Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer among men and the second most common cause of cancer
mortality in both sexes (1,2). Approximately 70–90% of HCC patients
have a background of chronic liver disease or liver cirrhosis.
Major risk factors for liver cirrhosis are chronic infection with
hepatitis B or C virus, alcoholic liver disease and non-alcoholic
steatohepatitis. The precise molecular mechanisms in
hepatocarcinogenesis have not been fully elucidated, however a
variety of organ microenvironmental factors during chronic
inflammation, such as viral proteins, cytokines and reactive oxygen
or nitrogen species, affect carcinogenesis through the genetic or
epigenetic activation of oncogenes and/or inactivation of tumor
suppressor genes. These genetic and epigenetic alterations work
synergistically, leading to a multistep developmental pathway for
HCC (3,4).
In regard to genetic alterations, whole-genome
sequencing has revealed that the genes involved in chromatin
regulation, such as ARID1B, ARID2, MLL3 and MLL, were
highly mutated in HCC, although no common somatic gene mutations
were found in multicentric tumor pairs from that study (5). Whole genome sequencing of hepatitis B
virus-associated HCC indicated β-catenin (CTNNB1) to be the
most frequently mutated oncogene (15.9%), with TP53 being
the most frequently mutated tumor-suppressor gene (35.2%) (6). On the contrary, the epigenetic
regulation of gene expression represented a complex crosstalk of
DNA methylation, histone modification, chromatin remodeling, and
non-coding RNAs. Of these, DNA methylation led to stable gene
silencing, primarily by the covalent modification of cytosine
residues within CpG dinucleotides.
In cancer cells, two kinds of aberrant methylation
have been observed. The first one is a genome-wide hypomethylation,
which causes chromosome instability, aberrant transcription or
transposable element reactivation and the other is a site-specific
hypermethylation, primarily of CpG islands located in or around,
gene promoter regions. Although most of the cytosine residues in
CpG islands were unmethylated (7,8), the
aberrant methylation of tumor-suppressor gene promoters has been
implicated in the development of a wide variety of cancers
(9). In HCC, gene silencing
associated with aberrant promoter hypermethylation was found in a
variety of genes, such as_CDKN2A (p16), RASSF1A
(Ras-association domain containing family 1), GSTP1
(glutathione S-transferase) and CDH1 (E-cadherin) (10,11).
Yoshikawa et al as well as other researchers have also
revealed, using restriction landmark genomic scanning, that the
suppressor of cytokine signaling-1 (SOCS-1), SOCS-3
and apoptotic speck protein-like (ASCL) genes are aberrantly
methylated (12–14). Our previous methylation analysis
(15) with methylation-specific PCR
revealed that Delta-like 3 (DLL3), a member of the
Delta/Serrate/Lag-2 family of ligands for the Notch receptor, was
frequently methylated in HCC cell lines. In the present study, we
investigated the expression of DLL3 in HCC tissues by
immunohistochemistry and confirmed the methylation status of the
DLL3 promoter in HCC cell lines using bisulfite sequencing.
Finally, we investigated the role of histone modification in the
regulation of DLL3 expression in HCC cell lines.
Materials and methods
Cell culture
The HuH2 and HuH4 HCC cell lines (generous gifts
from Dr. Hirohide Yoshikawa, Sasa Hospital, Tokyo, Japan) were
maintained in minimum essential medium (MEM; Sigma-Aldrich, St.
Louis, MO, USA) containing 10% fetal bovine serum (FBS; Invitrogen;
Thermo Fisher Scientific, Waltham, MA, USA) at 37°C under a 5%
CO2 atmosphere.
Tissue samples
Non-cancerous liver tissues were obtained from HCC
patients who underwent surgical resection in the Department of
General and Gastroenterological Surgery, Osaka Medical College
(Takatsuki, Japan). Informed written consents were obtained from
all patients and the study was conducted according to the
guidelines of the Ethics Committee of Osaka Medical College.
Immunohistochemical study of the DLL3
expression
Human tissue arrays containing HCC cells and
corresponding adjacent non-cancerous liver tissues (CSA4: Human,
liver cancer-metastasis-normal; SuperBioChips, Seoul, Korea) were
immunostained with anti-DLL3 antibody. Briefly, tissue array slides
were deparaffinized in xylene and hydrated using an ethanol series.
Endogenous peroxidase was blocked by incubating slides with 5 mM
periodic acid. Cell permeabilization was carried out by incubation
with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 5 min
at room temperature. After blocking non-specific binding with 10%
bovine serum albumin in PBS, the slides were incubated with rabbit
anti-human DLL3 polyclonal antibody (ARP47292_P050; Aviva Systems
Biology, San Diego, CA, USA), diluted 1:1,000, at 4°C overnight.
After washing with PBS, signals were detected using the Dako
EnVision™ Dual Link System-HRP kit (Agilent Technologies, Inc.,
Santa Clara, CA, USA), according to the manufacturer's protocol.
After counterstaining with hematoxylin, the slides were imaged by
microscopy (Eclipse E600; Nikon, Tokyo, Japan) and photographed
with a CCD camera (VB-7010; Keyence, Osaka, Japan). The staining
intensity of the DLL3 antibody was evaluated by three researchers
(YM, KM and AH) at separate times, and graded as either: (−),
Negative; (+), weak; (++), moderate; or (+++), strong.
Bisulfite sequencing of CpG islands in
the DLL3 promoter
Genomic DNA was purified from the HuH2 and HuH4 cell
lines and primary liver tissues were surgically resected, using the
QIAamp DNA Mini kit (Qiagen Inc., Valencia, CA, USA). Subsequently,
genomic DNA from each sample (1 µg) was subjected to bisulfite
treatment using the EpiTect Bisulfite kit (Qiagen), according to
the manufacturer's protocol. The target region of the DLL3
promoter, containing 26 CpG sites (nucleotides 103181-113850 in
AC011500), was divided into three parts, with each part amplified
twice by PCR using Platinum Taq DNA polymerase (Life
Technologies; Thermo Fisher Scientific, Inc.) and specific primers
(Fig. 1; Table I). PCR products were cloned into
pT7-Blue plasmid and 24 transformants were picked randomly. Colony
PCR was carried out using M13R and U19 primers. Eight
insert-positive clones were subsequently amplified in 1 ml of
growth media and plasmid DNA was purified for sequencing.
 | Table I.Primer sequences for bisulfite
sequencing. |
Table I.
