Introduction
The World Health Organization has reported that ~1.6
million people succumb to lung cancer annually, which accounts for
19.4% of the total cancer mortality (1). The 5-year survival rate ranges from
50% for stage IA to 2% for stage IV, and the average 5-year overall
survival rate of lung adenocarcinoma is ~15% (2,3).
Surgery, radiotherapy, chemoradiotherapy, molecular-targeted
therapy and immunotherapy have been successful in curing lung
cancer. However, most patients with advanced lung cancer have lost
the chance for surgery, and radiotherapy and chemotherapy are less
efficacious, leading to a very poor 5-year survival rate (<15%).
Non-small cell lung cancer (NSCLC) accounts for ~87% of lung
cancer; ~60% of NSCLC diagnoses are discovered at advanced stages
(4), with poor prognosis.
Therefore, deep investigation of NSCLC pathogenesis and
identification of new therapeutic targets is urgently needed.
Telomeres are protective structures at the ends of
chromosomes, which consist of random repeat DNA sequences (TTAGGG)
and associated proteins. The telomeres maintain chromosome length
and stability, and defend chromosome ends from deleterious
degradation and recombination or fusion. Telomerase, a key
component in the forming of telomeres, can extend telomere DNA
strands and repair broken ends of chromosomes, thereby extending,
and even immortalizing, the lifespan of cells. Human telomerase
reverse transcriptase (hTERT) mediates telomerase activation.
Telomerase activity is very low or at almost undetectable levels in
most normal cells, but may be overexpressed in tumor cells, which
proliferate unlimitedly.
Many factors are directly or indirectly involved in
the regulation of tumor growth and differentiation. Growth factors,
especially the epidermal growth factor (EGF) family, growth factor
receptor, oncogenes and anti-oncogenes are likely to affect cancer
cell proliferation. EGF consists of 53 amino acid residues, coded
on human chromosome 4 (4q25-27), and can be detected in most normal
cells, human cancer tissue, blood, urine, and saliva (5). EGF can promote cell proliferation in
tumors, and has an especially morphogenic effect on proliferation
and clonality of NSCLC cells (6).
Epidermal growth factor receptor (EGFR) is a member of the ErbB
family of tyrosine kinase receptors, and is highly expressed in
27–83% of NSCLC, including 83% of squamous cell carcinoma, ~40–50%
of adenocarcinoma and large-cell lung carcinoma, but not in small
cell lung carcinoma (7,8). Reportedly, EGF increases hTERT protein
expression in NSCLC cells, and can be blocked by EGFR tyrosine
kinase inhibitors (TKIs) (9).
Ret finger protein-like 3 (RFPL3) is a hTERT-induced
protein that helps regulate hTERT promoter activity and
hTERT expression, to enhance telomerase activity and induce cancer
cell proliferation (10). In the
present study, we investigated whether EGF could upregulate RFPL3
and hTERT expression in human lung adenocarcinoma A549 and H1299
cells, and identified the involvement of the MEK signaling pathway
in this process. We also investigated the relationship between
RFPL3 overexpression and related MEK signaling proteins.
Materials and methods
Cell culture
A549 human lung adenocarcinoma cells and H1299 human
NSCLC cells were kindly provided by the Regenerative Medicine
Center, First Affiliated Hospital of Dalian Medical University. The
cells were cultured in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA USA) supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 mg/ml streptomycin at 37°C in a 5%
CO2 humidified atmosphere. The cells were plated in
6-well plates for the activation of EGF (PeproTech, Inc., Rocky
Hill, NJ, USA) and transfection studies, and were plated in 96-well
plates for the MTT assay.
Cell treatment
We added EGF to the cells to final concentrations of
0 (control), 2.5, 5, 10, 20 or 50 ng/ml for 24 h or 48 h. To
inhibit EGFR, we added 10 µM AG1478 (Selleck Chemicals, Shanghai,
China) or 20 µM erlotinib (Selleck Chemicals) 4 h prior to EGF
treatment. To inhibit the MEK signaling pathway, we added 50 µM
PD98059 (Selleck Chemicals) 2 h prior to EGF treatment.
Western blot analysis
Cells were trypsinized and cell lysates were
harvested in RIPA-SDS buffer supplemented with protease inhibitors
and phosphatase inhibitors (Beijing Solarbio Science and Technology
Co., Ltd., Beijing, China). The lysates were centrifuged at 12,000
× g for 20 min, and the supernatants were then collected. Proteins
were quantified using the BCA kit (Beijing Solarbio Science and
Technology Co., Ltd.) according to the manufacturer's instructions.
An equivalent amount of protein extract from each sample was
electrophoresed by 12% SDS-PAGE and transferred to polyvinylidene
difluoride membranes (Millipore, Billerica, MA, USA). The membranes
were then blocked with 5% non-fat dried milk in PBS/0.1% Tween-20
for 1 h, and incubated overnight at 4°C with the anti-RFPL3 (1:500;
rabbit polyclonal; cat. no. 13215-1-AP; ProteinTech, Group, Inc.,
Chicago, IL, USA), anti-hTERT (1:1,000; cat. no. ARG54933; rabbit
polyclonal; Arigo, Shanghai, China), anti-pan-Ras (1:20,000; mouse
monoclonal; cat. no. 60309-1-lg; ProteinTech Group), anti-Raf1
(1:500; rabbit polyclonal; cat. no. 51140-1-AP; ProteinTech Group),
anti-ERK1/2 (1:10,000; rabbit monoclonal; cat. no. ab184699; Abcam,
Cambridge, MA, USA), anti-phospho-ERK1/2 (1:500; rabbit monoclonal;
cat. no. ab32538; Abcam) or anti-β-actin (1:1,500; rabbit
monoclonal; cat. no. bs0061R; Bioss, Shanghai, China),
respectively. Anti-β-actin was used as a loading control. The
membranes were then washed three times with PBS/0.1% Tween-20 (15
min each) and incubated with the corresponding secondary antibodies
(horseradish peroxidase-conjugated, goat antibodies to rabbit and
goat antibodies to mouse; 1:5,000; cat. nos. SA00001-2 and
SA00001-1; ProteinTech Group) for 1 h at room temperature. After
washing three times in PBS/0.1% Tween-20, the membranes were then
detected with ECL solution (Thermo Fisher Scientific). All the
protein bands were densitometrically scanned and analyzed with
ImageJ 1.44 software (National Institutes of Health, Bethesda, MD,
USA).
RNA extraction and real-time qPCR
assay
A549 and H1299 cells were treated with different
final concentrations of EGF (0, 2.5, 5, 10, 20 or 50 ng/ml) for 48
h. Total RNA was extracted from these A549 and H1299 cells using
RNAiso Plus (Takara Bio, Otsu, Japan) according to the
manufacturer's protocol and was quantified with NanoDrop 2000
(Thermo Fisher Scientific). RNA (1 µg) was used as the template for
cDNA synthesis; cDNA was reverse transcribed with the Primscript RT
Reagent kit (Takara Bio). RT-qPCR reactions were performed on ABI
StepOnePlus PCR instrument (Applied Biosystems; Thermo Fisher
Scientific) for 40 cycles at 95°C for 5 sec, and at 60°C for 30
sec. Comparative quantification was determined using the
2−ΔΔCT method. Expression levels of RFPL3 and hTERT mRNA
were standardized to GAPDH. Primer sequences are listed in Table I.
 | Table I.Sequences of all primers used in this
study. |
Table I.
