Introduction
Oral cancer is the fifth most common type of cancer
in the world (1). Despite modern
treatment modalities, improvement in survival rates over the past
decade has been minimal. The development of local recurrence, the
formation of second primary tumors and metastasis are the principal
reasons of this poor outcome (2).
Therefore, recent studies in this area have focused on potential
biomarkers for the early diagnosis of oral cancer and the
continuous monitoring and prediction of prognosis of patients with
oral cancer (3,4).
Epithelial-mesenchymal transition (EMT) is known to
be one of the key mechanisms leading to cancer metastasis (5). It is a process that causes epithelial
cells to lose their polarity and intercellular contacts and obtain
transport properties of mesenchymal cells. In addition, the
expression of mesenchymal markers is increased and changes in the
cytoskeleton occur. These changes render epithelial cells more
mobile and invasive as they acquire the appearance of mesenchymal
cells (6,7). Many signaling cascades, such as the
transforming growth factor-β (TGF-β) (8), wingless (Wnt) (9) and Notch pathway (10) can induce EMT. It is known that EMT
can also be regulated by several development factors, such as
Snail1, Snail2, zinc finger E-box binding homeobox 1 (ZEB1) and
forkhead box protein C2 (FOXC2) (11–13).
Although the correlation between EMT and cancer progression has
been established, the underlying molecular mechanism of this
relationship remains largely unclear.
Syndecan-1 (SDC1) is one of the key cell-surface
adhesion molecules that regulate cell and extracellular matrix
adhesion and cell migration. It also regulates tumor-cell survival,
proliferation, angiogenesis and metastasis and ultimately affects
tumorigenesis (14,15). Recent studies revealed that in a
number of cancers, such as head and neck, ovarian, breast and
colorectal carcinomas, the expression of SDC1 is dysregulated
(16–19). Despite a large number of published
studies, the precise mechanisms that explain the role of SDC1 in
these pathologies are still not fully elucidated.
In the present study, we observed that the
expression of SDC1 was frequently downregulated in oral cancer cell
lines. Exogenous overexpression of SDC1 led to the morphological
transformation of cells, attenuated the expression of EMT markers
and inhibited proliferation, migration and invasion of oral cancer
cells. Furthermore, the knockdown of endogenous SDC1 activated the
extracellular signal-regulated kinase (ERK) cascade, upregulated
the expression of Snail and inhibited the expression of E-cadherin.
These regulatory properties revealed novel mechanisms of the
involvement of SDC1 in EMT and the invasive phenotype of human oral
cancer cells. Our data demonstrated that SDC1 is a key negative
regulator of EMT and a potential target for therapeutic
intervention in oral cancer.
Materials and methods
Cell lines
The KB, Tca8113, ACC2 and CAL27 cell lines were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). Human periodontal ligament (hPDL) fibroblasts
were purchased from ScienCell Research Laboratories (Carlsbad, CA,
USA). KB cells were cultured in RPMI-1640 medium (Thermo Fisher
Scientific, Rockford, IL, USA) containing 10% fetal bovine serum
(FBS; Kang Yuan Biological Technology Co., Ltd., Tianjin, China).
Tca8113, ACC2 and CAL27 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Grand
Island, NY, USA) supplemented with 10% FBS (Kang Yuan Biological
Technology). The hPDLF cells were cultured in DMEM/F12 (Thermo
Fisher Scientific) supplemented with 10% FBS (Biological
Industries, Cromwell, CT, USA). The cell cultures were maintained
at 37°C in a humidified atmosphere of 95% air and 5%
CO2.
Quantitative PCR (qPCR)
Total RNA was reverse transcribed using the RT
system supplied by Promega (cat. no. A1702; Madison, WI, USA) and
qPCR was performed on a Mastercycler supplied by Eppendorf
(Hamburg, Germany). The PCR primer sequences were as follows: hSDC1
sense, AGGACGAAGGCAGCTACTCCT and antisense, TTTGGTGGGCTTCTGGTAGG;
β-actin sense, GAGCACAGAGCCTCGCCTTT and antisense,
ATCCTTCTGACCCATGCCCA; E-cadherin sense, GACAACAAGCCCGAATT and
antisense, GGAAACTCTCTCGGTCCA; vimentin sense, GAGAACTTTGCCGTTGAAGC
and antisense, GCTTCCTGTAGGTGGCAATC; Snail sense,
GCAAATACTGCAACAAGG and antisense, GCACTGGTACTTCTTGACA; Slug sense,
AGATGCATATTCGGACCCAC and antisense, CCTCATGTTTGTGCAGGAGA; Twist
sense GGAGTCCGCAGTCTTACGAG and antisense, TCTGGAGGACCTGGTAGAGG.
Plasmids and stable cell lines
The pTT5-hSDC1 plasmid (cat. no. 52326; Addgene,
Inc., Cambridge, MA, USA) was donated by Professor Gordon Laurie
(University of Virginia). Oral cancer cells were transfected with
pTT5-hSDC1 or control plasmids, using Lipofectamine™ LTX with PLUS™
reagent (cat. no. 15338100; Invitrogen; Thermo Fisher Scientific,
Carlsbad, CA, USA).
Short hairpin RNA (shRNA) plasmids were obtained
from Shanghai GeneChem (Shanghai, China). Lentiviruses were
produced by co-transfecting 293T cells with one of the shRNA
expression plasmids and the packaging plasmids (psPAX2 and pMD2.G).
The supernatants were collected 48 h after transfection, filtered
through 0.45-mm filters (cat. no. SLGV033RB; EMD Millipore,
Temecula, CA, USA) and concentrated using 100 kDa MWCO Amicon Ultra
centrifugal filters (cat. no. Z648043; EMD Millipore). Stable cells
infected with shSDC1 and shCtrl (control) were selected on 100
µg/ml hygromycin (Sigma-Aldrich, St. Louis, MO, USA; cat. no.
