Introduction
Breast cancer is one of the most common malignancies
in women. As a hormone-dependent tumor, its growth is regulated by
estrogen. When estrogen binds to the estrogen receptor-α (ER-α) and
estrogen receptor-β (ER-β), it activates the expression of genes
which include the estrogen responsive element (ERE) in the nucleus,
and consequently produces effector proteins and promotes the growth
of breast cancer cells. Therefore, anti-estrogen endocrine therapy
has become an important treatment for breast cancer (1–4). ER is
an important target in the endocrine therapy of breast cancer.
Tamoxifen (TAM), a selective ER regulator, is the most commonly
used anti-estrogen agent for patients with ER-positive breast
cancer and has been widely used clinically (5). However, ~40% of ER-positive breast
cancer patients are not sensitive to TAM, and the main reason is
the occurrence of drug resistance (6–8); yet,
the specific mechanism of TAM resistance is not very clear. In
recent years it has been reported that the increased expression of
ER-α36 is one of the mechanisms attributed to the acquired
resistance to TAM (9), since TAM
can also bind and stimulate membrane-related ERs (10).
ER is a member of the nuclear receptor superfamily
and has many subtypes, including the most common nuclear receptor
ER-α66, its splice variant ER-α46 and ER-β. We usually refer to
ER-α66 as positive when we say ER-α positive. The ER-α36 is a newly
discovered ER-α subtype, which is a unique variant of ER-α66.
Compared with ER-α66, ER-α36 lacks two transcription-activated
domains of activation function-1 and −2 (AF-1 and AF-2), but still
retains the DNA binding domain (DBD) and the partial dimerization
region and estrogen binding domain (11,12).
Different from ER-α66's nuclear localization, ER-α36 is located in
the cytomembrane and cytoplasm. In contrast to the classical
estrogen genome effect, the binding of ER-α36 to estrogen can
quickly activate estrogen non-genomic signaling pathways, such as
PI3K/Akt and MAPK/ERK signaling, increase the intracellular calcium
concentration, and regulate gene transcription, proliferation of
tumor cells and anti-estrogen drug resistance (13–15).
However, ER-α36 can be expressed in both ER-α66-negative and
-positive breast cancer tissues (11). The use of TAM in the treatment of
ER-α66-positive breast cancer with high expression of ER-α36 has no
clinical benefit (16). ER-α36 is
also highly expressed in triple-negative breast cancer (TNBC) cell
lines MDA-MB-231 and MDA-MB-436 (11,15).
The positive feedback loop of ER-α36 and epidermal growth factor
receptor (EGFR) can induce the growth of ER-α66-negative breast
cancer and TAM resistance (14,17).
Human protein arginine N-methyltransferase 2 (PRMT2;
HRMT1L1) is a protein that belongs to the arginine
methyltransferase family (18,19).
It is clearly involved in a variety of cellular processes,
including apoptosis promotion, lung function, Wnt signaling, the
inflammatory response and leptin signaling regulation (20–23)
indicating that PRMT2 has different roles in transcriptional
regulation through variant mechanisms depending on its binding
partners. In our previous study, we demonstrated the negative
effect of PRMT2 on breast cancer cell proliferation in vitro
and in vivo. PRMT2 was shown to inhibit the ER-α-binding
affinity to the activator protein-1 (AP-1) site in cyclin D1
promoter via indirect binding with the AP-1 site, leading to the
suppression of cyclin D1 promoter activity in MCF-7 cells (24). As an ER-α co-regulator, PRMT2 is
capable of binding to ER-α both in vitro and in vivo
(25,26). Thus, we speculated that PRMT2 also
interacts with ER-α36 and is associated with TAM resistance. In the
present study, we studied the relationship of PRMT2 and ER-α36 in
breast cancer cells, and investigated the contribution of the
MAPK/ERK and PI3K/Akt pathways mediated by PRMT2/ER-α36 to TAM
resistance in breast cancer.
Materials and methods
Cell culture
Human breast cancer MCF-7 and MDA-MB-231 cells
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA) were maintained in Dulbecco's modified Eagle's medium
(DMEM) supplemented with 10% heat-activated fetal bovine serum
(FBS; Biological Industries, Northern Kibbutz Beit Haemek, Israel),
100 U/ml penicillin and 100 µg/ml streptomycin at 37°C with 5%
CO2.
Lentiviral vector construction and
lentivirus infection
pGC-LV-GV308-PRMT2 (NM_001535) with the
wild-type PRMT2 (LV-Tet-on-PRMT2) gene were constructed by
the GeneChem Co., Ltd. (Shanghai, China). The pGC-LV-GV308
vector was used as a negative control. The packaging plasmid
pHelper 1.0 and pHelper 2.0 were purchased from GeneChem Co. Ltd.
We co-transfected the pGC-LV-GV308-PRMT2 vectors with the
pHelper 1.0 and pHelper 2.0 packaging plasmid into 293T cells to
generate recombinant lentiviruses. Culture medium was collected 72
h post-transfection, and MDA-MB-231 cells were then infected with
the aforementioned lentiviruses. A total of 5×105 MDA-MB-231 cells
were seeded into a 6-well cell plate and further incubated for 12 h
to reach 30% confluency, and then infected with LV-Tet-on-PRMT2
(PRMT2 overexpression group) for 48 h in the presence of 8 µg/ml of
Polybrene (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). A stable
cell line with the PRMT2-3Flag was obtained after infection with
Lv-tet-on-PRMT2 cells which were selected by puromycin for 2 weeks.
Western blot analysis was performed to verify the expression of
PRMT2-3Flag induced by 5 µg/ml of Dox in the infected MDA-MB-231
cells.
The lentivirus-based PRMT2 shRNA expression
plasmids, pYr-Lvsh-PRMT2, were purchased from Yingrun Biotechnology
Co. (Changsha, China). The pYr-Lvsh vector was used as a negative
control. The viral particles were produced and purified by
cotransfecting pYr-Lvsh-PRMT2 and the lentivirus packaging plasmids
(pVSVG, pLP1, pLP2) into 293FT cells. The stable clones with PRMT2
knockdown generated from the MCF-7 cells were selected in culture
medium containing 0.5 µg/ml puromycine. The selected stable clones
were verified by western blotting using the negative control cells
as control.
Western blot analysis
Total cell lysates were lysed on ice for 30 min.
Soluble proteins (40 µg) were probed with the anti-PRMT2 antibody
(1:500; cat. no. 3667; Abcam, Cambridge, MA, USA), anti-ER-α36
antibody (1:1,000; cat. no. CY1109; Cell Applications, San Diego,
CA, USA) and anti-p-AKT (cat. no. 4060), anti-AKT (cat. no. 4691),
anti-p-ERK1/2 (cat. no. 4370) and anti-ERK1/2 (cat. no. 4695)
(1:1,000; Cell Signaling Technology, Inc., Danvers MA, USA).