Sequences of all primers used in this
study.
| Target gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| GAPDH |
GCACCGTCAAGGCTGAGAAC |
TGGTGAAGACGCCAGTGGA |
| RFPL3 |
GCCCAATCGGCAGCTAGAGA |
CTGTGTCGGCATCCAAGGTC |
| hTERT |
CATCGCCAGCATCATCAAAC |
GCCACGAACTGTCGCATGTA |
Cell viability and proliferation
assay
For the MTT assay, A549 and H1299 cells were plated
onto a 96-well plate at a cell density of 1,500 cells (A549) or
3,000 cells (H1299) per well with 0.2 ml of supplemented RPMI-1640
per well. We treated the cells with different final concentrations
of EGF (0, 2.5, 5, 10, 20 or 50 ng/ml) for 48 h, and then added 20
µl of MTT (5 mg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
to each well and incubated them at 37°C for another 4 h. The
reaction was terminated by lysing the cells with 150 µl of DMSO
(Sigma-Aldrich; Merck KGaA), with gentle shaking for 10 min.
Optical density was measured at 490 nm in the spectrometer (BioTek
Instruments, Inc. USA) to determine the absorbance of each well.
Each separate treatment included five wells and was replicated
three times on the 96-well plate.
Apoptosis analysis via flow
cytometry
After treatment with different final concentrations
of EGF (0, 2.5, 5, 10, 20 or 50 ng/ml) for 48 h in 6-well plates,
A549 cells were trypsinized and harvested in cold PBS, collected by
centrifugation (5 min at 800 × g), resuspended at 1×106
cells/ml in 100 µl 1X binding buffer, and stained with 5 µl of
Annexin V-FITC for 10 min in room temperature, protected from
light. We then incubated them with 5 µl of PI solution for 5 min,
added PBS to a volume of 500 µl and immediately analyzed the cells
using a flow cytometer.
Transfection
The GFP-marked RFPL3-overexpression plasmid and its
control vector were designed and produced by Genepharma
Corporation, Suzhou, China. A549 and H1299 cells were plated in
6-well plates at 1.2×105 cells/well in 0.2 ml of medium
overnight. Transfection was performed when cells achieved 80–90%
confluency. Cells were assigned into blank control, negative