H0654) for about two weeks, as previously described (10).
Western blot analysis
The experiments were performed as previously
described (10). Τhe protein
lysates were resolved by SDS-PAGE, transferred to PVDF membranes
(cat. no. ISEQ00010; EMD Millipore), detected with primary antibody
overnight at 4°C and then incubated with HRP-conjugated secondary
antibodies for 90 min at 25°C. Western blots were visualized using
ECL reagents (cat. nos. RPN2232 and RPN2236; GE Healthcare,
Pittsburgh, PA, USA). The following antibodies were used: Rabbit
monoclonal anti-E-cadherin antibody (cat. no. 4065) and Rabbit
polyclonal anti-N-cadherin antibody (cat. no. 4061) (both 1:1,000
dilution; Cell Signaling Technology, Inc., Beverly, MA, USA);
rabbit monoclonal anti-SDC1 antibody (cat. no. ab128936) and rabbit
polyclonal anti-Snail antibody (cat. no. ab63371) (both 1:2,000
dilution; Abcam, Hong Kong, China); mouse monoclonal anti-vimentin
antibody (cat. no. 550513), mouse monoclonal anti-β-catenin
antibody (cat. no. 610153) (both 1:1,000 dilution; BD Biosciences,
San Jose, CA, USA); mouse monoclonal anti-β-actin antibody
(1:10,000, dilution; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany); HRP-conjugated secondary antibodies (1:1,000 dilution;
cat. no. SC-2048; ZSGB-BIO, Beijing, China).
Hoechst 33342 staining
The cells (1×104/well) were seeded in
96-well plates, stained with Hoechst 33342 (cat. no. B2261;
Sigma-Aldrich) and observed by fluorescence microscopy. The number
of apoptotic cells in five random fields of view was counted and
apoptotic characteristics were recorded.
Colony formation assay
The cells (1×103/well) were seeded in
6-well plates and selected with 0.1 mg/l puromycin for 14 days. The
colonies were stained with crystal violet (cat. no. C3886;
Sigma-Aldrich).
Proliferation assay
3-(4,5-Dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide (MTT) assays were carried out to
detect cell proliferation. Cells were plated on 96-well plates at a
density of 1×104 cells/well. The absorbance of each well
was measured at 492 nm using the Take3 microplate reader (Bio-Tek
Instruments, Inc., Winooski, VT, USA). The survival percentage (%)
was calculated relative to that observed in the control cells.
In vitro migration and invasion
assay
In vitro migration and invasion assays were
performed using 24-well Transwell plates with polycarbonate filters
(Costar; Corning Life Sciences, Cambridge, MA, USA), as previously
described (20). For the migration
assay, 2.5×104 cells were added to the upper insert. For
the invasion assay, 5×104 cells were seeded into the
upper insert coated with Matrigel (BD Biosciences). The cells were
observed with an optical microscope connected to a camera and the
number of migrated cells was assessed by randomly capturing ten
images from each membrane.
Immunofluorescence
Cells (1×105/well) were grown on glass
coverslips in a 6-well plate, washed three times with
phosphate-buffered saline (PBS), then fixed in 4% formaldehyde and
permeabilized with 0.1% Triton X-100 in PBS for 5 min. The cells
were blocked with 2% bovine serum albumin (BSA) in PBS for 30 min
at 20–25°C. Coverslips were incubated with the respective primary
antibodies at 1:100 dilution for 1 h and then washed with PBS and
incubated for 1 h with tetramethylrhodamine (TRITC)-conjugated
secondary antibodies at 1:50 dilution (Beijing Zhongshan Golden
Bridge Biotechnology Co., Ltd., Beijing, China). The cells were
further washed in PBS and mounted with Vectashield mounting medium
(Vector Laboratories, Inc., Burlingame, CA, USA) containing
4′,6-diamidino-2-phenylindole (DAPI) and were analyzed using
fluorescence microscopy. Images were captured under a Nikon (Tokyo,
Japan) microscope with a fluorescein isothiocyanate filter.
Statistical analysis
Statistical significance of differences between
experimental group values is assessed using Student's t-test. The
levels of statistical significance are set as follows, P<0.05
and P<0.01. Error bars denote the standard deviation (SD).
