Introduction
Hepatocellular carcinoma (HCC) is a commonly
diagnosed cancer, and its mortality rate was ranked second among
all cancers worldwide in 2012 (1).
However, therapy for HCC remains unsatisfactory. Patients commonly
experience relapse after surgery. Consequently, there is an urgent
need to identify new targets for HCC therapy.
Cyclin D1 and cyclin E proteins are key factors that
are involved in the transition from G1 phase to S phase of the cell
cycle. The aberrant expression of these proteins in G1 phase is a
marker of malignancy (2,3). It has been reported that, in breast
cancer cells, the upregulation of cyclin D1 shortens G1 phase and
promotes cell proliferation (4). In
head and neck squamous cell carcinoma cells, protein kinase C α and
microRNAs can upregulate the expression of cyclin E, resulting in
oncogenic dysregulation (5).
Metformin may induce G1 arrest by inhibiting E2F8 expression in
lung cancer cells, which can lead to improvement in the overall
survival of lung cancer patients (6).
DIDS (4,4′-diisothiocyanostilbene-2,2′-disulfonic
acid) is a chloride channel blocker that has low specificity. It
has been shown that DIDS can disrupt the glycolytic phenotype and
decrease cell viability, and also enhance cell death and inhibit
proliferation in colorectal cancer cells (7). In the human ovarian cancer cell line
A2780, DIDS has been shown to inhibit cell adhesion and migration
by regulatory volume decrease (RVD) and
[Ca2+]i changes (8). Furthermore, DIDS has demonstrated the
ability to sensitize cells to chemotherapy and induce apoptotic
cell death in resistant human glioblastoma multiforme cancer stem
cells by cell swelling-induced cell cycle arrest after chemotherapy
(9).
Chloride channels, one of the most important
negative ion channels, regulate the cell cycle and volume (10–12),
and the relationship between the volume-regulated regulated
channels and cancer is an area of active research at present.
ClC-3, one of the families of volume-regulated channels, is
involved in cell proliferation, apoptosis and migration (13). In human prostate cancer epithelial
cells, the inhibition of ClC-3 by an anti-ClC-3-specific antibody
was found to decrease cell swelling which was activated by chloride
currents (14). Some studies have
shown that ClC-3 promoted the resistance of cancer cells to
chemotherapeutic drugs, such as cisplatin and etoposide (15–17).
In osteosarcoma cells, ClC-3 induced G1 arrest by medicating cyclin
D1 and cyclin E to modulate the proliferation and migration
(18).
α-Fetoprotein (AFP), the most commonly used tumor
marker for the diagnosis of HCC, is included in international
guidelines for HCC surveillance (19–21). A
high expression level of AFP is considered to be an indicator that
a patient has HCC, and data show that AFP is elevated above the
normal range in 70% of patients with HCC (22). AFP may be used for prediction and
prognosis in HCC (23); when AFP
levels decrease, this is considered to be a favorable prognostic
indicator in HCC.
In the present study, we report a relationship
between chloride channels and the cell cycle in HCC cells, as well
as an association between chloride channels and AFP; to the best of
our knowledge, this is the first report of such findings. In
addition, the results suggested that ClC-3 had an impact on the
induction of G1 arrest and the levels of the prognostic indicator
AFP. These results suggest that ClC-3 may be a novel potential
candidate for the molecular-targeted treatment of HCC. Targeting
ClC-3 may effectively kill cancer cells and potentially improve
patient prognosis.
Materials and methods
Cell line
We acquired the human hepatocellular carcinoma Hep3B
cell line and human L-02 hepatocytes from the American Type Culture
Collection (ATCC; Manassas, VA, USA). We cultured cells in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS
in a humidified incubator containing 5% CO2 at 37°C. The
medium was replaced and the cells were passaged twice per week to
maintain logarithmic growth.
Reagent
DIDS was reconstituted in dimethyl sulfoxide (DMSO)
(both from Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
DIDS treatment of Hep3B cells in
vitro
In order to detect the function of DIDS by western
blot analysis and flow cytometric analysis, 5×104 Hep3B
cells were seeded into each well of 6-well plates overnight. The
cells were then treated with 0, 50, 100 or 200 µM DIDS in DMEM with
2.5% FBS for 24 h.
Western blot analysis
Hep3B cells were lysed in RIPA buffer with 1%
Nonidet P-40 (20 mM Tris, 150 mM NaCl, 0.1 mM EDTA). The amount of
total proteins was quantified using the bicinchoninic acid (BCA)
method. The proteins were separated according to molecular weight
by SDS-PAGE and then a semidry transfer instrument (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) was used to transfer the
proteins onto PVDF membranes. The membranes were blocked in 5% skim
milk, prior to incubation with primary and secondary antibodies
(1:5,000; Sigma-Aldrich; Merck KGaA). Finally, the PVDF membranes
were viewed with Molecular Analyst software (Bio-Rad Laboratories).
The primary antibodies we used were as follows: anti-ClC-3
(1:1,000; cat. no. 13359; Cell Signaling Technology, Inc., Danvers,
MA, USA), anti-cyclin D1 (1:1,000; cat. no. 2978; Cell Signaling
Technology), anti-cyclin E (1:1,000; cat. no. 2273964; EMD
Millipore, Billerica, MA, USA), anti-AFP (1:1,000; cat. no. 4448;
Cell Signaling Technology), anti-GAPDH (1:5,000; cat. no. AP0060;
Bioworld Technology, Inc., St. Louis Park, MN, USA) and
anti-β-actin (1:5,000; cat. no. AP0063; Bioworld Technology).
Cell transfection
Following the manufacturer's protocol, a Silencer
Small Interfering RNA (siRNA) Construction kit (Guangzhou RiboBio,
Co., Ltd., Guangzhou, China) was used to synthesize dsRNA-ClC-3.