control and transfected groups. The negative control was treated
with control vector. Cells were transfected with
RFPL3-overexpression plasmids or control vector, with serum-free
medium, using the Invitrogen™ Lipofectamine 2000 system (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. The medium was replaced with complete medium 6 h
later.
Statistical analysis
The experiments were repeated at least three times.
Statistical analyses were performed using the SPSS software
(version 22.0) (IBM Corp., Armonk, NY, USA), and values from three
independent experiments at least are represented as the mean ±
standard deviation. Unpaired Student's t-test and one-way analysis
of variance (ANOVA) was performed respectively for the two groups
and three or more than three groups to evaluate the statistical
significance. A value of P<0.05 was considered to indicate
statistical significance.
Results
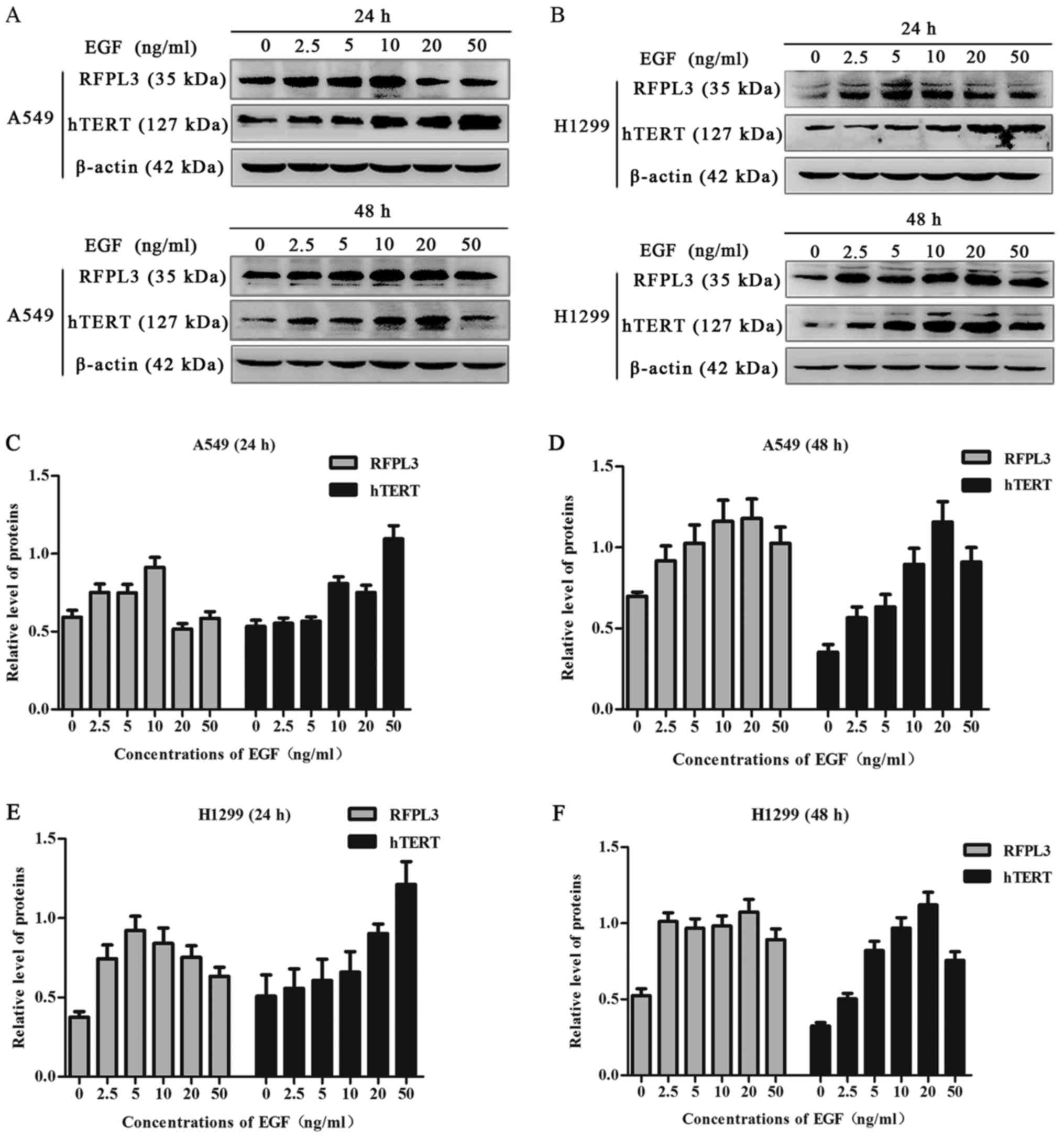
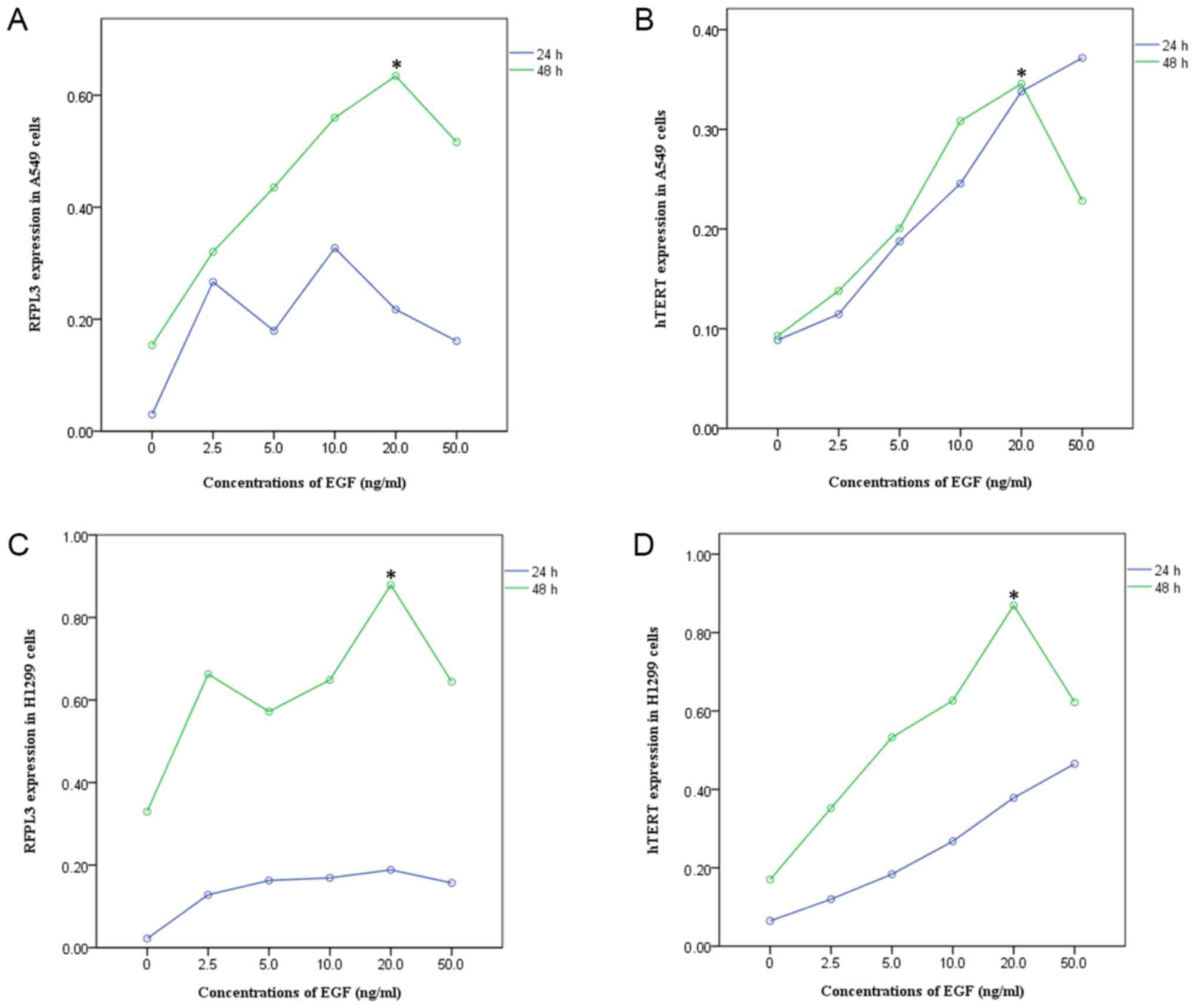
EGF upregulates expression of RFPL3
and hTERT proteins in NSCLC cells
A549 and H1299 cells were cultured in the presence
of EGF concentrations ranging from 0 to 50 ng/ml for 24 h or 48 h.
EGF stimulation significantly increased RFPL3 protein expression;
expression of hTERT exhibited a corresponding tendency (Figs. 1 and 2). This suggests a positive correlation
between expressions of RFPL3 and hTERT. Intriguingly, the protein
levels increased when stimulated for 24 h, but the activity
appeared to be unstable. When stimulated for 48 h, RFPL3 and hTERT
proteins were expressed incrementally in a dose-dependent manner,
with 20 ng/ml as the optimal concentration.
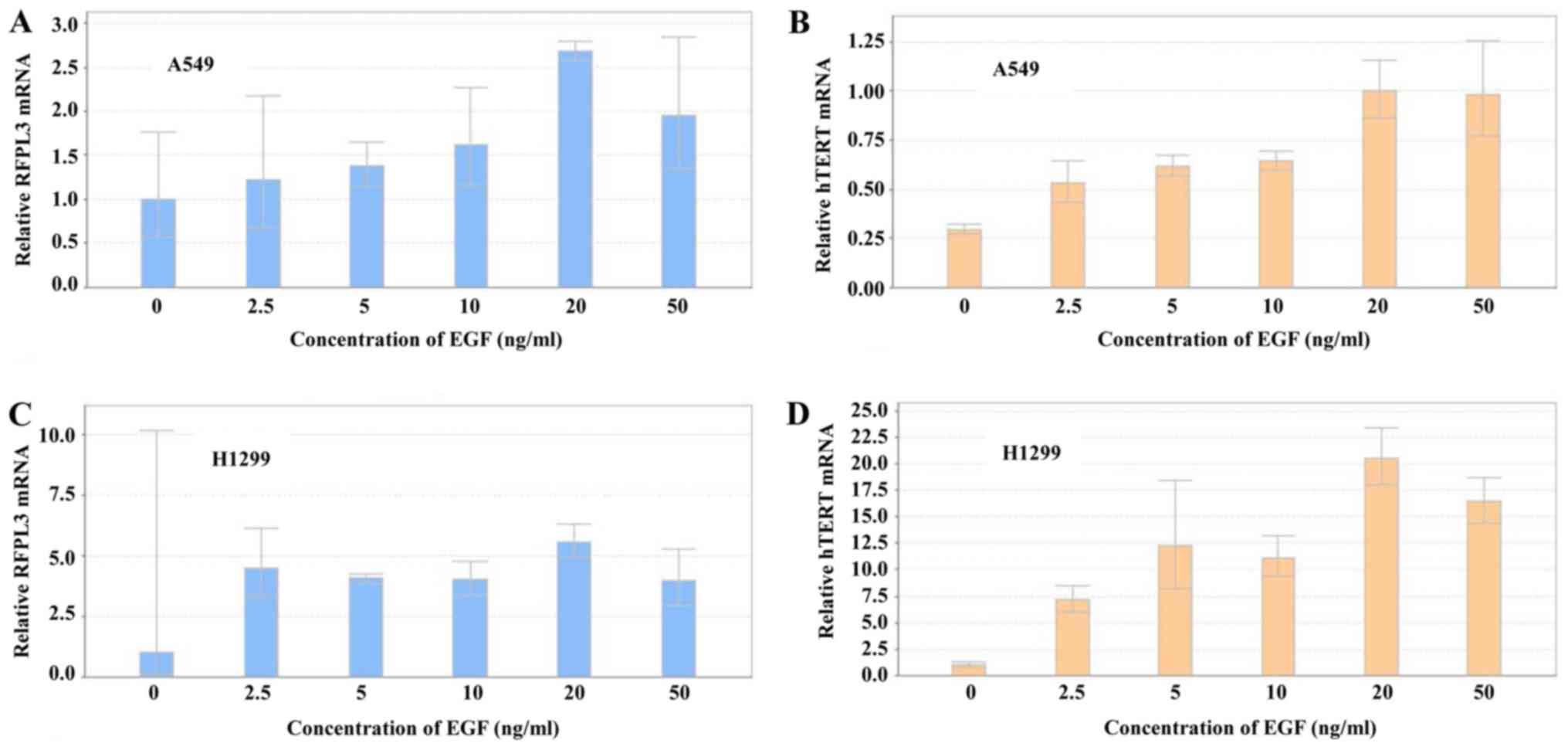
Expression of RFPL3 and hTERT mRNA was
analyzed by RT-qPCR in A549 and H1299 cells after treatment with
different concentrations of EGF for 48 h (Fig. 3). EGF induced a marked increase in
RFPL3 and hTERT mRNA expression, which was in accord
with their protein levels. The highest mRNA expression came at 20
ng/ml of EGF for 48 h.