Loading variations were normalized against β-actin, which was
identified by the anti-β-actin antibody (1:1,000; cat. no. 4970;
Cell Signaling Technology).
Cell cycle analysis by flow
cytometry
Cells were harvested, washed with phosphate-buffered
saline (PBS), and fixed with 66% ethanol overnight at −20°C. Cells
were centrifuged and washed with PBS, and then stained with
propidium iodide (PI; BD Biosciences, Franklin Lakes, NJ, USA) in
PBS solution for 1 h in the dark. Cell cycle distribution was
evaluated by use of a FACSCalibur flow cytometer equipped with
CellQuestPro software (BD Biosciences) (27).
Apoptosis assay by flow cytometry
Apoptosis was measured with use of an Annexin
V-fluoroisothiocyanate (Annexin V-FITC) or Annexin V-PE apoptosis
detection kit according to the manufacturer's instructions (BD
Biosciences) and analyzed with use of a FACSCalibur flow cytometer
and BD CellQuest Pro software (BD Biosciences) as previously
described (28). Briefly, cells
were harvested and washed, and incubated in binding buffer with
Annexin V-FTIC/PI or Annexin V-PE/7AAD for 15 min at room
temperature. The cells were washed and resuspended in binding
buffer before flow cytometric analysis.
Confocal microscopy
Transfections were performed using Lipofectamine
2000 transfection reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) according to the manufacturer's
instructions. Plasmid constructs expressing PRMT2 fused to an
N-terminal GFP tag and pcDNA3.1-Myc-His-ERα36 were generated by
Yingrun Biotechnologies Inc. (Hunan, China), and used to
transiently co-transfect the MCF-7 or MDA-MB-231 cells grown on
glass coverslips. At 48 h post-transfection, the cells were fixed
in paraformaldehyde, permeabilized with °Triton X-100 and incubated
with Cy3-conjugated secondary antibody. The cells were then stained
with 4,6-diamidino-2-phenylindole (DAPI) and viewed under a Zeiss
LSM 510 confocal microscope (Carl Zeiss, Oberkochem, Germany);
images were acquired from typical cells using a ×63 oil-immersion
lens.
Protein purification and GST pull-down
assay
GST and GST fusion proteins were expressed in
Escherichia coli BL-21 cells by induction with a final
concentration of 0.8 mM isopropyl-b-D-thiogalactopyranoside. Cells
were lysed by sonication in 10 ml of 1X PBS (NaCl/Pi) supplemented
with complete protease inhibitor tablets (Roche Applied Science,
Basel, Switzerland). GST fusion proteins with ER-α36 and ER-α66
were purified using glutathione-agarose beads (Sigma-Aldrich; Merck
KGaA). The recombinant human His-tag-PRMT2 protein (obtained from
Yingrun Biotechnologies Inc.) were mixed with 10 mg of GST
derivatives bound to glutathione-agarose beads in 0.5 ml of binding
buffer (50 mM Tris/HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.3 mM
dithiothreitol, 0.1% NP-40 and protease inhibitor tablets from
Roche Applied Science). The binding reaction was performed at 4°C
for 3 h and the beads were subsequently washed 4 times with the
washing buffer (the same as the binding buffer), 30 min each time.
The beads were eluted by boiling in SDS sample buffer and analyzed
by SDS/PAGE, and PRMT2, ER-α36 and ER-α66 were analyzed by
immunoblotting.
Co-immunoprecipitation
MCF-7 cells or MDA-MB-231 cells were transfected
with the indicated plasmids. At 48 h, cells were lysed (50 mM Tris
at pH 8.0, 500 mM NaCl, 0.5% NP-40, 1 mM dithiothreitol and
protease inhibitor tablets from Roche Applied Science). Five
hundred nanograms of lysate were precleared with 50 µl protein
A-Sepharose beads (Sigma-Aldrich; Merck KGaA) for 2 h at 4°C. The
PRMT2 antibody (Abcam) or ER-α36 antibody (Cell Applications) was
then added and incubated overnight at 4°C. One hundred microliters
of protein A agarose were then added to the antibody/lysate mixture
for another 2 h at 4°C, and the beads were pelleted and washed
thrice with lysis buffer. Bound proteins were eluted in SDS sample
buffer, subjected to SDS/PAGE and analyzed using an ER-α36 antibody
(Cell Applications) or PRMT2 antibody (Abcam).
Tissue microarray analysis
A tissue microarray (BR1921; US Biomax, Inc.,
Rockville, MD, USA) consisting of 160 breast cancer samples was
used. These samples were histologically interpretable and were
analyzed for the correlation between PRMT2 and ER-α36.
Immunohistochemical staining was performed as detailed in our
previous study (24). The tissues
were blocked with 1% bovine serum albumin (Roche Diagnostics,
Basel, Switzerland) at room temperature for 1 h. Antibodies against
PRMT2 (1:50; Abcam) and ER-α36 (1:50; Cell Applications) were used.
The correlation analysis was performed using SPSS software (version
18.0; SPSS, Inc., Chicago, IL, USA). Spearman's rank correlation
coefficients were generated to determine the degree of the
correlation.
Statistical analysis
All experiments were performed ≥3 times, and the
results are expressed as the mean ± standard deviation unless
otherwise stated. GraphPad Prism software (version 5.0; GraphPad
Software, Inc., La Jolla, CA, USA) was used for statistical
analysis. Comparisons between two groups were performed using the
two-tailed Student's t-test. Comparisons among multiple groups were
performed using one-way analysis of variance with post hoc
intergroup comparisons using the Tukey test. P<0.05 was
considered to indicate a statistically significant difference.