Hep3B cells (4×104) were seeded into each well of a
6-well plate overnight. Then in all of the wells, medium was
replaced with DMEM containing 1% FBS and serum-free Opti-MEM I
Medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
(without antibiotics or fungicides), and the different treatments,
including NC, RNA iMAX, siRNA-ClC-3-001, siRNA-ClC-3-002 and
siRNA-ClC-3-003, were applied for 48 h. There was no RNA sequence
or no lipidosome used in the control group. There was Random
sequence genome with lipidosome in the NC group. There was only
lipidosome in RNA iMAX. There were ClC-3 siRNA sequences with
lipidosome in the siRNA-ClC-3-002 or siRNA-ClC-3-003 group.
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) was
used to transfect the siRNA (10 nM) into the Hep3B cells. After 2
days, western blot analysis was conducted as described above. The
sequences of ClC-3 siRNA were: 5′-GAAGAGGUAUUGAAUGCUAdTdT-3′.
Quantitative real-time polymerase
chain reaction (qPCR)
Total RNA was extracted from the Hep3B cells with
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and
reverse-transcribed into cDNA using oligo(dT) primers (Stratagene;
Agilent Technologies, Inc., Santa Clara, CA, USA) and RevertAid
Reverse Transcriptase (Thermo Fisher Scientific, Inc.). qPCR was
performed with using an iCycler RT-PCR system (Bio-Rad
Laboratories, Munich, Germany) and SuperReal PreMix SYBR Green
(FP204-02; Tiangen Biotech, Co., Ltd., Beijing, China). We analyzed
the data using the Pfaffli method. The primers for ClC-1 to ClC-7
were as follows: ClC-1 forward, 5′-CAGCATCTGTGCTGCC-3′ and reverse,
5′-GTGCTTAGCAAGAAACTGGC-3′; ClC-2 forward,
5′-AGACAATCCCTACACCCTTCAA-3′ and reverse,
5′-TGTCGGTAGAACACCTTGTCAC-3′; ClC-3 forward,
5′-CAAUGGAUUUCCUGUCAUATT-3′ and reverse,
5′-UAUGACAGGAAAUCCAUUGTA-3; ClC-4 forward, 5′-GCGTCTCATCGGGTTTGC-3′
and reverse, 5′-TTGCTCACAATGCCCTCTTTG-3′; ClC-5 forward,
5′-CTGTGCCACTGCTTCAAC-3′ and reverse, 5′-CTGAGGGCAAATCCCACTAA-3′;
ClC-6 forward, 5′-GTCGCGCAAGACTGTAACCA-3′ and reverse,
5′-CGGCGAAATTCCATACCTG-3′; ClC-7 forward,
5′-CCCACACAACGAGAAGCTCC-3′ and reverse,
5′-ACTTGTCGATATTGCCCTTGATG-3′. The primer sequences for GAPDH are
were 5′-CTCATGACCACAGTCCATGC-3′ (forward) and
5′-CACATTGGGGGTAGGAACAC-3′ (reverse).
Flow cytometric analysis of cell cycle
distribution
Hep3B cells were treated with DIDS for 24 h or
transfected with ClC-3 siRNA for 48 h and then centrifuged at 1,000
× g for 5 min. Using phosphate-buffered saline (PBS), Hep3B cells
were washed three times and resuspended in 75% alcohol overnight.
The samples were stained by Triton-X 100 (Amresco, LLC, OH, USA), 1
mg/ml propidium iodide (PI; Thermo Fisher Scientific, Inc.),
DNase-free RNase (Thermo Fisher Scientific, Inc.) and PBS in dark
for 1 or 2 h. After staining the samples, we detected the cell
cycle distribution with a flow cytometer (FACSCalibur; BD
Biosciences, Franklin Lakes, NJ, USA).
Flow cytometric analysis of
apoptosis
Hep3B cells were treated with DIDS for 24 h and then
centrifuged at 1,000 × g for 5 min. The Hep3B cells were washed
three times in PBS and stained with propidium iodide (PI) and
Annexin V-FITC. After washing the samples and re-suspending them in
binding buffer, flow cytometry (FACSCalibur; BD Biosciences) was
used to assess the percentage of apoptotic cells. Fluorescence was
detected at an excitation wavelength of 488 nm and an emission
wavelength of 530 nm for Annexin V-FITC binding (as fluorescence
channel FL1), and 488 nm excitation and red emission for PI (as
fluorescence channel FL2).
MTT assay
For the MTT assay, 1×104 Hep3B cells or
L-02 cells were seeded in each well of a 24-well plate overnight.
Hep3B and L-02 cells were then treated with 0, 25, 50, 100, 200 and
400 µM DIDS in DMEM with 2.5% FBS for 24 h. Next, 50 µl MTT
(3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide)
solution was added to each well, and the plates were incubated for
3 h at 37°C. The supernatant was then discarded and 1 ml DMSO was
added to each well. The absorbance was read at 570 nm using an
automatic enzyme-linked immune detector (Bio-Rad Laboratories).
Statistical analysis
We analyzed all data by two-way ANOVA or
independent-samples t-tests as appropriate. The data are presented
as the mean ± SEM. Statistical analyses of all quantitative data
were performed with GraphPad software (GraphPad Software, Inc., La
Jolla, CA, USA).
