Introduction
Chloroquine (CQ), a 4-aminoquinoline compound, has
long been prescribed for the treatment of malaria (1), autoimmune disorders (2) and viral infections (3). It functions primarily by inhibiting
lysosomal proteases and blocking autophagosome-lysosome fusion
(4). In the range of neutral pH, CQ
can freely diffuse across the plasma membrane but can also get
protonated and trapped in acidic vesicles such as lysosomes. The
accumulation of protonated CQ results in less acidic conditions in
lysosomes, further preventing autophagosome-lysosome fusion and
autophagosome degradation. Nowadays, CQ is receiving more attention
from all over the world mostly due to its potent antitumor activity
as mono- or add-on therapy. It has been reported that CQ can
increase sensitization of radiation (5–7) and
chemotherapeutic agents including ABT-737, 5-fluorouracil and PI103
(8–10), and then induce cell death in a
subset of cancer cell lines. Recent evidence suggests that CQ alone
effectively suppressed the growth of pancreas, leukemia, lung,
colon and liver cancer cells (11–15),
and promoted apoptosis by increasing the expression of
pro-apoptotic protein Bim both in HepG2 and Huh7 cells (15), activating the p53 pathway in glioma
cells (16) and stabilizing the
BH3-only protein PUMA in melanoma cells (17). However, the mechanism underlying the
antitumor effect of CQ monotherapy in human A549 cells has not been
clearly investigated yet.
Autophagy is a self-defense event in all eukaryotic
cells triggered by internal needs or extracellular stressors. It
mainly serves as a mechanism for cell survival. Normally, autophagy
functions in the maintenance of cellular homeostasis by delivering
impaired organelles or unwanted cellular components to the
lysosomes for degradation and recycling (18). In recent years, there has been an
increasing amount of research on the relationship between autophagy
and cancer. The role of autophagy in cancer is highly complex and
still paradoxical. It appears to have the double-edged sword
effect: Under stress circumstances, autophagy can protect cancer
cells against apoptosis and/or other forms of cell death by
providing energy and essential macromolecules, whereas excessive
autophagy may cause irreversible self-destruction of cancer cells
and further induce autophagic cell death (19). Notably, several proteins functioning
in the process of apoptosis are also required for autophagy.
Autophagy and apoptosis can be triggered by the same stimuli. They
are closely interconnected at some special points of crosstalk in
different types of cancer (20).
Thus, thoroughly analyzing the mechanism of crosstalk between
autophagy and apoptosis is essential for successful anticancer
treatments as a potential therapeutic strategy.
Although CQ displays antitumor activity in human
A549 cells, there are very few studies that have described the
underlying mechanism of the antitumor effect of CQ as a monotherapy
and the relationship among autophagy, apoptosis and CQ. Therefore,
the present study was designed to investigate the antitumor effect
of CQ in human A549 cells, further analyze the possible molecular
mechanism of CQ monotherapy in the regulation of autophagy or
apoptosis, as well as the association between autophagy and
apoptosis when the A549 cells were exposed to CQ. Thus, it may
offer a solid experimental base for utilizing CQ as a monotherapy
agent in cancer therapy in the future.
Materials and methods
Cell line culture and reagents
Human lung A549 cells and human kidney 293T cells
were obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China), cultured in Ham's F-12 medium (Sigma-Aldrich
Chemie GmbH, Munich, Germany) and Dulbecco's modified Eagle's
medium (DMEM; HyClone Laboratories, Logan, UT, USA), respectively,
supplemented with 10% fetal bovine serum (FBS; HyClone,
Laboratories), 100 U/ml penicillin and 100 µg/ml streptomycin
(Gibco, Grand Island, NY, USA) at 37°C and 5% CO2 in a
humidified atmosphere. CQ was purchased from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany) and dissolved in deionized distilled
water (DDW) until the experiments.
Cell viability assay
The antiproliferative effect of CQ in A549 cells was
assessed by MTT assay. Briefly, A549 cells were seeded into 96-well
plates (5×103 cells/well) and treated with CQ at
different concentrations (1.25, 2.5, 5, 10, 20, 40, 80 and 160 µM)
for 24 or 48 h, respectively. A total of 20 µl methylthiazol
tetrazolium (MTT) solution (final concentration of 0.5 mg/ml) was
added and incubated at 37°C for another 4 h. Subsequently, 150 µl
dimethyl sulfoxide (DMSO) was added to dissolve the blue formazan
product. The absorbance values were measuring at 570 nm with an
enzyme-labeling instrument (Safire2; Tecan Group, Ltd., Männedorf,
Switzerland).
Immunofluorescence of LC3-II
The amount of LC3-II, an indicator of
autophagosomes, was detected following a standard procedure of
immunofluorescence. Firstly, A549 cells after being treated with or
without CQ for 24 h were grown on sterilized glass coverslips to
90–95% confluence and fixed with a 4% (w/v) paraformaldehyde
solution. After being washed 3 times with phosphate-buffered saline
(PBS; Sigma Chemical Co., St. Louis, MO, USA), the cells were
blocked with 0.1% Triton X-100 containing 1% bovine serum albumin
(BSA) in PBS for 1 h, and then incubated with LC3B primary antibody
(1:200; cat. no. 2775; Cell Signaling Technology, Inc., Danvers,
MA, USA) at 4°C overnight and subsequently with the corresponding
FITC-linked anti-rabbit secondary antibody (1:160; cat. no. F9887;
Sigma-Aldrich) for 40 min at room temperature. Finally, the cells
were rinsed with PBS and examined using a fluorescence microscope
(Zeiss Axio ObserveRA1; Carl Zeiss GmbH, Jena, German) in 3 random
fields.
Transmission electron microscopy (TEM)
observation
The ultrastructure of human A549 cells were observed
using a transmission electron microscope (HT7700; Hitachi, Tokyo,
Japan). After CQ administration, the cells were harvested and fixed
in 2.5% glutaraldehyde at 4°C overnight. After being washed 3 times
with a sucrose solution, the fixed cells were incubated with 1%
osmium tetroxide for 2 h at room temperature. Subsequently, the
cells were dehydrated with gradient ethanol and embedded in Spurr's
resin. Ultrathin sections (40–70 nm) were obtained using an
ultramicrotome and stained with lead citrate/uranyl acetate and
subsequently visualized with a transmission electron microscope at
80 kV.
Lentiviral shRNA vector construction
and transfection
The interfering sequence targeting the human
Beclin-1 gene (GeneBank accession: NM_003766.3) was designed as
follows: shRNA, 5′-GCTCAGTATCAGAGAGAATAC-3′. The sequence,
5′-TTCTCCGAACGTGTCACGT-3′, sharing no homology with any other human
gene was used as a negative control. The annealed, double-stranded
DNA was inserted into the lentiviral vector LV2-pGLV-u6-puro. The
recombinant plasmids were transformed into DH5α and the positive
colonies selected by PCR were sequenced. 293T cells were
co-transfected with 20 µg of lentiviral expression plasmid and
packaging plasmid (15 µg pHelper 1.0 and 10 µg pHelper 2.0) using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The recombinant lentiviral particles which were
obtained 48 h after transfection were harvested to infect human
A549 cells. The interference efficiency was assessed using western
blot analysis.