Primer sequences for bisulfite
sequencing.
| F1 |
5′-TTATTATTTTGTATAGTTTTTA-3′ |
| F2 |
5′-GTATAGAGTATAAAGTTATAGG-3′ |
| F3 |
5′-GGTAGTTTTGGTTTATATTA-3′ |
| F4 |
5′-GTATAATTTTATATATAGGT-3′ |
| F5 |
5′-TTAGATATAAGGTTTGGAAGTTAG-3′ |
| R1 |
5′-TAATATAAACCAAAACTACC-3′ |
| R2 |
5′-ACCTATATATAAAATTATAC-3′ |
| R3 |
5′-CTAACTTCCAAACCTTATATCTAA-3′ |
| R4 |
5′-AAAAACCATAACCTTCTAAT-3′ |
Treatment with demethylating agent and
histone methyltransferase inhibitors
HuH2 cells were incubated in media containing either
1, 3 or 10 µM of the demethylating agent 5-azadeoxycitidine
(5-Aza-dC; Sigma-Aldrich) for 72 h. The culture media was exchanged
to a new media containing 5-Aza-dC on day 3. For the inhibition of
histone deacetylation, 1 or 3 µM of trichostatin A (TSA)
(Sigma-Aldrich) was added to the medium on day 3. For the
inhibition of H3K9me2 dimethylation, the selective G9a and G9a-like
protein histone lysine methyltransferase inhibitor BIX 01294 (Enzo
Life Sciences, Inc., Farmingdale, NY, USA) was used (16). To block the trimethylation of lysine
27 on histone H3 and of lysine 20 on histone H4, the lysine
methyltransferase EZH2 inhibitor 3-deazaneplanocin A hydrochloride
(DZNep; R&D Systems, Minneapolis, MN, USA) was used (17). DZNep (30 or 50 µM final
concentration) or BIX 01294 (1, 10 or 30 µM final concentration)
was added to the culture media, either alone or together with
5-Aza-dC and TSA, on day 3. On day 4, total RNA was purified using
the NucleoSpin RNA kit (Machery-Nagel GmbH, Düren, Germany)
according to the manufacturer's instructions and subjected to
real-time PCR.
Real-time PCR
Real-time PCR was carried out with the StepOne Plus™
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific)
using the PrimeScript One Step RT-PCR kit (Takara Bio, Inc., Otsu,
Japan), according to the manufacturer's instructions. The final 20
µl reaction mixture consisted of: 10 µl of 2X One Step RT-PCR
buffer, 0.4 µl of Takara Ex Taq HS (5 U/µl), 0.4 µl of PrimeScript
RT enzyme Mix II, 1 µl of the 20X primer-probe set, 0.4 µl of ROX
Reference Dye, 2 µl of total RNA (100 ng/ml), and 5.8 µl of RNase
Free dH2O. Pre-made primer and probe sets for
DLL3 (Hs01085096-m1) and GAPDH (Hs032929097) as an
internal standard, were purchased from Applied Biosystems (Foster
City, CA, USA). The reaction was carried out at 42°C for 5 min,
then at 95°C for 10 sec, followed by 40 cycles at 95°C for 5 sec
and 60°C for 30 sec. Relative quantification of the DLL3
expression was analyzed using the ΔΔCq method and the StepOne
Software v2.3 (Applied Biosystems) (16).
Western blot analysis
Expression of DLL3 in HuH2 and HuH4 cells was
analyzed by western blotting. For the DLL3 reactivation assay, HuH2
cells were cultured in media containing 1 µM of 5-Aza-dC for 72 h,
replaced once on day 3 and TSA was added to the media on day 3 to
give a final concentration of 1 µM. On day 4, the cells were lysed
in lysis buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1%
NP-40, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate and
protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim,
Germany). The supernatant was then subjected to sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
electroblotted onto a polyvinylidene difluoride membrane (EMD
Millipore, Billerica, MA, USA). The blot was blocked with 5% skim
milk in TBS for 1 h at room temperature and incubated with
1:100-diluted anti-DLL3 antibody (cat. no. ab103102; Abcam,
Cambridge, MA, USA) overnight at 4°C. After several washes, the
blot was incubated with 1:1,000-diluted HRP-conjugated anti-rabbit
IgG (cat. no. 7074; Cell Signaling Technology, Beverly, MA, USA)
for 1 h at room temperature. Immunoreactive bands were detected
with the Fusion FX chemiluminescence imaging system (Vilber
Lourmat, Marne La Vallée, France), using Western Lightning ECL Pro
(PerkinElmer, Waltham, MA, USA).
Statistical analysis
In all statistical analyses, normality of the data
was assessed using the Shapiro-Wilk test. Differences between HuH2
and HuH4 cell lines with respect to DLL3 mRNA expression
were tested by Wilcoxon test. For the comparison of data among more
than three groups, the Tukey-Kramer Honest Significant Difference
test was applied when the data were normally distributed. Instead,
when the data were not normally distributed, Dunn's non-parametric
test was performed.