Results
Viability of Hep3B cells following
treatment with different concentrations of DIDS
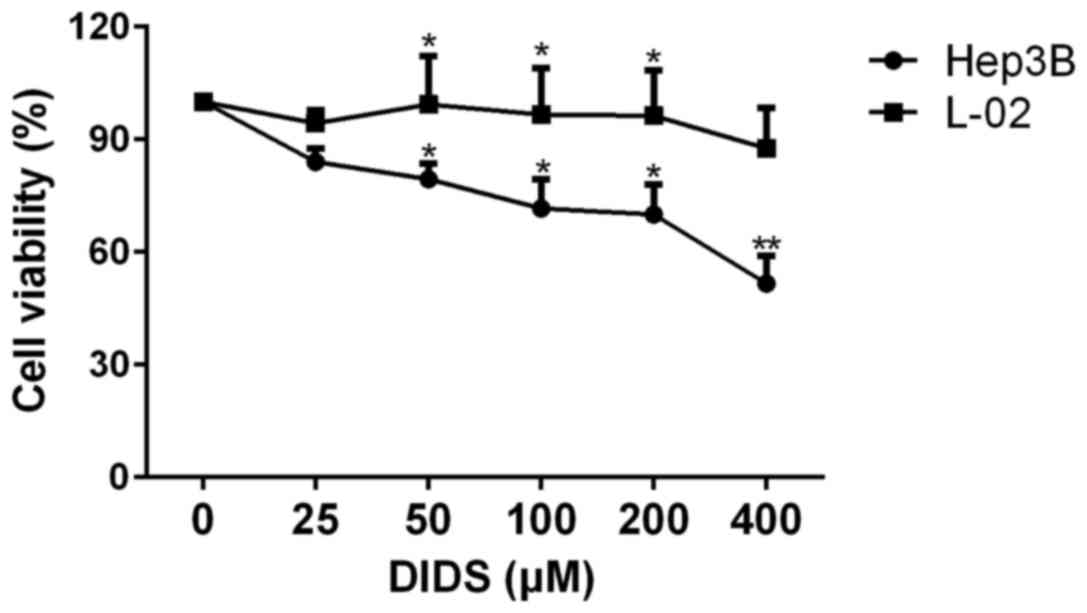
The effects of different concentrations of DIDS on
Hep3B and L-02 cells are shown in Fig.
1. We treated Hep3B and L-02 cells with 0, 25, 50, 100, 200 and
400 µM DIDS for 24 h. With increasing concentrations, the viability
of the Hep3B cells was decreased, whereas the viability of the L-02
cells remained almost constant. This indicated that DIDS has a
strong effect on HCC Hep3B cells, but had almost no effect on L-02
cells (a normal liver cell line). Based on the results shown in
Fig. 1, we determined the most
effective and suitable concentrations for use in the following
studies.
DIDS inhibits the proliferation of
Hep3B cells by G1 arrest
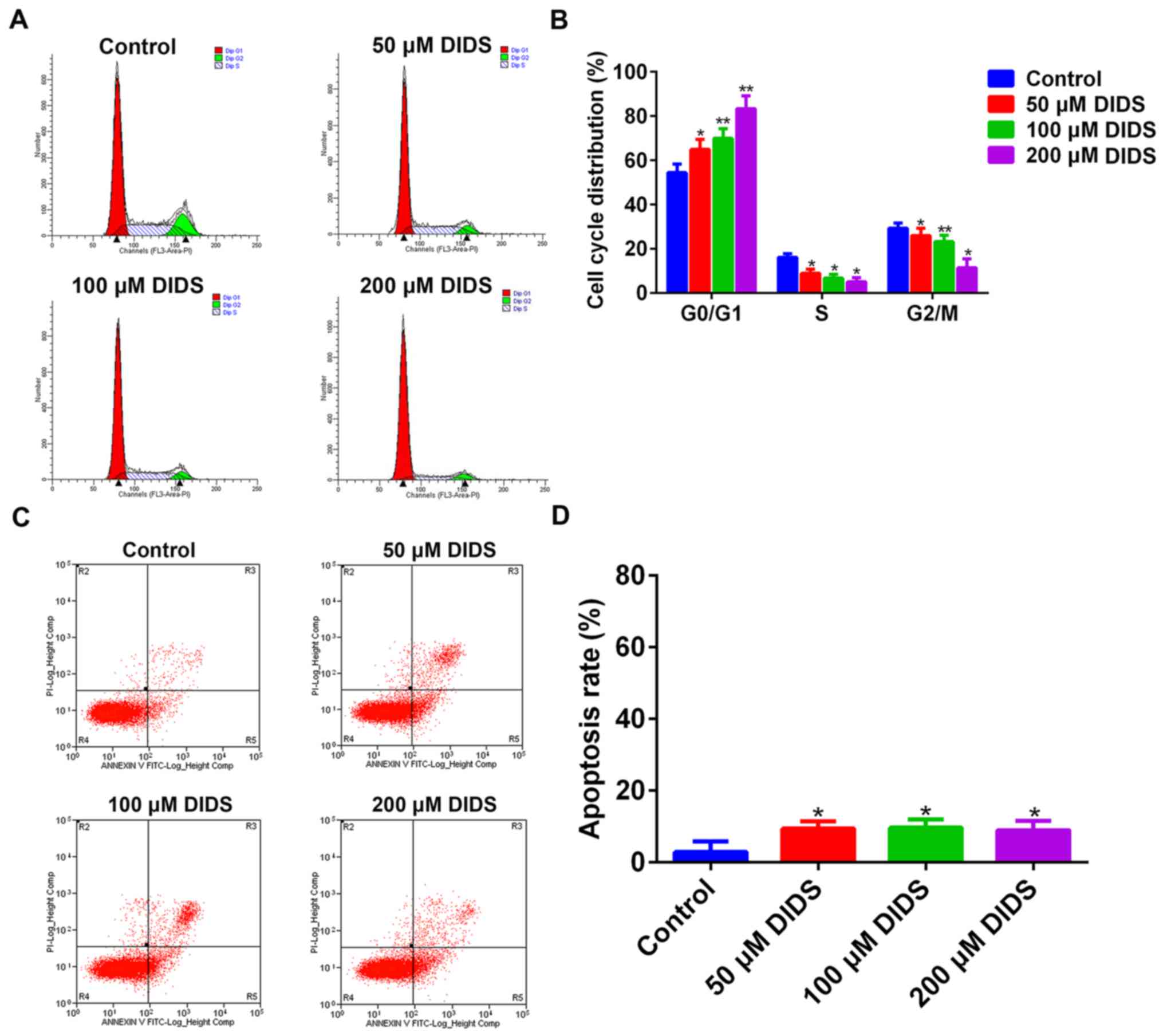
In order to elucidate how DIDS affects the viability
of Hep3B cells, we treated Hep3B cells with 0, 50, 100 and 200 µM
DIDS for 24 h prior to flow cytometry. It was demonstrated that
DIDS inhibited the proliferation of cells by arresting cells in the
G1 phase, preventing their transition into S phase. With increasing
concentrations of DIDS, the degree of inhibition was more obvious.
