Introduction
The tumor suppressor protein p53 plays a central
role in the cell cycle and maintenance of genomic integrity
(1–3). The p53 gene is frequently mutated in
human cancers, and germline mutations are the underlying cause of
Li-Fraumeni syndrome (4,5). The human p53 protein is comprised of
an N-terminal transactivation domain, a proline-rich region, a
structured DNA-binding domain connected to a tetramerization domain
(TD) via a flexible linker, and a C-terminal regulatory domain
(6). The p53 protein is active as a
homotetramer which adopts a dimer of dimer topology (7). The TD in human p53 (amino acid
residues 326–356) exhibits a dihedral symmetry of dimers (8). Two monomers interact with the
β-strands (Glu326-Arg333) to form a dimer, and the two dimers then
interact as a α-helix bundle (Arg335-Gly356) to form the tetramer
(9). Nine residues in the TD in
human p53 (Phe328, Leu330, Ile332, Arg337, Phe338, Met340, Phe341,
Leu344 and Leu348) are critical determinants in stabilizing the p53
tetramer (10). Leu344 mutants
lacking the ability to dimerize (L344P), or tetramerize (L344R),
have been previously reported (11), and notably, germline L344P mutation
has been found in a family with Li-Fraumeni syndrome (12). Leu344 is located in the α-helix
which forms the hydrophobic core of the tetramer interface.
Although, numerous cell lines containing mutated p53
genes have been established from cancer tissues, there are few cell
lines which contain a mutated p53 tetramerization domain (13,14).
Uyama et al established and characterized four pairs of
canine mammary tumor cell lines derived from either primary and
metastatic origin (15). One of
these cell lines, CTB-m, was passaged 50 times in our laboratory to
obtain a line containing a spontaneous L332Q mutation in p53
(corresponding to human L344); we designated this new cell line as
CTB-m2. In this study, we assessed the oligomerization abilities of
this canine p53 L332Q mutant and performed a tetramerization
reporter assay.
Materials and methods
Cell culture and transfection
The CIP-p, CIP-m, and CTB-m cell lines were kindly
provided by Dr N. Sasaki of the University of Tokyo. CIP-p, CIP-m,
CTB-m and CTB-m2 cells were cultured in RPMI-1640 medium (Wako,
Osaka, Japan) and HeLa cells (ATCC, Rockville, MD, USA) were
cultured in DMEM (Wako). All media were supplemented with 10% fetal
bovine serum (FBS) and penicillin-streptomycin (Wako) and cells
were cultured under a humidified atmosphere of 5% CO2 at
37°C. Cell transfection was performed using FuGENE HD (Promega,
Madison, WI, USA), as previously reported (16).
Establishment of the CTB-m2 cell
line
CTB-m2 cells were spontaneously established from the
CTB-m line after 50 passages in our laboratory.
Sequencing analysis and cloning of the
canine p53 gene
Total RNA from CTB-m and CTB-m2 cells was obtained
using a High Pure RNA Tissue Kit (Roche Diagnostics, Mannheim,
Germany), and reverse transcription conducted using a Reverse
Transcription System (Promega) according to the manufacturer's
protocol. Full length canine p53 cDNA was amplified by PCR using
the following primers: 5′-CTCGAGGACCACCATGCAAGAGCCACAGTCAGAGC-3′
and 5′-GAATTCCCGTCTGAGTCGGGCCCTTCTCTC-3′. Following digestion with
XhoI and EcoRI, the PCR product was cloned into the
XhoI and EcoRI sites of the plasmid pMACS KK HA(C)
(Miltenyi Biotec, Bergisch Gladbach, Germany). Samples containing
genomic DNA from cells were prepared using the ZR Genome DNATM
Tissue Miniprep (Zymo Research, Irvine, CA, USA). Exon 10–11 of p53
containing the sequence encoding L332 was amplified by PCR using
the following primers; 5′-GCACTTACACCTTAGTCTGAG-3′, and
5′-CGGAATAGGTGTGCTCAAGC-3′. The amplified canine p53 cDNA and exon
10–11 were directly sequenced using an ABI 3730 system (Applied
Biosystems, Waltham, MA, USA).
Cell proliferation analysis
A total of 20,000 cells were plated into each well
of a 24-well plate, and counted every 24 h up until 72 h using a
TC20™ Automated Cell Counter (Bio-Rad, Hercules, CA, USA).
p53 tetramerization reporter
assay
The p53 response element in p21WAF (GenBank
accession no. U24170, nucleotides 2303–2321) and human
cytomegalovirus in the pTet-splice vector (nucleotides 318–446;
Clontech Laboratories Inc., Palo Alto, CA, USA) (17) was inserted between the XhoI
and HindIII sites in pNL1.1[Nluc] (p53RE-pNL1.1) (Promega).