Results
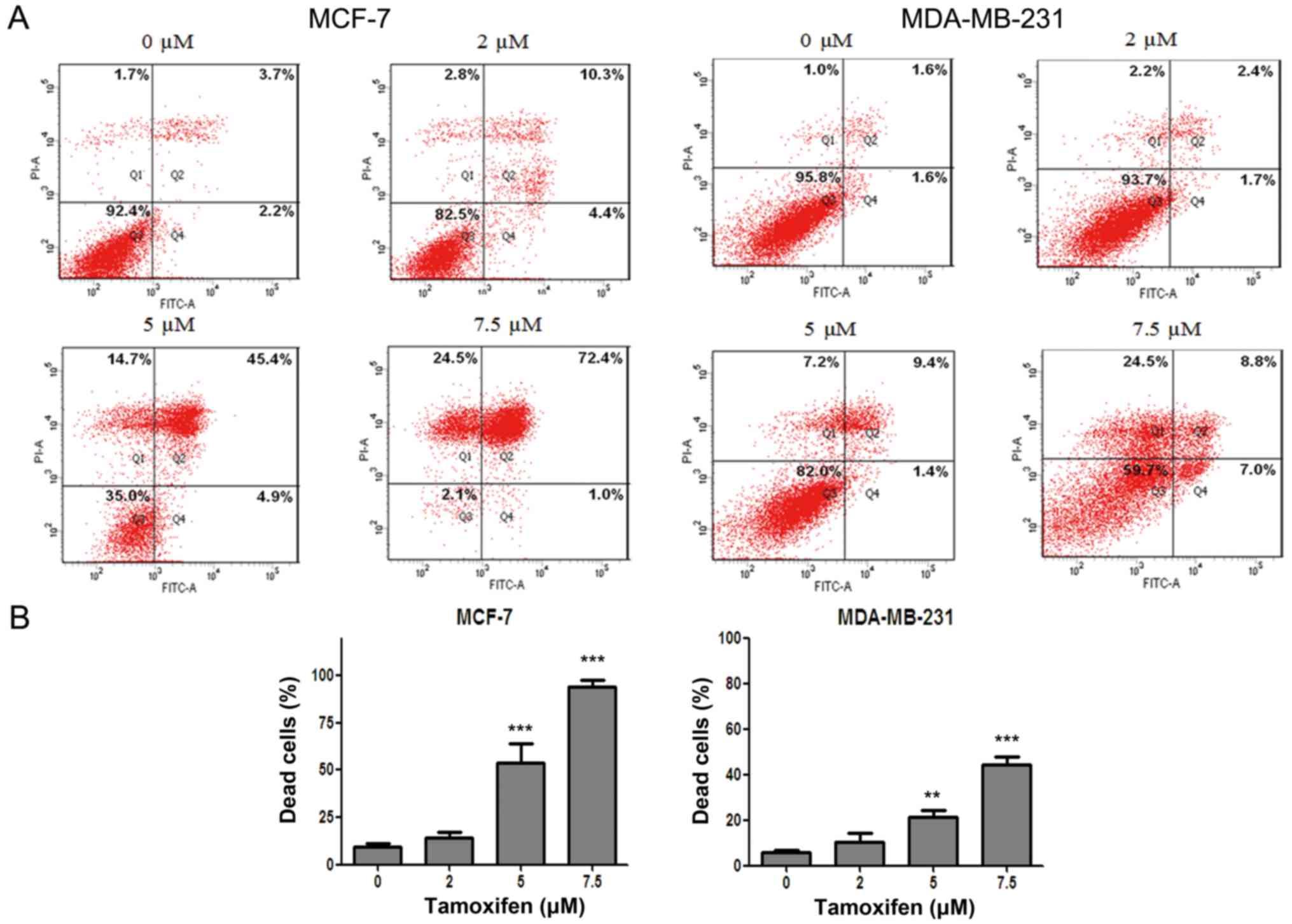
MDA-MB-231 cells are relatively
resistant to TAM by contrast to MCF-7 cells
In order to clarify the antitumor effect of
anti-estrogen drug TAM in different breast cancer cell lines, flow
cytometry after dual staining of Annexin V-FITC/PI was used to
evaluate the cell death rate of MCF-7 and MDA-MB-231 cells after
drug treatment. The results showed that the cell death of MCF-7 and
MDA-MB-231 cells were both induced by TAM in a dose-dependent
manner. However, the cell death rate of MDA-MB-231 cells was
significantly lower than that of MCF-7 cells for the same drug
concentration (Fig. 1), indicating
that MCF-7 cells were more sensitive to TAM but MDA-MB-231 cells
were relatively resistant to TAM.
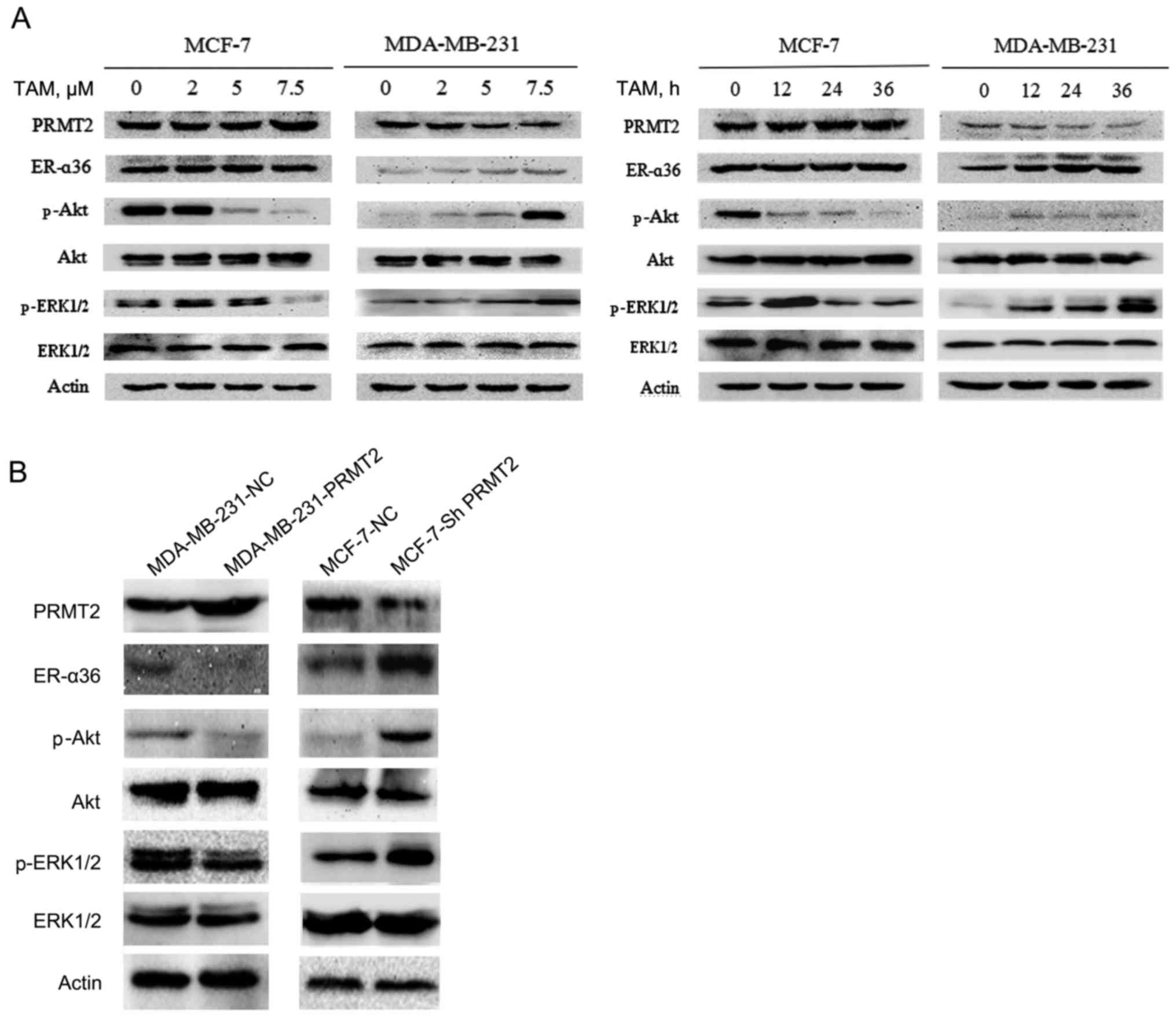
TAM resistance of MDA-MB-231 cells is
connected with the downregulation of PRMT2 and the upregulation of
ER-α36
To explore the mechanism of TAM resistance, we first
examined the expression of resistance-related potential proteins
using western blot analysis in MCF-7 and MDA-MB-231 cells. The
results revealed no obvious change in expression of PRMT2 and
ER-α36 in the MCF-7 cells, but a substantial decrease in PRMT2 and
an increase in ER-α36 level after treatment with TAM in the
MDA-MB-231 cells, and both proteins changed in a time- and
dose-dependent manner (Fig. 2A).