At the same time, the results suggested that the distribution of
cells was decreased in other phases of the cell cycle with
increasing DIDS concentrations (Fig. 2A
and B).
When apoptosis was analyzed by flow cytometry, it
was demonstrated that DIDS had less influence on the apoptosis of
Hep3B cells (Fig. 2C and D).
Therefore, DIDS predominantly affected the proliferation of Hep3B
cells by regulating cell cycle progression, but not through
apoptosis.
DIDS downregulates the proteins
associated with G0/G1 phase and AFP in Hep3B cells
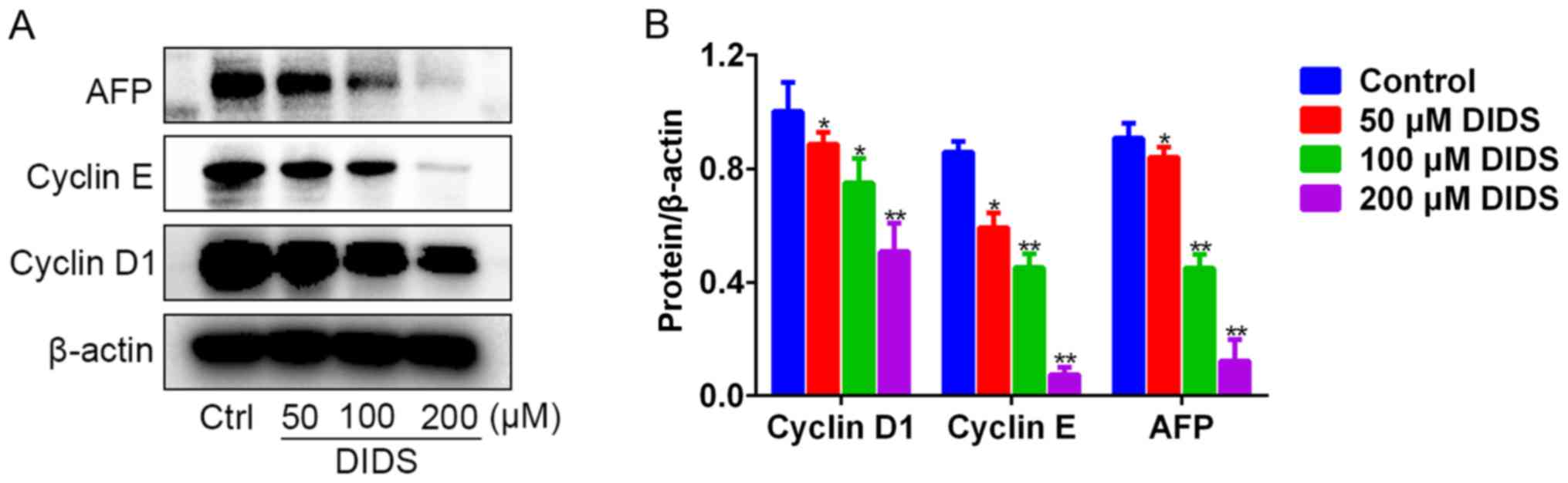
To observe the mechanism by which DIDS induces G1
arrest in Hep3B cells, we detected the levels of the proteins
responsible for G1 to S phase transition by western blot analysis.
The results indicated that, with the ascending concentrations of
DIDS, the protein levels of cyclin D1 and cyclin E were gradually
diminished in the Hep3B cells (Fig. 3A
and B). Cyclin D1 and cyclin E, the main G1-associated
proteins, are involved in driving the transition from G1 to S phase
of the cell cycle. Unexpectedly, we noted that DIDS also inhibited
the expression of AFP protein (Fig. 3A
and B). The results indicated that DIDS may downregulate the
protein levels of cyclin D1 and cyclin E and inhibit the protein
expression of AFP (a biomarker of Hep3B cells), resulting in the
inhibition of proliferation of Hep3B cells.
