yield from a
single rat liver by means of Percoll centrifugation and selective
adherence. J Leukoc Biol. 38:213–230. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Arteta B, Lasuen N, Lopategi A,
Sveinbjörnsson B, Smedsrød B and Vidal-Vanaclocha F: Colon
carcinoma cell interaction with liver sinusoidal endothelium
inhibits organ-specific antitumor immunity through
interleukin-1-induced mannose receptor in mice. Hepatology.
51:2172–2182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu Y, Zhao W, Xu J, Li J, Hong Z, Yin Z
and Wang X: Activated hepatic stellate cells promote liver cancer
by induction of myeloid-derived suppressor cells through
cyclooxygenase-2. Oncotarget. 7:8866–8878. 2016.PubMed/NCBI
|
|
22
|
Zhang SS, Han ZP, Jing YY, Tao SF, Li TJ,
Wang H, Wang Y, Li R, Yang Y, Zhao X, et al: CD133(+)CXCR4(+) colon
cancer cells exhibit metastatic potential and predict poor
prognosis of patients. BMC Med. 10:852012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kollmar O, Rupertus K, Scheuer C, Junker
B, Tilton B, Schilling MK and Menger MD: Stromal cell-derived
factor-1 promotes cell migration and tumor growth of colorectal
metastasis. Neoplasia. 9:862–870. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oh JW, Drabik K, Kutsch O, Choi C, Tousson
A and Benveniste EN: CXC chemokine receptor 4 expression and
function in human astroglioma cells. J Immunol. 166:2695–2704.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Odemis V, Moepps B, Gierschik P and Engele
J: Interleukin-6 and cAMP induce stromal cell-derived factor-1
chemotaxis in astroglia by up-regulating CXCR4 cell surface
expression. Implications for brain inflammation. J Biol Chem.
277:39801–39808. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hong F, Tuyama A, Lee TF, Loke J, Agarwal
R, Cheng X, Garg A, Fiel MI, Schwartz M, Walewski J, et al: Hepatic
stellate cells express functional CXCR4: Role in stromal
cell-derived factor-1alpha-mediated stellate cell activation.
Hepatology. 49:2055–2067. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun X, Cheng G, Hao M, Zheng J, Zhou X,
Zhang J, Taichman RS, Pienta KJ and Wang J: CXCL12/CXCR4/CXCR7
chemokine axis and cancer progression. Cancer Metastasis Rev.
29:709–722. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gassmann P, Haier J, Schlüter K,
Domikowsky B, Wendel C, Wiesner U, Kubitza R, Engers R, Schneider
SW, Homey B and Müller A: CXCR4 regulates the early extravasation
of metastatic tumor cells in vivo. Neoplasia. 11:651–661.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alsayed Y, Ngo H, Runnels J, Leleu X,
Singha UK, Pitsillides CM, Spencer JA, Kimlinger T, Ghobrial JM,
Jia X, et al: Mechanisms of regulation of CXCR4/SDF-1
(CXCL12)-dependent migration and homing in multiple myeloma. Blood.
109:2708–2717. 2007.PubMed/NCBI
|
|
30
|
Marra F and Tacke F: Roles for chemokines
in liver disease. Gastroenterology. 147:577–594.e1. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu Y, Peng H, Cui M, Whitney NP, Huang Y
and Zheng JC: CXCL12 increases human neural progenitor cell
proliferation through Akt-1/FOXO3a signaling pathway. J Neurochem.
109:1157–1167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Heinrich EL, Lee W, Lu J, Lowy AM and Kim
J: Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated
signaling pathways in pancreatic cancer cells. J Transl Med.