Results
DLL3 expression in HCC cells
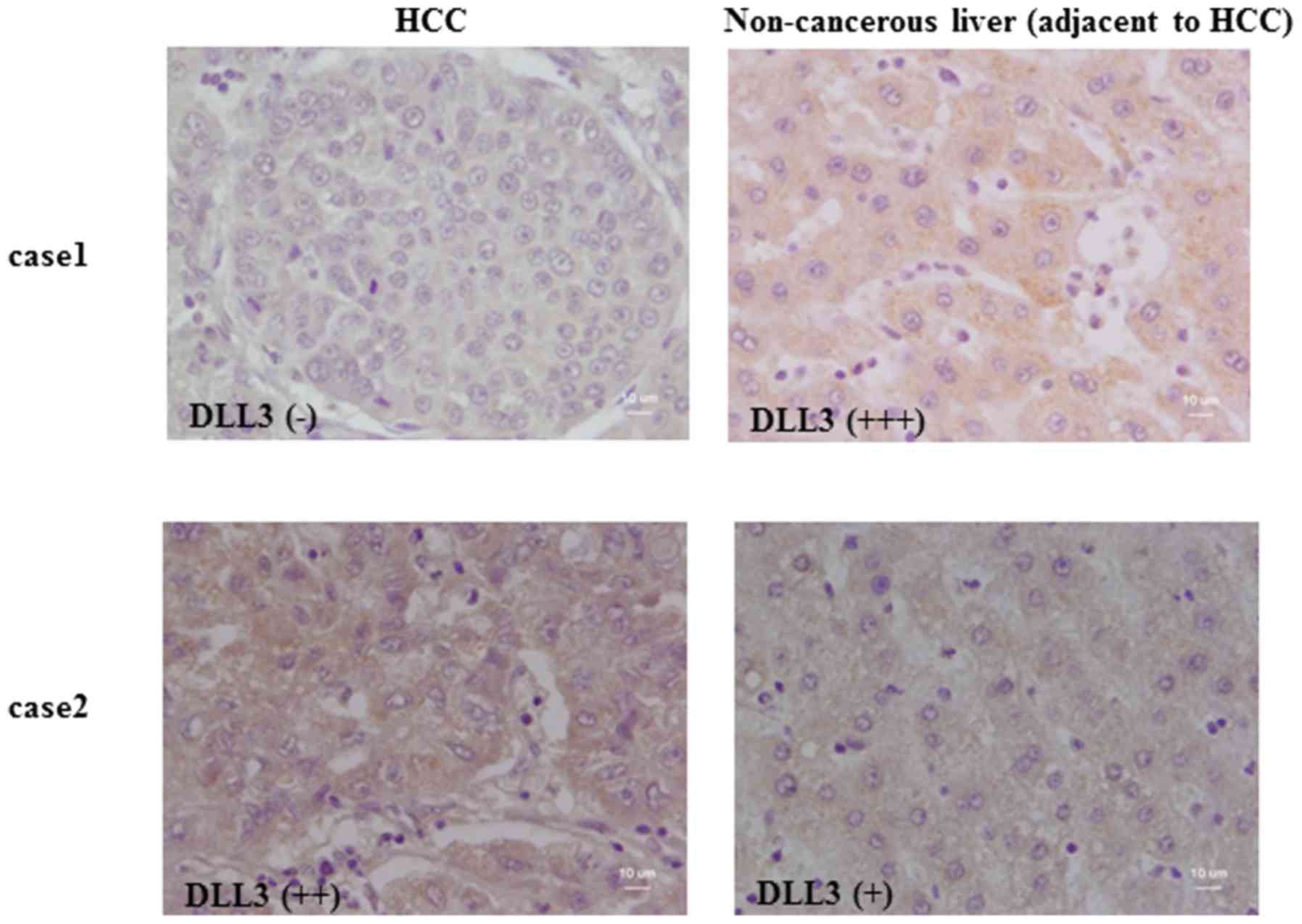
The expression of DLL3 in primary HCC samples and
adjacent liver tissue was investigated by immunohistochemistry of
tissue arrays. The results from two representative cases are
displayed in Fig. 2. In case 1,
strong immunoreactivity for DLL3 was detected in the cytoplasm of
non-cancerous hepatocytes, whereas no signal was detected in
adjacent cancer cells. In case 2, DLL3 was expressed both in
non-cancerous hepatocytes and the adjacent HCC cells. A summary of
the immunohistochemistry results from all cases is provided in
Table II. Moderate to strong
immunoreactivity against DLL3 was detected in non-cancerous
hepatocytes in all cases (9/9), whereas for half of the HCC cases
(18/36), no immunoreactivity could be detected. No DLL3
immunoreactivity was detected in two cases of cholangiocellular
carcinoma cells (data not shown).
| Table II.Summary of immunohistochemistry
results. |
Table II.
Summary of immunohistochemistry
results.
|
Immunoreactivity | Strong (+++) | Moderate (++) | Weak (+) | None (−) |
|---|
| Non-cancerous
liver | 5 | 4 | 0 | 0 |
| HCC | 0 | 9 | 9 | 18 |
Methylation analysis of the DLL3
promoter and expression of DLL3 in HCC cells
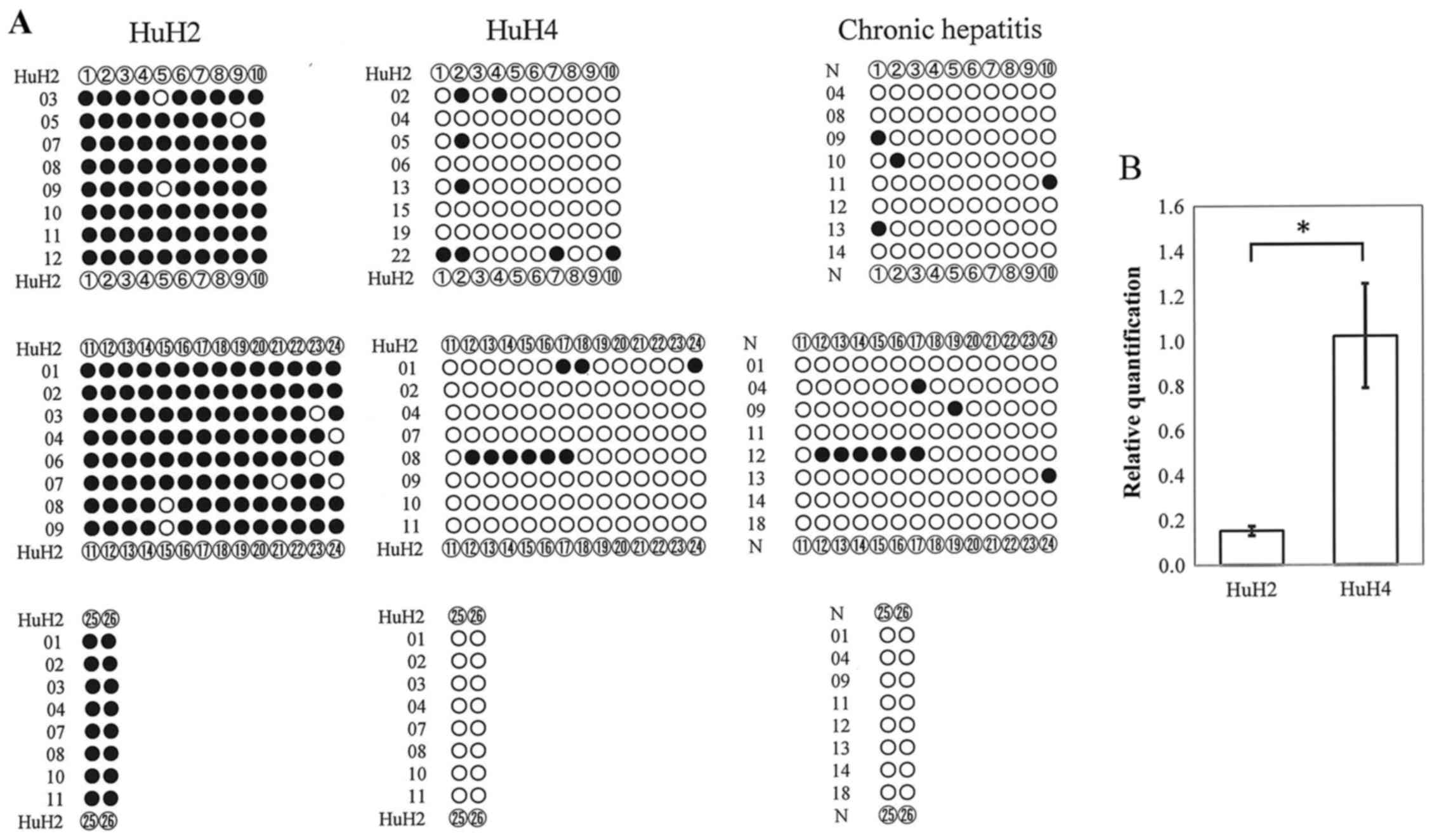
The methylation status of CpG sites presented in the
DLL3 promoter region was investigated by bisulfite
sequencing. In Fig. 1 tentative
nucleotide sequences of the target DLL3 promoter region after
bisulfite treatment are displayed. As displayed in Fig. 3A, CpG sites in the DLL3
promoter region were densely methylated (closed circles) in HuH2
cells in all randomly selected clones, whereas the CpG sites were
rarely methylated in clones derived from HuH4 cells or genomic DNA
from the liver tissues of chronic hepatitis patients (Fig. 3A). Real-time RT-PCR analysis
revealed that DLL3 mRNA expression in HuH4 cells was
significantly (P=0.005) higher than in HuH2 cells (Fig. 3B).