ER-α36 binds to estrogen and quickly activates estrogen non-genomic
signaling pathways, such as PI3K/Akt signaling and MAPK/ERK
signaling, and regulate anti-estrogen drug resistance (13–15).
Therefore, we also evaluated the expression of phosphorylated Akt,
ERK1/2 and total Akt, ERK1/2. The results demonstrated that the
levels of phosphorylated Akt and ERK1/2 were decreased in the MCF-7
cells but were increased in the MDA-MB-231 cells after exposure to
TAM, whereas no effects on total proteins were noted in the MCF-7
and MDA-MB-231 cells (Fig. 2A).
To confirm the relationship of PRMT2 and ER-α36, a
tetracycline (doxycycline hyclate; Dox)-inducible lentiviral system
was established to overexpress PRMT2, and the pGC-LV-GV308
vector was used as a negative control. Stable cell lines with the
PRMT2-3Flag were obtained after selection with puromycin for 2
weeks (MDA-MB-231-PRMT2 cells). Western blot analysis verified the
overexpression of PRMT2-3Flag induced by Dox in the infected
MDA-MB-231 cells. Moreover, as shown in Fig. 2B, with treatment of 5 µg/ml of Dox,
MDA-MB-231 cells carrying lentivirus PRMT2 expression exhibited
markedly decreased ER-α36 and phosphorylated Akt, phosphorylated
ERK1/2 compared to negative control cells. In turn, the
lentivirus-based shRNA expression plasmids, pYr-Lvsh-PRMT2, was
used to knockdown PRMT2. The pYr-Lvsh vector was used as a negative
control. The stable clones of PRMT2 knockdown generated from MCF-7
cells were selected in the culture medium containing puromycine
(MCF-7-shPRMT2 cells). The selected stable clones were verified by
immunoblotting using the negative control cells as control.
Knockdown of PRMT2 in MCF-7 cells increased the expression of
ER-α36 and phosphorylated Akt and phosphorylated ERK1/2 compared to
the negative control cells (Fig.
2B). The results in these two cell lines demonstrated that
PRMT2 inhibited ER-α36 and its estrogen non-genomic signaling
pathways, PI3K/Akt and MAPK/ERK.
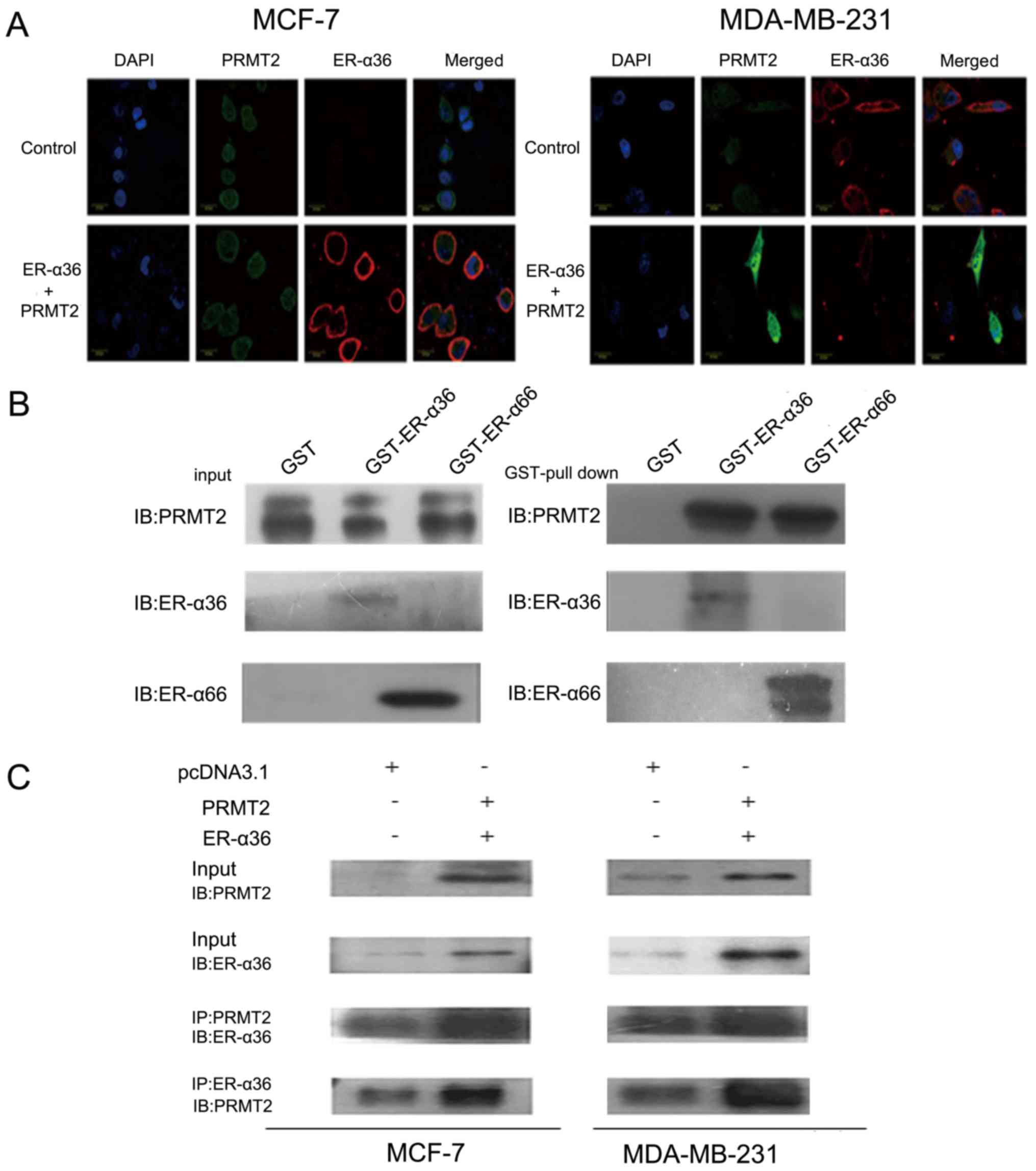
Interaction of PRMT2 and ER-α36
To further study the interaction of PRMT2 and
ER-α36, we first examined the subcellular localization of PRMT2 and
ER-α36 by confocal microscopy. As shown in Fig. 3A, PRMT2 appeared to be largely
localized to the nucleus excluding the nucleoli and a weak
fluorescence was detected in the cytosol as described previously
(29–31), whereas ER-α36 was predominantly
localized to the cell membrane and cytoplasm. In addition, the
fluorescence signals related to ER-α36 and PRMT2 overlapped,
indicating co-localization of PRMT2 and ER-α36 inside MCF-7 cells.