Results
SDC1 expression is downregulated in
oral cancer cells
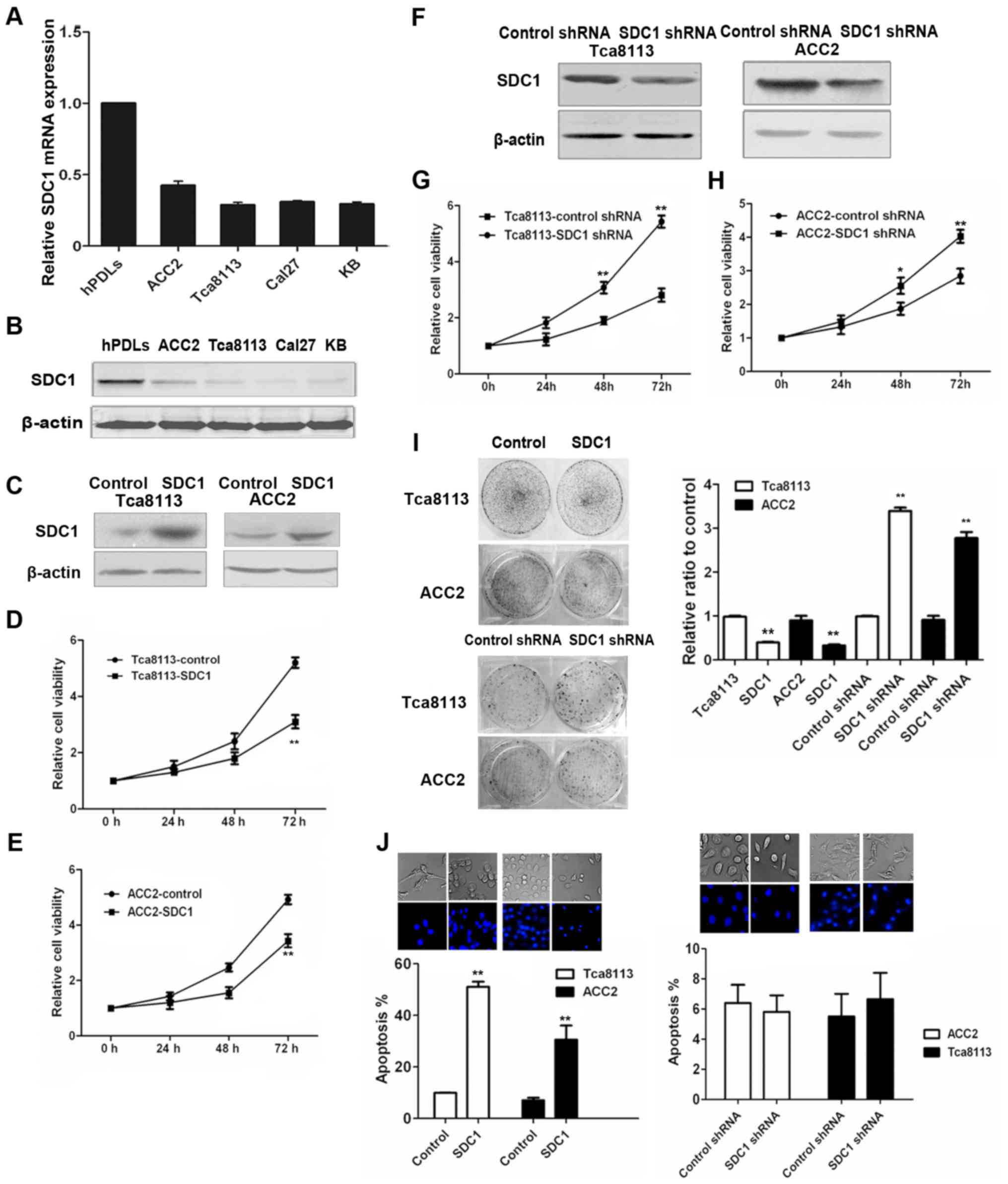
We first evaluated SDC1 expression in four human
oral carcinoma cell lines, KB (oral epidermoid carcinoma cells),
Tca8113 (tongue squamous cells), ACC2 (adenoid cystic carcinoma)
and CAL27 (tongue squamous cells), by qPCR and western blotting. As
displayed in Fig. 1A and B, all
four oral cancer cell lines exhibited lower levels of SDC1 mRNA and
protein, when compared with hPDL fibroblasts.
SDC1 induces apoptosis and inhibits
growth and colony formation in oral cancer cells
To study the functional significance of low SDC1
expression in oral cancer, we tested the effect of SDC1 on
proliferation and apoptosis in oral cancer cells. Firstly, we
confirmed the expression levels of SDC1 in Tca8113-SDC1 and
ACC2-SDC1 cells, both overexpressing SDC1, by immunoblotting
(Fig. 1C). Subsequently, we carried
out MTT assays to study the effect of SDC1 on the proliferation of
oral cancer cells. We found that the overexpression of SDC1
significantly inhibited cell proliferation, particularly 48 and 72
h after transfection (Fig. 1D and
E). To further assess the role of SDC1 in oral cancer cells, we
knocked down endogenous SDC1 by shRNA interference in Tca8113 and
ACC2 cells (Fig. 1F). MTT assays
showed that SDC1 knockdown promoted the growth of Tca8113 and ACC2
cells (Fig. 1G and H). These
results indicated that SDC1 can inhibit cell growth in oral cancer
cells.
Furthermore, colony formation assays confirmed that
overexpression of SDC1 inhibited cell proliferation while
interfering with endogenous SDC1 expression significantly increased
the number of cell colonies (Fig.
1I). These results indicated that SDC1 expression inhibited
colony formation in oral cancer cells. Hoechst 33342 staining
revealed that overexpression of SDC1 promoted apoptosis in oral
cancer cells (Fig. 1J).
Fluorescence microscopy revealed that the nuclei of the control
cells were round and stained evenly. At the same time,
SDC1-overexpressing cells exhibited typical morphological features
of apoptotic cells, such as cell shrinkage, chromatin condensation
and apoptotic bodies as shown by the arrows. However, when we
knocked down endogenous SDC1 by shRNA interference, we found that
the nuclei of both SDC1-knockdown and control group cells had
regular morphology and nuclear membrane integrity and did not
reveal significant apoptosis.