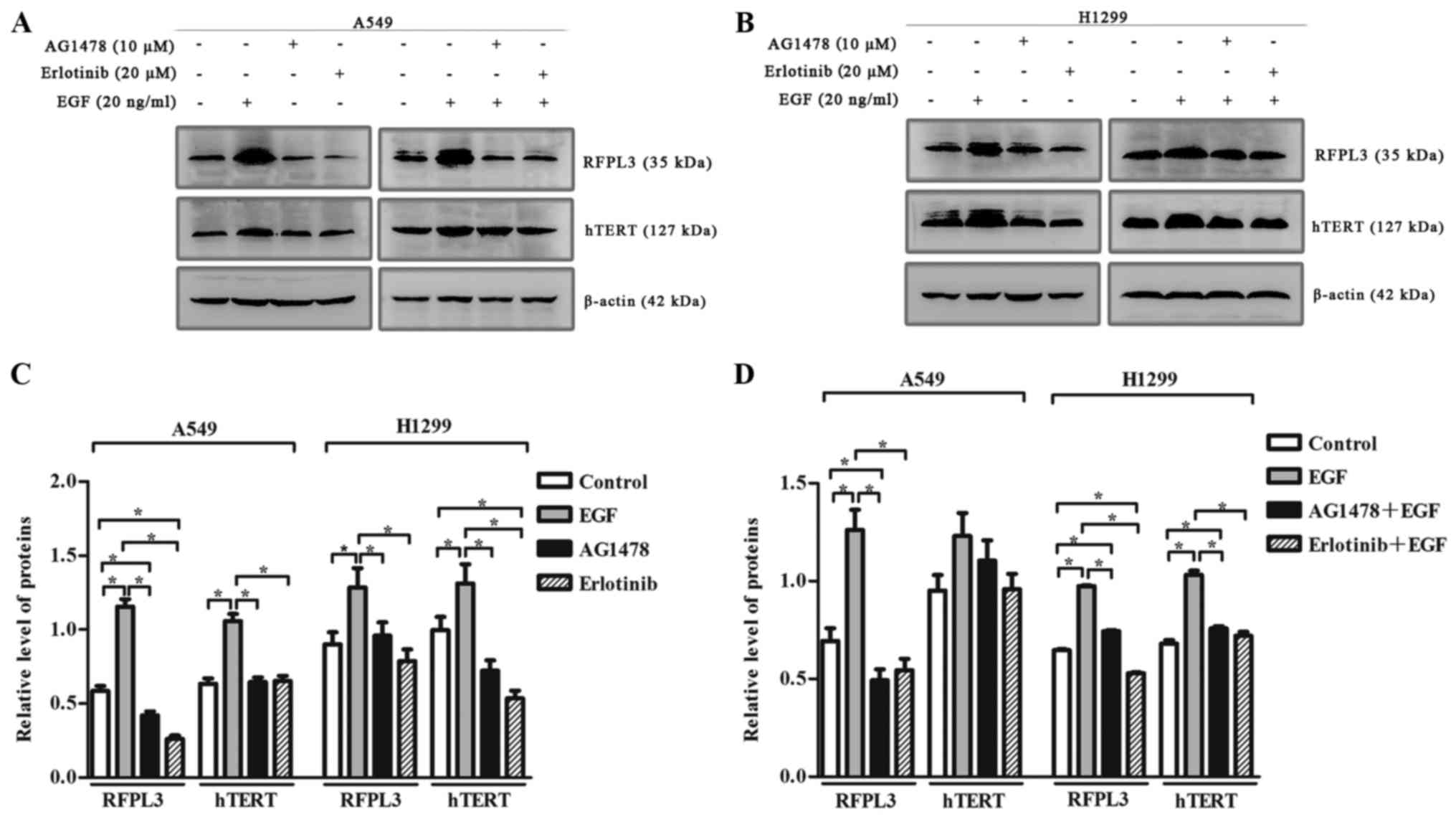
EGF-induced upregulation of RFPL3 and
hTERT is attenuated by pretreatment with EGFR inhibitors, AG1478
and erlotinib
To clarify the effect of EGF on RFPL3 and hTERT
expression, we incubated A549 and H1299 cells with or without
AG1478, and with or without erlotinib, in the presence of EGF. EGF
(20 ng/ml) markedly upregulated RFPL3 and hTERT (Fig. 4). When treated with EGFR-TKIs
[AG1478 (10 µM) or erlotinib (20 µM)] alone, RFPL3 and hTERT
expression was attenuated. When treated with a combination of
EGFR-TKIs and EGF, EGF-induced upregulation of RFPL3 and hTERT was
also attenuated, but was slightly higher than the levels in cells
treated with AG1478 or erlotinib alone. These findings suggest that
EGFR-TKIs partially and reversibly inhibit RFPL3 and hTERT
expression.
EGF significantly promotes cell
proliferation and affects apoptosis in NSCLC
To investigate the effect of EGF on cell growth, we
detected the viability of A549 and H1299 cells using the MTT assay.
EGF significantly enhanced cell proliferation, with maximum
increased growth at 20 ng/ml EGF, which was consistent with the
other results in this study (Fig.
5). Compared with the control group, EGF doses of 2.5, 5, 10
and 20 ng/ml inhibited apoptosis, whereas 50 ng/ml increased
apoptosis.