Similar results were observed in the MDA-MB-231 cells (Fig. 3A). PRMT2 is able to bind to ER-α66
both in the presence and absence of estrogen but can enhance ER-α66
activity only with estrogen as previously reported (25). To understand the molecular
mechanisms of the ER-α36 dependence of PRMT2, we performed a
protein-protein interaction study in vitro and inside the
cells. As shown in Fig. 3B, PRMT2
displayed similar binding affinity to ER-α36 and ER-α66 in
vitro. This binding activity of PRMT2 to ER-α36 was also
confirmed inside MCF-7 and MDA-MB-231 cells. PRMT2 was
co-immunoprecipitated by the PRMT2 antibody, and ER-α36 could be
captured by the ER-α36 antibody, and vice versa (Fig. 3C). These results showed that PRMT2
directly associates with ER-α36.
PRMT2 regulates the response of breast
cancer cells to TAM
Given that PRMT2 was significantly decreased after
TAM treatment in MDA-MB-231 cells and was closely related to
ER-α36, we reasoned that PRMT2 may be a critical mediator for
regulating the response of breast cancer cells to TAM. To test this
hypothesis, we evaluated the impact of silencing PRMT2 on the
sensitivity of MCF-7 cells to TAM and the impact of overexpression
of PRMT2 on the resistance of MDA-MB-231 cells to TAM. The stable
MCF-7-PRMT2 cells exhibited sufficient upregulation of PRMT2 and
the MDA-MB-231-shPRMT2 cells displayed marked downregulation of
PRMT2 by western blot analysis (Fig.
2B). PRMT2 knockdown in MCF-7 cells obviously attenuated
TAM-induced cell death when compared with the MCF-7-NC cells, as
analyzed by flow cytometry (Fig.
4A). In turn, PRMT2 overexpression in MDA-MB-231 cells notably
increased TAM-induced cell death when compared with the
MDA-MB-231-NC cells (Fig. 4B).
Moreover, MDA-MB-231-PRMT2 cells exhibited increased G1 arrest and
decreased S phase (to reflect cell proliferation status) when
compared with the MDA-MB-231-NC cells, whether or not with TAM
treatment (Fig. 4C). These results
suggest that PRMT2 is a critical mediator for regulating the
response of breast cancer cells to TAM, and PRMT2 could reverse TAM
resistance in breast cancer cells.
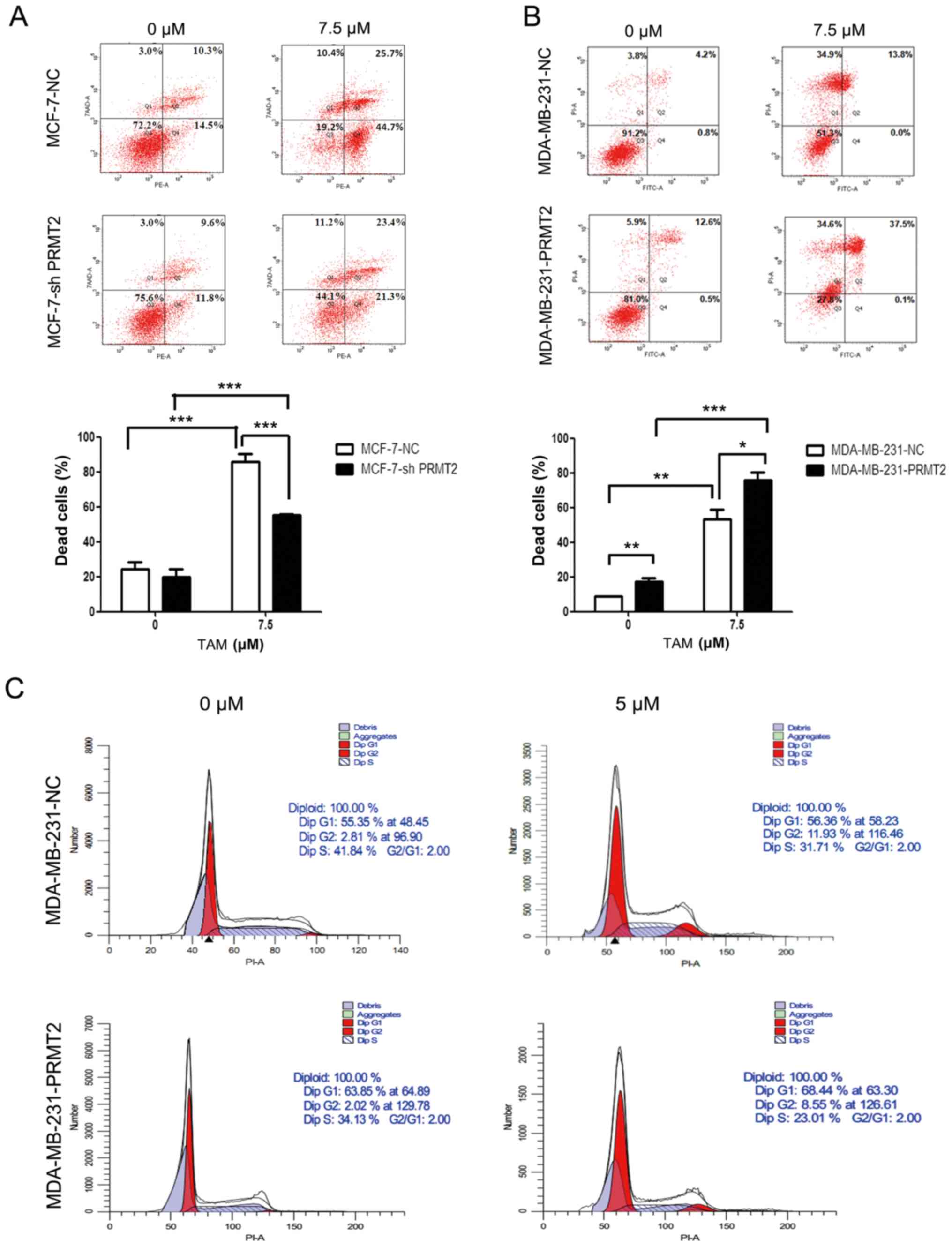 | Figure 4.PRMT2 regulates the response of
breast cancer cells to tamoxifen (TAM). (A) Knockdown of PRMT2
reduces TAM sensitivity in MCF-7 cells. MCF-7-NC and MCF-7-shPRMT2
cells were treated with or without 7.5 µM TAM for 36 h, and then
cells were collected, washed, fixed and stained with Annexin
V-PE/7-AAD to detect the cell death with flow cytometry. (B) PRMT2
overexpression reverses TAM resistance in MDA-MB-231 cells.