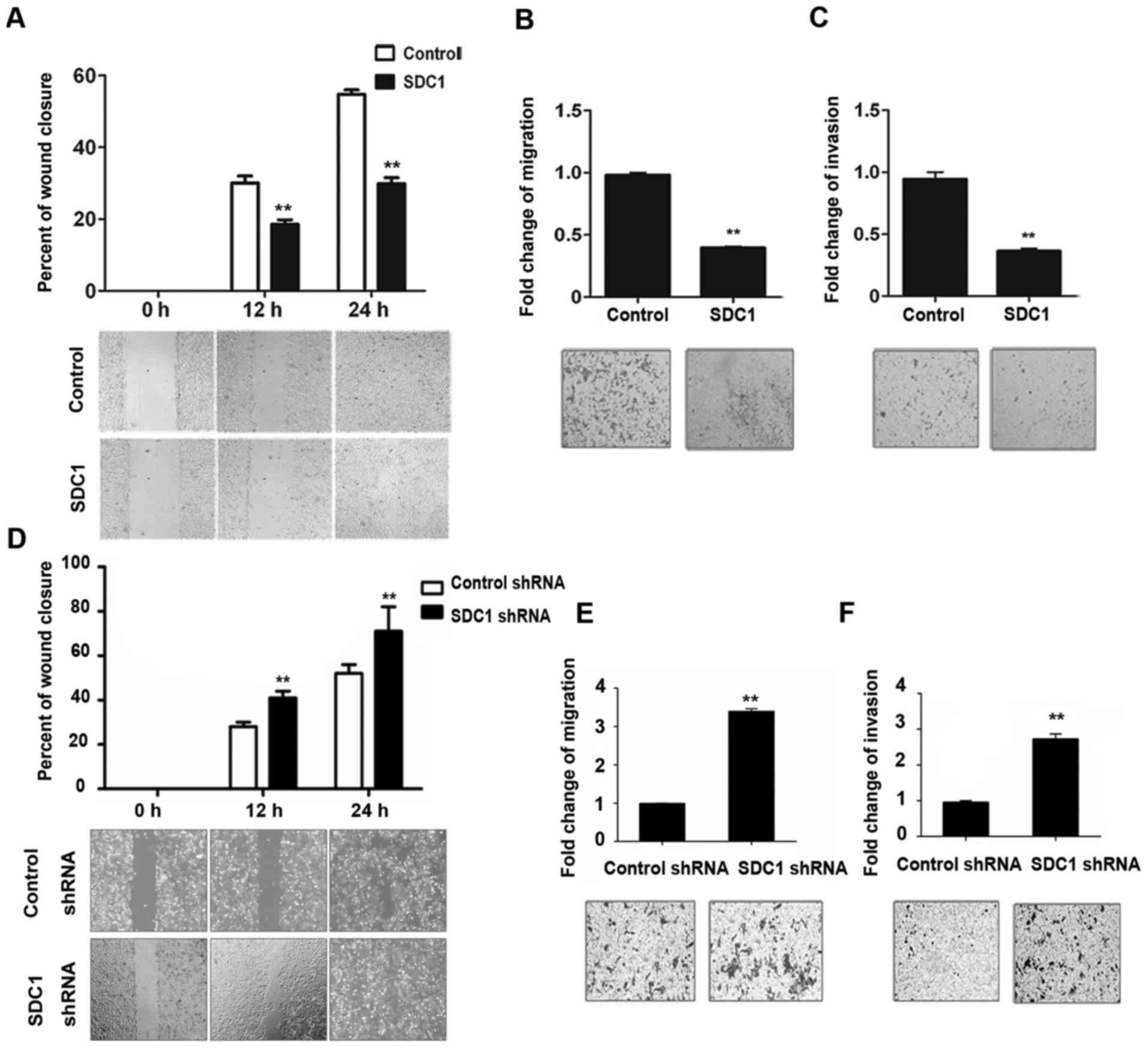
SDC1 suppresses migration and invasion
in oral cancer cells
Subsequently, we explored the role of SDC1 in cell
migration and invasion. As displayed in Fig. 2A, the wound healing rate of
Tca8113-SDC1 cells was only half of that observed in the control
group 24 h after the generation of the wound. Transwell migration
and invasion assays revealed that the abilities of Tca8113-SDC1
cells to migrate and invade were much lower than those of the
control cells (Fig. 2B and C).
Opposite results were obtained upon SDC1 RNA interference in oral
cancer cells: As displayed in Fig.
2D, the wound healing rate of SDC1-knockdown cells was higher
than that of the control group, indicating that SDC1-knockdown
cells had greater migratory ability. In addition, SDC1 knockdown in
ACC2 cells enhanced the migratory and invasive properties of the
cells (Fig. 2E and F).
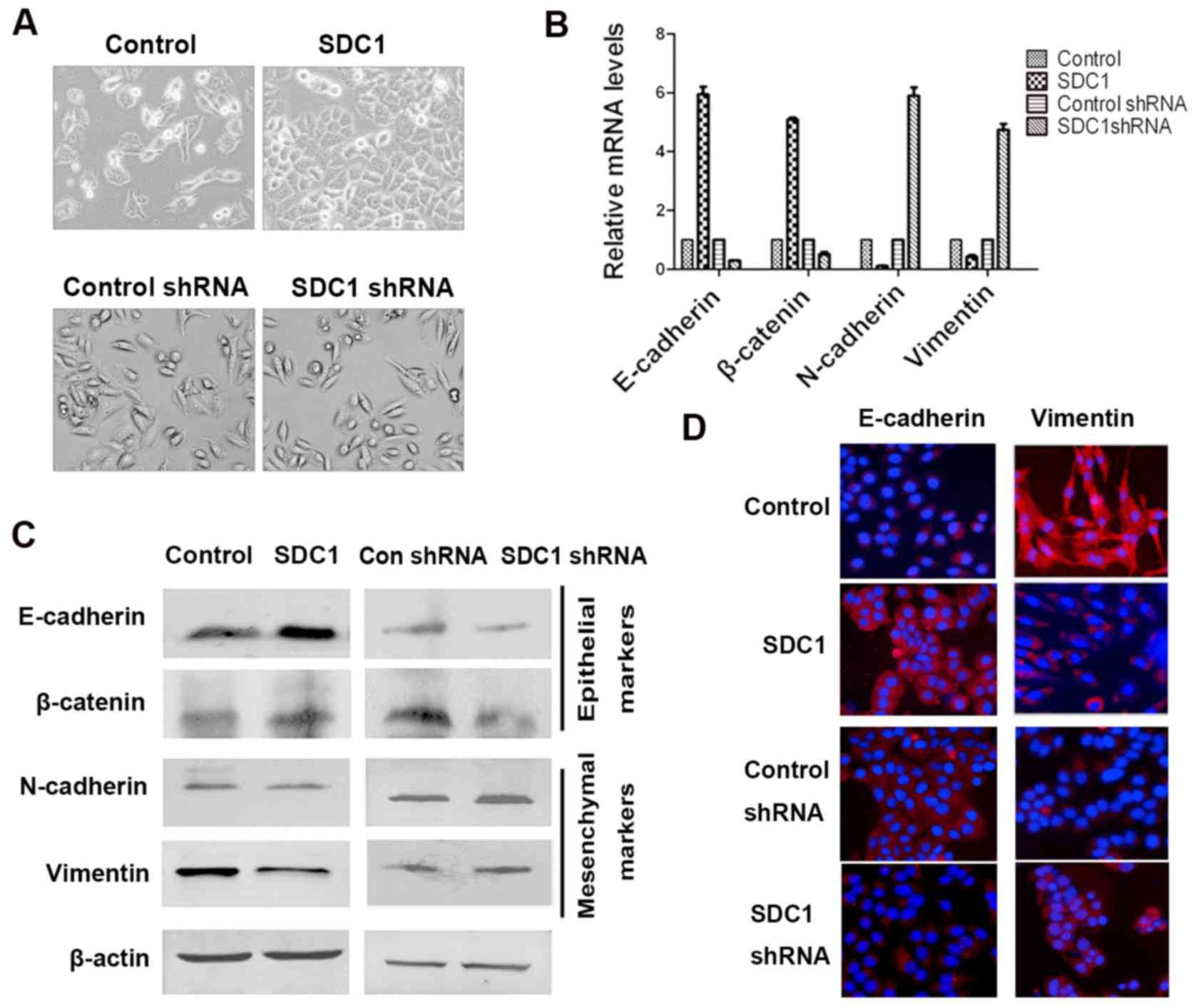
SDC1 regulates EMT
We intended to clarify the biological role of SDC1
imbalance in the development of oral cancer. We first examined
whether the overexpression of SDC1 inhibited EMT in Tca8113 human
tongue squamous cell carcinoma cells, which express low levels of
SDC1. Tca8113 cells expressing a control vector displayed a
spindle-like, fibroblastic phenotype. In contrast, Tca8113-SDC1
cells, overexpressing SDC1, demonstrated significantly polarized
epithelial cell morphology with strong intercellular cell adhesion
(Fig. 3A, upper panel). In addition
to morphological changes, overexpression of SDC1 caused changes in
the expression of epithelial and mesenchymal markers. Specifically,
both at the mRNA and protein levels, the expression of E-cadherin,
an epithelial marker, in Tca8113-SDC1 cells was significantly
upregulated, whereas the expression levels of N-cadherin and
vimentin, mesenchymal markers, were greatly reduced (Fig. 3B and C). The observed change was
further validated by the examination of the subcellular
localization of proteins by immunofluorescent staining. As
expected, immunofluorescence microscopy revealed high E-cadherin
staining intensity in the cell membrane of Tca8113-SDC1 cells,
whereas only weak membrane staining was noted in the control cells.
Staining for vimentin exhibited an opposite pattern (Fig. 3D).
Subsequently, we examined whether the inhibition of
endogenous SDC1 expression induced EMT progression in oral cancer
cells. ACC2 cells expressing shSDC1 displayed spindle-like,