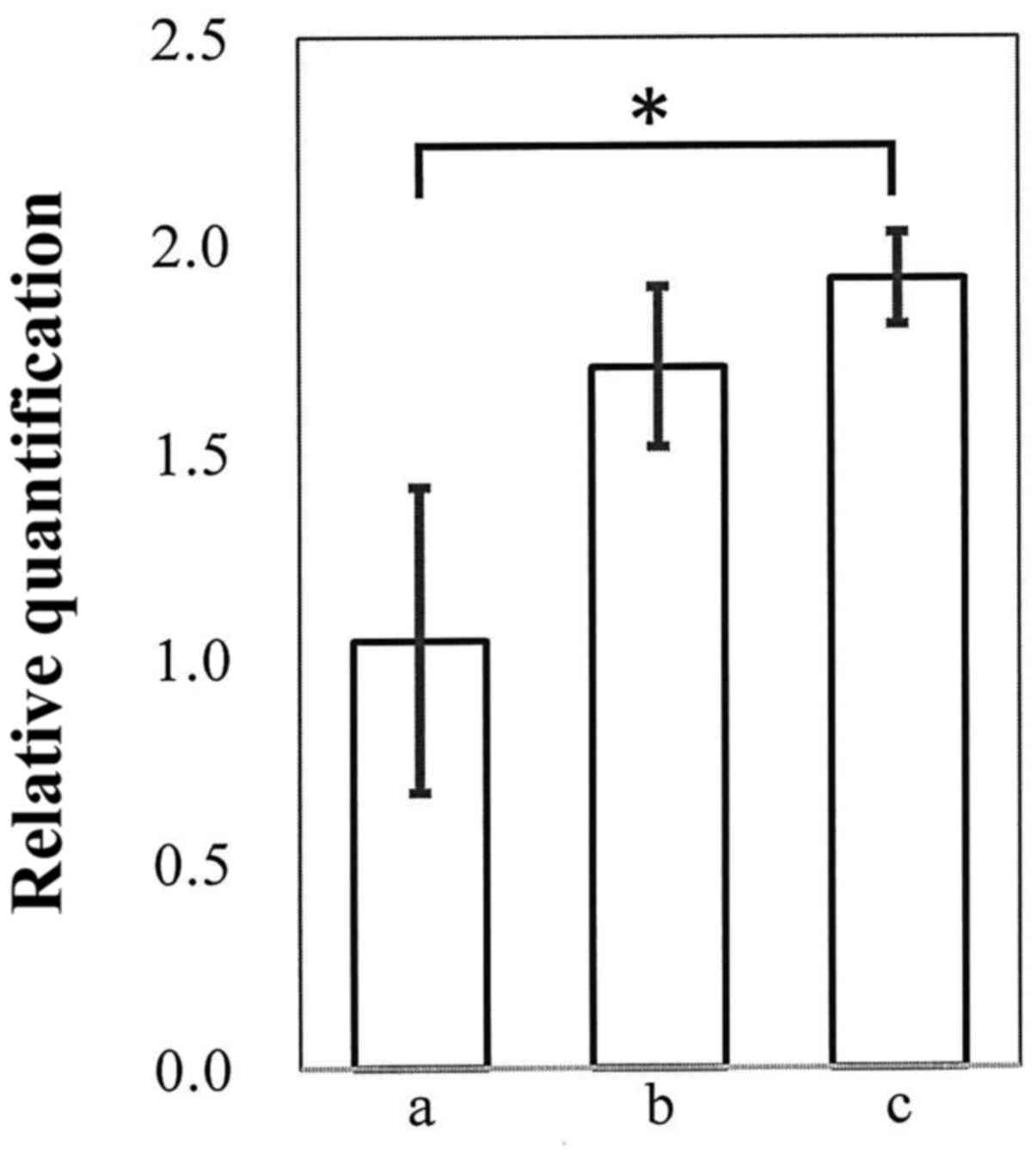
DLL3 reactivation using the
demethylating agent 5-Aza-dC
When the deoxycytidine analogue 5-Aza-dC is taken up
by the cells, it is incorporated into DNA and inhibits
methyltransferase activity, resulting in DNA demethylation.
Although demethylation by 5-Aza-dC induces reactivation of silent
genes, it can be toxic to cultured cells (17). Thus, we first investigated the
survival of HCC cells after administration of 5-Aza-dC to determine
the optimal dose for subsequent DLL3 reactivation assays. As
summarized in Table III, 5-Aza-dC
was cytotoxic to HCC cells in a dose-dependent manner. HuH2 cell
confluency was ~25% or less after 4 days of treatment with either 3
or 10 µM 5-Aza-dC, whereas it was 75% when HuH2 cells were treated
with 1 µM 5-Aza-dC. Thus, we evaluated the effect of 5-Aza-dC on
DLL3 reactivation at the concentration of 1 or 3 µM. We found that
when the HuH2 cells were treated with 1 or 3 µM 5-Aza-dC for 4
days, the expression of DLL3 mRNA was reactivated up to
two-fold. Treatment with 3 µM 5-Aza-dC induced a significant
(p=0.03) increase in DLL3 mRNA expression, although it also
had a cytotoxic effect (Fig.