MDA-MB-231-NC and MDA-MB-231-PRMT2 cells were treated with or
without 7.5 µM TAM for 36 h, and then cells were collected, washed,
fixed and stained with Annexin V-FITC/PI to detect the cell death
with flow cytometry. Upper panel, representative graphs of 3
independent experiments; lower panel, statistical charts. Columns,
mean; error bars, SD. Student's t-test; *P<0.05, **P<0.01,
***P<0.0001. (C) PRMT2 overexpression induces G1 arrest in
MDA-MB-231 cells. MDA-MB-231-NC and MDA-MB-231-PRMT2 cells were
treated with or without 5 µM TAM for 36 h; and then cells were
collected, washed, stained with propidium iodide (PI) and analyzed
using flow cytometry. PRMT2, protein arginine N-methyltransferase
2. |
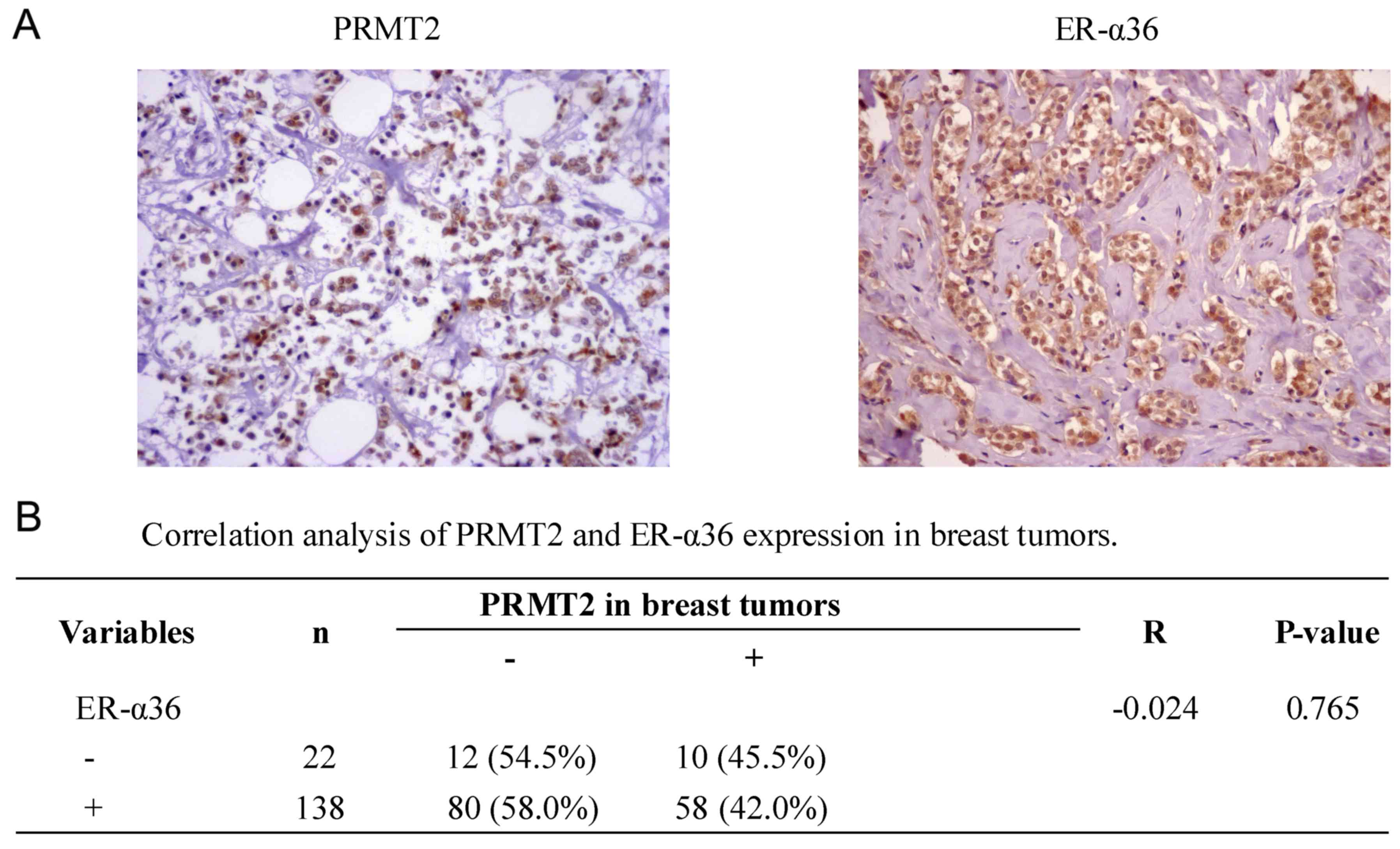
Correlation between PRMT2 and ER-α36
expression in breast cancer tissues
To further identify the association between PRMT2
and ER-α36 expression in breast cancer, a tissue microarray
(BR1921), consisting of 160 breast cancer cases, was used. No
significant correlation (r=−0.024; P=0.765; Fig. 5) was identified between the
expression of PRMT2 and ER-α36 upon analysis of the human breast
cancer tissue microarray, regardless of breast cancer types. The
data suggest that there was no significant correlation between the
expression of PRMT2 and ER-α36 in breast cancer tissues, which was
not consistent with the results observed in the cells.
Discussion
Tamoxifen (TAM) is a selective estrogen receptor
modulator (SERM) that has been widely used to treat advanced
ER-positive breast cancer, and to prevent breast cancer in
high-risk pre- and post-menopausal women as a chemopreventive
agent. It competes with estrogens for the ligand binding domain of
ER, hence inhibiting ER-mediated mitogenic estrogen signaling
(13). However, the major obstacle
to TAM usage is TAM resistance, which occurs de novo or can
be acquired after its use (32).
The specific mechanism of TAM resistance remains unclear.
Clinical data show that up to 40% of ER-positive
breast cancer patients are insensitive to TAM, while some
ER-negative patients respond well to TAM therapy (33,34),
suggesting that there are other ER subtypes involved in TAM
resistance in addition to ER-α66. ER-α36, a novel variant of ER-α
(ER-α66), can be expressed both in ER-α-positive and ER-α-negative
breast cancer tissues (11). It is
highly expressed on the plasma membrane and cytoplasm of cancer
cells. Moreover, the activation of the PI3K/Akt and MAPK/ERK
pathways mediated by ER-α36 contributes to the TAM resistance,
which is the non-genomic effect of ER-α36 (13,35–37).