fibroblastic morphology (Fig. 3A,
bottom panel). Additionally, using qPCR and western blotting, we
demonstrated that after SDC1 knockdown in ACC2 cells, the
expression of mesenchymal markers such as N-cadherin and vimentin
was higher, whereas the expression of the epithelial markers
E-cadherin and occludin (data not shown) was lower than that in
control cells (Fig. 3B and C). We
obtained similar results in immunofluorescence experiments. We
observed that, upon SDC1 knockdown, E-cadherin staining intensity
was attenuated, whereas staining for vimentin was significantly
enhanced (Fig. 3D). These results
indicated that inhibition of SDC1 expression in oral cancer cells
increased cell migration and invasion and induced EMT.
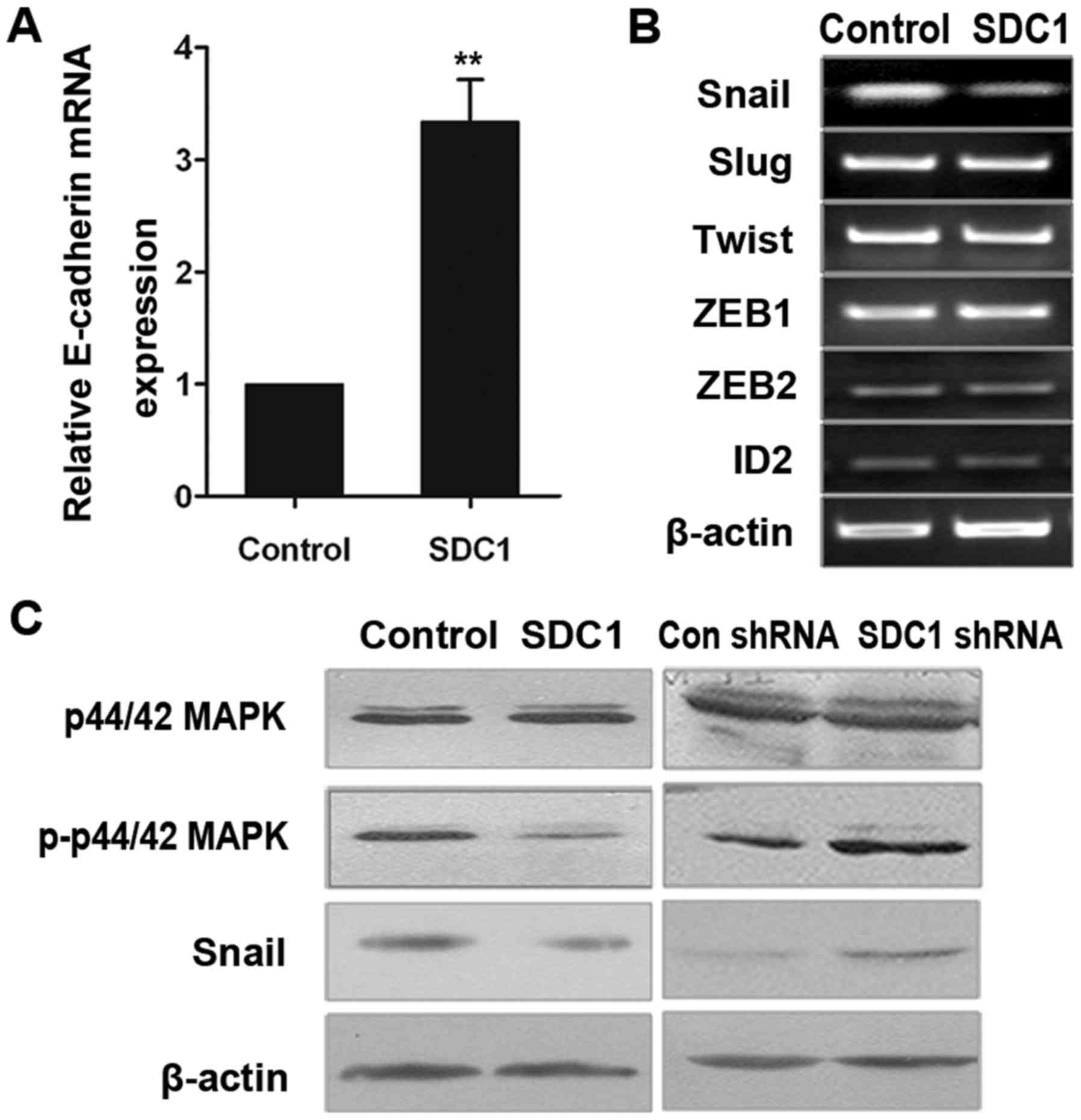
SDC1 regulates EMT through the
ERK-Snail signaling
We found that the overexpression of SDC1 increased
the levels of E-cadherin mRNA expression (Fig. 4A). Snail, Slug, Twist and ZEB are
transcription factors that control the expression of E-cadherin in
EMT (7,12,13).
Therefore, we examined whether the expression of these factors is
affected by SDC1 levels in oral cancer cells. qPCR analysis
revealed that the expression levels of ZEB1, ZEB2, inhibitor of DNA
binding (ID)2, Slug and Twist were similar in Tca8113-SDC1 and
control cells (Fig. 4B), whereas
Snail expression level was significantly lower in Tca8113-SDC1
cells. Western blot analysis confirmed these results (Fig. 4C, third line).
It is known that the activation of the ERK signaling
pathway induces Snail expression (21,22).
Therefore, we examined whether the ERK1 signaling pathway is
involved in the regulation of Snail expression mediated by SDC1.
Western blot analysis revealed that the overexpression of SDC1
inhibited the phosphorylation of ERK and suppressed Snail
expression (Fig. 4C, left panel).
In contrast, the levels of phosphorylated ERK and Snail in
shSDC1-ACC2 cells were significantly higher than those in the
control cells. These results indicate that the ERK signaling
pathway may function downstream of SDC1 (Fig. 4C, right panel).
Collectively, our data indicated that downregulation
of endogenous SDC1 activated the ERK cascade, upregulated Snail
expression and inhibited E-cadherin expression. These changes may
enhance the understanding of the relationship between EMT and human
oral cancer invasion.
Discussion
It has been previously demonstrated that the levels