AO/EB dual fluorescence staining
Acridine orange/ethidium bromide (AO/EB) staining
was performed for the observation of morphological changes in
cultured cells. Human A549 cells were seeded in a 6-well plate at a
density of 5×105 cells/ml and incubated overnight before
treatment. Then, the cells were exposed to CQ at final
concentrations of 0, 10, 20, 40 and 80 µM for 24 h. Untreated cells
were used as the negative control. In subsequent assays, the cells
were harvested and stained with AO/EB solution (mixture of AO 100
µg/ml and EB 100 µg/ml prepared in PBS) at room temperature for 15
min. The morphological changes of A549 cells were observed
immediately under a fluorescence microscope (Zeiss Axio Observer
A1; Carl Zeiss).
Annexin V binding assay
The rate of apoptosis induced by CQ was quantified
using the Annexin V-FITC/PI kit (Nanjing KeyGen Biotech, Co., Ltd.,
Nanjing, China) following the manufacturer's instructions. A549
cells (5×105 cells/well) in the exponential phase were
treated with different concentrations of CQ (0, 10, 20, 40 and 80
µM) for 24 h. After harvesting, the cells were resuspended with
Annexin binding buffer and then incubated with 5 µl Annexin V-FITC
and 5 µl of propodium iodine (PI) solutions at room temperature for
15 min in the dark. The early apoptotic (Annexin V-FITC-positive)
and necrotic/late apoptotic cells (Annexin V-FITC-positive,
PI-positive) were quantified by BD FACSCalibur Flow Cytometer (BD
Biosciences, San Jose, CA, USA).
Mitochondrial membrane potential
assay
Mitochondrial membrane potential (ΔΨm)
changes after CQ exposure were detected using the JC-1
Mitochondrial Potential Detection kit (Nanjing KeyGen Biotech) and
flow cytometry (FCM). After being treated with CQ, A549 cells were
trypsinized and washed twice with cold PBS, and then were stained
using JC-1 in PBS for 15 min at room temperature in the dark,
followed by FCM analysis.
Western blot analysis
Human A549 cells were treated with CQ in designated
concentrations for 24 h. Total protein was isolated from the
control and treated cells using RIPA lysis buffer (Beyotime
Institute of Biotechnology, Shanghai, China). The bicinchoninic
acid (BCA) protein assay was employed to determine the protein
concentration. For western blot analysis, equal amounts of protein
were denatured, separated by 10% sodium dodecyl sulfate-acrylamide
gel, and transferred onto nitrocellulose membranes (Pall Life
Sciences, Ann Arbor, MI, USA). The non-specific protein bands were
blocked in BSA blocking buffer for 1 h at room temperature.
Subsequently, the membranes were incubated with primary antibodies
respectively against GAPDH (1:2,500; cat. no. 2118), LC3B (1:1,000;
cat. no. 2775), Beclin-1 (1:1,000; cat. no. 3495), p62 (1:1,000;
cat. no. 88588), p-AKT (1:2,000; cat. no. 4060), AKT (1:1,000; cat.
no. 9272), p-PI3K (1:1,000; cat. no. 4228), PI3K (1:1,000; cat. no.
4292), Bcl-2 (1:1,000; cat. no. 2872), Bax (1:1,000; cat. no.
2774), cytochrome c (1:1,000; cat. no. 11940), c-caspase-3
(1:1,000; cat. no. 9664) and poly(ADP-ribose) polymerase (PARP)
(1:1,000; cat. no. 9542) (Cell Signaling Technology, Inc.) at 4°C
overnight followed by either a goat anti-rabbit (1:2,500; cat. no.
7074; Cell Signaling Technology, Inc.), or goat anti-mouse
HRP-conjugated secondary antibody (1:2,500; cat. no. 7076; Cell
Signaling Technology, Inc.) for another 2 h at room temperature.
Finally, the reactive bands were visualized using an enhanced
chemiluminescent substrate to HRP (Pierce, Woburn, MA, USA). GAPDH
was used as the internal control.
Statistical analysis
All experiments were run independently in
triplicate. The data were presented as the mean ± standard
deviation (SD). One-way analysis of variance (ANOVA) was employed
to determine the differences between the control and treated groups
followed by a Student's t-test for multiple comparisons. A
P<0.05 indicated a statistically significant result.
Results
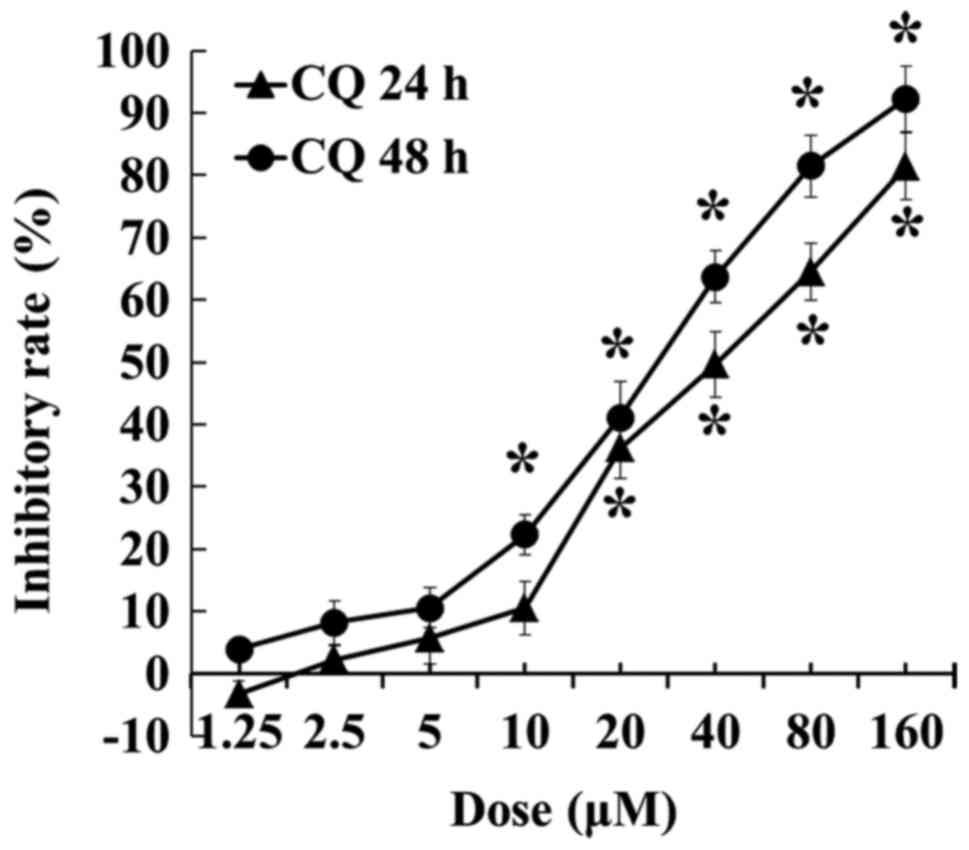
CQ inhibits the viability of A549
cells
The inhibitory effect of CQ on A549 cells was
detected by MTT assay. As shown in Fig.
1, CQ inhibited the viability of A549 cells in a dose-and
time-dependent manner in CQ concentrations between 2.5 and 160 µM,
and a significant decrease was recorded after the concentration
reached 20 µM at 24 h and 10 µM at 48 h, respectively (P<0.05).
CQ inhibited cell proliferation to ~80% at a concentration of 160
µM at 24 h and 92.28% at a concentration of 160 µM at 48 h,
respectively.