To measure the endogenous p53 tetramerization activities, CTB-m and
CTB-m2 cells were transfected with the p53RE-pNL1.1 reporter
plasmid. The cells were harvested 48 h after transfection, and
luciferase activity was measured using the Nano-Glo Dual-Luciferase
Reporter Assay System (Promega). The luciferase activity was
normalized to the value of the luc2 activity from co-transfected
pGL4.51[luc2/CMV/Neo] (Promega). To measure the exogenous p53
tetramerization activities, HeLa cells were co-transfected with
p53RE-pNL1.1 and either WT canine p53 or its L332Q mutant in pMACS
KK HA (C).
Sample preparation and cross-linking
procedure
HeLa cells in 6-well plates were transfected with
either WT or L332Q p53 expression plasmids (1 µg/well). After 48 h
of transfection, the cells were treated with doxorubicin at a final
concentration of 0.5 µM for 6 h. Cells were lysed with mammalian
lysis buffer (Promega) supplemented with a protease inhibitor
cocktail (Promega). After lysis, the samples were centrifuged
(15,000 × g for 15 min at 4°C) to obtain the supernatant. Total
protein levels were measured using BCA (Nacalai Tesque, Kyoto,
Japan). Equal amounts of proteins (100 µg/condition) were incubated
with glutaraldehyde at different concentrations (0, 0.02 or 0.04%)
and incubated on ice for 30 min. To make a working solution of
glutaraldehyde, the commercially available 25% glutaraldehyde
solution was diluted in PBS and discarded after use. To quench the
reaction, sample buffer was added to obtain the following final
concentrations: 250 mM Tris-HCl, pH 8.5; 2% lithium dodecyl
sulfate; 100 mM DTT; 0.4 mM EDTA; 10% glycerol; 0.2 mM bromophenol
blue. Samples were then separated by polyacrylamide gel
electrophoresis (18).
Electrophoresis and
immunoblotting
Samples were resolved on a 5–12.5% Tris-glycine
polyacrylamide gradient gel using a Perfect Cell B from DRC (Tokyo,
Japan) (∆V = 150 V, 50 min at 25°C), transferred to a PVDF membrane
using a standard semi-dry apparatus from Bio-Rad (∆V = 15 V, 1 h at
25°C), and probed with an anti-HA specific mouse monoclonal
antibody (M180-3, MBL, Aichi, Japan), and then incubated with
HRP-conjugated secondary anti-mouse IgG (7076; Cell Signaling
Technology, Beverly, MA, USA). Immunoreactive bands were visualized
with a gel documentation system (LAS-4000 mini; Fujifilm, Tokyo,
Japan).
Crystal structure modeling
We retrieved the crystal structure of the human p53
tetramerization domain from the Research Collaboratory for
Structural Bioinformatics Protein Data Bank at http://www.rcsb.org/ (PDB ID: 3FAK) and analyzed it
using the University of California, San Francisco (UCSF) Chimera
software (http://www.cgl.ucsf.edu/chimera/) (18).
Microsatellite genotyping of canine
cell lines by polymerase chain reaction (PCR) and amplified
fragment length polymorphism analysis
Thirteen microsatellite loci were compared between
the CTB-m and CTB-m2 or CIP-p and CIP-m cell lines by PCR and
amplified fragment length polymorphism analysis in a genetic
diagnosis laboratory (http://www.kahotechno.co.jp/clinic/index.html,
Kahotechno, Fukuoka, Japan).
Statistical analysis
Data are expressed as mean ± standard deviation
(SD). Differences between two groups were analyzed using the
Student's t-test. Analysis of variance (ANOVA) with a Tukey's post
hoc test was used when multiple comparisons were required.
Significance was assessed at the 0.05 (or lower) level for all
tests.