ClC-3 may serve a main role in G1
arrest induced by DIDS
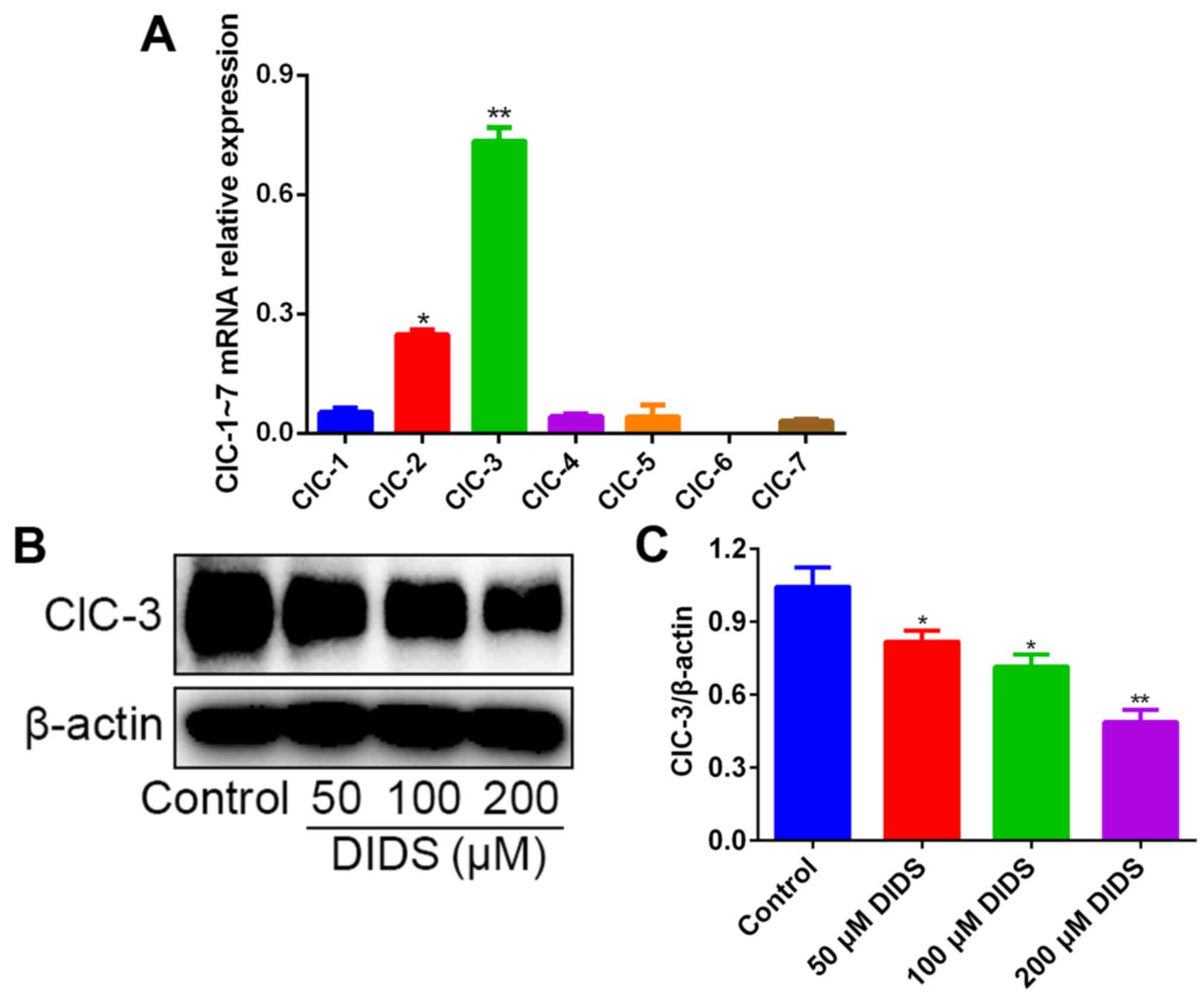
We detected the relative mRNA expression levels of
the chloride channel family, ClC-1 to ClC-7, in Hep3B cells by
qPCR. The results revealed that, in contrast to the mRNA levels of
ClC-2 and ClC-1, the mRNA levels of ClC-3 were distinctly much
higher (Fig. 4A).
Subsequently we detected the expression levels of
ClC-3 by western blotting in Hep3B cells treated with 0, 50, 100
and 200 µM DIDS for 24 h, in order to confirm whether DIDS could
block ClC-3 in Hep3B cells. As expected, the protein levels of
ClC-3 were inhibited by DIDS treatment, with a higher concentration
of DIDS resulting in more marked inhibition (Fig. 4B and C). These results suggest that
ClC-3, one member of the chloride channel family, may play a key
role in G1 arrest and in inhibiting the protein expression of AFP
in response to DIDS treatment.