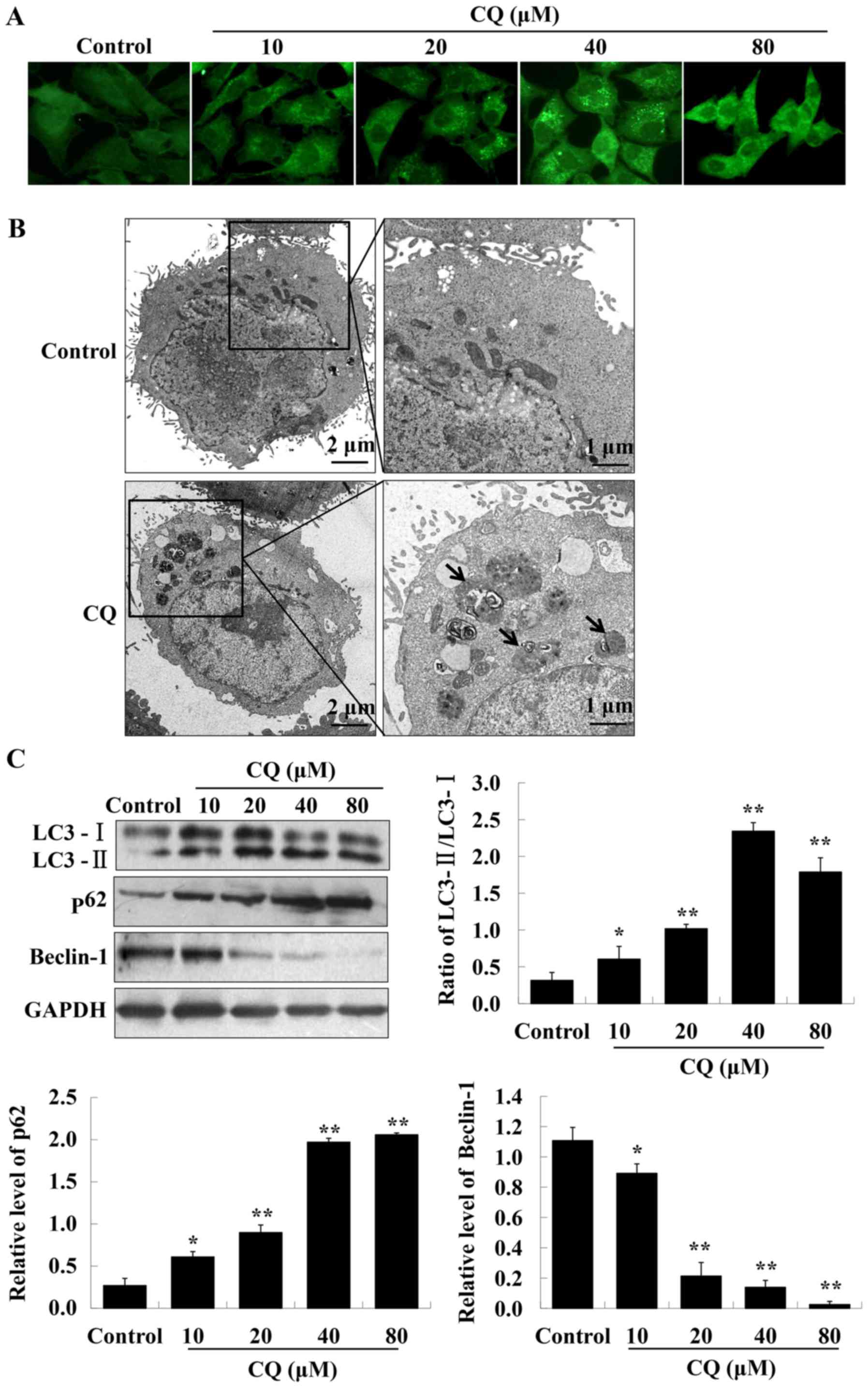
CQ inhibits autophagic flux in A549
cells
CQ, a typical autophagy inhibitor, contributes to
the inhibition of late-stage autophagy by blocking
autophagosome-lysosome fusion. We hypothesized that CQ may target
autophagy to induce the cell death of A549 cells. LC3
immunofluorescence results indicated that LC3 fluorescence dots
were accumulated in A549 cells of the experimental groups, reaching
a maximum at a concentration of 40 µM of CQ (Fig. 2A). The western blot analysis further
confirmed the enhanced expression of LC3-II, whereas that of LC3-I
was reduced, resulting in an increased ratio of LC3-II/LC3-I, with
the highest ratio reached at 40 µM of CQ treatment (Fig. 2C). It is well known that LC3-II
specifically associates with autophagosome membranes. A cellular
level of LC3-II can be used as an autophagosome formation marker
(21). TEM-based analysis further
confirmed that treatment with CQ markedly increased the number of
autophagosomes in A549 cells (Fig.
2B).
The accumulation of autophagosomes may be due to an
induction of autophagy or the inhibition of the autophagic flux
(22). Beclin-1 has an essential
role in autophagy initiation and regulates autophagy positively.
p62 is generally used as an autophagic flux marker. The expression
of Beclin-1 and p62, were analyzed by western blot analysis. The
results demonstrated that exposure of A549 cells to CQ
significantly reduced the expression level of Beclin-1. The
expression of p62 increased in a dose-dependent manner after
treatment with CQ (Fig. 2C).
Therefore, these findings revealed that CQ blocks autophagic flux
in human A549 cells.
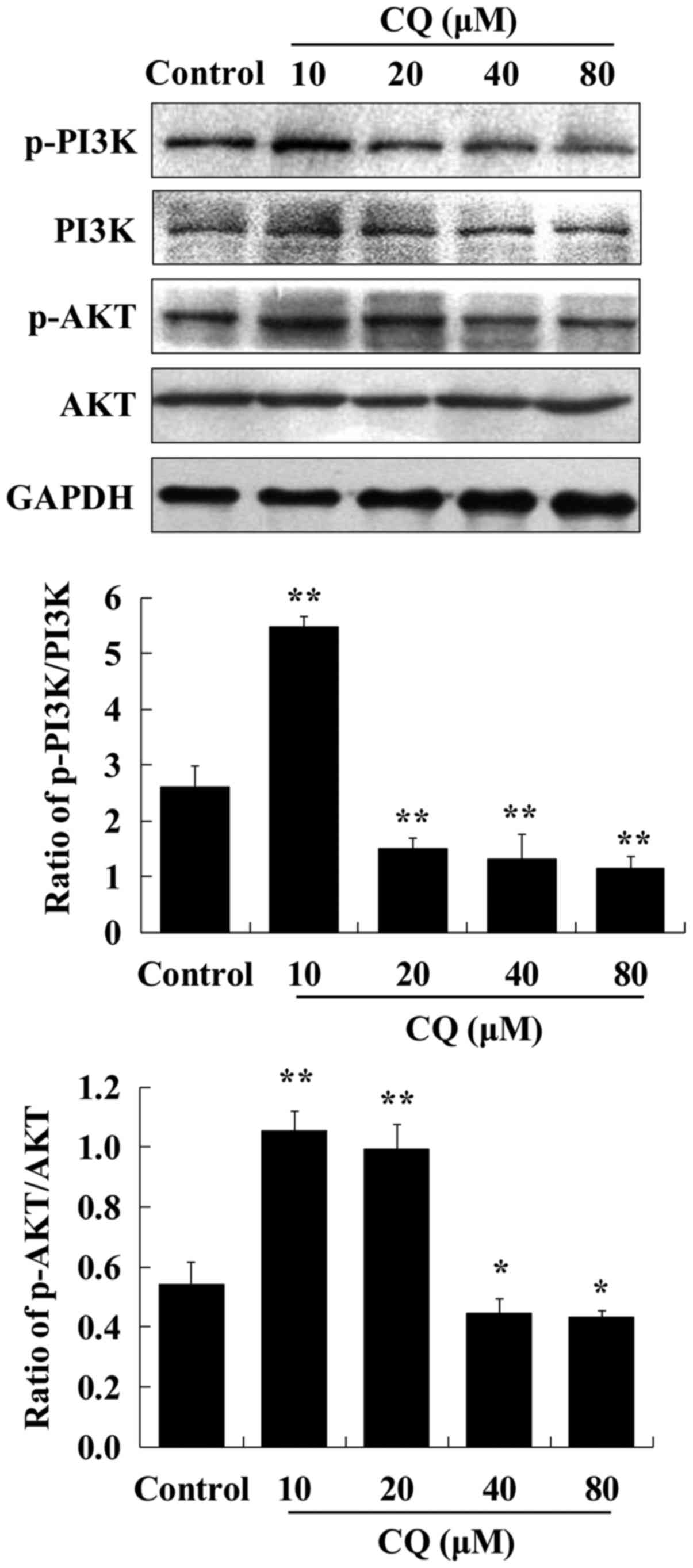
CQ inhibits autophagy by targeting the
PI3K/AKT pathway
The process of autophagy is tightly regulated by
autophagy-related (ATG) proteins. Beclin-1, the mammalian ortholog
of ATG6, governs most autophagic processes and can regulate
autophagy positively (23). It was
confirmed that the expression levels of Beclin-1 in human A549
cells were downregulated in dose-dependent manner after exposure to
CQ. The PI3K/AKT signaling pathway is well known as a regulator of
various cellular processes, such as cell survival (24). To further explore the mechanisms
underlying the autophagy-inhibiting effect of CQ, the expression of
critical proteins associated with the PI3K/AKT pathway was examined
by western blot analysis in human A549 cells treated with CQ. We
observed that the expression of p-PI3K and p-AKT was enhanced after
10 µM of CQ treatment compared with the control group.