Results
p53 in CTB-m2 cells contains a
heterozygous mutation at L332Q
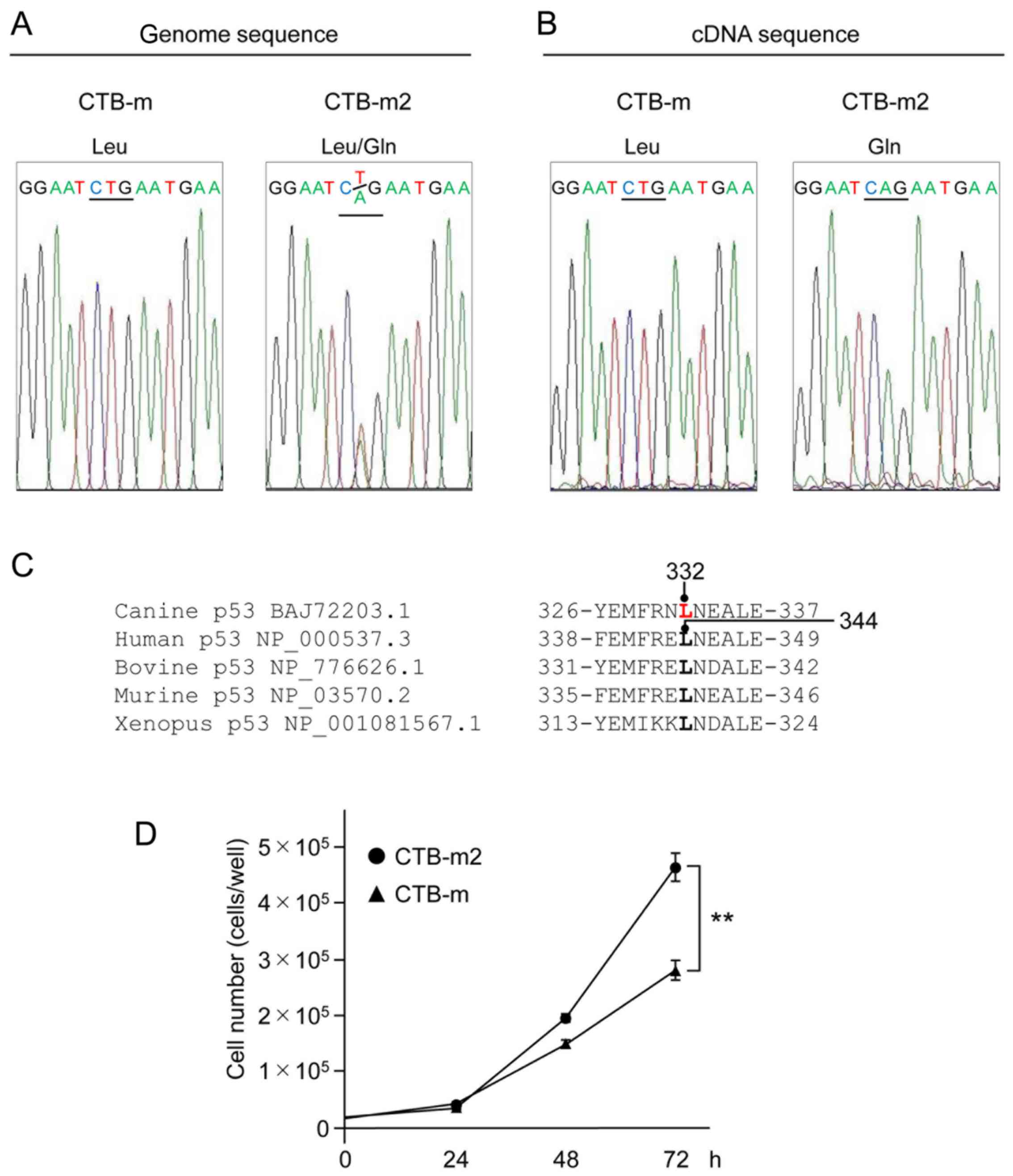
Sequencing analysis of genomic p53 from the
canine mammary gland tumor cell line CTB-m2 showed the presence of
a heterozygous missense mutation L332Q, compared to CTB-m cells,
which expressed homozygous L332 (Fig.
1A). However, the sequence of p53 mRNA derived from
CTB-m2 cells showed almost exclusively the mutated allele (L332Q)
(Fig. 1B). A comparison of a part
of the tetramerization domain of the canine p53 protein (GenBank
accession: BAJ72203.1) with human, bovine, murine and Xenopus p53
protein (NP_000537.3; NP_776626.1; NP_03570.2; NP_001081567.1)
showed that canine Leu332 corresponds to human Leu344, and that
this sequence is completely conserved in other species (Fig. 1C).
Cell viability of CTB-m2 cells is
higher than CTB-m
A cell proliferation analysis was performed using an
automatic cell counter every 24 h after cultivation. Seventy-two
hours after cultivation, CTB-m2 cell proliferation was found to be
significantly higher than CTB-m cell proliferation (Fig. 1D).