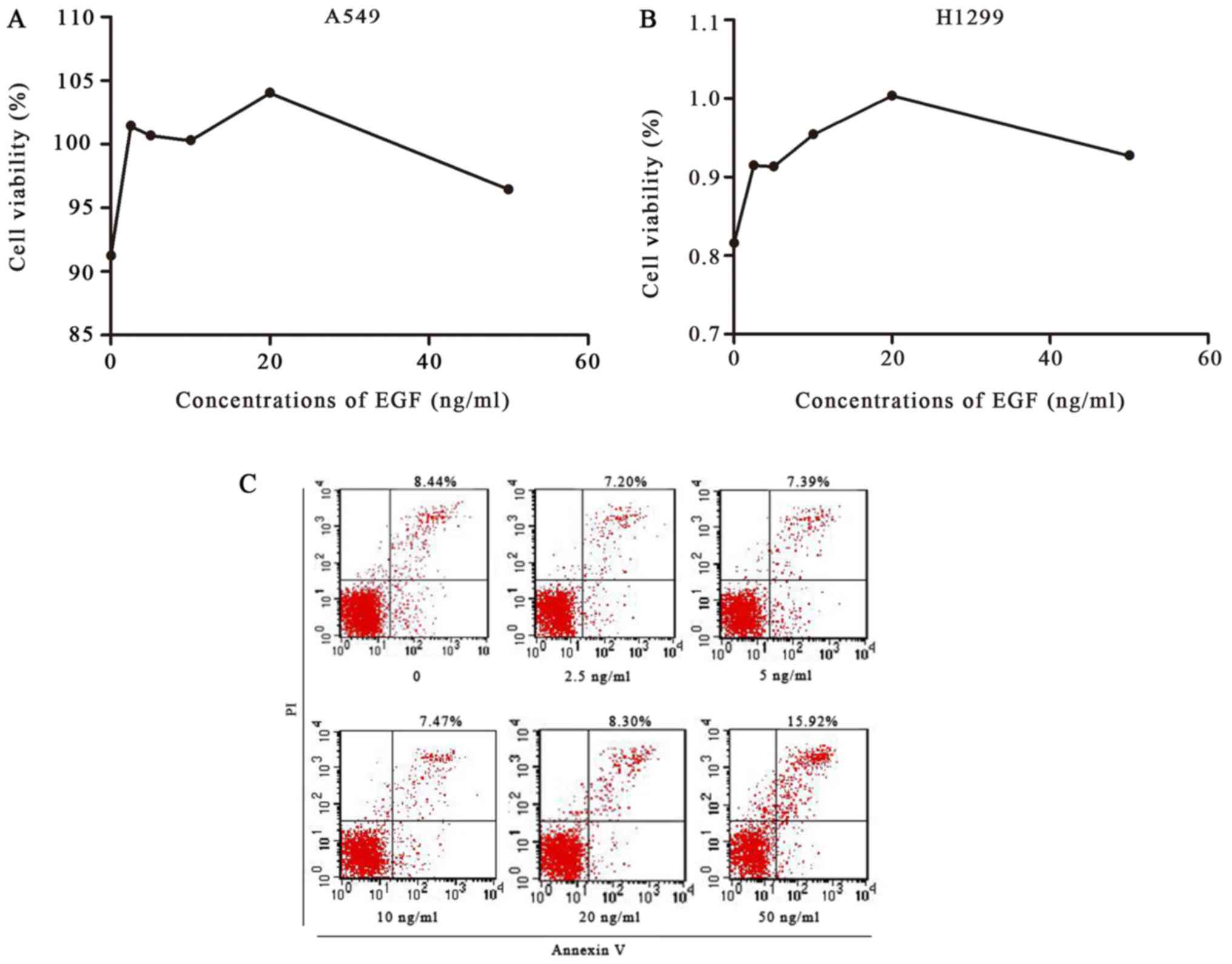 | Figure 5.Cell viability was detected by MTT
assay, and apoptosis of A549 cells was detected by flow cytometry.
(A and B) A549 and H1299 cells were treated with 0, 2.5, 5, 10, 20
or 50 ng/ml of EGF for 48 h. An MTT assay showed that EGF
significantly enhanced the proliferation of A549 and H1299 cells,
with a maximum increase at 20 ng/ml EGF. (C) A549 cells were
treated with 0, 2.5, 5, 10, 20 or 50 ng/ml of EGF for 48 h;
apoptosis was then detected by flow cytometry. Compared with the
control group, 2.5, 5, 10 and 20 ng/ml of EFG inhibited apoptosis,
whereas 50 ng/ml of EGF increased apoptosis. EGF, epidermal growth
factor. |
EGF upregulates expression of RFPL3
and hTERT via the MEK signaling pathway
To determine whether EGF-induced upregulation of
RFPL3 and hTERT proteins in NSCLC cells was related to the MEK
signaling pathway, we pretreated A549 and H1299 cells with an
MEK-specific inhibitor [PD98059 (50 µM)]. Expression of RFPL3 and
hTERT was significantly increased by EGF treatment (Fig. 6). The treatment of A549 and H1299
cells with PD98059 decreased expression of p-ERK protein, and also
markedly decreased EGF-induced upregulation of RFPL3 and hTERT,
without affecting total ERK protein.
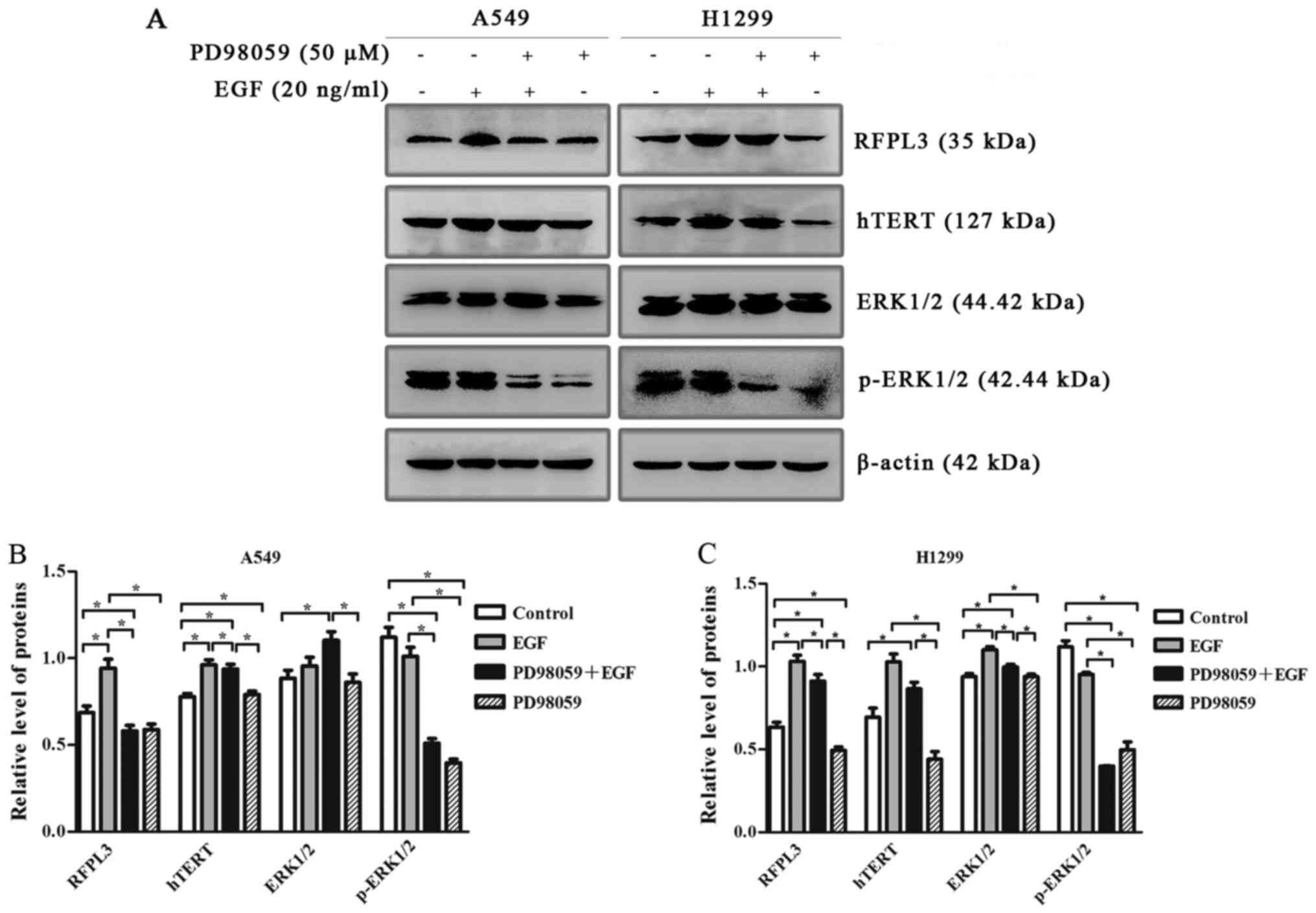 | Figure 6.Western blot analysis of RFPL3, hTERT,
ERK and p-ERK protein expression in A549 and H1299 cells, treated
with or without MEK-specific inhibitor (PD98059). (A) A549 and
H1299 cells were treated with PD98059 (50 µM) for 2 h, and then
with 20 ng/ml EGF for 48 h; expression levels of RFPL3, hTERT, ERK
and p-ERK proteins were then detected by western blot analysis.