Subsequently, a dose-dependent decrease in the expression levels of
p-PI3K and p-AKT was observed during exposure to increasing CQ
concentrations from 20 to 80 µM (Fig.
3). It was speculated that CQ inhibited autophagy at a low
concentration (10 µM) and induced apoptosis at higher
concentrations (20, 40 and 80 µM).
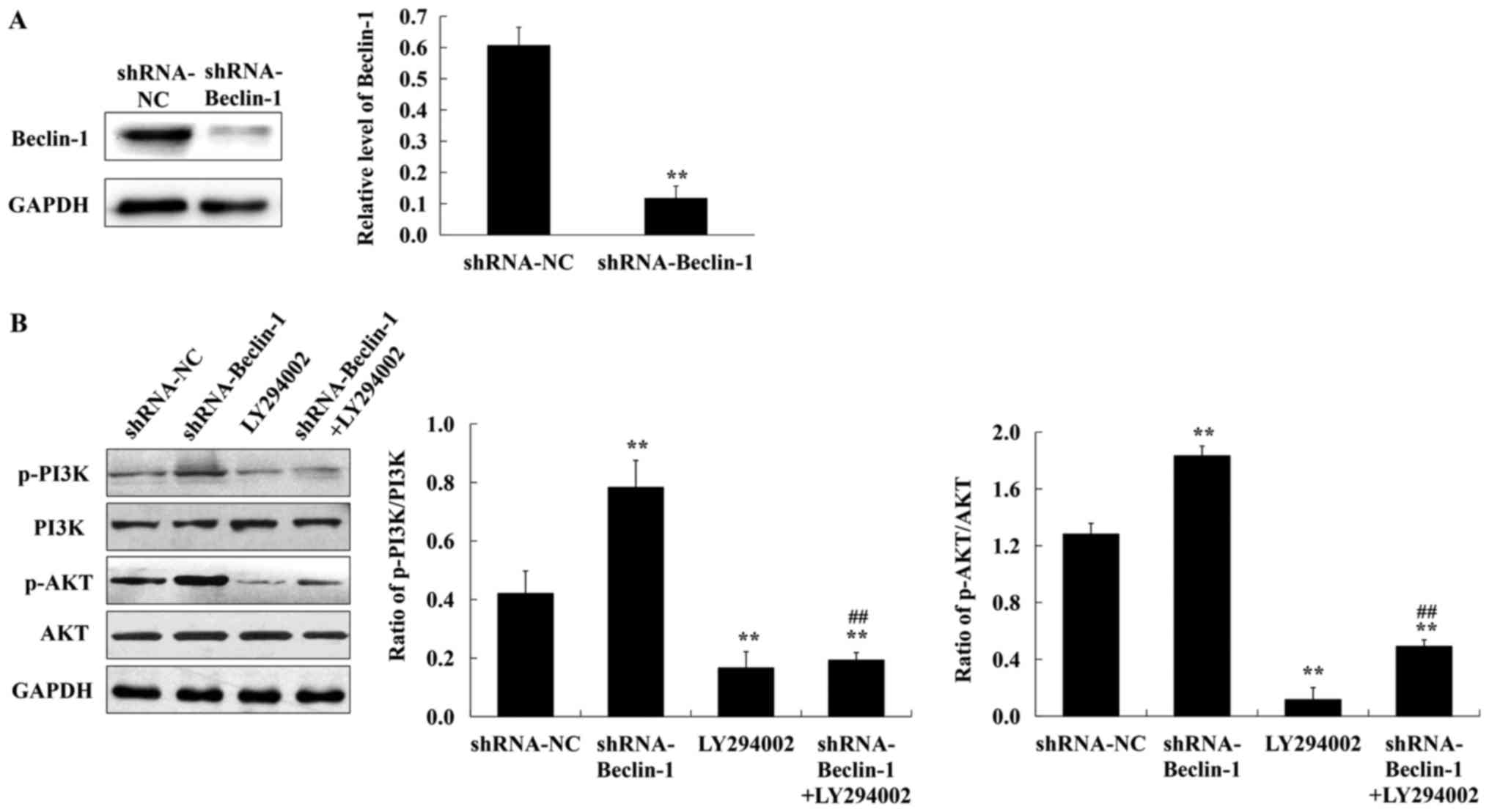
It was surmised that CQ-inhibited autophagy in A549
cells occurred mainly by reducing Beclin-1 and targeting the
PI3K/AKT signaling pathway. To ascertain this conclusion, the
lentiviral shRNA-Beclin-1 vector was constructed and the expression
of p-PI3K and p-AKT was assessed in A549 cells which were lacking
in the Beclin-1 gene in the presence of 50 µM LY294002 (a PI3K
inhibitor). As revealed in Fig. 4A,
the expression of Beclin-1 was significantly reduced after
lentivirus shRNA interference. Compared with the negative control,
the expression of p-PI3K and p-AKT was increased in the absence of
the Beclin-1 gene. LY294002 treatment decreased the expression of
p-PI3K and p-AKT in A549 cells transfected with shRNA-Beclin-1
(Fig. 4B). Collectively, these
findings confirmed that CQ inhibited autophagy in human A549 cells
by downregulating Beclin-1 and activating the PI3K/AKT signaling
pathway.
