Introduction
GTPase-activating proteins (GAPs) function as a
deactivator of G-protein signaling by accelerating GTP hydrolysis.
Regulator of G-protein signaling (RGS) proteins are GAPs for Gα
subunits (1). RGS1 was first
identified as an immediate early gene responsive to several B-cell
activation signals (2), and it has
been shown to be related to the regulation of chemokine-induced
signaling in B cells (3). The RGS1
gene resides at 1q31, which is involved in several malignancies by
gains or amplifications in certain subtypes of melanoma (4), non-Hodgkin lymphoma (5), retinoblastoma (6), pancreatic cancer (7) and nasopharyngeal carcinoma (8). RGS1 has been shown to be upregulated
by gene expression profiling in several different tumor model
systems. For example, RGS1 has been shown to be overexpressed in
the more aggressive (blastoid) variant of mantle cell lymphoma
(9), the tumorigenic variant of
adult T-cell leukemia (10), and in
late-stage cervical cancer (11).
RGS1 plays an important role in melanoma
progression. Researchers analyzed gene profiling from 34
melanocytic neoplasms and found that RGS1 was differentially
overexpressed in primary melanomas vs. benign nevi (12). Another analysis of a tissue
microarray containing 301 primary melanomas showed a close
relationship between RGS1 expression and the clinical outcomes
associated with melanoma (13).
Furthermore, RGS1 expression was shown to be an independent
predictor of recurrence-free survival (RFS) and disease-specific
survival (DSS) when the six factors listed by the AJCC melanoma
analysis were all included. Intriguingly, in the analysis of DSS,
RGS1 emerged as the top factor predicting DSS, other than tumor
thickness or ulceration (13).
However, none of these studies on RGS1 and melanoma revealed any
hidden mechanisms.
The Gαs pathway is one of the earliest G-protein
signaling pathways to be studied, and many vital concepts including
that of second messengers (14),
protein phosphorylation (15), and
signal transducers (16,17) have come from this pathway. Gαs is a
tumor suppressor in neural and epidermal progenitor-derived
malignancies such as medulloblastoma, basal cell carcinoma,
neuroblastoma, and melanoma (originates from neural progenitors)
(18,19). In these stem cell compartments,
signaling through Gαs causes GTP hydrolysis that activates the
cAMP-dependent protein kinase A (PKA) signaling pathway (20), inhibits the Sonic Hedgehog (SHH) and
Hippo pathways (19), and finally
suppresses cell self-renewal. The loss of Gαs leads to activation
of these pathways, over-proliferation of progenitor cells, and
tumor formation. Thus, Gαs acts as a brake on excessive
self-renewal or proliferation of progenitor cells.
In the present study, we explored RGS1 expression in
40 melanoma and 18 nevus samples from 58 different patients. Then,
we investigated the role of RGS1 in melanoma progression using cell
viability and Matrigel-based assays. Further immunoprecipitation
and rescue experiments were performed to investigate the mechanism
utilized by RGS1 to regulate melanoma progression.
Materials and methods
Immunohistochemistry
To prepare tissue sections of 40 melanoma and 18
nevus from patients for immunohistochemistry, sections from each
patient were deparaffinized with xylene (3×5 min) followed by
treatment with serial dilutions of ethanol (100, 100, 95 and 95%,
10 min each) and by two changes of ddH2O. Antigen
unmasking was conducted by boiling the slides (95–99°C) for 10 min.
Sections were rinsed three times with ddH2O, immersed in
3% H2O2 for 20 min, washed twice with
ddH2O and once with TBS-T (TBS, 0.1% Tween-20) and
blocked for 1 h with blocking solution (5% normal goat serum in
TBS-T). Antibody of RGS1 (cat. no. PA5-29579; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) was diluted according to the
manufacturer instructions and the sections were incubated overnight
at 4°C. Then, the sections were washed three times, 5 min each,
with TBS-T and incubated for 1 h at room temperature with Signal
Stain Boost (Cell Signaling Technology, Inc., Danvers, MA, USA).
The negative control used for immunohistochemistry included the use
of phosphate-buffered saline instead of the primary antibody.
Finally, tissues were dehydrated. Images were captured with an
Olympus microscope (Olympus DP80; Olympus Corp., Tokyo, Japan). All
images were captured and processed using identical settings. During
evaluation, for each sample, five horizons were randomly chosen to
calculate the average positive ratio.
The immunostaining scores were calculated using an
approved standard (13). The
regions of most uniform staining were scored for each tissue array
core, which included the entire midportion of the core, to exclude
any ‘edge effect’ of increased staining. Expression of RGS1 protein
was graded combining two factors. One factor was the staining
intensity: 0, no staining; 1, weak staining; 2, moderate staining;
and 3, intense staining. The other factor included the proportion
of positive-staining cells. In all target cells of one region, the
proportion of ‘no staining’ cells was considered ‘A’, and ‘weak
staining’ was ‘B’, and by this analogy, the final score of this
region was equal to: (0 × A) + (1 × B) + (2 × C) + (3 × D). This
score was categorized into 3 grades: ≤1.0 (+); >1.0 but ≤1.5
(++); >1.5 (+++). The arrays were scored by a pathologist
blinded to the identity of the patients, and each score was
replicated by a separate, independent scoring trial by the study
pathologist. For the melanoma patients, we divided them into high
and low expression groups, and were followed up to determine their
disease-related survival, and analyzed it using the Kaplan-Meier
curve.
Cell culture
The A375 human melanoma cell line (Cell Bank in
Shanghai, Chinese Academy of Sciences), RGS1-knockdown (KD) A375
cells and RGS1-overexpression cells were incubated at 37°C in a
humidified 5% CO2 enriched atmosphere. These cells were
cultured with Dulbecco's modified Eagle's medium with high glucose
(DMEM; Gibco; Thermo Fisher Scientific, Inc.), supplemented with
10% heat inactivated fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.), 1% fungi zone (Invitrogen; Thermo Fisher
Scientific, Inc.), and 1% penicillin, twice weekly, at every change
in media, for normal growth by phase contrast microscopy. The
cultures were grown to confluence and passaged by treatment with
0.25% trypsin-EDTA (Gibco; Thermo Fisher Scientific, Inc.) at 37°C
and washed in 7 ml DMEM media before being centrifuged at 120 × g
for 10 min to form a pellet. The lentivirus base RGS1
overexpression system and RGS1 knockdown system (sequence of
shRGS1,
5′-GATCCGCCCTGTAAAGCAGAAGAGATTTCAAGAGAATCTCTTCTGCTTTACAGGGCTTTTTTG-3′)
were purchased from Hanyin Biotechology (Shanghai, China) and used
to infect cells as described in a previous study (21).