4).
| Table III.Summary of cell viability. |
Table III.
Summary of cell viability.
| Treatment | Viability (%) |
|---|
| No treatment | 100 |
| 1 µM 5-Aza-dC | 75 |
| 3 µM 5-Aza-dC | 25 |
| 10 µM 5-Aza-dC | 10 |
| 1 µM TSA | 95 |
| 3 µM TSA | 95 |
| 1 µM 5-Aza-dC + 1
µM TSA | 55 |
| 1 µM 5-Aza-dC + 3
µM TSA | 75 |
| 30 µM DZNep | 100 |
| 50 µM DZNep | 100 |
| 1 µM 5-Aza-dC + 1
µM TSA + 30 µM DZNep | 25 |
| 1 µM 5-Aza-dC + 1
µM TSA + 50 µM DZNep | 30 |
| 1 µM BIX 01294 | 100 |
| 10 µM BIX
01294 | 100 |
| 1 µM 5-Aza-dC + 1
µM TSA +1 µM BIX 01294 | 40 |
| 1 µM 5-Aza-dC + 1
µM TSA +10 µM BIX 01294 | 30 |
DLL3 reactivation using inhibitors of
histone modification
The effects of histone deacetylation, histone H3
lysine 9 dimethylation and lysine 27 trimethylation on DLL3
expression were investigated using specific inhibitors. Treatment
with 1 or 3 µM of the histone deacetylase (HDAC) inhibitor TSA
alone, did not cause cellular toxicity in HuH2 cells (Table III). DLL3 mRNA expression
was significantly (P=0.0005) upregulated in the presence of 1 µM of
TSA, whereas 3 µM TSA had only a slight effect. When cells were
treated with 1 µM of TSA together with 1 or 3 µM (data not shown)
of 5-Aza-dC, cell confluency was around 50–70%, indicating that
cell damage was less serious. Notably, 1 µM of TSA in combination
with 5-Aza-dC revealed a marked synergistic effect on DLL3
mRNA expression (P<0.0001, compared to no treatment controls),
without inducing serious cytotoxicity (Fig. 5A).
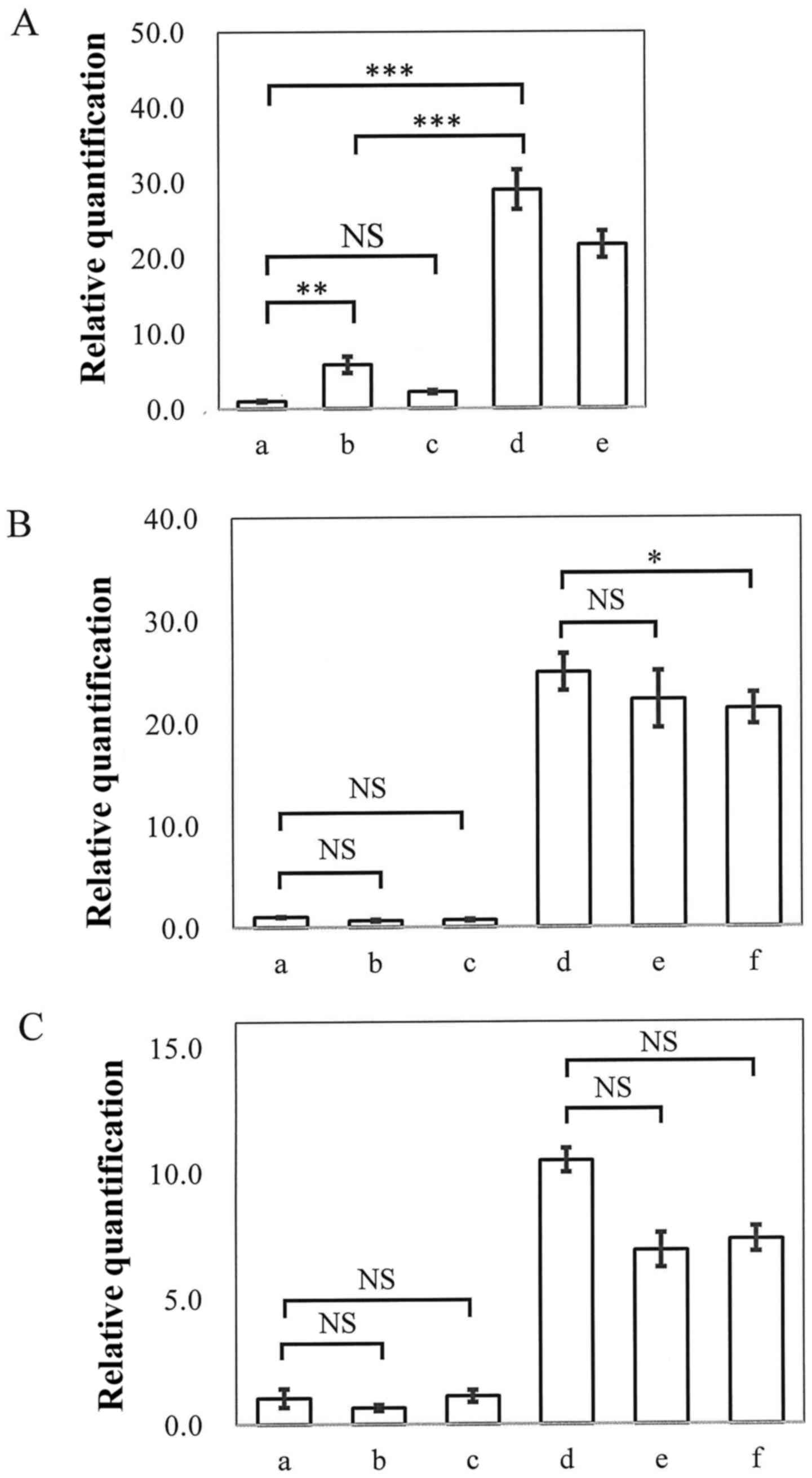 | Figure 5.DLL3 mRNA expression.