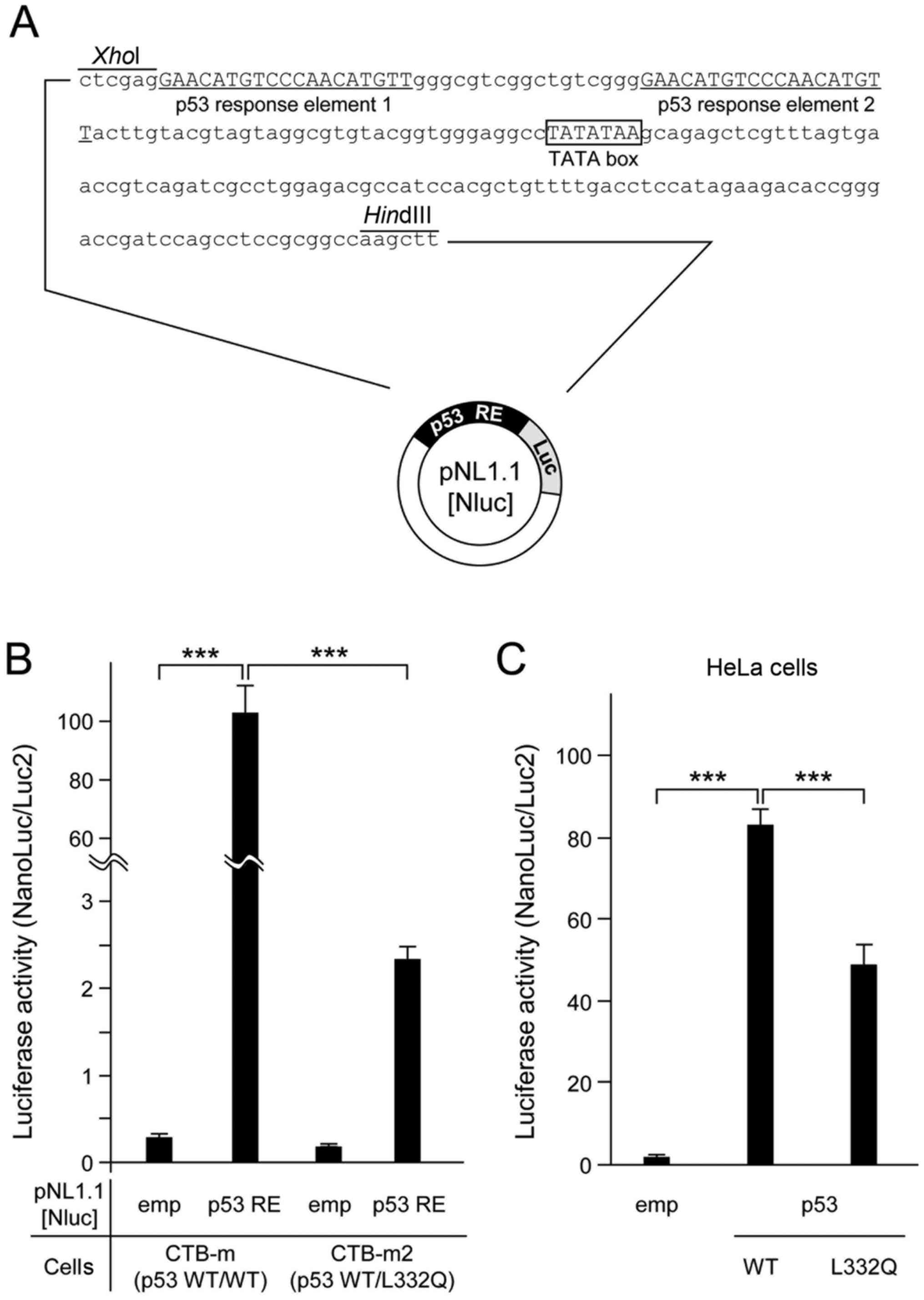
L332Q mutant in p53 reduces the
tetramerization ability compared with WT p53
The p53 tetramer response element, along with the
human cytomegalovirus minimal promoter sequence, were cloned
upstream of the sequence encoding Nano Luc (Fig. 2A), and the resulting plasmid was
transfected into CTB-m or CTB-m2 cells, or into HeLa cells also
expressing exogeneous p53 WT (wild-type) or its L332Q mutant, in
order to assess endogenous or exogenous p53 tetramerization. The
L332Q heterozygous mutant CTB-m2 had an approximately 50-fold
reduction in luciferase activity compared to WT CTB-m cells
(Fig. 2B). Forced expression of the
canine p53 L332Q mutant in HeLa cells also showed significantly
lower luciferase activity compared to forced expression of WT p53
(Fig. 2C).