Cell proliferation assay
Cells were seeded into 96-well plates (Corning Inc.,
Corning, NY, USA) at a density of 2×103 cells/well. Cell
viability was assessed using Cell Counting Kit-8 assay (CCK-8;
Dojindo Molecular Technologies, Inc., Rockville, MD, USA). The
absorbance of each well was read on a spectrophotometer (Thermo
Fisher Scientific, Inc.) at 450 nm (OD450). Three independent
experiments were performed in quintuplicate.
Cell invasion assays
For the determination of cell invasion, Transwell
chambers were coated with 30 µl Matrigel (Merck KGaA, Darmstadt,
Germany), and incubated at 37°C for 40 min. In the Transwell assays
with and without Matrigel, the cells were trypsinized and then
seeded in chambers at a density of 1×104 cells/well at
48 h after transfection. The cells were then cultured in DMEM with
2% serum. Meanwhile 600 µl of medium supplemented with 10% FBS was
injected into the lower chambers. After cell harvest, the inserts
were fixed and stained in a dye solution containing 1% crystal
violet and 20% methanol. Cells adhering to the lower membrane of
the inserts were imaged with a microscope (Olympus DP80; Olympus
Corp.). Six views are randomly picked for each well.
Apoptosis assay
A375 cells in the three groups were measured by
FACS. Annexin V-PE/7-AAD (cat# 559763; eBioscience; Thermo Fisher
Scientific, Inc.) double staining was used to identify the
apoptosis rate of the A375 cells. The cells (1×106
cells/ml) were harvested, washed twice with 4 centigrade PBS, and
incubated for 15 min in 1X Annexin V binding buffer containing 10
µl 7-AAD and 5 µl Annexin V-PE. Finally, apoptosis was detected by
FACS and analyzed using FlowJo software (Tree Star Inc., Ashland,
OR, USA). Experiments were carried out in triplicate.
Immunoprecipitation
The FLAG-tag RGS1 and HA-Gαs plasmids were instantly
transferred into the 293T cells in the three groups and named
293T-RGS1, 293T-Gαs-GDP, 293T-Gαs-GDP-AlF4−,
which was without tetrafluoroaluminate (AlF4). The cells
were collected and lysed with 200 µl cold RIPA buffer (RIPA
buffer:PMSF = 100:1; Beyotime Institute of Biotechnology, Haimen,
China) for 30 min, followed with centrifugation at 13,200 × g, at
4°C for 10 min. HA-G proteins, (Gαs-GDP,
Gαs-GDP-AlF4−) were added into the cell lysis
supernatant liquor separately and mixed. Each blend was divided
into ‘total’ and ‘co-IP’ parts. The protein A agarose was prepared
and washed using Lysis buffer B (pH 7.6) 4 times, 2,000 g. This was
diluted by half with Lysis buffer B (pH 7.6). Protein A agarose was
added into each ‘co-IP’ portion and was agitated slowly at room
temperature for 2 h. Then, 1 µg of the Gαs flag antibody was added
into the ‘co-IP’ parts, and swayed slowly at 4°C overnight. Then
centrifugation was carried out instantaneously at 3,000 rpm, and
the precipitate was collected and washed with cold Lysis buffer B
(pH 7.6) 3 times. The samples were boiled for 5 min at 100°C, for
immunoprecipitation.
Spontaneously, a group of absolute exogenous co-IP
was prepared. The FLAG-tag RGS1 and HA-Gαs proteins were expressed
and purified, and the binding experiment procedure was carried out
as in Watson et al (1). The
reaction buffer consisted of a solution of 50 mM Tris-HCI, pH 8.0,
0.1 M NaCl, 1 mM MgS04, 20 mM imidazole, 0.025%
polyoxyethylene 10-lauryl ether (C12E10), 10
mM β-mercaptoethanol and 10% glycerol. Protein immunoprecipitation
(IP) was performed, respectively using Chromatin ChIP Kits (EMD
Millipore, Billerica, MA, USA). Antibody of HA-Gαs and FLAG-tag
RGS1 were used.
GTPase activity
The HA-Gαi and HA-Gαs were purified and extracted
using the method in the binding process. Then this was proceeded
according to the ATPase/GTPase Activity Assay kit (MAK113;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) instructions. First,
the phosphate standards were set as indicated in the kit
instructions. Second, a series of dilutions of enzyme were
performed in assay buffer. The sample reactions and the control
well were set up according to the scheme. The reaction was
incubated for the desired period of time (in our research, 1, 3, 5
and 10 min) at room temperature. Reagent (200 ml) was added to each
well and incubation was carried out for an additional 30 min at
room temperature to terminate the enzyme reaction and generate the
colorimetric product separately. Absorbance at 600–660 nm [maximum
absorbance at 620 nm (A620)] was read. We calculated the change in
absorbance values (DA620) for the samples by subtracting the A620
of the control well (A620) control from the A620 of the sample well
(A620) sample. The concentration (mM) of free phosphate
[Pi] was computed in the sample from the standard curve.
The formula was: Enzyme activity (units/l) = [Pi] (mM) ×
40 ml ÷ [10 µl × reaction time (min)]. One unit is the amount of
enzyme that catalyzes the production of 1 mmol of free phosphate
per minute under the assay conditions.