of SDC1 expression in cancer cells inversely correlate with the
extent of tumor differentiation and prognosis. In the present
study, we investigated the role of SDC1 in the progression of oral
cancer and found that SDC1 inhibited EMT and thereby affected the
infiltration of tumor cells in the oral cavity tissues.
SDC1 basal expression level and cell location are
significant for understanding the occurrence, development,
diffusion and infiltration of tumors. Although SDC1 has multiple
functions, its mechanism of action and precise role in oral cancer
has so far remained unclear. An important finding of the present
study was that the oral cancer cell lines KB, Tca8113, ACC2 and
CAL-27 exhibited low levels of SDC1 expression. As displayed in
Fig. 1, the expression of SDC1 in
all four oral cancer cell lines was significantly lower than that
in functionally similar hPDL fibroblasts at both mRNA and protein
levels. Ectopic overexpression of SDC1 led to the suppression of
migration, invasion and proliferation of oral cancer cells. Our
results were consistent with the observations of Kurokawa et
al (23), who reported that
decreased expression of SDC1 could be an effective marker of the
histological grade of malignancy before deep tumor invasion of oral
squamous cell carcinoma. Similarly, Muramatsu et al
(24) examined the expression of
SDC1 in seven different human oral cancer cell lines (HSC2, HSC3,
HSC4, Ca9-22, SAS, KB and BSC-OF) and found that SDC1 was involved
in the growth and invasiveness of tumor cells (24). These studies indicated that
manipulating SDC1 levels may be useful to inhibit the progression
of oral cancer. However, the functions of SDC1 in patients and
mouse models still need to be further investigated.
EMT has been demonstrated to promote tumor invasion
and metastasis by conferring a mesenchymal cell phenotype to cancer
cells. It has been previously demonstrated that EMT is important
for oral cancer progression (25–27).
In the present study, we observed that overexpression of SDC1
resulted in changes in cell morphology and attenuated the molecular
manifestations of EMT. In contrast, suppression of endogenous SDC1
expression inhibited the expression of E-cadherin and promoted EMT
progression in oral cancer cells. These findings revealed that SDC1
may be a key negative regulator of EMT. Consistent with our
results, previous studies have reported the role of SDC1 in EMT.