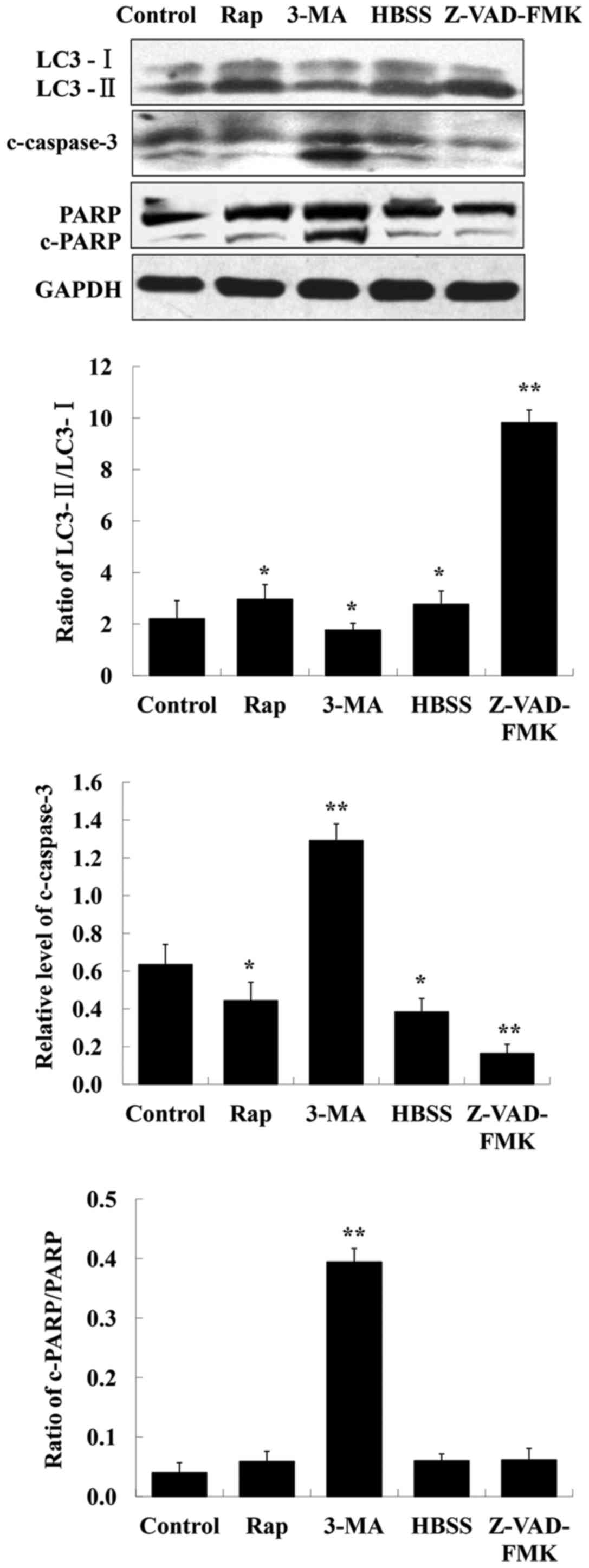
CQ induces apoptosis in human A549
cells
To determine whether apoptosis induction is involved
in the CQ-mediated inhibitory effects on cell growth, rapamycin and
HBSS (two autophagy inducers), 3-MA (an autophagy inhibitor),
Z-VAD-FMK (a caspase inhibitor) were used in human A549 cells, and
their effects on the levels of LC3-II, c-caspase-3 and PARP were
assessed by western blot analysis. The results indicated that the
level of c-caspase-3 was markedly enhanced after inhibition of
autophagy using 3-MA (Fig. 5). PARP
is a specific substrate of caspase-3. If caspase-3 is activated,
cleaved PARP can be observed. In the present study, detection of
cleaved PARP was possibly due to its faster expression. In
addition, it was observed that inhibition of apoptosis by Z-VAD-FMK
increased the level of LC3-II whereas that of LC3-I decreased
(Fig. 5).
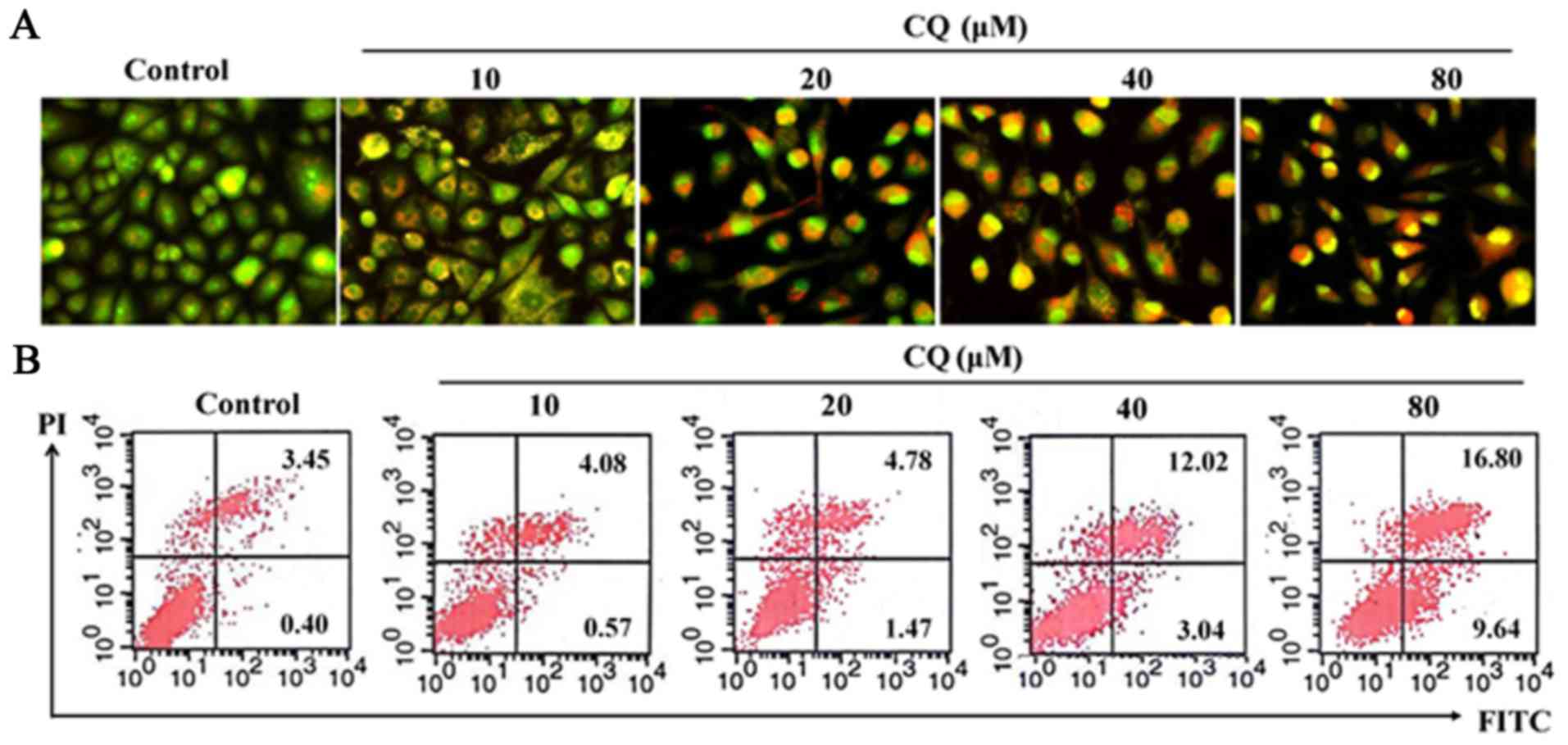
AO/EB staining was performed in human A549 cells. As
it is well known, AO penetrates the membranes of all cells,
fluorescing green when bound to DNA. EB can only enter cells when
their membranes are damaged, appearing as orange-red fluorescence
when bound to concentrated DNA fragments or apoptotic bodies.