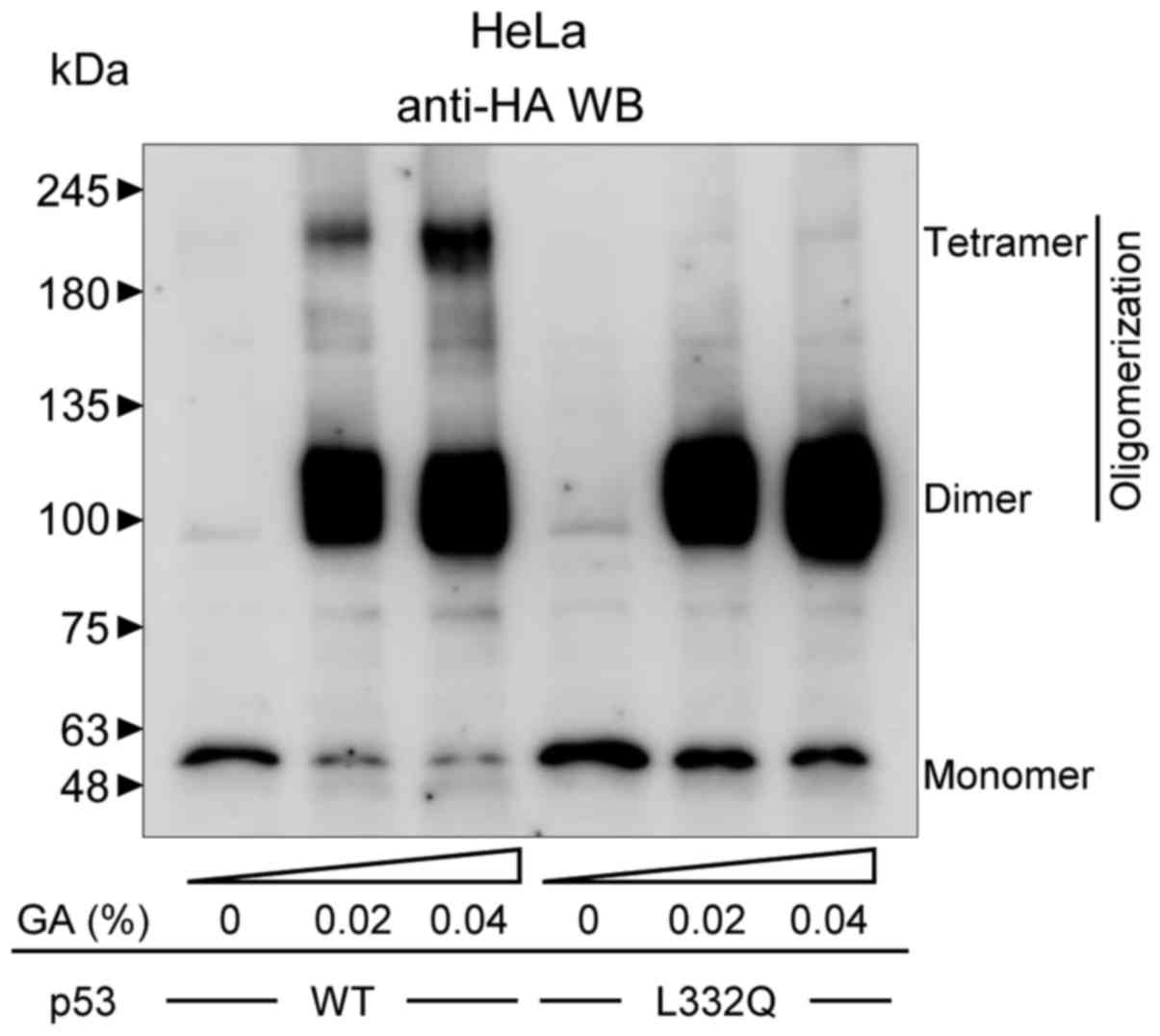
The p53 L332Q mutant lacks the ability
to tetramerize but not the ability to dimerize
The oligomerization ability of p53 transfected into
HeLa cells was assessed using a glutaraldehyde cross-linking
assays. Cell lysates from HeLa cells expressing HA-tagged WT p53 or
HA-tagged p53 L332Q were treated with 0.02 and 0.04% glutaraldehyde
crosslinker and then analyzed by western blotting. The HA-tagged WT
p53 formed both dimers and tetramers following glutaraldehyde
treatment. On the other hand, the L332Q mutant formed only a dimer
(Fig. 3).