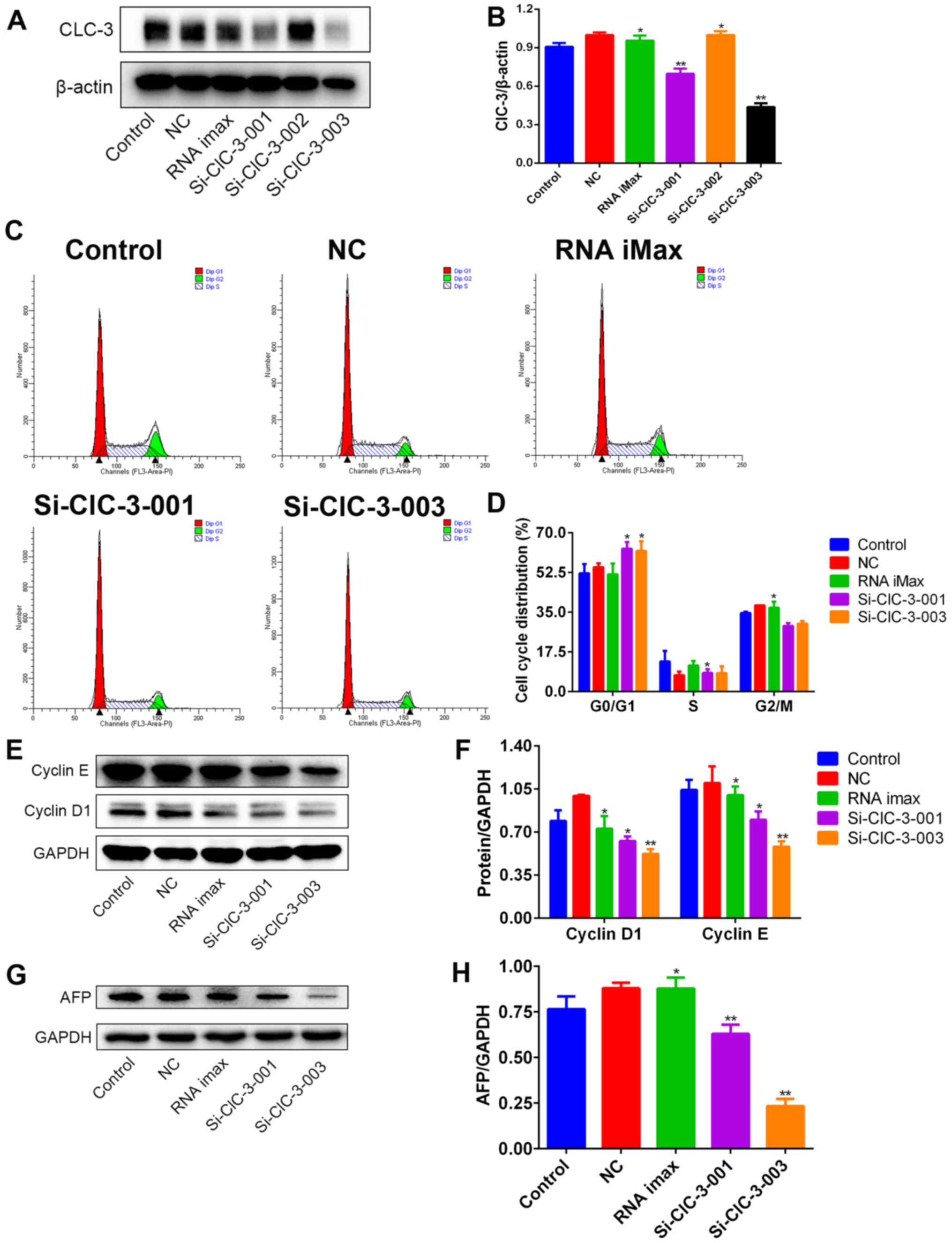
ClC-3 siRNA transfection induces G1
arrest and AFP protein in Hep3B cells
In order to confirm whether ClC-3 is involved in
inducing G1 arrest and inhibiting the protein expression of AFP in
Hep3B cells, we used transfection to silence the expression of
ClC-3 in Hep3B cells. First, we selected the most effective ClC-3
siRNA from si-ClC-3-001, si-ClC-3-002 and si-ClC-3-003 by detecting
the expression levels of ClC-3 protein through western blot
analysis after transfection. The silencing effects of si-ClC-3-001
and si-ClC-3-003 were found to be markedly greater (Fig. 5A and B).
Next, we detected the cell cycle distribution
following transfection using flow cytometry. After silencing ClC-3,
cell cycle progression was arrested at G1 phase and transition into
S phase was suppressed (Fig. 5C and
D). Finally, we confirmed that the levels of G1 phase-related
proteins after the silencing of ClC-3 were the same as those after
DIDS treatment. Cyclin D1 and cyclin E protein were both decreased
in Hep3B cells treated with si-ClC-3-001 and si-ClC-3-003 (Fig. 5E and F). Simultaneously, the
expression of AFP protein was also decreased (Fig. 5G and H). These results are
consistent with the results obtained in DIDS-treated cells,
indicating that DIDS targets ClC-3 to induce G1 arrest and to
inhibit the expression of AFP in Hep3B cells, as predicted.
Discussion
Ion channels are potential novel targets for
improving the treatment of cancer. Chloride channels, including
ClC-3, are the most important negative ion channels. ClC-3 plays a
significant role in regulating the cell cycle, and can result in G1
arrest in nasopharyngeal carcinoma and osteosarcoma cells (18,24).
However, to the best of our knowledge, there has been no report of
similar findings in HCC cells to date. Consequently, we researched
ClC-3 protein and the cell cycle in Hep3B cells (a HCC cell line),
and found associations among ClC-3, the cell cycle and the level of
AFP.
First, we demonstrated that DIDS had a
concentration-dependent effect on Hep3B cells, but had hardly any
effect on L-02 cells. This supported our hypothesis that DIDS can
inhibit the proliferation of HCC cells effectively, and also has
little disadvantage to normal liver cells. It was reported that 100
and 250 µM DIDS had no effect on normal liver cells at day 2
following treatment (25), which
was consistent with our results that 25–200 µM DIDS had no effect
on L-02 cells at 24 h (Fig. 1).
This is a huge benefit in cancer therapy, as such a treatment could
target HCC while having a limited effect on normal liver cells.
Second, in order to determine how DIDS affects the proliferation of
Hep3B cells, we used flow cytometry to detect the cell cycle
distribution and cell apoptosis rate. It was observed that DIDS
inhibited the proliferation of Hep3B cells through G1 arrest but
not by promoting apoptosis. These results are consistent with
previous research in other tumor cells (18,24).
Then, we detected the expression of the main proteins associated
with G1 phase by western blot analysis, in order to demonstrate the
mechanism of cell cycle regulation by DIDS. The data suggested that
DIDS induced G1 arrest by downregulating the protein expression
levels of cyclin D1 and cyclin E. Meanwhile, we observed that DIDS
also reduced the protein level of AFP, which indicates that it may
be able to improve the prognosis of HCC in patients.