10:682012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Valcárcel M, Arteta B, Jaureguibeitia A,
Lopategi A, Martínez I, Mendoza L, Muruzabal FJ, Salado C and
Vidal-Vanaclocha F: Three-dimensional growth as multicellular
spheroid activates the proangiogenic phenotype of colorectal
carcinoma cells via LFA-1-dependent VEGF: Implications on hepatic
micrometastasis. J Transl Med. 6:572008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Saiman Y, Agarwal R, Hickman DA, Fausther
M, El-Shamy A, Dranoff JA, Friedman SL and Bansal MB: CXCL12
induces hepatic stellate cell contraction through a
calcium-independent pathway. Am J Physiol Gastrointest Liver
Physiol. 305:G375–G382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yuan X, Xiao T, Dai F, Yu Y and Lou J:
CXCL12α and CXCL12β stimulate the migration of cultured LX-2
hepatic stellate cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi.
31:293–296. 2015.(In Chinese). PubMed/NCBI
|
|
36
|
Zhang J, Sarkar S and Yong VW: The
chemokine stromal cell derived factor-1 (CXCL12) promotes glioma
invasivenessthrough MT2-matrix metalloproteinase. Carcinogenesis.
26:2069–2077. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Teng F, Tian WY, Wang YM, Zhang YF, Guo F,
Zhao J, Gao C and Xue FX: Cancer-associated fibroblasts promote the
progression of endometrial cancer via the SDF-1/CXCR4 axis. J
Hematol Oncol. 9:82016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun X, Cheng G, Hao M, Zheng J, Zhou X,
Zhang J, Taichman RS, Pienta KJ and Wang J: CXCL12/CXCR4/CXCR7
chemokine axis and cancer progression. Cancer Metastasis Rev.
29:709–722. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ma L, Qiao H, He C, Yang Q, Cheung CH,
Kanwar JR and Sun X: Modulating the interaction of CXCR4 and CXCL12
by low-molecular-weight heparin inhibits hepatic metastasis of
colon cancer. Invest New Drugs. 30:508–517. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Heckmann D, Maier P, Laufs S, Li L,
Sleeman JP, Trunk MJ, Leupold JH, Wenz F, Zeller WJ, Fruehauf S and
Allgayer H: The disparate twins: A comparative study of CXCR4 and
CXCR7 in SDF-1α-induced gene expression, invasion and
chemosensitivity of colon cancer. Clin Cancer Res. 20:604–616.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liang Z, Brooks J, Willard M, Liang K,
Yoon Y, Kang S and Shim H: CXCR4/CXCL12 axis promotes VEGF-mediated
tumor angiogenesis through Akt signaling pathway. Biochem Biophys
Res Commun. 359:716–722. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lang A, Sakhnini E, Fidder HH, Maor Y,
Bar-Meir S and Chowers Y: Somatostatin inhibits pro-inflammatory
cytokine secretion from rat hepatic stellate cells. Liver Int.
25:808–816. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chou HS, Hsieh CC, Yang HR, Wang L,
Arakawa Y, Brown K, Wu Q, Lin F, Peters M, Fung JJ, et al: Hepatic
stellate cells regulate immune response by way of induction of
myeloid suppressor cells in mice. Hepatology. 53:1007–1019. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sawanobori Y, Ueha S, Kurachi M, Shimaoka
T, Talmadge JE, Abe J, Shono Y, Kitabatake M, Kakimi K, Mukaida N
and Matsushima K: Chemokine-mediated rapid turnover of
myeloid-derived suppressor cells in tumor-bearing mice. Blood.
111:5457–5466. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang L, Huang J, Ren X, Gorska AE, Chytil
A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC and Moses
HL: Abrogation of TGF beta signaling in mammary carcinomas recruits
Gr-1+CD11b+ myeloid cells that promote
metastasis. Cancer Cell. 13:23–35. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ba H, Li B, Li X, Li C, Feng A, Zhu Y,
Wang J, Li Z and Yin B: Transmembrane tumor necrosis factor-α
promotes the recruitment of MDSCs to tumor tissue by upregulating
CXCR4 expression via TNFR2. Int Immunopharmacol. 44:143–152. 2017.
View Article : Google Scholar : PubMed/NCBI
|