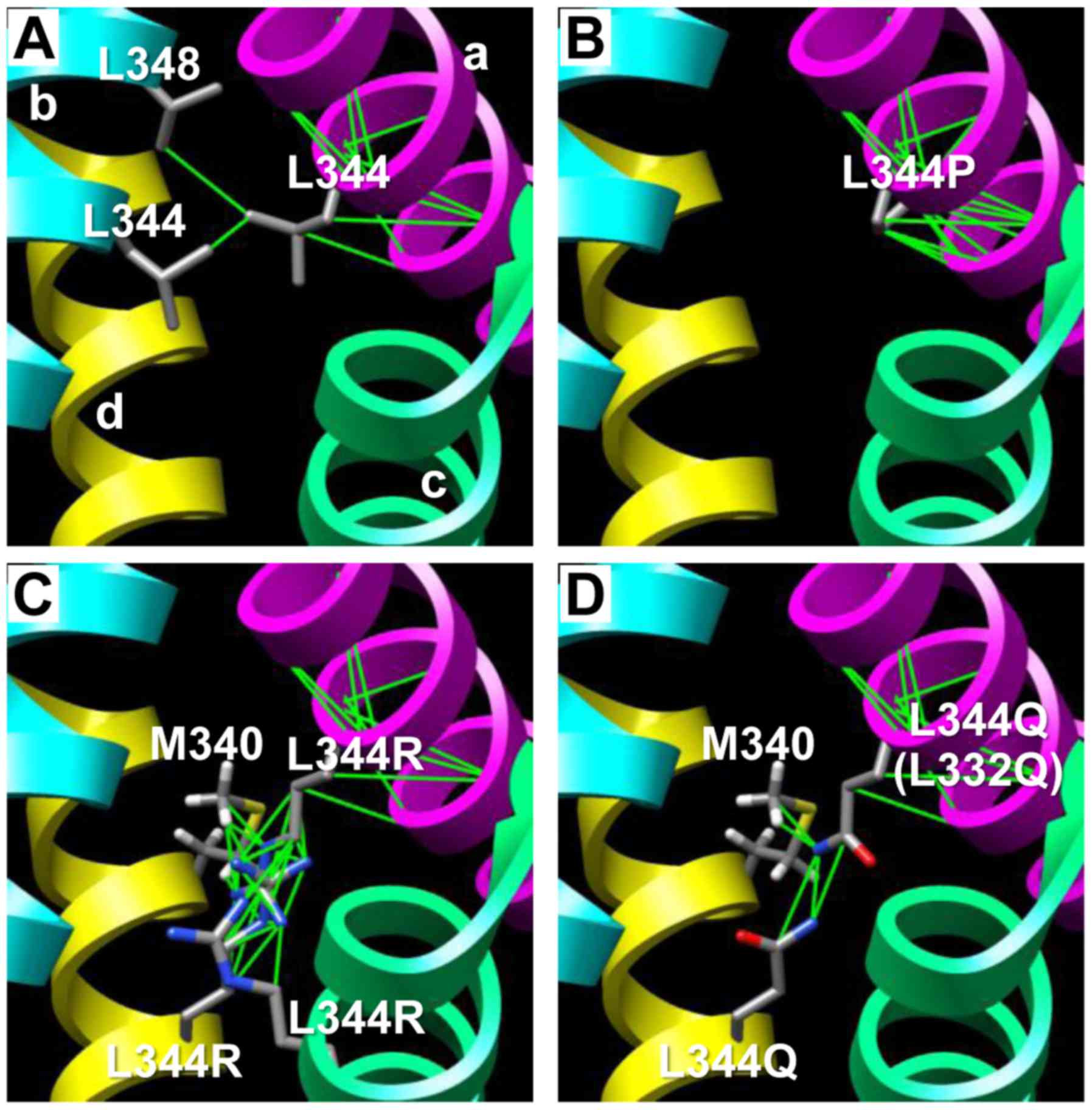
The L332Q mutation results in a
conformational change similar to that observed for the L344R
mutation in human p53
To verify the results of the functional assay, the
protein structure editing tool in the UCSF Chimera software package
was used to analyze the possible structural outcomes of L332Q
substitutions and L344P(R) mutations based on previous reports
(11). The L344 of chain a showed
hydrogen bonds with amino acids belonging to the intra-strand and
L344/L348 in chain b (Fig. 4A). The
Rotamers tool allows amino acid side chain rotamers to be viewed
and evaluated (19). The best
rotamers for Arg, Pro, or Gln were selected based on their
side-chain torsion, as well as probability values in the rotamer
library and in the context of the structural environment. These
calculations revealed that the substitution L344P disrupted the
inter-strand hydrogen bond required for tetramerization (Fig. 4B). The L344R and L344Q substitutions
resulted in a disruption of the hydrogen bonds to chain b, and with
appearance of hydrogen bonds to the substituted R344 or Q344 and
M340 in chain c. The L344R mutant also showed contact with R344 in
chain d (Fig. 4C and D).
Microsatellite analysis of cell lines
established from the same origins
Thirteen microsatellite loci were compared among the
CTB or CIP canine mammary gland tumor cell lines. Two
microsatellite loci between the CTB-m and CTB-m2 cell lines were
different, while the other 11 were identical. Five microsatellite
loci between the CIP-p and CIP-m cell lines were different, while
the other 8 were identical (Fig. 5A and
B).
Discussion
The functions of the tetramerization domain in the
p53 protein and its inhibitory and stabilizing ligands have been
well investigated using biochemical, cell biological and in
silico analysis (20–22). However, there were no suitable
models expressing a mutant of p53 containing a dysfunction in the
tetramerization domain. The NCI-H1299 cell line, which does not
express the p53 protein, has typically been used to investigate the
effect of exogenous p53 mutant expression (23,24).
Here, we established the cell line CTB-m2 which expresses the p53
L332Q (which corresponds to L344 in human p53) mutant.