However, DIDS is a broad-spectrum and non-specific
blocker of volume-regulated chloride channels. In order to
determine which volume-regulated chloride channels were affected,
we performed qPCR to detect the expression levels of the chloride
channel protein family in Hep3B cells. The mRNA expression of ClC-3
was found to be the highest, while the second highest was ClC-2 in
Hep3B cells. The results of the western blotting suggested that the
expression of ClC-3 protein was obviously decreased with increasing
concentrations of DIDS. Next, we silenced the expression of ClC-3
specifically to confirm this hypothesis. The results showed that,
when the protein expression of ClC-3 was silenced, G1 arrest
occurred and the protein levels of cyclin D1 and cyclin E were
decreased. Meanwhile, the expression of AFP protein was markedly
decreased following ClC-3 siRNA transfection. We believe that the
siRNA sequences may have influence on Hep3B cells so that in NC
(negative control), AFP expression was increased. Therefore, we
compared the results of siRNA-ClC-3 with NC but Control to
attenuate the influence of interference sequences in Hep3B cells.
These results were consistent with the results obtained after
treatment of the cells with DIDS.
As has been proposed in certain previous reports,
ClC-3, a volume-activated chloride channel, is an important
regulator at the plasma membrane (24). DIDS may inhibit the protein levels
of ClC-3 at the plasma membrane to downregulate the expression of
cyclin D1 and cyclin E, causing G1 arrest, potentially resulting in
the improvement of prognosis. In this way, DIDS could prevent the
growth of HCC cells and effectively kill them, while having a very
limited effect on the growth of normal liver cells.
To the best of our knowledge, we are the first to
report that chloride channels are associated with the HCC marker
AFP. It has been reported that anti-AFP single-chain variable
fragments can induce growth inhibition through the induction of G1
arrest and apoptosis in AFP-expressing HCC cell lines (26). Silencing of AFP was shown to induce
G1 arrest in the HCC cell line EGHC-9901 (27). Therefore, it is possible that ClC-3
could mediate AFP to induce G1 arrest in Hep3B cells. We will
continue researching the mechanism of how ClC-3 mediates AFP and
how AFP induces G1 arrest in HCC cells in our further studies,
through siRNA transfection, co-immunoprecipitation assay and
western blot analysis.
Blocking chloride channels can lead to the
inhibition of cell proliferation and the arrest of cell cycle
progression in human laryngeal cancer cells (28). It was reported that ClC-3, the
inhibition of which was shown to decrease the aggressiveness of
neuroglioma cells by inhibiting the NF-κB pathway, may be a novel
therapeutic target and prognostic biomarker in neuroglioma
(29). ClC-3 is a potential target
for cancer therapy, and we found that it was highly expressed in
Hep3B cells. This result supported our hypothesis that ClC-3 may be
an extremely valuable biotherapeutic target in HCC. Our research
mainly focused on ClC-3 protein, and we did not perform an in-depth
study on the change of the Cl− current. Other mechanisms
may also be responsible for our findings. We will conduct research
on the Cl− current in our subsequent study. For further
studies, we will continue our experiments using more HCC cell
lines, such as HepG2 and Huh-7 cells, and we will demonstrate our
findings in vivo.
Acknowledgements
We thank Dr Xi Lin (Department of Pharmacology,
Medical College, Jinan University, Guangzhou; Department of Key
Laboratory for Environmental Exposure and Health, Environment
College, Jinan University, Guangzhou, China) and Dr Jichen Du
(Department of Neurology, Aerospace Center Hospital, Peking
University Aerospace Clinical College, Beijing, China) for the
helpful discussion.
Funding
The present study was funded by the Science and
Technology Planning Project of Guangdong Province (no.
2014A020211022), and the Guangdong Province Ordinary University
Innovation Team Project and Science and Technology Planning Project
of Guangzhou Canton (no. 201510010074).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
LX, DJ and WR and conceived and designed the study.
WR, KB, HR and HY performed the experiments. WR and KB wrote the
paper. HR and QZ reviewed and edited the manuscript. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
The study was conducted using cell lines and no
human tissues were used. Thus no ethical approval and patient
consent were required.
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Zhang Y, Ren JS, Shi JF, Li N, Wang YT, Qu
C, Zhang Y and Dai M: International trends in primary liver cancer
incidence from 1973 to 2007. BMC Cancer. 15:942015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Park GH, Song HM and Jeong JB: The coffee
diterpene kahweol suppresses the cell proliferation by inducing
cyclin D1 proteasomal degradation via ERK1/2, JNK and
GKS3β-dependent threonine-286 phosphorylation in human colorectal
cancer cells. Food Chem Toxicol. 95:142–148. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yuan C, Zhu X, Han Y, Song C, Liu C, Lu S,
Zhang M, Yu F, Peng Z and Zhou C: Elevated HOXA1 expression
correlates with accelerated tumor cell proliferation and poor
prognosis in gastric cancer partly via cyclin D1. J Exp Clin Cancer
Res. 35:152016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Quelle DE, Ashmun RA, Shurtleff SA, Kato
JY, Bar-Sagi D, Roussel MF and Sherr CJ: Overexpression of mouse
D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes
Dev. 7:1559–1571. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cohen EE, Zhu H, Lingen MW, Martin LE, Kuo
WL, Choi EA, Kocherginsky M, Parker JS, Chung CH and Rosner MR: A
feed-forward loop involving protein kinase Calpha and microRNAs
regulates tumor cell cycle. Cancer Res. 69:65–74. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jin DH, Kim Y, Lee BB, Han J, Kim HK, Shim
YM and Kim DH: Metformin induces cell cycle arrest at the G1 phase
through E2F8 suppression in lung cancer cells. Oncotarget.
8:101509–101519. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Amorim R, Pinheiro C, Miranda-Gonçalves V,
Pereira H, Moyer MP, Preto A and Baltazar F: Monocarboxylate
transport inhibition potentiates the cytotoxic effect of
5-fluorouracil in colorectal cancer cells. Cancer Lett. 365:68–78.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li M, Wang Q, Lin W and Wang B: Regulation
of ovarian cancer cell adhesion and invasion by chloride channels.