PRMT2, a member of the human protein arginine
methyltransferase family whose effects are not fully known, has
been shown to regulate factors that influence cell activation and
apoptosis. PRMT2 inhibits NF-κB-dependent transcription and renders
cells more susceptible to apoptotic stimuli. PRMT2 also affects
transcriptional regulation through its effect on transcription
coactivators or coinhibitors, which is involved in chromatin
remodeling (23). Furthermore,
PRMT2 has been reported to be a coactivator of ER-α, but its
mechanism of action is unclear (25). In our previous study, PRMT2 was
capable of binding to ER-α both in vitro and in vivo
(25,26) and regulating cyclin D1 (24); thus, we reasoned that PRMT2 also
interacts with ER-α36 and is associated with the TAM resistance. To
verify our hypothesis, we first evaluated TAM-induced apoptosis in
different breast cancer cell lines. We chose MCF-7 and MDA-MB-231
cells as the cell models since ER-α66 is positive and ER-α36 is
lowly expressed in MCF-7 cells, while ER-α66 is negative and ER-α36
is highly expressed in MDA-MB-231 cells (17). As expected, MDA-MB-231 cells were
resistant to TAM in contrast to MCF-7 cells. Obviously, the absence
of ER-α66 is an important reason. Notably, we found that p-Akt and
p-ERK1/2 were decreased with the absence of changes in ER-α36 and
PRMT2 after TAM treatment in the MCF-7 cells, while p-Akt, p-ERK1/2
and ER-α36 were all increased with a decrease in PRMT2 following
TAM treatment in the MDA-MB-231 cells. This indicated that the
downregulation of PRMT2 as well as the upregulation of ER-α36 and
its non-genomic effect may result in the resistance of MDA-MB-231
cells to TAM. The p-Akt and p-ERK1/2 levels in the MCF-7 cells may
be downregulated by other factors such as ER-α66. To further
understand the relationship of PRMT2 and ER-α36, we knocked down
PRMT2 in MCF-7 cells or overexpressed PRMT2 in MDA-MB-231 cells
since PRMT2 is highly expressed in MCF-7 cells and lowly expressed
in MDA-MB-231 cells, relatively (26). The results revealed that PRMT2
inhibited ER-α36 and its non-genomic signaling pathways, PI3K/Akt
and MAPK/ERK. Moreover, the direct interaction of PRMT2 and ER-α36
was confirmed by immunofluorescence, GST pull-down and Co-IP assay.
Nevertheless, the specific methods by which PRMT2 suppresses ER-α36
remains to be resolved in future research. Finally, we found that
PRMT2 overexpression in MDA-MB-231 cells notably increased
TAM-induced cell death and the G1 arrest, which suggested that
PRMT2 could reverse the TAM resistance in MDA-MB-231 cells.
However, no significant correlation between the expression of PRMT2
and ER-α36 were identified in breast cancer tissues using a tissue
microarray assay, which was not consistent with the results
observed in the cells. We speculated that this is because the human
tumor microenvironment is more complex than the cellular
microenvironment, and individual differences in humans may also
have a great impact on the results. Therefore, we will use an
increased number of tissue specimens, and analyze the correlation
of PRMT2 and ER-α36 in different pathological types or molecular
subtypes of breast cancer tissues in further research.
In summary, PRMT2 was able to reverse the TAM
resistance in breast cancer cells through suppression of ER-α36 and
its non-genomic effect. Therefore, PRMT2 may be a new target with
which to overcome TAM resistance and is a valuable prognostic
marker in breast cancer treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81502276, 81372824,
81472608 and 81773294), the Major Projects of Science and
Technology of Health and Family Planning Commission of Hunan
Province (grant no. A2017013), the Natural Science Foundation of
Hunan Province (grant no. 2016JJ4077), the Key Project of the
Education Department of Hunan Province (grant no. 16A189) and the
Young Talents Program of University of South China.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
YS, JZ, XZ and RC conceived and designed the study.
YS, JL, KL, JZ, TX, TZ, ZL, YC, WD and GW performed the
experiments. YS and ZJ wrote the paper. XZ and RC reviewed and
edited the manuscript. All authors read and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Institutional Review Board of the Department of Laboratory Animal
Science of University of South China (Hengyang, China).
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Lupien M, Eeckhoute J, Meyer CA, Krum SA,
Rhodes DR, Liu XS and Brown M: Coactivator function defines the
active estrogen receptor alpha cistrome. Mol Cell Biol.
29:3413–3423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Holz MK, Digilova A, Yamnik RL, Davis DC,
Murphy CJ and Brodt N: Estrogen receptor α is a target of mTOR/S6K1
signaling in control of breast cancer cell proliferation. Cancer
Res. 69 Suppl 2:S40622009. View Article : Google Scholar
|
|
3
|
Laws MJ, Das A, Li Q, et al: The Estrogen
Receptor Alpha Plays a Central Role in Controlling Stromal
Differentiation and Angiogenesis in the Mouse and Human Endometria
During Early Pregnancy. Meeting of the
Society-For-The-Study-Of-Reproduction. 55. 2009.https://doi.org/10.1093/biolreprod/81.s1.32
|
|
4
|
Sayeed A, Konduri SD, Liu W, Bansal S, Li
F and Das GM: Estrogen receptor α inhibits p53-mediated
transcriptional repression: Implications for the regulation of
apoptosis. Cancer Res. 67:7746–7755. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lewis JS and Jordan VC: Selective estrogen
receptor modulators (SERMs): Mechanisms of anticarcinogenesis and
drug resistance. Mutat Res. 591:247–263. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meijer D, van Agthoven T, Bosma PT, Nooter
K and Dorssers LC: Functional screen for genes responsible for
tamoxifen resistance in human breast cancer cells. Mol Cancer Res.
4:379–386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ignatov A, Ignatov T, Roessner A, Costa SD
and Kalinski T: Role of GPR30 in the mechanisms of tamoxifen
resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat.
123:87–96. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zheng Y, Zhang J, Xu ZZ, Sheng JM, Zhang
XC, Wang HH, Teng XD, Liu XJ, Cao J and Teng LS: Quantitative
profiles of the mRNAs of ER-α and its novel variant ER-α36 in
breast cancers and matched normal tissues. J Zhejiang Univ Sci B.
11:144–150. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gu W, Dong N, Wang P, Shi C, Yang J and
Wang J: Tamoxifen resistance and metastasis of human breast cancer
cells were mediated by the membrane-associated estrogen receptor
ER-α36 signaling in vitro. Cell Biol Toxicol. 2016.
|
|
10
|
Liang J and Shang Y: Estrogen and cancer.
Annu Rev Physiol. 75:225–240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee LMJ, Cao J, Deng H, Chen P, Gatalica Z
and Wang ZY: ER-α36, a novel variant of ER-α, is expressed in
ER-positive and -negative human breast carcinomas. Anticancer Res.