Moreover, the fluorescence intensity of EB is stronger than that of
AO. This method allows us to distinguish normal cells, early and
late apoptotic cells and necrotic cells (25). As demonstrated in Fig. 6A, compared with the negative control
group, treatment with CQ in A549 cells induced evident
morphological changes associated with apoptosis, such as adherent
cell detachement from the culture surface, cell shrinkage and
vacuolization gradually as the concentration of CQ increased. The
early apoptotic cells were marked by green AO nuclear staining.
Late-stage apoptotic cells revealed concentrated and asymmetrical
orange fluorescence. The apoptotic cells were increased by CQ in a
concentration-dependent manner.
Apoptosis was also detected using Annexin V-FITC and
PI dual staining in A549 cells which can quantitatively distinguish
normal cells (Annexin V-FITC−/PI−), early
apoptotic cells (Annexin V-FITC+/PI−), late
apoptotic cells (Annexin V-FITC+/PI+) and
necrotic cells (Annexin V-FITC−/PI+). As
shown in Fig. 6B, there was a
marked increase of cells in early-stage apoptosis (from 0.40% in
the untreated cells to 1.47, 3.04 and 9.64% respectively, in the
20, 40 and 80-µM CQ-treated cells for 24 h) and late-stage
apoptosis (from 3.45% in the untreated cells to 4.78, 12.02 and
16.80% respectively, in the 20, 40 and 80-µM CQ-treated cells for
24 h). No obvious cell apoptosis was observed in the 10-µM
CQ-treated A549 cells. These results confirmed that CQ treatment
induced apoptosis at higher concentrations (20, 40 and 80 µM) in
A549 cells. The proportion of apoptotic cells increased
dose-dependently when they were exposed to different concentrations
of CQ.
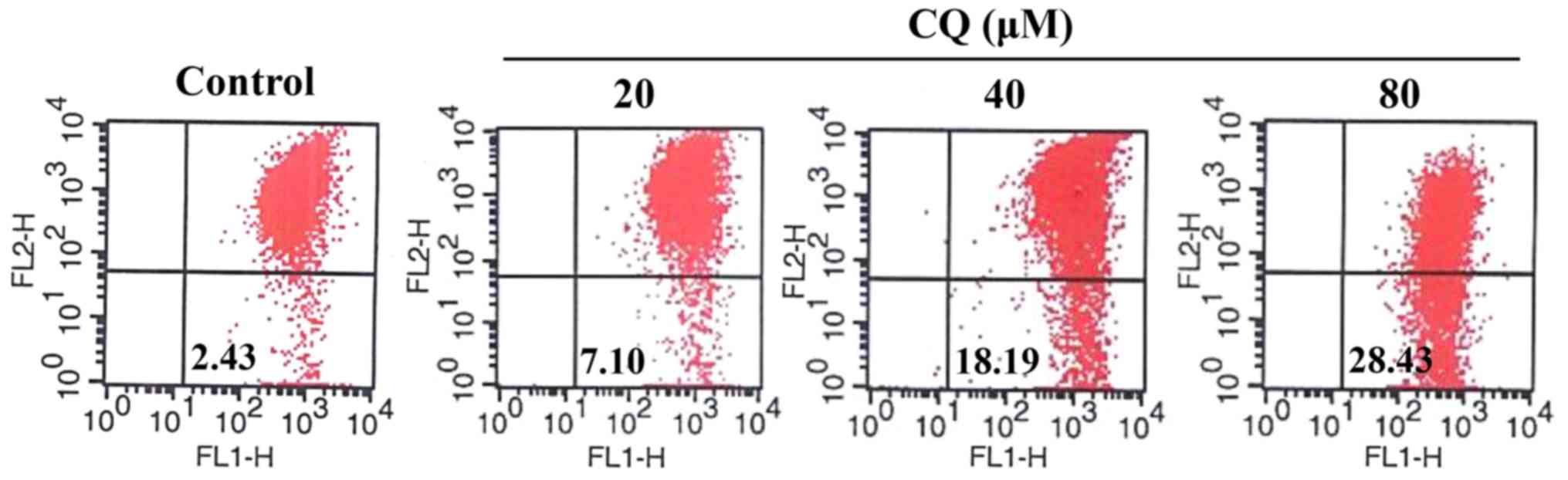
CQ activates mitochondrial-dependent apoptosis in
human A549 cells. Apoptosis is generally associated with the
activation of caspases but it is also accompanied by a loss of
mitochondrial membrane potential and the release of proapoptotic
proteins from the intermembrane space of the mitochondria.
Mitochondrial membrane potential in A549 cells with or without CQ
treatment was detected using JC-1 Mitochondrial Potential Detection
kit. As shown in Fig. 7, compared
with the control group, CQ treatment caused a drop in mitochondrial
membrane potential in a concentration-dependent manner which was
observed by an increase in green fluorescent probe JC-1 (from 2.43%
in the untreated cells to 7.10, 18.19 and 28.43%, respectively in
the 20, 40 and 80-µM CQ-treated cells).
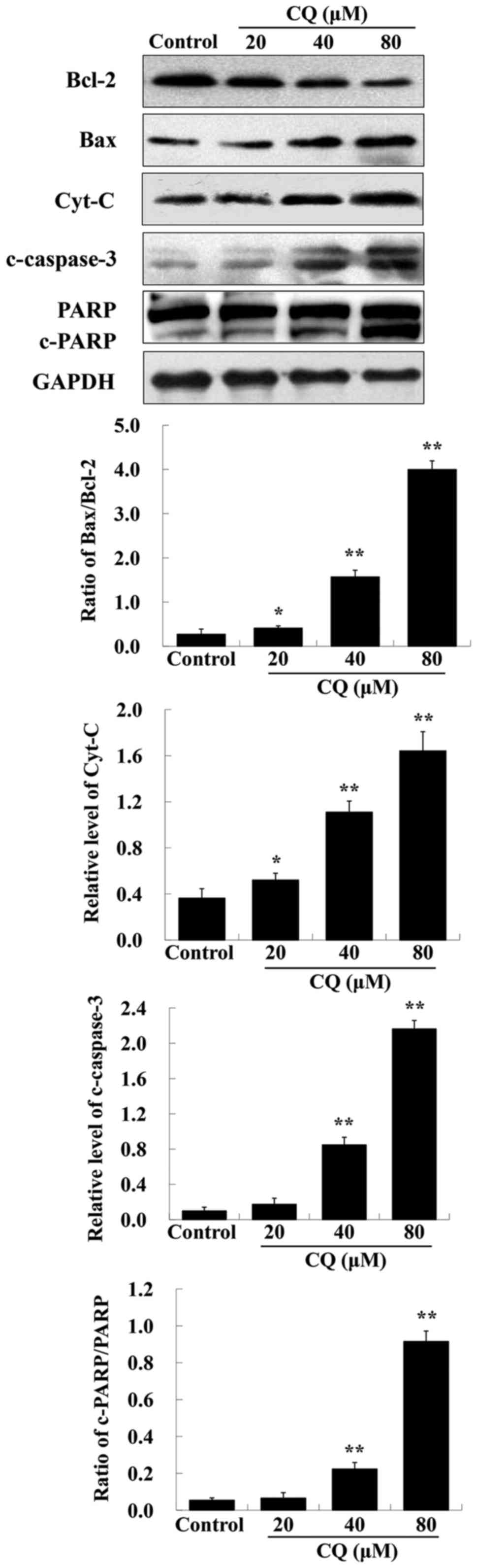
It is known that proteins of the Bcl-2 family and
caspase family, along with PARP and cytochrome c, play vital
roles in the mitochondrial apoptotic pathway (26). To further ascertain whether
CQ-induced apoptosis was mitochondrial-dependent, we also
investigated the expression of apoptosis-related proteins including
Bcl-2, Bax, cytochrome c, c-caspase-3 and PARP after
treatment with various concentrations of CQ. As depicted in
Fig. 8, upregulation of Bax,
cytochrome c, c-caspase-3 and downregulation of Bcl-2 and
PARP was induced by CQ in a dose-dependent manner. The expression
of Bcl-2 was downregulated, whereas that of Bax was upregulated,
suggesting that CQ induced an increase in the Bax/Bcl-2 ratio. We
speculated that CQ treatment reduced the level of Bcl-2, activated
the expression of proapoptotic factor Bax, reducing mitochondria
membrane potential which led to cytochrome c escape from the
mitochondria into the cytosol, further activating caspase-3 and
inducing apoptosis. Collectively, CQ induced cell apoptosis of
human A549 cells through the mitochondrial-dependent pathway.