Western blotting
Protein was extracted from the cultured cells and
dissolved, homogenized, and quantified using the BCA Protein Assay
kit (Pierce; Thermo Fisher Scientific, Inc.). Sample buffer was
then added and the prepared samples were stored at −80°C after
boiling. During the western blotting, protein samples were
subjected to SDS-PAGE and transferred to polyvinylidene difluoride
membranes and placed in 25 mM Tris and 192 mM glycine. The
membranes were blocked with 5% non-fat dry milk in PBS, 0.05%
Tween-20 and probed with P-AKT (CY6569; Abways, Shanghai, China),
P-ERK (CY5277; Abways), AKT (CY5551; Abways), ERK (CY5487; Abways),
Gas (ab83735; Abcam, Cambridge, UK according to the manufacturers'
instructions. Blots were developed with ECL reagent (Thermo Fisher
Scientific, Inc.) and exposed using the FC2 Image Station (Alpha,
Bellingham, WA, USA).
Statistical analysis
All of the statistical analyses were performed by
SPSS (version 19.0) software (IBM Corp., Armonk, NY, USA) and all
of the data are represented as the mean ± standard deviations (SD).
Student's t-tests and analysis of variance (ANOVA) were performed
to compare the differences between groups. P<0.05 was considered
to be indicative of statistical significance. Three and more
independent experiments were performed in each group.
Results
RGS1 is highly expressed in melanoma
and is inversely associated with disease-specific survival
(DSS)
RGS1 expression has been detected throughout the
cell (22). In the present study,
we analyzed RGS1 expression by immunohistochemical staining in
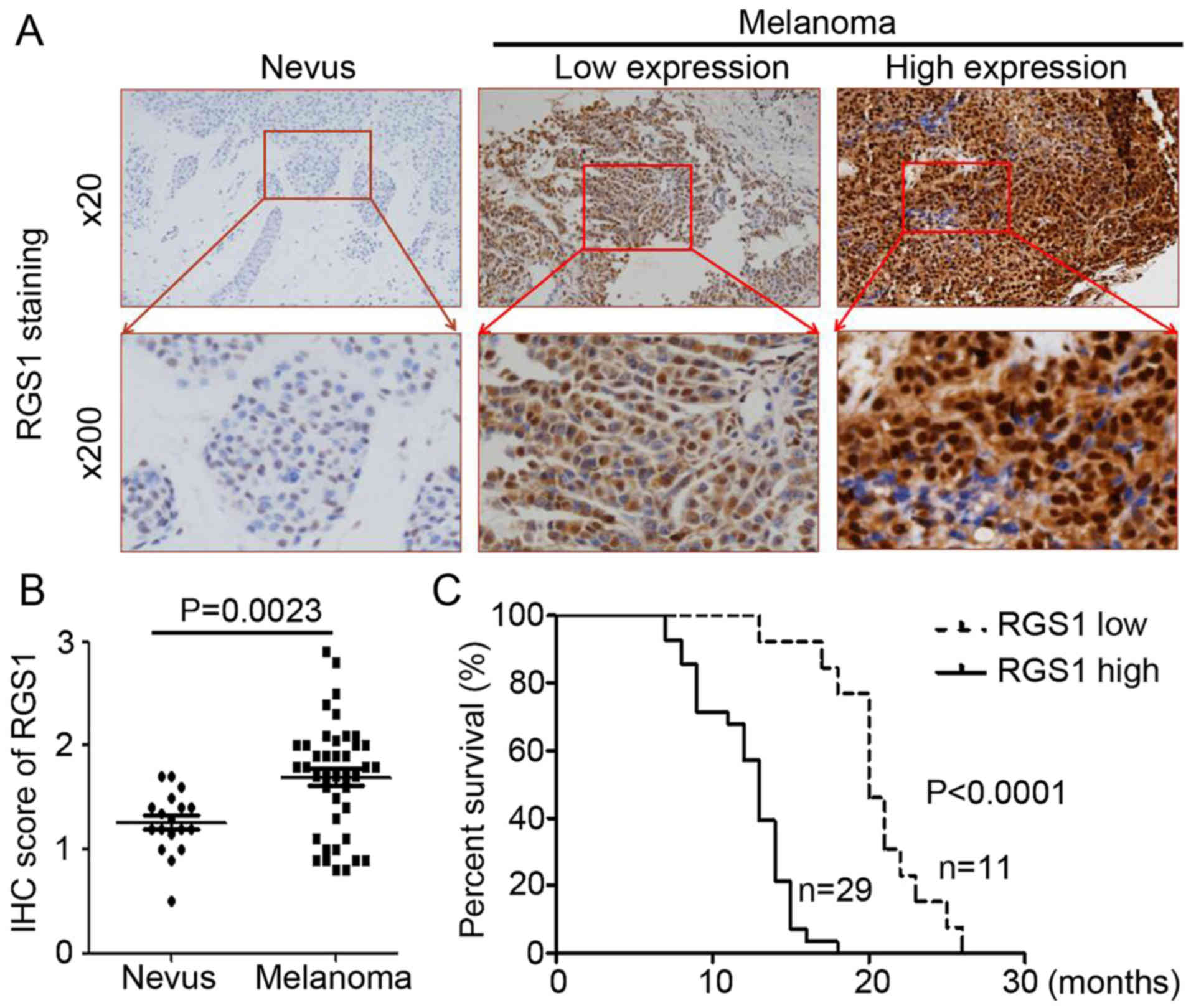
nevus and melanoma tissues. As shown in Fig. 1A, the RGS1 antibody staining
intensity was darker and stained more target cells in the melanoma
samples when compared to the nevus samples (Fig. 1A). Compared with the nevus tissue,
RGS1 expression was significantly upregulated in the melanoma
tissues (Fig. 1B, P=0.0023).
Furthermore, we collected the DSS data and performed the
Kaplan-Meier estimation. The results demonstrated that high RGS1
expression was inversely correlated with overall survival (Fig. 1C, P<0.0001). Collectively, RGS1
is highly expressed in melanoma and is inversely associated with
DSS.