PD98059 decreased expression of p-ERK without affecting total ERK
protein, and also markedly decreased EGF-induced upregulation of
RFPL3 and hTERT in H1299 cells, but did not significantly decrease
hTERT expression. (B and C) Histograms indicate relative
quantitative expression of these proteins. Data are expressed as
the mean ± standard deviation of three independent experiments.
*P<0.05 EGF, epidermal growth factor; RFPL3, Ret finger protein
like 3; hTERT, human telomerase reverse transcriptase. |
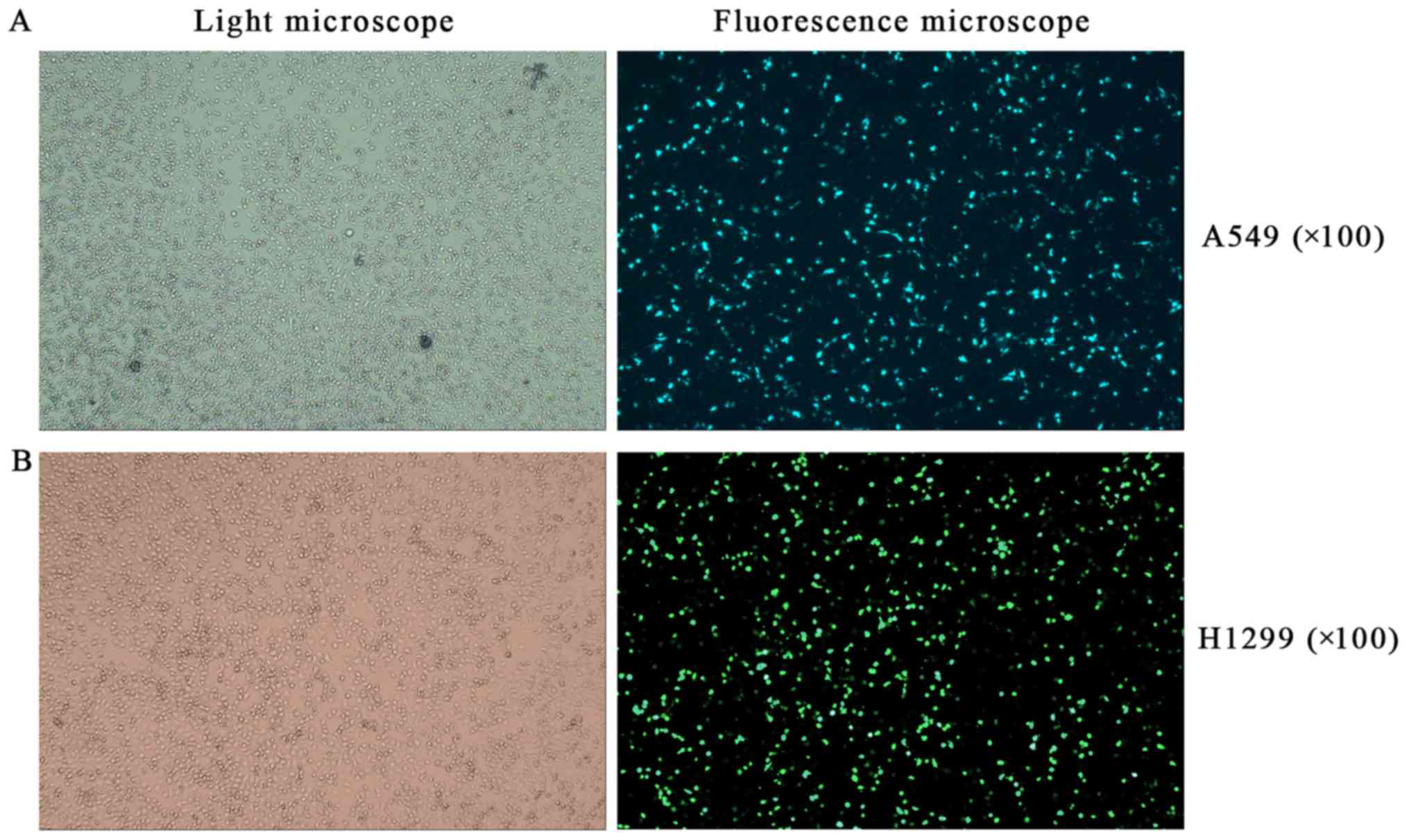
Effects of RFPL3 overexpression on MEK
signaling proteins
To further explore the effects of RFPL3
overexpression in NSCLC cells on related proteins in the MEK
pathway, we transfected A549 and H1299 cells with a
RFPL3-overexpression plasmid, and then detected the transfection
efficiency by fluorescence microscope and examined the expression
of RFPL3, hTERT, Ras, Raf, ERK, and p-ERK by western blot analysis.
After a successful transfection (>80% of cells transfected;
Fig. 7), RFPL3 protein was
overexpressed, which promoted hTERT expression and also increased
expression of the related MEK pathway proteins (Fig. 8). These results imply that control
for expression of RFPL3 and hTERT, and for these MEK signaling
proteins, may be inter-related.
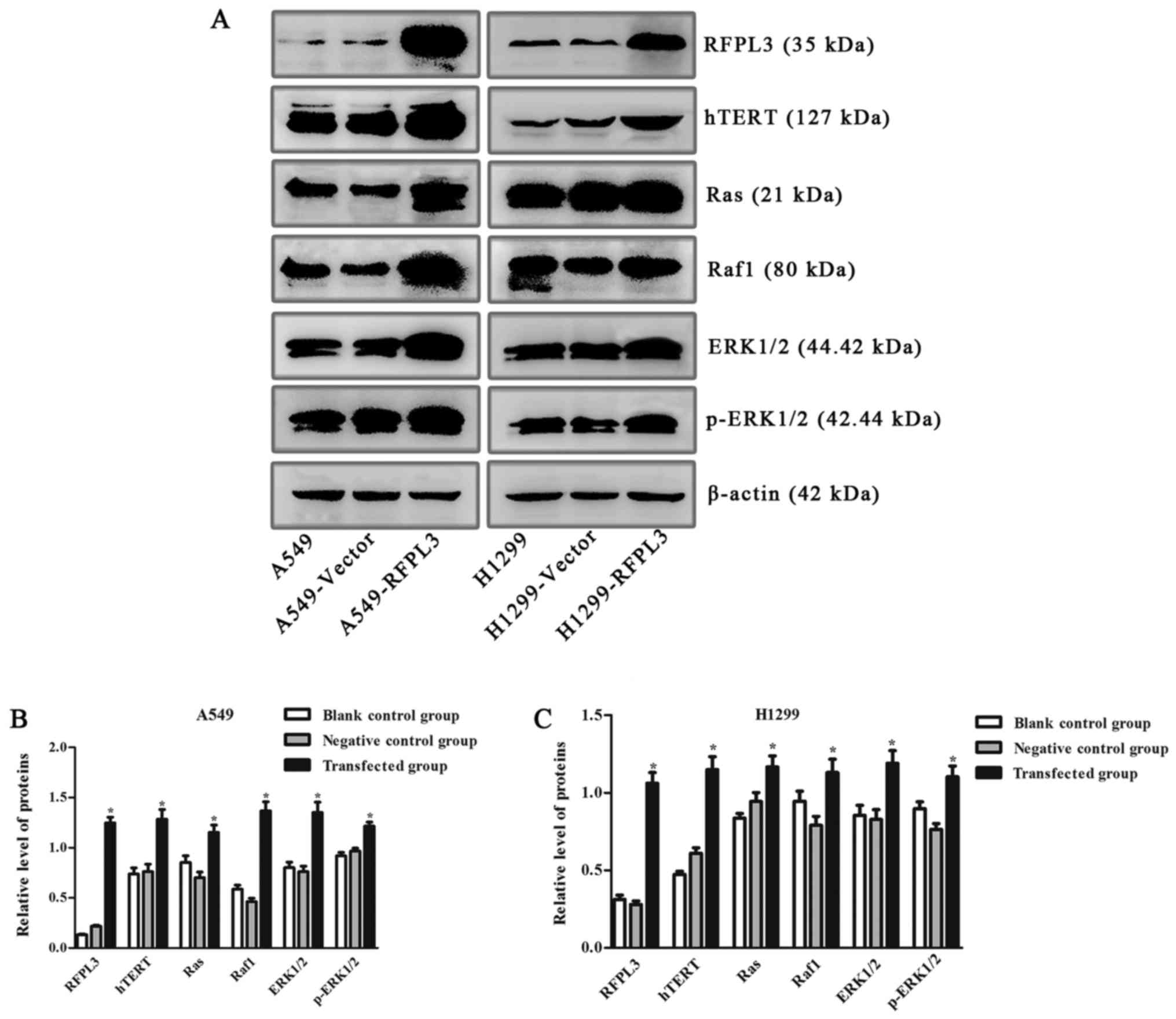 | Figure 8.Expression of hTERT and related
proteins in the MEK signaling pathway in A549 and H1299 cells
transfected with an RFPL3-overexpression plasmid. (A)
Western blot analysis was used to detect expression of RFPL3,
hTERT, Ras, Raf, ERK and p-ERK. Compared with the blank control and
negative control groups, RFPL3 in transfected cells was
overexpressed, and expression of hTERT, Ras, Raf, ERK and p-ERK
also increased. (B and C) Histograms indicate relative quantitative
expression of these proteins. Data are expressed as the mean ±
standard deviation of three independent experiments. *P<0.05,
compared with the negative control group. RFPL3, Ret finger protein
like 3; hTERT, human telomerase reverse transcriptase. |
Discussion
EGFR is an important therapeutic target in lung and
other cancers. Therapies targeted to EGFR, including
low-molecular-weight TKIs and monoclonal antibodies, have been
successfully implemented in the clinic for more than a decade.
First-generation TKIs include gefinitib, erlotinib and icotinib;
second-generation TKIs include afatinib and dacomitinib.
NSCLC tissues with EGFR mutations are more
sensitive to EGFR-TKIs. Reportedly, the NSCLC response rate to
EGFR-TKIs is 70–80%, but only 10–20% for NSCLC tumors with
wild-type EGFR (11), which
restricts the use of this treatment. In addition, EGFR-TKI
resistance, including both primary and acquired resistance, is
increasingly serious. Primary resistance may be caused by oncogenic
K-ras mutation, MET amplification, BIM gene
expression and polymorphism, and PTEN absence. EGFR
T790M secondary mutations may cause acquired resistance.
RAF/MEK/ERK signaling pathway inhibitors, such as respective
inhibitors of Raf, MEK, and ERK, have been investigated in clinical
trials; among these, MEK inhibitors have the strongest cytotoxic
effects, but cancer cells with K-ras mutations are less
sensitive to MEK inhibitors. Our present results showed that, when
treated with a MEK-specific inhibitor (PD98059), hTERT expression
in A549 cells did not decrease significantly.
Binding by EGF changes the intramolecular
conformation of EGFR, which activates intracellular tyrosine
kinase, and sequentially triggers several signaling pathways,
including RAS-MAPK, PI3K-Akt, PLCγ and JAKs-STATs. Thus, EGF