Microsatellite analysis using 11 markers showed that in CTB-m
cells, which are the parental cells for CTB-m2, are nearly
identical compared with another pair of canine mammary gland tumor
cell lines, namely CIP-p and CIP-m (Fig. 5A and B) (15). These data suggest that the L332Q
mutation in CTB-m2 is a post-oncogenic transformation. Despite
being genetically heterozygous at the nucleotides encoding L332,
CTB-m2 cells expressed almost only the mutated L332Q p53 mRNA
suggesting that in these cells the wild-type p53 allele is
inactive. The expression of p53 mutants has been shown to be
heterogenous and gene methylation has also been observed in human
cancers (25,26). CTB-m2 cells may therefore have a
unique expression status among p53 mutant alleles. Leu332 in
canine p53 corresponds to Leu344 in human p53, which is widely
recognized as being an essential residue for tetramerization
(10). A cell proliferation
analysis of the mutated p53 showed enhanced viability of CTB-m2
cells compared with CTB-m cells. The p53 R280T mutant, which is
located in the DNA binding domain also promoted cell proliferation
in a human glioma cell line (27).
Ablation of p53 tetramerization as a result of the L332Q mutation
would be expected to change the conformation of the DNA binding
domains, which may then result in enhancement of cell proliferation
(28).
We examined the tetramerization ability of the p53
L332Q mutant using a functional reporter assay. Two p53 response
elements and the minimal human cytomegalovirus promoter sequence,
cited by a previous study (17),
were modified and cloned into a vector containing a luciferase
reporter. The large difference in luciferase activities observed
between CTB-m and CTB-m2 cells, which reflects the endogenous
tetramerization abilities of p53 in these two cells, suggest that
the L332Q mutation is critical for tetramerization, and also
confirms that almost only the mutated allele is expressed in CTB-m2
cells. Although there was a significant attenuation in
tetramerization for the p53 L332Q mutant, the dimer forming ability
of this mutant was retained, as shown by electrophoresis and
immunoblotting of p53 cross-linked with glutaraldehyde. The in
silico simulation of amino acid substitution supports this
retention of dimerization ability, and L332Q (L344Q) mutant showed
a greater degree of similarity to L344R, which dimerizes, than to
L344P, which can only form monomers. The mutated residues in the
L332Q (L344Q), L344P, and L344R mutants could not form bonds with
Leu344 and Leu348 of the neighboring chain in the tetramerization
domain. However, both L332Q (L344Q) and L344R mutants showed
contacts with Met340 and L344Q (L344R) of the oblique neighboring
chain.
In summary, we established and characterized a new
cell line, CTB-m2, which expresses p53 L332Q, a mutant in the p53
tetramerization domain. As a result of this mutation, p53 L332Q
lacked tetramerization but not dimerization ability. These CTB-m2
cells can be used to investigate p53 pathogenesis and to evaluate
new strategies to restore p53 function (29).
Acknowledgements
Not applicable.
Funding
The present study was supported by the KAKENHI
Scientific Research grants from the Ministry of Education, Culture,
Sports, Science and Technology of Japan (nos. 15K07754 and
26450400).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author upon reasonable
request.
Authors' contributions
KO, MM1 and TO designed the research. KO and DA
mainly did the research. HH, SK, MM2, AE and TS also performed some
experiments. TN and MW analyzed the data. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the study are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Meek DW: Tumour suppression by p53: A role
for the DNA damage response? Nat Rev Cancer. 9:714–723. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lane D and Levine A: p53 Research: The
past thirty years and the next thirty years. Cold Spring Harb
Perspect Biol. 2:a0008932010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bieging KT, Mello SS and Attardi LD:
Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev
Cancer. 14:359–370. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: Lessons from recent developments in the IARC TP53
database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Olivier M, Hollstein M and Hainaut P: TP53
mutations in human cancers: Origins, consequences, and clinical
use. Cold Spring Harb Perspect Biol. 2:a0010082010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Joerger AC and Fersht AR: Structural
biology of the tumor suppressor p53. Annu Rev Biochem. 77:557–582.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rajagopalan S, Huang F and Fersht AR:
Single-molecule characterization of oligomerization kinetics and
equilibria of the tumor suppressor p53. Nucleic Acids Res.
39:2294–2303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jeffrey PD, Gorina S and Pavletich NP:
Crystal structure of the tetramerization domain of the p53 tumor
suppressor at 1.7 angstroms. Science. 267:1498–1502. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kamada R, Toguchi Y, Nomura T, Imagawa T
and Sakaguchi K: Tetramer formation of tumor suppressor protein
p53: Structure, function, and applications. Biopolymers.