RGS1 promotes melanoma cell
proliferation and invasion
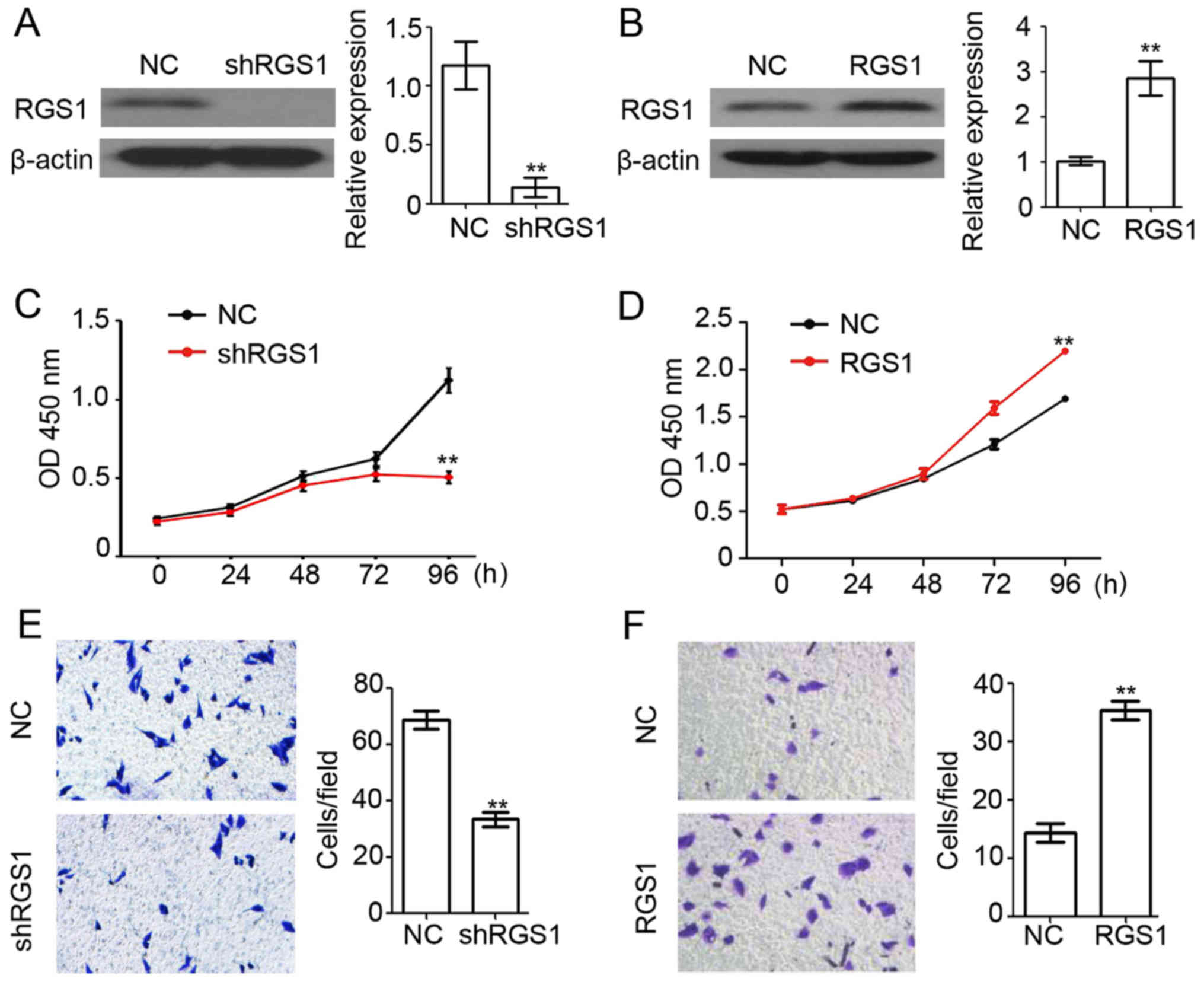
To study the function of RGS1 in the melanoma cell
line A375, the plasmid-based RGS1 overexpression and knockdown
systems (shRGS1) were used for transfection. A western blot assay
showed that RGS1 expression was efficiently downregulated in the
shRGS1-transfected A375 cells (Fig.
2A), while expression was significantly upregulated in the
RGS1-transfected overexpressing A375 cells (Fig. 2B). Next, cell viability in the
shRGS1-transfected, RGS1-transfected, and negative control
(NC)-transfected A375 cells were determined using the CCK-8 assay.
We found that knockdown of RGS1 significantly inhibited A375 cell
proliferation (Fig. 2C), and
overexpression of RGS1 significantly promoted A375 cell
proliferation (Fig. 2D).
Furthermore, a Matrigel-based invasion assay indicated that
knockdown of RGS1 significantly inhibited A375 cell invasion
(Fig. 2E) and overexpression of
RGS1 significantly promoted A375 cell invasion (Fig. 2F). These results demonstrated the
stimulatory role of RGS1 in melanoma proliferation and
invasion.