28:479–483. 2008.PubMed/NCBI
|
|
12
|
Rao J, Jiang X, Wang Y and Chen B:
Advances in the understanding of the structure and function of
ER-α36, a novel variant of human estrogen receptor-alpha. J Steroid
Biochem Mol Biol. 127:231–237. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin SL, Yan LY, Zhang XT, Yuan J, Li M,
Qiao J, Wang ZY and Sun QY: ER-alpha36, a variant of ER-alpha,
promotes tamoxifen agonist action in endometrial cancer cells via
the MAPK/ERK and PI3K/Akt pathways. PLoS One. 5:e90132010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang XT, Kang LG, Ding L, Vranic S,
Gatalica Z and Wang ZY: A positive feedback loop of ER-α36/EGFR
promotes malignant growth of ER-negative breast cancer cells.
Oncogene. 30:770–780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang X, Ding L, Kang L and Wang ZY:
Estrogen receptor-alpha 36 mediates mitogenic antiestrogen
signaling in ER-negative breast cancer cells. PLoS One.
7:e301742012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J,
Wang T, Fan Z, Fan T, Lin B, et al: Expression of ER-{α}36, a novel
variant of estrogen receptor {α}, and resistance to tamoxifen
treatment in breast cancer. J Clin Oncol. 27:3423–3429. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Honma N, Horii R, Iwase T, Saji S, Younes
M, Takubo K, Matsuura M, Ito Y, Akiyama F and Sakamoto G: Clinical
importance of estrogen receptor-β evaluation in breast cancer
patients treated with adjuvant tamoxifen therapy. J Clin Oncol.
26:3727–3734. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Katsanis N, Yaspo ML and Fisher EMC:
Identification and mapping of a novel human gene, HRMT1L1,
homologous to the rat protein arginine N-methyltransferase 1
(PRMT1) gene. Mamm Genome. 8:526–529. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scott HS, Antonarakis SE, Lalioti MD,
Rossier C, Silver PA and Henry MF: Identification and
characterization of two putative human arginine methyltransferases
(HRMT1L1 and HRMT1L2). Genomics. 48:330–340. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Blythe SA, Cha SW, Tadjuidje E, Heasman J
and Klein PS: beta-Catenin primes organizer gene expression by
recruiting a histone H3 arginine 8 methyltransferase, Prmt2. Dev
Cell. 19:220–231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iwasaki H, Kovacic JC, Olive M, Beers JK,
Yoshimoto T, Crook MF, Tonelli LH and Nabel EG: Disruption of
protein arginine N-methyltransferase 2 regulates leptin
signaling and produces leanness in vivo through loss of STAT3
methylation. Circ Res. 107:992–1001. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yildirim AO, Bulau P, Zakrzewicz D,
Kitowska KE, Weissmann N, Grimminger F, Morty RE and Eickelberg O:
Increased protein arginine methylation in chronic hypoxia: Role of
protein arginine methyltransferases. Am J Respir Cell Mol Biol.
35:436–443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ganesh L, Yoshimoto T, Moorthy NC, Akahata
W, Boehm M, Nabel EG and Nabel GJ: Protein methyltransferase 2
inhibits NF-kappaB function and promotes apoptosis. Mol Cell Biol.
26:3864–3874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhong J, Cao RX, Liu JH, Liu YB, Wang J,
Liu LP, Chen YJ, Yang J, Zhang QH, Wu Y, et al: Nuclear loss of
protein arginine N-methyltransferase 2 in breast carcinoma is
associated with tumor grade and overexpression of cyclin D1
protein. Oncogene. 33:5546–5558. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qi C, Chang J, Zhu Y, Yeldandi AV, Rao SM
and Zhu YJ: Identification of protein arginine methyltransferase 2
as a coactivator for estrogen receptor α. J Biol Chem.
277:28624–28630. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhong J, Cao RX, Zu XY, Hong T, Yang J,
Liu L, Xiao XH, Ding WJ, Zhao Q, Liu JH, et al: Identification and
characterization of novel spliced variants of PRMT2 in breast
carcinoma. FEBS J. 279:316–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen Y, Shi X and Pan J: The
conformational control inhibitor of tyrosine kinases DCC-2036 is
effective for imatinib-resistant cells expressing T674I
FIP1L1-PDGFRα. PLoS One. 8:e730592013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shen Y, Ren X, Ding K, Zhang Z, Wang D and
Pan J: Antitumor activity of S116836, a novel tyrosine kinase
inhibitor, against imatinib-resistant FIP1L1-PDGFRα-expressing
cells. Oncotarget. 5:10407–10420. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Herrmann F, Pably P, Eckerich C, Bedford
MT and Fackelmayer FO: Human protein arginine methyltransferases in
vivo - distinct properties of eight canonical members of the PRMT
family. J Cell Sci. 122:667–677. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Frankel A, Yadav N, Lee J, Branscombe TL,
Clarke S and Bedford MT: The novel human protein arginine
N-methyltransferase PRMT6 is a nuclear enzyme displaying unique
substrate specificity. J Biol Chem. 277:3537–3543. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kzhyshkowska J, Schütt H, Liss M, Kremmer
E, Stauber R, Wolf H and Dobner T: Heterogeneous nuclear
ribonucleoprotein E1B-AP5 is methylated in its Arg-Gly-Gly (RGG)
box and interacts with human arginine methyltransferase HRMT1L1.
Biochem J. 358:305–314. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY,
Zhu Y, Skaar TC, Gomez B, O'Brien K, Wang Y, et al: Antiestrogen
resistance in breast cancer and the role of estrogen receptor
signaling. Oncogene. 22:7316–7339. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jacquemier JD, Hassoun J, Torrente M and
Martin PM: Distribution of estrogen and progesterone receptors in
healthy tissue adjacent to breast lesions at various stages -
immunohistochemical study of 107 cases. Breast Cancer Res Treat.
15:109–117. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Clarke R, Leonessa F, Welch JN and Skaar
TC: Cellular and molecular pharmacology of antiestrogen action and
resistance. Pharmacol Rev. 53:25–71. 2001.PubMed/NCBI
|
|
35
|
Dontu G, Abdallah WM, Foley JM, Jackson
KW, Clarke MF, Kawamura MJ and Wicha MS: In vitro propagation and
transcriptional profiling of human mammary stem/progenitor cells.
Genes Dev. 17:1253–1270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ginestier C, Hur MH, Charafe-Jauffret E,
Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG,
Liu S, et al: ALDH1 is a marker of normal and malignant human
mammary stem cells and a predictor of poor clinical outcome. Cell
Stem Cell. 1:555–567. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Piva M, Domenici G, Iriondo O, Rábano M,
Simões BM, Comaills V, Barredo I, López-Ruiz JA, Zabalza I, Kypta
R, et al: Sox2 promotes tamoxifen resistance in breast cancer
cells. EMBO Mol Med. 6:66–79. 2014. View Article : Google Scholar : PubMed/NCBI
|