regulates transcription factors and activates gene transcription or
related protein expression by adapter proteins and enzyme cascade
reactions, ultimately regulating cell proliferation,
differentiation, apoptosis, and angiogenesis (12–14).
The activation of the Ras/MAPK (MEK) signaling transduction pathway
is associated with cell proliferation. The MEK signaling pathway
mainly regulates cell proliferation and differentiation through
phosphorylation and dephosphorylation. The underlying mechanisms of
the MEK pathway in promoting tumorigenesis include increasing tumor
cell proliferation, inhibiting apoptosis, promoting angiogenesis
and inducing tissue invasion (15).
Human lung adenocarcinoma A549 cells contain
wild-type EGFR and mutant-type K-ras, whereas H1299
cells have no detected mutation. Mutation of oncogene K-ras
is the major driver of growth and proliferation in lung cancer
cells. About 25% of patients with NSCLC have K-ras mutations
which are associated with poor prognoses (16). The oncogene K-ras is
associated with multiple signaling pathways, such as Raf/MEK/ERK,
PI3K/AKT/mTOR, and RALGDS/RAL, that affect tumor development and
progression.
Telomerase contains three important components:
Human telomerase RNA (hTR), telomerase TP1 and human telomerase
reverse transcriptase (hTERT). hTERT is the key component that
determines telomerase activity. Although expression of hTERT is low
or non-existent in normal cells, it is overexpressed in tumor
cells, which suggests that hTERT activation is a key step in the
somatic cell transformation to tumor cells. hTERT is the catalytic
subunit of telomerase responsible for telomerase activation,
telomere elongation, or even cell immortality. Activation of
telomerase due to activation of hTERT would be a pivotal step in
tumor occurrence and development. An in vitro study
(17,18) has indicated that drug resistance in
lung cancer may be directly related to activation of telomerase.
Ectopic overexpression of TERT in cancer cell lines makes them
insensitive to radiotherapy and chemotherapy drugs.
The RFPL3 gene (located at human 22q12.3) has
not been widely studied. RFPL3 belongs to the RFPL protein family
(RFPL1, RFPL2 and RFPL3), and is homologous to RFP, which is a
nucleoprotein that helps regulate cell proliferation,
differentiation and tumorigenesis. RFPL1, RFPL2 and RFPL3 present
high transcriptional levels in the human cerebral cortex, where the
highest concentration is in the developing cerebrum frontal lobe,
due to the effect of transcription factor Pax6 (19). RFPL3 is reported to clearly enhance
integration activity of the human immunodeficiency virus
pre-integration complex protein in infected cells in vitro,
which implies that RFPL3 overexpression would reinforce HIV
infectivity (20). RFPL3 protein
has been recently associated with occurrence and development of
lung cancer. RFPL3 can regulate expression of the hTERT gene
in NSCLC cells and drive hTERT promoter transcription, thus
increasing telomerase activity and length, and promoting
proliferation of tumor cells.
In the present study, we found that EGF stimulation
significantly increased the levels of RFPL3 and hTERT proteins in
NSCLC cells, and increased cell proliferation while inhibiting
apoptosis; but EGFR TKIs could block its function.
Furthermore, we present novel evidence that EGF
activates MEK signaling pathway; that EGF upregulates expression of
RFPL3 and hTERT proteins through the activation of MEK signaling
pathway; and that overexpression of RFPL3 protein increases
expression of hTERT and related proteins in MEK pathway.