Real-time PCR was carried out and evaluated using the ΔΔCT method,
with GAPDH as an internal control. RNA was purified from HuH2 cells
that had been treated as follows: (A) Without TSA (a) or with 1 µM
TSA (b), 3 µM TSA (c), 1 µM 5-Aza-dC + 1 µM TSA (d), 1 µM 5-Aza-dC
+ 3 µM TSA (e). (B) Without treatment (a), or with 1 µM BIX 01274
(b), 10 µM BIX 01274 (c), 1 µM 5-Aza-dC + 1 µM TSA (d), 1 µM
5-Aza-dC + 1 µM TSA + 1 µM BIX 01274 (e), 1 µM 5-Aza-dC + 1 µM TSA
+ 10 µM BIX 01274 (f). (C) Without treatment (a), or with 30 µM
DZNep (b), 50 µM DZNep (c), 1 µM 5-Aza-dC + 1 µM TSA (d), 1 µM
5-Aza-dC + 1 µM TSA + 30 µM DZNep (e), 1 µM 5-Aza-dC + 1 µM TSA +
50 µM DZNep (f). Representative data, indicating relative mRNA
expression vs. untreated samples are shown; data represent the
means ± SD (n=5; ***P<0.001; *P<0.05; NS, not
significant). |
The H3 lysine 9 methyltransferase inhibitor BIX
01294 at 1 or 10 µM did not induce cell damage in HuH2 cells
(Table III), whereas the higher
dose (30 µM) was cytotoxic, with cell confluency being around 5% on
day 4 (data not shown). Treatment with BIX 01294 alone did not
affect DLL3 mRNA expression however and no synergistic
effect was observed when the cells were treated with BIX 01294 (1
or 3 µM) together with both 5-Aza-dC and TSA (Fig. 5B). Similarly, the inhibitor of
trimethylation of lysine 27 on histone H3 and lysine 20 on histone
H4 (DZNep) did not induce DLL3 reactivation in HCC cells. When
DZNep (30 or 50 µM) was administered together with both 5-Aza-dC (1
µM) and TSA (1 µM), DLL3 expression was not reactivated, but
rather suppressed, without affecting cytotoxicity (Fig. 5C; Table III).
Discussion
In the present study it was demonstrated that
downregulation of the expression of DLL3 occured in
hepatocarcinogenesis through epigenetic mechanisms, predominately
DNA methylation and histone acetylation. In mammals, DNA
methylation primarily occurs by covalent addition of a methyl-group
at the C5 position of cytosine residues in CpG dinucleotides.
Dinucleotide clusters of CpGs or CpG islands, are typically found
in, or near, the promoter regions of genes. Although most CpGs in
genomic DNA are methylated, CpG islands in gene promoter regions
remain unmethylated, allowing transcription to occur (18). In the present study, we detected
densely-methylated CpG sites in the DLL3 promoter region of
HuH2 HCC cells using bisulfite sequencing and demonstrated that
DLL3 expression could be reactivated by treatment with the
demethylating agent 5-Aza-dC. These results supported the data of
our previous study (15), generated
using methylation-specific PCR, that DLL3 expression was
silenced by DNA methylation in several HCC cell lines. Methylated
DNA itself inhibited transcriptional activation by blocking the
access of transcription factors to their binding sites (19). In addition, methylated DNA may be
specifically recognized by a set of methyl-CpG binding domain
proteins, leading to gene supression by the recruitment of
additional proteins, such as HDACs, to these loci (7).
DNA is packaged in the form of chromatin, with
nucleosomes (a basic structural unit of chromatin) being comprised
of histone octamers (each consists of two copies of each of the
four core histone proteins H2A, H2B, H3 and H4) that are wrapped by
a 146 bp length of DNA. The N- and C-terminal histone tails
protrude from the nucleosome and provide sites on specific residues
for covalent modification, such as acetylation, methylation,
phosphorylation, sumoylation and ubiquitylation (20). These modifications regulate
chromatin structure to provide binding sites for the recruitment of
other molecules (21). Among these
modifications, histone acetylation has been the most extensively
studied and it has been demonstrated that histone
acetylation/deacetylation is involved in transcriptional activation
- serving as a switch between supressive and permissive chromatin
states (22,23). Histone acetylation is enriched in
transcriptionally-active regions and is typically found on histone
H3 and H4 tails (21). Histone
acetylation and deacetylation is catalyzed by histone
acetyl-transferases (HATs) and HDACs, respectively and acetylation
status is thus regulated by a balance in the activities of these
enzymes. Acetylation of the histone N-terminus removes positive
charge and the overall charge of the histone tail thus becomes
neutral, reducing the affinity between DNA and histone and allowing
transcription factors to access promotor regions (23).
Acetylation of specific lysine residues on
particular histones is involved in the promotion of gene
transcription. For example, acetylation of lysine 9 on histone H3
(H3K9ac), mainly catalyzed by GCN5/PCAF acetyltransferase and/or
Tip 60 and Lysine 14 (H3K14ac), catalyzed by GCN5/PCAF, p300/CBP
and/or Myst3, can be observed (24). Conversly, deacetylation of histones
by HDACs has the opposite effect and negatively regulates
transcriptional activity. Thus far, 18 HDAC enzymes (HDACs 1–11,
and sirtuin 1–7) have been identified in mammals and can be
classified into four main Zn2+-dependent groups (classes
I, IIa, IIb and IV), as well as a group of nicotinamide
dehydrogenase-dependent class III HDACs (25,26).
Although histones are the most studied substrate for HDACs, there
are a wide variety of non-histone targets, including DNA-binding
transcription factors (p53, c-Myc and NF-κB), steroid receptors
(androgen receptor and estrogen receptor α), chaperone proteins
(HSP90), structural proteins (α-tubulin) and signaling mediators
(STAT3, Smad7 and β-catenin) (27).
The expression levels of HDACs vary by cell type and since they
play pivotal roles in cell proliferation and cell death, these
enzymes have become important therapeutic targets for treating
various types of cancer (28,29).
Thus far, a range of HDAC inhibitors have been identified, which
are either natural products or synthetic compounds and which have
differing target-specificities and activities. Although the precise
mechanism is unclear, some HDAC inhibitors are effective in
treating malignant diseases. Vorinostat (suberoylanilide hydroxamic
acid or SAHA) binds to the catalytic domain of HDACs and has been
demonstrated to inhibit the activity of HDAC1, 2, 3 and 6 (27,30).
Vorinostat induces apoptosis and has anti-proliferative effects
against cancer cell lines and was approved by the FDA in 2006 to be
used in the management of cutaneous T-cell lymphoma. TSA,
conversly, an antifungal antibiotic derived from
Streptomyces, is a potent reversible inhibitor of class I,
II and IV HDACs (27). TSA inhibits
both G1- and G2-phases of the mammalian cell cycle and has been
tested as a potential anticancer agent (31,32).