Int J Gynecol Cancer. 19:526–530. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kang MK and Kang SK: Pharmacologic
blockade of chloride channel synergistically enhances apoptosis of
chemotherapeutic drug-resistant cancer stem cells. Biochem Biophys
Res Commun. 373:539–544. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kunzelmann K: Ion channels and cancer. J
Membr Biol. 205:159–173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Arcangeli A, Crociani O, Lastraioli E,
Masi A, Pillozzi S and Becchetti A: Targeting ion channels in
cancer: A novel frontier in antineoplastic therapy. Curr Med Chem.
16:66–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Conti M: Targeting ion channels for new
strategies in cancer diagnosis and therapy. Curr Clin Pharmacol.
2:135–144. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guan YY, Wang GL and Zhou JG: The ClC-3
Cl− channel in cell volume regulation, proliferation and
apoptosis in vascular smooth muscle cells. Trends Pharmacol Sci.
27:290–296. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lemonnier L, Shuba Y, Crepin A, Roudbaraki
M, Slomianny C, Mauroy B, Nilius B, Prevarskaya N and Skryma R:
Bcl-2-dependent modulation of swelling-activated Cl−
current and CLC-3 expression in human prostate cancer epithelial
cells. Cancer Res. 64:4841–4848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weylandt KH, Nebrig M, Jansen-Rosseck N,
Amey JS, Carmena D, Wiedenmann B, Higgins CF and Sardini A: ClC-3
expression enhances etoposide resistance by increasing
acidification of the late endocytic compartment. Mol Cancer Ther.
6:979–986. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Su J, Xu Y, Zhou L, Yu HM, Kang JS, Liu N,
Quan CS and Sun LK: Suppression of chloride channel 3 expression
facilitates sensitivity of human glioma U251 cells to cisplatin
through concomitant inhibition of Akt and autophagy. Anat Rec.
296:595–603. 2013. View
Article : Google Scholar
|
|
17
|
Xu Y, Zheng H, Kang JS, Zhang L, Su J, Li
HY and Sun LK: 5-Nitro-2-(3-phenylpropylamino) benzoic acid induced
drug resistance to cisplatin in human erythroleukemia cell lines.
Anat Rec. 294:945–952. 2011. View
Article : Google Scholar
|
|
18
|
Du S and Yang L: ClC-3 chloride channel
modulates the proliferation and migration of osteosarcoma cells via
AKT/GSK3β signaling pathway. Int J Clin Exp Pathol. 8:1622–1630.
2015.PubMed/NCBI
|
|
19
|
Bruix J and Sherman M: Practice Guidelines
Committee, American Association for the Study of Liver Diseases:
Management of hepatocellular carcinoma. Hepatology. 42:1208–1236.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bruix J and Sherman M: American
Association for the Study of Liver Diseases: Management of
hepatocellular carcinoma: An update. Hepatology. 53:1020–1022.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
European Association For The Study Of The
LiverEuropean Organisation For Research And Treatment Of Cancer:
EASL-EORTC clinical practice guidelines: Management of
hepatocellular carcinoma. J Hepatol. 56:908–943. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chan SL, Mo FK, Johnson PJ, Hui EP, Ma BB,
Ho WM, Lam KC, Chan AT, Mok TS and Yeo W: New utility of an old
marker: Serial alpha-fetoprotein measurement in predicting
radiologic response and survival of patients with hepatocellular
carcinoma undergoing systemic chemotherapy. J Clin Oncol.
27:446–452. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Keam B, Oh DY, Lee SH, Kim DW, Im SA, Kim
TY, Heo DS and Bang YJ: A phase II study of 5-fluorouracil and
cisplatin systemic chemotherapy for inoperable hepatocellular
carcinoma with α fetoprotein as a predictive and prognostic marker.
Mol Med Rep. 1:415–422. 2008.PubMed/NCBI
|
|
24
|
Ye D, Luo H, Lai Z, Zou L, Zhu L, Mao J,
Jacob T, Ye W, Wang L and Chen L: ClC-3 Chloride Channel Proteins
Regulate the Cell Cycle by Up-regulating cyclin D1-CDK4/6 through
Suppressing p21/p27 Expression in Nasopharyngeal Carcinoma Cells.
Sci Rep. 6:302762016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wondergem R, Gong W, Monen SH, Dooley SN,
Gonce JL, Conner TD, Houser M, Ecay TW and Ferslew KE: Blocking
swelling-activated chloride current inhibits mouse liver cell
proliferation. J Physiol. 532:661–672. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ji X, Shen Y, Sun H and Gao X: A novel
anti-alpha-fetoprotein single-chain variable fragment displays
anti-tumor effects in HepG2 cells as a single agent or in
combination with paclitaxel. Tumour Biol. 37:10085–10096. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang L, He T, Cui H, Wang Y, Huang C and
Han F: Effects of AFP gene silencing on apoptosis and proliferation
of a hepatocellular carcinoma cell line. Discov Med. 14:115–124.
2012.PubMed/NCBI
|
|
28
|
Yu WF, Zhao YL, Wang K and Dong MM:
Inhibition of cell proliferation and arrest of cell cycle
progression by blocking chloride channels in human laryngeal cancer
cell line Hep-2. Neoplasma. 56:224–229. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang B, Xie J, He HY, Huang EW, Cao QH,
Luo L, Liao YS and Guo Y: Suppression of CLC-3 chloride channel
reduces the aggressiveness of glioma through inhibiting nuclear
factor-κB pathway. Oncotarget. 8:63788–63798. 2017.PubMed/NCBI
|