Leppa et al (28) found that
SDC1 overexpression imparted epithelial-like morphology on
tumorigenic mammary cell lines. Transfection of mammary cell lines
with antisense RNA specific for E-cadherin suppressed SDC1
expression. Conversely, transfection with antisense SDC1 led to
downregulation of the expression of E-cadherin. Simultaneous loss
of SDC1 and E-cadherin expression was observed in the embryonic
palate during EMT (29). Masola
et al (30) reported that
the interplay between heparanase and SDC1 is important fibroblast
growth factor-2-induced EMT in renal tubular cells. Additionally,
SDC1 expression pattern in prostate cancer indicated the
involvement of this protein in EMT and tumor progression (31). The aforementioned data indicated
that SDC1 expression levels negatively correlated with EMT
progression and our results are in agreement with these
findings.
Furthermore, the findings of the present study
demonstrated that SDC1 knockdown activated the ERK signaling
pathway, upregulated Snail expression and inhibited E-cadherin
expression. These changes ultimately led to the occurrence of EMT
in human oral cancer cells, thereby making tumor cells more
aggressive. Several studies revealed that SDC1 and ERK/Snail
signaling have a close relationship in tumor development process.
Poblete et al (32) analyzed
the expression of Snail, SDC1 and other EMT markers in a tissue
microarray of prostate cancer samples and prostate cancer cell
lines and, consistent with our results, demonstrated that increased
Snail expression and low SDC1 levels were associated with high
Gleason grade. Additionally, it is known that activation of the ERK
signaling pathway can induce Snail expression in human breast and
gastric cancer, and lung adenocarcinoma (21,22).
In contrast, some previous studies have indicated that increased
ERK activity enhanced SDC1 expression. Heidari-Hamedani et
al (33) reported that ERK1/2
activity was enhanced six-fold upon SDC1 overexpression in
malignant mesothelioma. Ju et al (34) found that SDC1/integrin interaction
was essential in the activation of ERK I/II by insulin in
osteoblast cells. These discrepancies may be due to distinct
functions of SDC1 in different types of tumors. SDC1 may play an
oncogenic function in breast cancer, lung cancer and glioma,
whereas may be a tumor suppressor in prostate cancer and oral
cancer. Further investigations to address these issues are
required.
In conclusion, we demonstrated that SDC1 expression
is downregulated in oral cancer cell lines. Overexpression of SDC1
reduced the expression of mesenchymal markers in oral cancer cells,
increased the expression of epithelial markers and inhibited
invasion, migration and proliferation of oral cancer cells.
Knockdown of endogenous SDC1 led to morphological transformation of
cells to mesenchymal phenotype, increased the expression levels of
mesenchymal markers and enhanced cell migration, invasion and
proliferation. Furthermore, knockdown of SDC1 in oral cancer cells
activated the ERK signaling pathway, upregulated the expression of
the EMT-inducing transcription factor Snail and inhibited the
expression of E-cadherin. The results of the present study will
help to elucidate the mechanism by which SDC1 affects EMT in tumor
progression. In addition, the present study provided useful
insights for the potential use of SDC1 as a molecular target for
the treatment of oral cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by a grant
from National Natural Science Foundation of China (nos. 81402395,
31401086 and 81402338), Jilin Province Natural Science Foundation
(no. 2012150780).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
TZ and CK conceived and designed the study. TQ
participated in the concept of the study providing experimental
ideas for the mechanism research part. XW, CK, JH and XZ performed
the experiments. TQ provided some financial support for the
experiment. CK and XW wrote the paper. CK, TZ, TQ and XW reviewed
and edited the manuscript. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The present study does not contain any studies with
human participants or animals performed by any of the authors.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang B, Zhang S, Yue K and Wang XD: The
recurrence and survival of oral squamous cell carcinoma: A report
of 275 cases. Chin J Cancer. 32:614–618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Exarchos KP, Goletsis Y, Poli T and
Fotiadis DI: Gene expression profiling towards the prediction of
oral cancer reoccurrence. Conf Proc IEEE Eng Med Biol Soc. 2011:pp.
8307–8310. 2011; PubMed/NCBI
|
|
4
|
Saintigny P, Zhang L, Fan YH, El-Naggar
AK, Papadimitrakopoulou VA, Feng L, Lee JJ, Kim ES, Ki Hong W and
Mao L: Gene expression profiling predicts the development of oral
cancer. Cancer Prev Res. 4:218–229. 2011. View Article : Google Scholar
|
|
5
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thiery JP: Epithelial-mesenchymal
transitions in cancer onset and progression. Bull Acad Natl Med.
193:1969–1979. 2009.(In French). PubMed/NCBI
|
|
7
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
David CJ, Huang YH, Chen M, Su J, Zou Y,
Bardeesy N, Iacobuzio-Donahue CA and Massagué J: TGF-beta Tumor
suppression through a Lethal EMT. Cell. 164:1015–1030. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vincan E and Barker N: The upstream
components of the Wnt signalling pathway in the dynamic EMT and MET
associated with colorectal cancer progression. Clin Exp Metastasis.