Our results suggest that stimulation of A549 and
H1299 cells with EGF at 24 h increased RFPL3 and hTERT expression
but the activity appeared to be unstable-probably due to the lack
of enough affinity. At 48 h, expression of RFPL3 and hTERT
increased progressively, and was time- and concentration-dependent
within concentrations of 0–20 ng/ml EGF. When treated with EGF at
50 ng/ml, production of RFPL3 and hTERT proteins declined, cell
viability decreased and apoptosis increased. Accordingly, we
conjectured that constant EGF stimulation decreases cell
sensitivity to EGFR. We also found that EGFR TKIs (AG1478,
erlotinib) partially and reversibly inhibited expression of RFPL3
and hTERT proteins, but did not completely stop their expression,
which may reflect decreased sensitivity to EGFR TKIs and increased
drug resistance.
A previous study showed that RFPL3 could drive
hTERT promoter transcriptional activity, thus increasing its
expression and contributing to development of NSCLC (10). Our results showed that RFPL3
overexpression increased hTERT expression; expression of RFPL3 and
hTERT were positively correlated. The MEK signaling pathway is
pivotal in this process. EGF binds with a specific cytomembrane
receptor (EGFR), activates the downstream MEK signaling proteins
(RAS/RAF/MEK/ERK), and then increases expression of ERK, and
upregulates RFPL3 and hTERT proteins. Overexpressed RFPL3 protein
also increased expression of upstream molecules (Ras, Raf) in the
MEK signaling pathway, and sequentially increased ERK expression,
which activates expression of growth-regulated oncoproteins.
In conclusion, our results indicate that EGF
upregulates expression of RFPL3 and hTERT proteins in NSCLC cells
via the MEK signaling pathway, promotes cell proliferation and
inhibits apoptosis. Our experiments demonstrated the link between
EGFR and hTERT, indicating that EGF may upregulate hTERT expression
and activity through RFPL3. Moreover, RFPL3 overexpression
increases related MEK-signaling proteins (Ras, Raf, ERK, p-ERK),
and promotes cell proliferation and differentiation. RFPL3 could
thus be a therapeutic target in NSCLC, and have a role in NSCLC
drug resistance.
Acknowledgements
We are very thankful to the Regenerative Medicine
Center, First Affiliated Hospital of Dalian Medical University for
providing us with two human NSCLC cell lines, A549 and H1299.
Funding
No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
CL, YQ, YFW and MYG conceived and designed the
study. CL performed the experiments. CL and YQ wrote the
manuscript. CL, YQ, HZ, YFW and MYG reviewed and edited the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
This article contains no studies with human
participants performed by any of the authors.
Consent for publication
No human participants were involved in this
study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
World Health Organization, . International
Agency for Research on Cancer. Press Release No. 224. Lyon/London:
3–February. 2014
|
|
2
|
Minna JD and Schiller JH: Lung Cancer:
Harrison's Principles of Internal Medicine (17th edition). 551–562.
2008.
|
|
3
|
Goldstraw P, Crowley J, Chansky K, Giroux
DJ, Groome PA, Rami-Porta R, Postmus PE, Rusch V and Sobin L; and
International Association for the Study of Lung Cancer
International Staging Committee; Participating Institutions, : The
IASLC lung cancer staging project: Proposals for the revision of
the TNM stage groupings in the forthcoming (seventh) edition of the
TNM classification of malignant tumours. J Thorac Oncol. 2:706–714.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
DeSantis CE, Lin CC, Mariotto AB, Siegel
RL, Stein KD, Kramer JL, Alteri R, Robbins AS and Jemal A: Cancer
treatment and survivorship statistics, 2014. CA Cancer J Clin.
64:252–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hirata Y, Moore GW, Bertagna C and Orth
DN: Plasma concentrations of immunoreactive human epidermal growth
factor (urogastrone) in man. J Clin Endocrinol Metab. 50:440–444.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiqing Li, Zunlan Zhao, Mingyue Liu, et
al: Epidermal growth factor affects the growth and proliferation of
non-small cell lung cancer A549 and H23 cells. Chinese Journal of
applied diagnosis and treatment. 29:562–564. 2015.(In Chinese).
|
|
7
|
Bethune G, Bethune D, Ridgway N and Xu Z:
Epidermal growth factor receptor (EGFR) in lung cancer: An overview
and update. J Thorac Dis. 2:48–51. 2010.PubMed/NCBI
|
|
8
|
Lee HJ, Xu X, Choe G, Chung DH, Seo JW,
Lee JH, Lee CT, Jheon S, Sung SW and Chung JH: Protein
overexpression and gene amplification of epidermal growth factor
receptor in nonsmall cell lung carcinomas: Comparison of four
commercially available antibodies by immunohistochemistry and
fluorescence in situ hybridization study. Lung Cancer. 68:375–382.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsu CP, Lee LW, Tang SC, Hsin IL, Lin YW
and Ko JL: Epidermal growth factor activates telomerase activity by
direct binding of Ets-2 to hTERT promoter in lung cancer cells.
Tumour Biol. 36:5389–5398. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen W, Lu J, Qin Y, Wang J, Tian Y, Shi
D, Wang S, Xiao Y, Dai M, Liu L, et al: Ret finger protein-like 3
promotes tumor cell growth by activating telomerase reverse
transcriptase expression in human lung cancer cells. Oncotarget.
5:11909–11923. 2014.PubMed/NCBI
|
|
11
|
Mitsudomi T and Yatabe Y: Mutations of the
epidermal growth factor receptor gene and related genes as
determinants of epidermal growth factor receptor tyrosine kinase
inhibitors sensitivity in lung cancer. Cancer Sci. 98:1817–1824.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murray SA, Yang S, Demicco E, Ying H,
Sherr DH, Hafer LJ, Rogers AE, Sonenshein GE and Xiao ZX: Increased
expression of MDM2, cyclin D1, and p27Kip1 in
carcinogen-induced rat mammary tumors. J Cell Biochem. 95:875–884.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Anderson D, Koch CA, Grey L, Ellis C,
Moran MF and Pawson T: Binding of SH2 domains of phospholipase C
gamma 1, GAP, and Src to activated growth factor receptors.
Science. 250:979–982. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yuqin Hao and Huixia Zheng: The mechanism
research of MAPK/ERK signaling pathways in cancer treatment. China
J Leprosy and Skin Diseases. 7:490–493. 2012.(In Chinese).
|
|
16
|
Roberts PJ and Stinchcombe TE: KRAS
mutation: Should we test for it, and does it matter? J Clin Oncol.
31:1112–1121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Loboda A, Nebozhyn M, Klinghoffer R,
Frazier J, Chastain M, Arthur W, Roberts B, Zhang T, Chenard M,
Haines B, et al: A gene expression signature of RAS pathway
dependence predicts response to PI3K and RAS pathway inhibitors and
expands the population of RAS pathway activated tumors. BMC Med
Genomics. 3:262010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Matallanas D and Crespo P: New druggable
targets in the Ras pathway? Curr Opin Mol Ther. 12:674–683.
2010.PubMed/NCBI
|
|
19
|
Bonnefont J, Nikolaev SI, Perrier AL, Guo
S, Cartier L, Sorce S, Laforge T, Aubry L, Khaitovich P, Peschanski
M, et al: Evolutionary forces shape the human RFPL1,2,3
genes toward a role in neocortex development. Am J Hum Genet.
83:208–218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tan BH and Suzuki Y, Takahashi H, Ying PH,
Takahashi C, Han Q, Chin WX, Chao SH, Sawasaki T, Yamamoto N and
Suzuki Y: Identification of RFPL3 protein as a novel E3 ubiquitin
ligase modulating the integration activity of human
immunodeficiency virus, type 1 preintegration complex using a
microtiter plate-based assay. J Biol Chem. 289:26368–26382. 2014.
View Article : Google Scholar : PubMed/NCBI
|