Discussion
Chloroquine (CQ), is widely used as an anti-malarial
and anti-rheumatoid drug (27).
Recently, it has been reported that CQ, either alone, or in
combination with other agents, displayed antitumor activity,
including growth inhibition and/or induction of apoptosis in
various types of cancer. The antitumor effect of CQ appears to
depend on the tumor type, stage and genetic context (28). Previously, we also revealed that CQ
administered as a mono-drug therapy effectively suppressed the
growth of S180 sarcoma in vivo (29). Although previous studies have
examined the antitumor effect of CQ, only a few studies have
focused on the mechanism underlying the effect of CQ and the cause
and effect relationship among autophagy, apoptosis and CQ in human
A549 cells. Therefore, in the present study, we focused on the
antitumor effects of CQ and its possible mechanism. We clearly
demonstrated that in vitro, CQ effectively inhibited the
proliferation of A549 cells. The inhibitory effect of CQ on
proliferation was characterized by the blockage of autophagy
through targeting of the PI3K/AKT pathway, coupled with the
induction of mitochondrial-mediated apoptosis at relatively higher
concentrations.
Conversion of LC3-I into LC3-II is widely used as a
marker for autophagosome formation (30). p62 is degraded following an increase
in autophagic flux for which it serves as an indicator. The
increase of LC3 conversion and p62 abundance suggests the
inhibition of autophagic flux. Our results indicated that, marked
LC3 conversion and induced p62 accumulation was detected in
CQ-treated A549 cells, suggesting that CQ inhibits the autophagic
flux. Beclin-1, as a multifaceted protein, is crucial in several
cellular processes, such as autophagy, endocytosis, phagocytosis,
cytokinesis and pollen germination. Beclin-1 can intervene at every
major step in autophagic processes. It functions in the recruitment
of ATGs which are essential for autophagosome formation (31,32).
The expression level of Beclin-1 can determine the autophagic
response. The decreased protein level of Beclin-1 after CQ
treatment demonstrated that CQ inhibited autophagy in A549 cells.
The results were further confirmed by TEM-based analysis by
observing the ultrastructural changes of A549 cells.
The PI3K/AKT pathway is an important signaling
pathway in the regulation of autophagy (33). Autophagy was impaired by activation
of the PI3K/AKT pathway (34). It
has been reported that PI3K causes the phosphorylation and
activation of AKT. In the present study, it was revealed that CQ at
a low concentration induced the phosphorylation of PI3K and AKT,
indicating that CQ at a low concentration mainly inhibits autophagy
via the activation of the PI3K/AKT pathway in A549 cells. A
dose-dependent decrease in the expression levels of p-PI3K and
p-AKT during exposure to increasing CQ concentrations from 20 to 80
µM revealed that CQ-mediated growth inhibition in A549 cells may be
characterized by the inhibition of autophagy and induction of
apoptosis.
Previously, research has demonstrated that the
biological effect of CQ is concentration- and time-dependent. At
low concentrations, CQ inhibits cell proliferation by increasing
the volume of lysosomes; at high concentrations, or over longer
periods, CQ induces apoptosis and necrosis (12). In this study, CQ effectively
inhibited A549 cell proliferation in both a dose- and
time-dependent manner. Orange vacuoles were observed when cells
were exposed to CQ. Accumulation of orange vacuoles was caused by
CQ-mediated autophagy inhibition. CQ induced cell apoptosis at
relatively higher concentrations. The number of early and late
apoptotic cells increased following an increase in CQ
concentration. These findings are consistent with results from
aforementioned research. Further experiments illustrated that
CQ-induced apoptosis was mitochondrial-dependent in A549 cells by
downregulating the expression of the anti-apoptotic factor Bcl-2,
increasing proapoptotic factor Bax expression, reducing
mitochondrial membrane potential, and triggering cytochrome c
release into the cytosol, followed by caspase-3 activation and
cleavage of PARP.
Autophagy and apoptosis are both cellular catabolic
processes essential for organism homeostasis. Many stimuli elicit
autophagy and apoptosis within the same cell. In many cases,
autophagy before apoptosis dismantles the cell (19). In special cases, autophagy or
autophagy-relevant proteins sensitize cells to apoptosis or
necrosis, leading to autophagic cell death. Autophagy and apoptosis
may be triggered by common upstream signals. The PI3K-AKT axis
exhibits the dual capacity to regulate both autophagy and
apoptosis. AKT can phosphorylate Beclin-1 and BAD, further
inhibiting their pro-autophagic and pro-apoptotic activity,
respectively (35). Beclin-1 and
Bcl-2 have also been implicated in bridging autophagy and
apoptosis. The PI3Kc3 complex controls autophagy by regulating
autophagosome formation. Beclin-1, as a key component of the PI3Kc3
complex, works as the platform for assembly and triggers its
activity (36). Notably, Beclin-1
can be cleaved in apoptosis by caspases, such as, caspase-3,
caspase-7 and caspase-8. The cleavage of Beclin-1 loses its
capacity to induce autophagy and generates N- and C-terminal
fragments. The C-terminal fragments are able to sensitize cells to
apoptotic signals. Bcl-2 is a direct binding partner of Beclin-1.
In cells, Bcl-2 is constitutively bound to Beclin-1, leading to
decreased autophagic activity. However, Bcl-2 does not lose its
anti-apoptotic potential (37).
In summary, the present study demonstrated that CQ
at a low concentration inhibited autophagy by targeting the
PI3K/AKT pathway. With increased concentration of CQ, apoptosis was
markedly triggered through the activation of the mitochondrial
pathway and CQ effectively inhibited human A549 cell proliferation
in vitro. The present study may provide new theoretical and
experimental evidence for a clinical trial on CQ in lung cancer
patients.
Acknowledgements
We thank the Molecular Biology Experiment Center
(MBEC) at Qiqihar Medical University for the use of shared
facilities.
Funding
The present study was sponsored by the Youth Special
Purpose Foundation (grant no. 1253G066) from the Education
Department of Heilongjiang Province, the Qiqihar Municipal Science
and Technology Project of China (no. SFGG-201556), the Natural
Science Foundation of Heilongjiang Province for Returned Scholars
of China (no. LC2011C34) and the Key Program from Qiqihar Medical
University of China (no. QY2013ZD-02).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
LY conceived and designed research. LL analyzed data
and wrote the paper. CH, WZ, HC and CZ performed the experiments.
LY, HY and LZ reviewed and edited the manuscript. HY and LZ were
also involved in the conception of the study. All authors read and
approved in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interest.