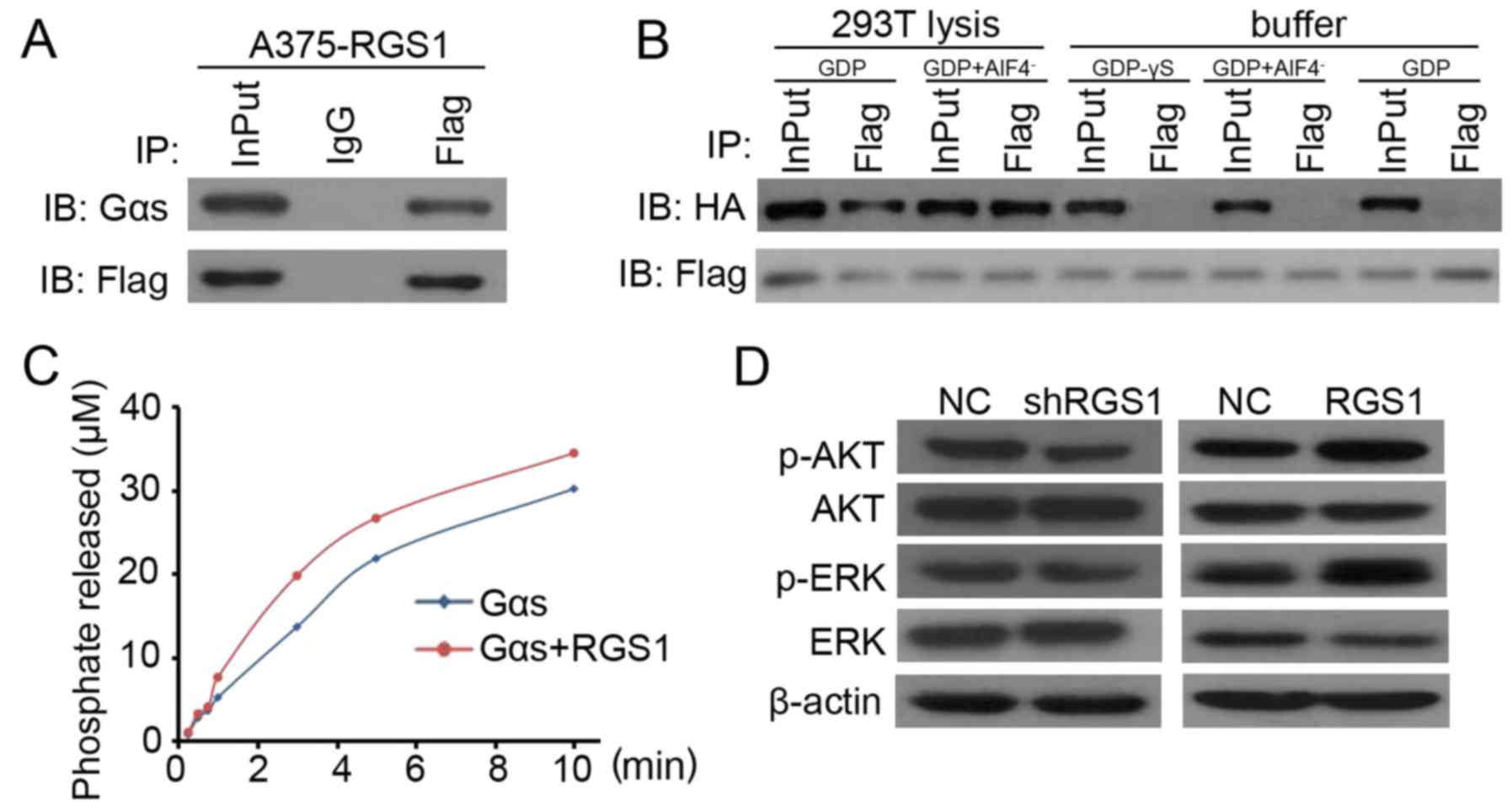
RGS1 binds to Gαs in an endogenous
environment and regulates AKT and ERK activation
Previous research has demonstrated that Gαs is a
tumor suppressor in neural and epidermal progenitor-derived
malignancies (18,19). In the present study,
co-immunoprecipitation (Co-IP) was performed to determine the
potential for direct targeting of RGS1 to Gαs. As shown in Fig. 3A, RGS1 was found to directly target
Gαs in the RGS1-overexpressing A375 cells. Next, the binding of
RGS1 protein and Gαs was assessed in the following three types of
exogenous environments: Gαs-GDP,
Gαs-GDP-AlF4−, and nothing added to the
buffer. The result indicated that RGS1 did not bind to Gαs in any
state (Fig. 3B). In the
circumstance of the 293T cell lysis with added GDP or
GDP+AlF4−, RGS1 binding to Gαs was detected
in both environments (Fig. 3B).
Binding was also detected in the endogenous experiment performed
with A375 cells (Fig. 3B). The
above results demonstrated the direct targeting of RGS1 and Gαs in
the endogenous environment.
GTPase activity was evaluated using a specific kit
testing GTPase activity in the exogenous environment (no other
molecules added). The result showed that the GTPase activity was
not significantly elevated for Gαs after the combination with RGS1
(Fig. 3C). This indicated that the
binding of RGS1 to Gαs might not accelerate the GTP hydrolysis
process (Fig. 3C). Further western
blotting demonstrated the stimulatory role of RGS1 on AKT and ERK
activation in A375 cells (Fig. 3D).
Collectively, the above results suggest that RGS1-induced AKT and
ERK phosphorylation is dependent on the non-GAP function of
Gαs.
Gαs plays a necessary role during
RGS1-mediated promotion of melanoma proliferation and
migration
To confirm that Gαs regulates the RGS1-mediated
promotion of AKT and ERK activation involved in A375 cell
proliferation and invasion, Gαs overexpression and knockdown
systems were utilized. As shown in Fig.
3D, the increased expression of RGS1 significantly promoted AKT
and ERK phosphorylation. Using the Gαs overexpression system, the
phosphorylation of AKT and ERK was reduced (Fig. 4A). In contrast to RGS1
overexpression, knockdown of RGS1 significantly decreased AKT and
ERK phosphorylation (Fig. 3D).
Using the knockdown system to reduce the expression of Gαs, the
phosphorylation of AKT and ERK was enhanced (Fig. 4A). The above results demonstrated
that Gαs plays a critical role during RGS1-mediated promotion of
AKT and ERK phosphorylation in melanoma.