In the present study, as displayed in Figs. 3 and 4, treatment with 5-Aza-dC alone exerted
only a slight effect on DLL3 reactivation, whereas TSA
together with 5-Aza-dC exhibited a much more potent effect. We
further investigated the effect of siRNAs for HDAC1, 2 and 3 on
DLL3 reactivation, but neither individual nor synergistic
effects were observed (data not shown). These results indicated
that class II and/or class III HDACs may play an important role in
DLL3 activation.
Histone methylation, catalyzed by histone
methyltransferases, is another major chromatin modification and
causes transcriptional supression or activation depending on which
amino acids are methylated and the degree of methylation. Lysine is
able to be mono-, di- or trimethylated, with a methyl group
replacing each hydrogen atom of its NH3+
group. With a free NH2 and NH3+
group, arginine is able to be mono- or dimethylated (21). Active genes are typically associated
with di- or tri-methylation of H3 lysine 4 (H3K4me2/3) and H3K79me3
and supressed genes typically carry H3K9me2 and H3K27me3 (21,33).
Therefore, we investigated the involvement of H3K9me2 and H3K27me3
in silencing of the DLL3 expression. As displayed in
Fig. 4, neither DZNep, an inhibitor
of histone methyltransferase EZH2-mediated trimethylation of H3K27,
nor BIX 01294, an inhibitor of G9a histone methyltransferase with a
strong activity towards H3K9, had individual or synergistic effects
on DLL3 expression.
It has been reported that DLL3 is detected in the
Golgi apparatus and rarely on the cell surface (34). Thus, DLL3 does not bind cell surface
Notch receptors to activate Notch signaling, however, DLL3
suppressed Notch signaling in a cell-autonomous manner (34). Notch signaling plays an important
role in liver development by regulating a fate choice between
hepatocytes and biliary epithelial cells and in the regulation of
hepatoblast differentiation (35–37).
In addition, it has been reported that Notch signaling is activated
in human HCC and hepatocyte-specific expression of intracellular
domain of Notch promotes formation of liver tumors in mice
(38). Thus, silencing of DLL3 in
HCC hampers inactivation of Notch signaling, which may contribute
to hepatocarcinogenesis.
Although natural inducers of epigenetic aberrations
have not yet been fully elucidated, it has been reported that age
(39), chronic inflammation
(40–42) and viral (43) or bacterial infections (44) may induce such changes. As detailed
clinical information in regard to such factors such as the presence
of chronic inflammation or viral infections was not available for
the patient material included in the present study, further
investigation using clinical specimens will be necessary to clarify
the precise mechanisms that contribute to the epigenetic regulation
of DLL3 expression. Our preliminary data revealed that
infection of hepatitis virus affected the silencing of DLL3
expression in HCC (unpublished data), thus the levels of DLL3
expression in HCC in the present study may have been influenced by
the status of the viral infection (Fig.
1; cases 1 and 2).
In conclusion, both our previous and current results
indicated that in addition to DNA methylation, acetylation of
lysine residues, rather than the methylation of lysine, played a
pivotal role in post-transcriptional downregulation of DLL3
expression during hepatocarcinogenesis. Regulation of DLL3
expression through epigenetic mechanisms may thus be associated
with the development of Notch-dependent HCC, with DLL3
representing a potential molecular target for therapy.
Acknowledgements
The authors wish to thank Rintaro Oide for technical
assistance and Tadashi Kanayama for writing assistance.
References
|
1
|
Ryerson AB, Eheman CR, Altekruse SF, Ward
JW, Jemal A, Sherman RL, Henley SJ, Holtzman D, Lake A, Noone AM,
et al: Annual report to the nation on the status of cancer,
1975–2012, featuring the increasing incidence of liver cancer.
Cancer. 122:1312–1337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
de Martel C, Maucort-Boulch D, Plummer M
and Franceschi S: World-wide relative contribution of hepatitis B
and C viruses in hepatocellular carcinoma. Hepatology.
62:1190–1200. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Herath NI, Leggett BA and MacDonald GA:
Review of genetic and epigenetic alterations in
hepatocarcinogenesis. J Gastroenterol Hepatol. 21:15–21. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kondoh N, Wakatsuki T, Hada A, Shuda M,
Tanaka K, Arai M and Yamamoto M: Genetic and epigenetic events in
human hepatocarcinogenesis. Int J Oncol. 18:1271–1278.
2001.PubMed/NCBI
|
|
5
|
Fujimoto A, Totoki Y, Abe T, Boroevich KA,
Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al:
Whole-genome sequencing of liver cancers identifies etiological
influences on mutation patterns and recurrent mutations in
chromatin regulators. Nat Genet. 44:760–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kan Z, Zheng H, Liu X, Li S, Barber TD,
Gong Z, Gao H, Hao K, Willard MD, Xu J, et al: Whole-genome
sequencing identifies recurrent mutations in hepatocellular
carcinoma. Genome Res. 23:1422–1433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fatemi M, Pao MM, Jeong S, Gal-Yam EN,
Egger G, Weisenberger DJ and Jones PA: Footprinting of mammalian
promoters: Use of a CpG DNA methyltransferase revealing nucleosome
positions at a single molecule level. Nucleic Acids Res.
33:e1762005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Calvisi DF, Ladu S, Gorden A, Farina M,
Lee JS, Conner EA, Schroeder I, Factor VM and Thorgeirsson SS:
Mechanistic and prognostic significance of aberrant methylation in
the molecular pathogenesis of human hepatocellular carcinoma. J
Clin Invest. 117:2713–2722. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pogribny IP and Rusyn I: Role of
epigenetic aberrations in the development and progression of human
hepatocellular carcinoma. Cancer Lett. 342:223–230. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wahid B, Ali A, Rafique S and Idrees M:
New insights into the epigenetics of hepatocellular carcinoma.