25:657–663. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou J, Jain S, Azad AK, Xu X, Yu HC, Xu
Z, Godbout R and Fu Y: Notch and TGFβ form a positive regulatory
loop and regulate EMT in epithelial ovarian cancer cells. Cell
Signal. 28:838–849. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kong C, Wang C, Wang L, Ma M, Niu C, Sun
X, Du J, Dong Z, Zhu S, Lu J and Huang B: NEDD9 is a positive
regulator of epithelial-mesenchymal transition and promotes
invasion in aggressive breast cancer. PLoS One. 6:e226662011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during Snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shih JY and Yang PC: The EMT regulator
slug and lung carcinogenesis. Carcinogenesis. 32:1299–1304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Harada K, Masuda S, Hirano M and Nakanuma
Y: Reduced expression of syndecan-1 correlates with histologic
dedifferentiation, lymph node metastasis, and poor prognosis in
intrahepatic cholangiocarcinoma. Hum Pathol. 34:857–863. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khotskaya YB, Dai Y, Ritchie JP, MacLeod
V, Yang Y, Zinn K and Sanderson RD: Syndecan-1 is required for
robust growth, vascularization, and metastasis of myeloma tumors in
vivo. J Biol Chem. 284:26085–26095. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Anttonen A, Kajanti M, Heikkilä P,
Jalkanen M and Joensuu H: Syndecan-1 expression has prognostic
significance in head and neck carcinoma. Br J Cancer. 79:558–564.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maeda T, Alexander CM and Friedl A:
Induction of syndecan-1 expression in stromal fibroblasts promotes
proliferation of human breast cancer cells. Cancer Res. 64:612–621.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo Q, Yang X, Ma Y and Ma L: Syndecan-1
serves as a marker for the progression of epithelial ovarian
carcinoma. Eur J Gynaecol Oncol. 36:506–513. 2015.PubMed/NCBI
|
|
19
|
Giordano RJ: Heparanase-2 and syndecan-1
in colon cancer: The ugly ducklings or the beautiful swans? Eur J
Gastroenterol Hepatol. 20:716–718. 2008.PubMed/NCBI
|
|
20
|
Moon A, Kim MS, Kim TG, Kim SH, Kim HE,
Chen YQ and Kim HR: H-ras, but not N-ras, induces an invasive
phenotype in human breast epithelial cells: A role for MMP-2 in the
H-ras-induced invasive phenotype. Int J Cancer. 85:176–181. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hsu YL, Hou MF, Kuo PL, Huang YF and Tsai
EM: Breast tumor-associated osteoblast-derived CXCL5 increases
cancer progression by ERK/MSK1/Elk-1/snail signaling pathway.
Oncogene. 32:4436–4447. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li S, Lu J, Chen Y, Xiong N, Li L, Zhang
J, Yang H, Wu C, Zeng H and Liu Y: MCP-1-induced ERK/GSK-3β/Snail
signaling facilitates the epithelial-mesenchymal transition and
promotes the migration of MCF-7 human breast carcinoma cells. Cell
Mol Immunol. 14:621–630. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kurokawa H, Zhang M, Matsumoto S,
Yamashita Y, Tanaka T, Takamori K, Igawa K, Yoshida M, Fukuyama H,
Takahashi T and Sakoda S: Reduced syndecan-1 expression is
correlated with the histological grade of malignancy at the deep
invasive front in oral squamous cell carcinoma. J Oral Pathol Med.
35:301–306. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Muramatsu T, Saitoh M, Ro Y, Uekusa T,
Iwamura E, Ohta K, Kohno Y, Abiko Y and Shimono M: Inhibition of
syndecan-1 expression and function in oral cancer cells. Oncol Rep.
20:1353–1357. 2008.PubMed/NCBI
|
|
25
|
Patel S, Shah K, Mirza S, Daga A and Rawal
R: Epigenetic regulators governing cancer stem cells and
epithelial-mesenchymal transition in oral squamous cell carcinoma.
Curr Stem Cell Res Ther. 10:140–152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vered M, Dayan D, Yahalom R, Dobriyan A,
Barshack I, Bello IO, Kantola S and Salo T: Cancer-associated
fibroblasts and epithelial-mesenchymal transition in metastatic
oral tongue squamous cell carcinoma. Int J Cancer. 127:1356–1362.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chang CJ, Hsu CC, Chang CH, Tsai LL, Chang
YC, Lu SW, Yu CH, Huang HS, Wang JJ, Tsai CH, et al: Let-7d
functions as novel regulator of epithelial-mesenchymal transition
and chemoresistant property in oral cancer. Oncol Rep.
26:1003–1010. 2011.PubMed/NCBI
|
|
28
|
Leppä S, Vleminckx K, Van Roy F and
Jalkanen M: Syndecan-1 expression in mammary epithelial tumor cells
is E-cadherin-dependent. J Cell Sci. 109:1393–1403. 1996.PubMed/NCBI
|
|
29
|
Sun D, McAlmon KR, Davies JA, Bernfield M
and Hay ED: Simultaneous loss of expression of syndecan-1 and
E-cadherin in the embryonic palate during epithelial-mesenchymal
transformation. Int J Dev Biol. 42:733–736. 1998.PubMed/NCBI
|
|
30
|
Masola V, Gambaro G, Tibaldi E, Brunati
AM, Gastaldello A, D'Angelo A, Onisto M and Lupo A: Heparanase and
syndecan-1 interplay orchestrates fibroblast growth
factor-2-induced epithelial-mesenchymal transition in renal tubular
cells. J Biol Chem. 287:1478–1488. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Contreras HR, Ledezma RA, Vergara J,
Cifuentes F, Barra C, Cabello P, Gallegos I, Morales B, Huidobro C
and Castellón EA: The expression of syndecan-1 and −2 is associated
with Gleason score and epithelial-mesenchymal transition markers,
E-cadherin and beta-catenin, in prostate cancer. Urol Oncol.
28:534–540. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Poblete CE, Fulla J, Gallardo M, Muñoz V,
Castellón EA, Gallegos I and Contreras HR: Increased SNAIL
expression and low syndecan levels are associated with high Gleason
grade in prostate cancer. Int J Oncol. 44:647–654. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heidari-Hamedani G, Vivès RR, Seffouh A,
Afratis NA, Oosterhof A, van Kuppevelt TH, Karamanos NK, Metintas
M, Hjerpe A, Dobra K and Szatmári T: Syndecan-1 alters heparan
sulfate composition and signaling pathways in malignant
mesothelioma. Cell Signal. 27:2054–2067. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ju Ha H and Kim SJ: Association of insulin
receptor and syndecan-1 by insulin with activation of ERK I/II in
osteoblast-like UMR-106 cells. J Recept Signal Transduct Res.
33:37–40. 2013. View Article : Google Scholar : PubMed/NCBI
|