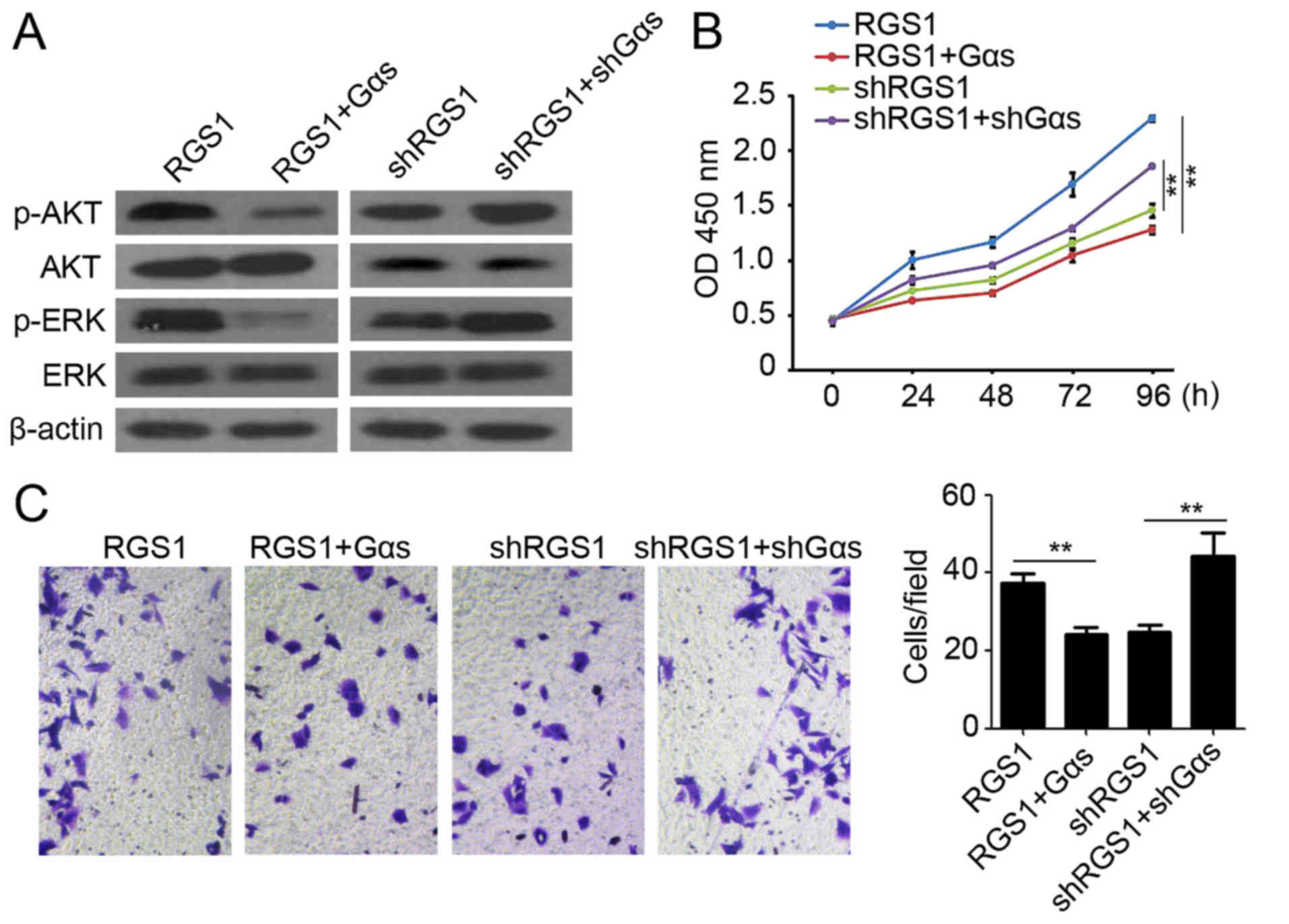 | Figure 4.Gαs plays a necessary role during
RGS1-mediated promotion of melanoma proliferation and migration.
(A) Western blot analysis of p-AKT, AKT, p-ERK and ERK expression
in A375 cells after RGS1, RGS1+Gαs, shRGS1 and shRGS1+shGαs
transfection. β-actin was used as a loading control. (B) CCK-8
assay was utilized to analyze A375 cell proliferation at 0, 24, 48,
72 and 96 h post RGS1, RGS1+Gαs, shRGS1 and shRGS1+shGαs
transfection. **P<0.01. (C) Matrigel-based invasion assay was
performed to determine A375 cell invasion ability post RGS1,
RGS1+Gαs, shRGS1 and shRGS1+shGαs transfection. **P<0.01. |
Our CCK-8 assay also demonstrated the stimulatory
role of RGS1 in melanoma proliferation (Fig. 2C), but increased expression of Gαs
reduced the cell proliferation (Fig.
4B). In contrast, knockdown of Gαs reversed the shRGS1-mediated
inhibition of proliferation (Figs.
2D and 4B). Furthermore, RGS1
was found to function as a promoter of melanoma invasion (Fig. 2E and F). Overexpression of Gαs
abrogated RGS1-mediated promotion of A375 invasion (Fig. 4C), while knockdown of Gαs promoted
A375 invasion of the shRGS1-transfected cells (Fig. 4C). Collectively, the above results
suggest that Gαs plays a critical role during RGS1-mediated
promotion of melanoma proliferation and migration.
Discussion
In the present study, we identified differential
RGS1 expression levels between melanoma and nevus samples, as well
as a significant role for RGS1 in promoting melanoma cell invasion
and proliferation. In addition, RGS1 expression was found to be
negatively correlated with patient disease-specific survival (DSS).
Further mechanistic investigation indicated that RGS1 directly
targets Gαs in the endogenous environment and promotes AKT and ERK
activation through the non-GAP function of Gαs. Rescue experiments
established the critical role of Gαs during RGS1-mediated promotion
of melanoma proliferation and invasion.
We found that the two recombinant proteins did not
bind in pure buffer with either GDP+AlF4− or
GDP added, while they did bind in 293T cells lysed with the
addition of either GDP+AlF4− or GDP. We also
detected the binding in A375 cells lysed with nothing added. These
results indicated that their binding requires an environment
containing specific molecules. Previous studies demonstrated that
the binding of Gαs to RGS proteins is controversial and varies in
different conditions. Gαs binds directly to the RGS domain of axin
in its transition-state in human colon cancer cells (23). Its inactive state also binds PX1
(RGS domain) (23), albeit to a
much lesser extent, as observed for other RGS proteins in
vitro (1). In
Magnaporthe pathogenesis, RGS1 regulates MagA, the Gαs
subunit, during surface signaling (24), and this result was based on a cell
function experiment instead of a binding assay. Gαs does not bind
to any RGS in its GTP-bound active state (23). The binding of RGS1 to Gαs in
different states was not fully investigated in previous studies
(1,25). Our exogenous binding assay in buffer
agrees with the previous research, but the ‘half-exogenous’ binding
assay, the recombinant proteins in 293T cell lysis, showed a
positive result. This is not necessarily that different from the
negative result found by Moratz et al (25), for in that research the total Gαs
expression in HS-Sultan cells was extremely low. Our half-exogenous
binding assay showed a much higher expression of total HA-Gαs
protein. The binding was not based on the state of Gαs. Considering
all these binding assays, RGS1 is able to bind to Gαs in a
different way from the traditional RGS-Gα binding pattern involving
specific and indispensable molecules in cells.