Biomed Res Int. 2017:16095752017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshikawa H, Matsubara K, Qian GS, Jackson
P, Groopman JD, Manning JE, Harris CC and Herman JG: SOCS-1, a
negative regulator of the JAK/STAT pathway, is silenced by
methylation in human hepatocellular carcinoma and shows
growth-suppression activity. Nat Genet. 28:29–35. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Niwa Y, Kanda H, Shikauchi Y, Saiura A,
Matsubara K, Kitagawa T, Yamamoto J, Kubo T and Yoshikawa H:
Methylation silencing of SOCS-3 promotes cell growth and migration
by enhancing JAK/STAT and FAK signalings in human hepatocellular
carcinoma. Oncogene. 24:6406–6417. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kubo T, Yamamoto J, Shikauchi Y, Niwa Y,
Matsubara K and Yoshikawa H: Apoptotic speck protein-like, a highly
homologous protein to apoptotic speck protein in the pyrin domain,
is silenced by DNA methylation and induces apoptosis in human
hepatocellular carcinoma. Cancer Res. 64:5172–5177. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maemura K, Yoshikawa H, Yokoyama K, Ueno
T, Kurose H, Uchiyama K and Otsuki Y: Delta-like 3 is silenced by
methylation and induces apoptosis in human hepatocellular
carcinoma. Int J Oncol. 42:817–822. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tachibana M, Sugimoto K, Fukushima T and
Shinkai Y: Set domain-containing protein, G9a, is a novel
lysine-preferring mammalian histone methyltransferase with
hyperactivity and specific selectivity to lysines 9 and 27 of
histone H3. J Biol Chem. 276:25309–25317. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Glazer RI, Knode MC, Tseng CK, Haines DR
and Marquez VE: 3-Deazaneplanocin A: A new inhibitor of
S-adenosylhomocysteine synthesis and its effects in human colon
carcinoma cells. Biochem Pharmacol. 35:4523–4527. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jüttermann R, Li E and Jaenisch R:
Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated
primarily by covalent trapping of DNA methyltransferase rather than
DNA demethylation. Proc Natl Acad Sci USA. 91:11797–11801. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Choy MK, Movassagh M, Goh HG, Bennett MR,
Down TA and Foo RS: Genome-wide conserved consensus transcription
factor binding motifs are hyper-methylated. BMC Genomics.
11:5192010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang T, Cooper S and Brockdorff N: The
interplay of histone modifications-writers that read. EMBO Rep.
16:1467–1481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin B, Li Y and Robertson KD: DNA
methylation: Superior or subordinate in the epigenetic hierarchy?
Genes Cancer. 2:607–617. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eberharter A and Becker PB: Histone
acetylation: A switch between repressive and permissive chromatin.
Second in review series on chromatin dynamics. EMBO Rep. 3:224–229.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Farria A, Li W and Dent SY: KATs in
cancer: Functions and therapies. Oncogene. 34:4901–4913. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
de Ruijter AJ, van Gennip AH, Caron HN,
Kemp S and van Kuilenburg AB: Histone deacetylases (HDACs):
Characterization of the classical HDAC family. Biochem J.
370:737–749. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Delcuve GP, Khan DH and Davie JR: Roles of
histone deacetylases in epigenetic regulation: emerging paradigms
from studies with inhibitors. Clinical Epigenetics. 4:52012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: Molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eckschlager T, Plch J, Stiborova M and
Hrabeta J: Histone deacetylase inhibitors as anticancer drugs. Int
J Mol Sci. 18:E14142017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Richon VM: Cancer biology: Mechanism of
antitumour action of vorinostat (suberoylanilide hydroxamic acid),
a novel histone deacetylase inhibitor. Br J Cancer. 95(S1): S2–S6.
2006. View Article : Google Scholar
|
|
33
|
Li B, Carey M and Workman JL: The role of
chromatin during transcription. Cell. 128:707–719. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Geffers I, Serth K, Chapman G, Jaekel R,
Schuster-Gossler K, Cordes R, Sparrow DB, Kremmer E, Dunwoodie SL,
Klein T, et al: Divergent functions and distinct localization of
the Notch ligands DLL1 and DLL3 in vivo. J Cell Biol. 178:465–476.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kodama Y, Hijikata M, Kageyama R,
Shimotohno K and Chiba T: The role of notch signaling in the
development of intrahepatic bile ducts. Gastroenterology.
127:1775–1786. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ader T, Norel R, Levoci L and Rogler LE:
Transcriptional profiling implicates TGFbeta/BMP and Notch
signaling pathways in ductular differentiation of fetal murine
hepatoblasts. Mech Dev. 123:177–194. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zong Y, Panikkar A, Xu J, Antoniou A,
Raynaud P, Lemaigre F and Stanger BZ: Notch signaling controls
liver development by regulating biliary differentiation.
Development. 136:1727–1739. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Villanueva A, Alsinet C, Yanger K, Hoshida
Y, Zong Y, Toffanin S, Rodriguez-Carunchio L, Solé M, Thung S,
Stanger BZ, et al: Notch signaling is activated in human
hepatocellular carcinoma and induces tumor formation in mice.
Gastroenterology. 143:1660–1669.e7. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Issa JP, Ottaviano YL, Celano P, Hamilton
SR, Davidson NE and Baylin SB: Methylation of the oestrogen
receptor CpG island links ageing and neoplasia in human colon. Nat
Genet. 7:536–540. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hsieh CJ, Klump B, Holzmann K, Borchard F,
Gregor M and Porschen R: Hypermethylation of the p16INK4a promoter
in colectomy specimens of patients with long-standing and extensive
ulcerative colitis. Cancer Res. 58:3942–3945. 1998.PubMed/NCBI
|
|
41
|
Kondo Y, Kanai Y, Sakamoto M, Mizokami M,
Ueda R and Hirohashi S: Genetic instability and aberrant DNA
methylation in chronic hepatitis and cirrhosis - A comprehensive
study of loss of heterozygosity and microsatellite instability at
39 loci and DNA hypermethylation on 8 CpG islands in microdissected
specimens from patients with hepatocellular carcinoma. Hepatology.
32:970–979. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang JS, Guo M, Montgomery EA, Thompson
RE, Cosby H, Hicks L, Wang S, Herman JG and Canto MI: DNA promoter
hypermethylation of p16 and APC predicts neoplastic progression in
Barrett's esophagus. Am J Gastroenterol. 104:2153–2160. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang MS, Uozaki H, Chong JM, Ushiku T,
Sakuma K, Ishikawa S, Hino R, Barua RR, Iwasaki Y, Arai K, et al:
CpG island methylation status in gastric carcinoma with and without
infection of Epstein-Barr virus. Clin Cancer Res. 12:2995–3002.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Park SY, Yoo EJ, Cho NY, Kim N and Kang
GH: Comparison of CpG island hypermethylation and repetitive DNA
hypomethylation in premalignant stages of gastric cancer,
stratified for Helicobacter pylori infection. J Pathol.
219:410–416. 2009. View Article : Google Scholar : PubMed/NCBI
|