References
|
1
|
Homewood CA, Warhurst DC, Peters W and
Baggaley VC: Lysosomes, pH and the anti-malarial action of
chloroquine. Nature. 235:50–52. 1972. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rainsford KD, Parke AL, Clifford-Rashotte
M and Kean WF: Therapy and pharmacological properties of
hydroxychloroquine and chloroquine in treatment of systemic lupus
erythematosus, rheumatoid and related diseases.
Inflammopharmacology. 23:231–269. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Savarino A, Di Trani L, Donatelli I, Cauda
R and Cassone A: New insights into the antiviral effects of
chloroquine. Lancet Infect Dis. 6:67–69. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Poole B and Ohkuma S: Effect of weak bases
on the intralysosomal pH in mouse peritoneal macrophages. J Cell
Biol. 90:665–669. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim SH, Kim JH and Fried J: Enhancement of
the radiation response of cultured tumor cells by chloroquine.
Cancer. 32:536–540. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Djordjevic B, Lange CS, Austin JP and
Rotman M: Potentiation of radiation lethality in HeLa cells by
combined mild hyperthermia and chloroquine. Radiat Res.
130:267–270. 1992. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Djordevic B, Lange CS and Rotman M:
Potentiation of radiation lethality in mouse melanoma cells by mild
hyperthermia and chloroquine. Melanoma Res. 2:321–326. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zinn RL, Gardner EE, Dobromilskaya L,
Murphy S, Marchionni L, Hann CL and Rudin CM: Combination treatment
with ABT-737 and chloroquine in preclinical models of small cell
lung cancer. Mol Cancer. 12:162013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sasaki K, Tsuno NH, Sunami E, Tsurita G,
Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, Kaneko
M, et al: Chloroquine potentiates the anti-cancer effect of
5-fluorouracil on colon cancer cells. BMC Cancer. 10:3702010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Enzenmüller S, Gonzalez P, Debatin KM and
Fulda S: Chloroquine overcomes resistance of lung carcinoma cells
to the dual PI3K/mTOR inhibitor PI103 by lysosome-mediated
apoptosis. Anticancer Drugs. 24:14–19. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang S, Wang X, Contino G, Liesa M, Sahin
E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al:
Pancreatic cancers require autophagy for tumor growth. Genes Dev.
25:717–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fan C, Wang W, Zhao B, Zhang S and Miao J:
Chloroquine inhibits cell growth and induces cell death in A549
lung cancer cells. Bioorg Med Chem. 14:3218–3222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng Y, Zhao Y, Deng X, Yang S, Mao Y, Li
Z, Jiang P, Zhao X and Wei Y: Chloroquine inhibits colon cancer
cell growth in vitro and tumor growth in vivo via induction of
apoptosis. Cancer Invest. 27:286–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang PD, Zhao YL, Yang SY, Mao YQ, Zheng
YZ, Li ZG and Wei YQ: Effects of chloroquine diphosphate on
proliferation and apoptosis of human leukemic K562 cells. Zhongguo
Shi Yan Xue Ye Xue Za Zhi. 16:768–771. 2008.(In Chinese).
PubMed/NCBI
|
|
15
|
Hu T, Li P, Luo Z, Chen X, Zhang J, Wang
C, Chen P and Dong Z: Chloroquine inhibits hepatocellular carcinoma
cell growth in vitro and in vivo. Oncol Rep. 35:43–49. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim EL, Wüstenberg R, Rübsam A,
Schmitz-Salue C, Warnecke G, Bücker EM, Pettkus N, Speidel D, Rohde
V, Schulz-Schaeffer W, et al: Chloroquine activates the p53 pathway
and induces apoptosis in human glioma cells. Neruo-Oncol.
12:389–400. 2010. View Article : Google Scholar
|
|
17
|
Lakhter AJ, Sahu RP, Sun Y, Kaufmann WK,
Androphy EJ, Travers JB and Naidu SR: Chloroquine promotes
apoptosis in melanoma cells by inhibiting BH3 domain-mediated PUMA
degradation. J Invest Dermatol. 133:2247–2254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kroemer G and Levine B: Autophagic cell
death: The story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kaminskyy V, Abdi A and Zhivotovsky B: A
quantitative assay for the monitoring of autophagosome accumulation
in different phases of the cell cycle. Autophagy. 7:83–90. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yan Y, Jiang K, Liu P, Zhang X, Dong X,
Gao J, Liu Q, Barr MP, Zhang Q, Hou X, Meng S and Gong P:
Bafilomycin A1 induces caspase-independent cell death in
hepatocellular carcinoma cells via targeting of autophagy and MAPK
pathways. Sci Rep. 6:370522016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang L, Gao C, Yao S and Xie S: Blocking
autophagic flux enhances matrine-induced apoptosis in human
hepatoma cells. Int J Mol Sci. 14:23212–23230. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sheppard KE, Kinross KM, Solomon B,
Pearson RB and Phillips WA: Targeting PI3 kinase/Akt/mTOR signaling
in cancer. Crit Rev Oncog. 17:69–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu K, Liu P, Liu R and Wu X: Dual AO/EB
staining to detect apoptosis in osteosarcoma cells compared with
flow cytometry. Med Sci Monit Basic Res. 21:15–20. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao X, Ma S, Liu N, Liu J and Wang W: A
polysaccharide from Trametes robiniophila Murrill induces apoptosis
through intrinsic mitochondrial pathway in human osteosarcoma (U-2
OS) cells. Tumor Biol. 36:5255–5263. 2015. View Article : Google Scholar
|
|
27
|
Thomé R, Lopes SC, Costa FT and Verinaud
L: Chloroquine: Modes of action of an undervalued drug. Immunol
Lett. 153:50–57. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao XG, Sun RJ, Yang XY, Liu DY, Lei DP,
Jin T and Pan XL: Chloroquine-enhanced efficacy of cisplatin in the
treatment of hypopharyngeal carcinoma in xenograft mice. PLoS One.
10:e01261472015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yue L, Mei Q, Zhe W, Zhang W, Liu D and
Liu Y: Study of chloroquine on anti-tumor effects of
S180 tumor-bearing mice and its mechanisms. Acta
Anatomica Sinica. 6:779–784. 2016.
|
|
30
|
Mizushima N: Methods for monitoring
autophagy. Int J Biochem Cell Biol. 36:2491–2502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maiuri MC, Criollo A and Kroemer G:
Crosstalk between apoptosis and autophagy within the Beclin 1
interactome. EMBO J. 29:515–516. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heras-Sandoval D, Pérez-Rojas JM,
Hernández-Damián J and Pedraza-Chaverri J: The role of
PI3K/AKT/mTOR pathway in the modulation of autophagy and the
clearance of protein aggregates in neurodegeneration. Cell Signal.
26:2694–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Datta K, Suman S and Fornace AJ Jr:
Radiation persistently promoted oxidative stress, activated mTOR
via PI3K/Akt, and downregulated autophagy pathway in mouse
intestine. Int J Biochem Cell Biol. 57:167–176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yue Z and Zhong Y: From a global view to
focused examination: Understanding cellular function of lipid
kinase VPS34-Beclin 1 complex in autophagy. J Mol Cell Biol.
2:305–307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ciechomska IA, Goemans GC, Skepper JN and
Tolkovsky AM: Bcl-2 complexed with Beclin-1 maintains full
anti-apoptotic function. Oncogene. 28:2128–2141. 2009. View Article : Google Scholar : PubMed/NCBI
|