The acceleration of GTP hydrolysis by RGS occurs
through the stabilization of Gα proteins' transition state upon
binding. We did not find clear evidence of RGS1 accelerating the
GTPase activity of Gαs. From current information, the GAP function
of RGS upon binding to Gαs is also controversial. The RGS domain in
RGS-PX1 acts as a Gαs-specific GAP, which is the only example of
RGS promoting GTPase activity (26). In another study, neither the RGS
domain of axin nor the full-length axin purified from
baculovirus-infected Sf9 cells demonstrated the GTPase activity of
Gαs (23). In the case mentioned
above, additional accessory molecules or other modifications of
axin could be required for its GAP activity, as is the case for
other RGS proteins (27).
Similarly, our GTPase activity experiment was performed in a pure
chemical environment containing only artificial buffer, Gαs
protein, and RGS1 protein. Due to the limits of the method, the
real interaction and effects are difficult to confirm. Not merely
accessory molecules or modifications need to be taken into
consideration. It is also possible the RGS domain of axin is used
as a scaffold protein that can interact with and act as an effector
for Gαs, as do the RGS domain-containing RhoGEFs, which are
effectors for G proteins of the Ga12/13 family (27). Therefore, RGS1 could either act as
an effector, antagonize the effector of Gαs, or potentially target
PKA and receptor kinases (28).
Further rescue experiments confirmed the critical role of the
function of RGS1 through the interaction with Gαs in melanoma
progression.
We found that RGS1 promoted the activation of AKT
and ERK by regulating the non-GAP function of Gαs. Previous
research has demonstrated that all G-protein pathways may either
stimulate or inhibit one or more of the MAPK signaling pathways
(29). For example, in Gαs
signaling pathways, MEK can be stimulated or inhibited through
different paths in different conditions (29). A possible mechanism is that RGS1
enhances some receptor signaling through RGS or non-RGS domains and
motifs (28). These functions
depend on intact cells and physiological systems. Another potential
mechanism worth noting is that RGS1 may have promoted melanoma
progression with the heterotrimeric G-protein derived Gβγ-mediated
signaling or protein-protein interactions upon the binding of Gαs
and RGS1 proteins. According to the competitive mechanism in which
GAP (including RGS1) competes with Gβγ, the two surfaces of Gα that
interact with Gβγ and RGS proteins overlap substantially. High
expression of RGS1 may influence the binding of Gβγ to Gαs,
therefore causing some downstream effects (30). For example, among the downstream
effectors of Gβγ are the class I PI 3-kinases, PI3Kβ and PI3Kγ
(31–33). Gβγ activates these PI3K isoforms by
directly binding to the p110β and p110γ catalytic subunits
(34,35). It is possible that the binding of
RGS1 to Gαs maintained the function of Gβγ by activating PI3K,
which consequently increased the phosphorylation of AKT. In our
rescue experiment, the increased and decreased expression of Gαs
may have abolished and elevated the AKT and ERK expression,
respectively.
Our study offers a novel finding to explain the
tumor-enhancing mechanism of RGS1 in melanoma progression. The
binding with Gαs in melanoma is confirmed and meaningful. When RGS
binds to Gαs, it carries with it other functional units providing a
great diversity of protein-protein interactions (28), which may also influence the
downstream effectors of Gαs. The present study provides new
insights into the regulation and functional diversity of G-protein
signaling in tumor progression.
Acknowledgements
The authors thank the support of the Department of
Central Laboratory of Changhai Hospital.
Funding
The present study was supported by the Natural
Science Foundation of Shanghai (13JC1401403).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SMY and WYC conceived and designed the study. SMY,
WYC and ZJ performed the experiments and wrote the paper. LC, WK,
WXW and XCY reviewed and edited the manuscript. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Medical Ethics Committee of Second Military Medical University
(Shanghai, China).
Consent for publication
Not applicable.
Competing interests
All the authors declare that there are no conflicts
of interest.
References
|
1
|
Watson N, Linder ME, Druey KM, Kehrl JH
and Blumer KJ: RGS family members: GTPase-activating proteins for
heterotrimeric G-protein alpha-subunits. Nature. 383:172–175. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Newton JS, Deed RW, Mitchell EL, Murphy JJ
and Norton JD: A B cell specific immediate early human gene is
located on chromosome band 1q31 and encodes an alpha helical basic
phosphoprotein. Biochim Biophys Acta. 1216:314–316. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reif K and Cyster JG: RGS molecule
expression in murine B lymphocytes and ability to down-regulate
chemotaxis to lymphoid chemokines. J Immunol. 164:4720–4729. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Curtin JA, Fridlyand J, Kageshita T, Patel
HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, et
al: Distinct sets of genetic alterations in melanoma. N Engl J Med.
353:2135–2147. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mehra S, Messner H, Minden M and Chaganti
RS: Molecular cytogenetic characterization of non-Hodgkin lymphoma
cell lines. Genes Chromosomes Cancer. 33:225–234. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen D, Gallie BL and Squire JA: Minimal
regions of chromosomal imbalance in retinoblastoma detected by
comparative genomic hybridization. Cancer Genet Cytogenet.
129:57–63. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tirado CA, Sandberg AA and Stone JF:
Identification of a novel amplicon at 1q31 in pancreatic cancer
cell lines. Cancer Genet Cytogenet. 113:110–114. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chien G, Yuen PW, Kwong D and Kwong YL:
Comparative genomic hybridization analysis of nasopharygeal
carcinoma: Consistent patterns of genetic aberrations and
clinicopathological correlations. Cancer Genet Cytogenet.
126:63–67. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu Y, Hollmén J, Räty R, Aalto Y, Nagy B,
Elonen E, Kere J, Mannila H, Franssila K and Knuutila S:
Investigatory and analytical approaches to differential gene
expression profiling in mantle cell lymphoma. Br J Haematol.
119:905–915. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koga H, Imada K, Ueda M, Hishizawa M and
Uchiyama T: Identification of differentially expressed molecules in
adult T-cell leukemia cells proliferating in vivo. Cancer Sci.