106:598–612. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mateu MG and Fersht AR: Nine hydrophobic
side chains are key determinants of the thermodynamic stability and
oligomerization status of tumour suppressor p53 tetramerization
domain. EMBO J. 17:2748–2758. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kamada R, Nomura T, Anderson CW and
Sakaguchi K: Cancer-associated p53 tetramerization domain mutants:
Quantitative analysis reveals a low threshold for tumor suppressor
inactivation. J Biol Chem. 286:252–258. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Varley JM, Thorncroft M, McGown G, Tricker
K, Birch JM and Evans DG: A novel deletion within exon 6 of TP53 in
a family with Li-Fraumeni-like syndrome, and LOH in a benign lesion
from a mutation carrier. Cancer Genet Cytogenet. 90:14–16. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Atema A and Chène P: The gain of function
of the p53 mutant Asp281Gly is dependent on its ability to form
tetramers. Cancer Lett. 185:103–109. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Uyama R, Nakagawa T, Hong SH, Mochizuki M,
Nishimura R and Sasaki N: Establishment of four pairs of canine
mammary tumour cell lines derived from primary and metastatic
origin and their E-cadherin expression. Vet Comp Oncol. 4:104–113.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kato Y, Ochiai K, Kawakami S, Nakao N,
Azakami D, Bonkobara M, Michishita M, Morimatsu M, Watanabe M and
Omi T: Canine REIC/Dkk-3 interacts with SGTA and restores androgen
receptor signalling in androgen-independent prostate cancer cell
lines. BMC Vet Res. 13:1702017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Imagawa T, Terai T, Yamada Y, Kamada R and
Sakaguchi K: Evaluation of transcriptional activity of p53 in
individual living mammalian cells. Anal Biochem. 387:249–256. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cubillos-Rojas M, Schneider T,
Sánchez-Tena S, Bartrons R, Ventura F and Rosa JL: Tris-acetate
polyacrylamide gradient gel electrophoresis for the analysis of
protein oligomerization. Anal Bioanal Chem. 408:1715–1719. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pettersen EF, Goddard TD, Huang CC, Couch
GS, Greenblatt DM, Meng EC and Ferrin TE: UCSF Chimera - a
visualization system for exploratory research and analysis. J
Comput Chem. 25:1605–1612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Salvatella X, Martinell M, Gairí M, Mateu
MG, Feliz M, Hamilton AD, De Mendoza J and Giralt E: A
tetraguanidinium ligand binds to the surface of the tetramerization
domain of protein P53. Angew Chem Int Ed Engl. 43:196–198. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Martinell M, Salvatella X,
Fernández-Carneado J, Gordo S, Feliz M, Menéndez M and Giralt E:
Synthetic ligands able to interact with the p53 tetramerization
domain. Towards understanding a protein surface recognition event.
ChemBioChem. 7:1105–1113. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gordo S, Martos V, Santos E, Menéndez M,
Bo C, Giralt E and de Mendoza J: Stability and structural recovery
of the tetramerization domain of p53-R337H mutant induced by a
designed templating ligand. Proc Natl Acad Sci USA.
105:16426–16431. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Osaki S, Nakanishi Y, Takayama K, Pei XH,
Ueno H and Hara N: Alteration of drug chemosensitivity caused by
the adenovirus-mediated transfer of the wild-type p53 gene in human
lung cancer cells. Cancer Gene Ther. 7:300–307. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yoshikawa K, Hamada J, Tada M, Kameyama T,
Nakagawa K, Suzuki Y, Ikawa M, Hassan NM, Kitagawa Y and Moriuchi
T: Mutant p53 R248Q but not R248W enhances in vitro invasiveness of
human lung cancer NCI-H1299 cells. Biomed Res. 31:401–411. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tornaletti S and Pfeifer GP: Complete and
tissue-independent methylation of CpG sites in the p53 gene:
Implications for mutations in human cancers. Oncogene.
10:1493–1499. 1995.PubMed/NCBI
|
|
26
|
Soussi T and Lozano G: p53 mutation
heterogeneity in cancer. Biochem Biophys Res Commun. 331:834–842.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin C, Liang Y, Zhu H, Zhang J and Zhong
X: R280T mutation of p53 gene promotes proliferation of human
glioma cells through GSK-3β/PTEN pathway. Neurosci Lett. 529:60–65.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Retzlaff M, Rohrberg J, Küpper NJ,
Lagleder S, Bepperling A, Manzenrieder F, Peschek J, Kessler H and
Buchner J: The regulatory domain stabilizes the p53 tetramer by
intersubunit contacts with the DNA binding domain. J Mol Biol.
425:144–155. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Joerger AC and Fersht AR: The p53 Pathway:
Origins, inactivation in cancer, and emerging therapeutic
approaches. Annu Rev Biochem. 85:375–404. 2016. View Article : Google Scholar : PubMed/NCBI
|