95:411–417. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wong YF, Cheung TH, Tsao GS, Lo KW, Yim
SF, Wang VW, Heung MM, Chan SC, Chan LK, Ho TW, et al: Genome-wide
gene expression profiling of cervical cancer in Hong Kong women by
oligonucleotide microarray. Int J Cancer. 118:2461–2469. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haqq C, Nosrati M, Sudilovsky D, Crothers
J, Khodabakhsh D, Pulliam BL, Federman S, Miller JR III, Allen RE,
Singer MI, et al: The gene expression signatures of melanoma
progression. Proc Natl Acad Sci USA. 102:6092–6097. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rangel J, Nosrati M, Leong SP, Haqq C,
Miller JR III, Sagebiel RW and Kashani-Sabet M: Novel role for RGS1
in melanoma progression. Am J Surg Pathol. 32:1207–1212. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sutherland EW Jr and Wosilait WD:
Inactivation and activation of liver phosphorylase. Nature.
175:169–170. 1955. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krebs EG and Fischer EH: The phosphorylase
b to a converting enzyme of rabbit skeletal muscle. Biochim Biophys
Acta. 20:150–157. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rodbell M, Birnbaumer L, Pohl SL and Krans
HM: The glucagon-sensitive adenyl cyclase system in plasma
membranes of rat liver. V. An obligatory role of guanylnucleotides
in glucagon action. J Biol Chem. 246:1877–1882. 1971.PubMed/NCBI
|
|
17
|
Ross EM and Gilman AG: Resolution of some
components of adenylate cyclase necessary for catalytic activity. J
Biol Chem. 252:6966–6969. 1977.PubMed/NCBI
|
|
18
|
Martin BR and Lambert NA: Activated G
protein Gαs samples multiple endomembrane compartments.
J Biol Chem. 291:20295–20302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rao R, Salloum R, Xin M and Lu QR: The G
protein Gαs acts as a tumor suppressor in sonic hedgehog
signaling-driven tumorigenesis. Cell Cycle. 15:1325–1330. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen J, Zhang R, Chen X, Wang C, Cai X,
Liu H, Jiang Y, Liu C and Bai B: Heterodimerization of human orexin
receptor 1 and kappa opioid receptor promotes protein kinase
A/cAMP-response element binding protein signaling via a
Gαs-mediated mechanism. Cell Signal. 27:1426–1438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dai L, Cui X, Zhang X, Cheng L, Liu Y,
Yang Y, Fan P, Wang Q, Lin Y, Zhang J, et al: SARI inhibits
angiogenesis and tumour growth of human colon cancer through
directly targeting ceruloplasmin. Nat Commun. 7:119962016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chatterjee TK and Fisher RA: Cytoplasmic,
nuclear, and golgi localization of RGS proteins. Evidence for
N-terminal and RGS domain sequences as intracellular targeting
motifs. J Biol Chem. 275:24013–24021. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Castellone MD, Teramoto H, Williams BO,
Druey KM and Gutkind JS: Prostaglandin E2 promotes colon
cancer cell growth through a Gs-axin-β-catenin signaling
axis. Science. 310:1504–1510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu H, Suresh A, Willard FS, Siderovski
DP, Lu S and Naqvi NI: Rgs1 regulates multiple Gα subunits in
Magnaporthe pathogenesis, asexual growth and thigmotropism.
EMBO J. 26:690–700. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moratz C, Kang VH, Druey KM, Shi CS,
Scheschonka A, Murphy PM, Kozasa T and Kehrl JH: Regulator of G
protein signaling 1 (RGS1) markedly impairs Gi alpha signaling
responses of B lymphocytes. J Immunol. 164:1829–1838. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zheng B, Ma YC, Ostrom RS, Lavoie C, Gill
GN, Insel PA, Huang XY and Farquhar MG: RGS-PX1, a GAP for GalphaS
and sorting nexin in vesicular trafficking. Science. 294:1939–1942.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ingi T, Krumins AM, Chidiac P, Brothers
GM, Chung S, Snow BE, Barnes CA, Lanahan AA, Siderovski DP, Ross
EM, et al: Dynamic regulation of RGS2 suggests a novel mechanism in
G-protein signaling and neuronal plasticity. J Neurosci.
18:7178–7188. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhong H and Neubig RR: Regulator of G
protein signaling proteins: Novel multifunctional drug targets. J
Pharmacol Exp Ther. 297:837–845. 2001.PubMed/NCBI
|
|
29
|
Neves SR, Ram PT and Iyengar R: G protein
pathways. Science. 296:1636–1639. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tang W, Tu Y, Nayak SK, Woodson J, Jehl M
and Ross EM: Gbetagamma inhibits Galpha GTPase-activating proteins
by inhibition of Galpha-GTP binding during stimulation by receptor.
J Biol Chem. 281:4746–4753. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kurosu H, Maehama T, Okada T, Yamamoto T,
Hoshino S, Fukui Y, Ui M, Hazeki O and Katada T: Heterodimeric
phosphoinositide 3-kinase consisting of p85 and p110beta is
synergistically activated by the betagamma subunits of G proteins
and phosphotyrosyl peptide. J Biol Chem. 272:24252–24256. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Murga C, Fukuhara S and Gutkind JS: A
novel role for phosphatidylinositol 3-kinase beta in signaling from
G protein-coupled receptors to Akt. J Biol Chem. 275:12069–12073.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Leopoldt D, Hanck T, Exner T, Maier U,
Wetzker R and Nürnberg B: Gbetagamma stimulates phosphoinositide
3-kinase-gamma by direct interaction with two domains of the
catalytic p110 subunit. J Biol Chem. 273:7024–7029. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dbouk HA, Vadas O, Shymanets A, Burke JE,
Salamon RS, Khalil BD, Barrett MO, Waldo GL, Surve C, Hsueh C, et
al: G protein-coupled receptor-mediated activation of p110beta by
Gbetagamma is required for cellular transformation and
invasiveness. Sci Signal. 5:ra892012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vadas O, Dbouk HA, Shymanets A, Perisic O,
Burke JE, Saab Abi WF, Khalil BD, Harteneck C, Bresnick AR,
Nürnberg B, et al: Molecular determinants of PI3Kgamma-mediated
activation downstream of G-protein-coupled receptors (GPCRs). Proc
Natl Acad Sci USA. 110:18862–18867. 2013. View Article : Google Scholar : PubMed/NCBI
|