Introduction
Glioblastoma multiforme (GBM), with a 5-year
survival rate of 5.1% (1), has a
high fatality rate. The standard therapy of maximal safe resection
or subtotal resection followed by concurrent chemoradiation and
adjuvant temozolomide (TMZ) is recommended by the Clinical Practice
Guidelines for Oncology (2) in the
National Comprehensive Cancer Network® (NCCN). This
therapeutic strategy is widely carried out all over the world.
However, the lifespan of GBM patients varies by relative increments
from less than 6 months to more than 3 years. Furthermore, few
studies or drugs have been designed to improve the survival time of
GBM patients who have undertaken standard therapy.
Cancer cell genotypes, in combination with
expression programmes, govern tumour immune fitness, evolution and
resistance to therapy (3). In
recent years, studies such as those of The Cancer Genome Atlas
(TCGA), have charted the genetic landscape and derived from it the
bulk expression states of GBM, identifying driver mutations and
defining tumour subtypes based on specific transcriptional profiles
(4,5). Currently, there is a lack of knowledge
concerning the reasons for the variable survival times of GBM
patients who have undertaken standard therapy. Therefore,
systematic screening of differentially expressed genes (DEGs) and
cellular pathways in patients with different survival periods may
be of great significance for the development of more effective
diagnostic and therapeutic strategies.
In the present study, 136 eligible patients who had
undertaken standard therapy were selected from TCGA profiles. DEGs
correlating with survival time were identified from 12,042 genes.
The most important hub genes and signalling pathways were screened
by Gene Ontology (GO) analysis, pathway enrichment analysis,
protein-protein interaction (PPI) network and module analysis. In
addition, the importance of the hub genes was validated by an
additional glioma dataset. Finally, evidence in the published
literature is discussed to illustrate the importance of these genes
and pathways. The recognition of the pivotal genes and pathways
that affect the survival of GBM patients who had undertaken
standard therapy may be a potential benefit for developing
therapeutic strategies.
Materials and methods
Microarray data
The gene expression (AffyU133a) and phenotypes of
the TCGA GBM data were downloaded from UCSC Xena (https://xenabrowser.net/datapages), which is a
hub for gene data deposit and retrieval. The gene expression
profile was evaluated experimentally using the Affymetrix HT Human
Genome U133a microarray platform by the Broad Institute of MIT and
Harvard University Cancer Genomic Characterization Center. Level 3
interpreted level data was originated from the TCGA data
coordination center. This dataset showed the gene-level
transcription estimates and data were in log space. The profile of
phenotypes included detailed clinical information corresponding to
each of the samples. The profile of Tumor
Glioma-French-284-MAS5.0-u133p2 was chosen to validate the
importance of the hub genes. It included 8 control samples, 24
grade II glioma samples, 85 grade III glioma samples and 159 grade
IV GBM samples. And 272 samples were recorded with their exact
survival time. Hub gene expression profiles, historical profiles
and survival profiles were downloaded from R2 (http://r2platform.com), a web-based genomic analysis
and visualization application.
Sample screening and group
assignment
The included samples were from patients who had
undertaken surgical resection of the tumor, followed by TMZ
chemoradiation and long-term therapy with TMZ. Homogeneity was of
great importance for this study, so exclusion criteria were
established: i) treated primary GBM; ii) recurrent tumor and normal
tissue; iii) Karnofsky performance scores <60; iv) no exact
survival time; and v) no gene expression files. A total of 136
eligible samples were selected according to the above criteria.
General characteristics of samples were analyzed with SPSS software
(version 22; IBM Corp., Armonk, NY, USA). Median survival time ±10
percentiles was set as a boundary of group assignment. There were
54 samples in each group.
Identification of DEGs
DEGs were analyzed through the online tool Morpheus
(https://software.broadinstitute.org/morpheus/), a
versatile matrix visualization and analysis software. Based on the
Morpheus analysis, the dataset was adjusted by robust z-score. A
total of 383 DEGs were identified through t-test with a P-value
cut-off of ≤0.05; of which, 259 were upregulated and 124 were
downregulated. P-value cut-off of ≤0.01 was indispensable for hub
gene candidates. Both the heat map and dataset of results were
downloaded.
Gene Ontology and pathway enrichment
analysis of DEGs
Gene Ontology analysis (GO) and KEGG pathway
analysis were conducted through DAVID (https://david.ncifcrf.gov/home.jsp), a database for
annotation, visualization and integrated discovery. Upregulated
genes and downregulated genes were analyzed respectively and P≤0.05
was considered as statistically significant. Annotation tables were
downloaded.
Hub gene screening and module
analysis
Search tool for the retrieval of interacting genes
(STRING) (https://string-db.org/) is a database of
prediction of protein-protein interaction (PPI). DEGs were mapped
to STRING to conduct PPI analysis, and a minimum required
interaction score ≥0.4 was set as significant. A simple tabular
text was downloaded. With a differentially expressed P≤0.01, the
most active 11 genes (the last two genes had the same nodes) in PPI
networks were considered as hub genes. The relationships of hub
genes between expression and glioma histology or survival were
analyzed with GraphPad Prism software (version 6). The RNA levels
of the 11 hub genes in 136 eligible samples were analyzed with the
online tool Morpheus. PPI networks were constructed using the
Cytoscape software (version 3.5.1). The modules of PPI networks
were filtered with the plug-in Molecular Complex Detection (MCODE)
in Cytoscape. MCODE scores >3 and number of nodes >5 were
necessary for modules. Moreover, the function and pathway
enrichment analysis were conducted for DEGs in the modules. P≤0.05
was considered to be significant.
Results
Eligible patients
From the inclusion and exclusion criteria described
above, 136 eligible patients who had undertaken surgical resection
for GBM followed by TMZ chemoradiation and long-term therapy with
TMZ were selected. The median survival rate for the patients was
448 days [95% confidence interval (CI), 421 to 475 days]. To obtain
more significant differences and to reduce sample capacity loss,
patients with survival times that were shorter or longer than the
median survival time by ±10 percentiles were set as short and long
survival groups, respectively. Each group had 54 patients, and
differences in age, sex and subtypes were not significant (Table I).
 | Table I.Clinical and subtype features of the
GBM sample groups. |
Table I.
Clinical and subtype features of the
GBM sample groups.
| Variables | Short survival | Long survival | P-value |
|---|
| No. of
patients | 54 | 54 |
|
| Survival, in
days |
|
|
P<0.001a |
| Median
(range) | 250 (47–394) | 681 (502–1977) |
|
| Age, in years |
|
| 0.080b |
| Mean
(range) | 52.9 (18–77) | 57.6 (17–83) |
|
| Sex, n (%) |
|
| 0.425c |
|
Male | 36 (66.7) | 32 (59.3) |
|
|
Female | 18 (33.3) | 12 (40.7) |
|
| GBM subtype, n |
|
| 0.782c |
|
Classical | 13 | 17 |
|
|
Mesenchymal | 20 | 16 |
|
|
Neural | 7 | 8 |
|
|
Proneural | 14 | 13 |
|
Identification of DEGs
The TCGA gene expression profile identified 12,042
genes. Based on t-test analysis with P≤0.05 criterion in Morpheus,
a total of 383 genes (259 upregulated and 124 downregulated) were
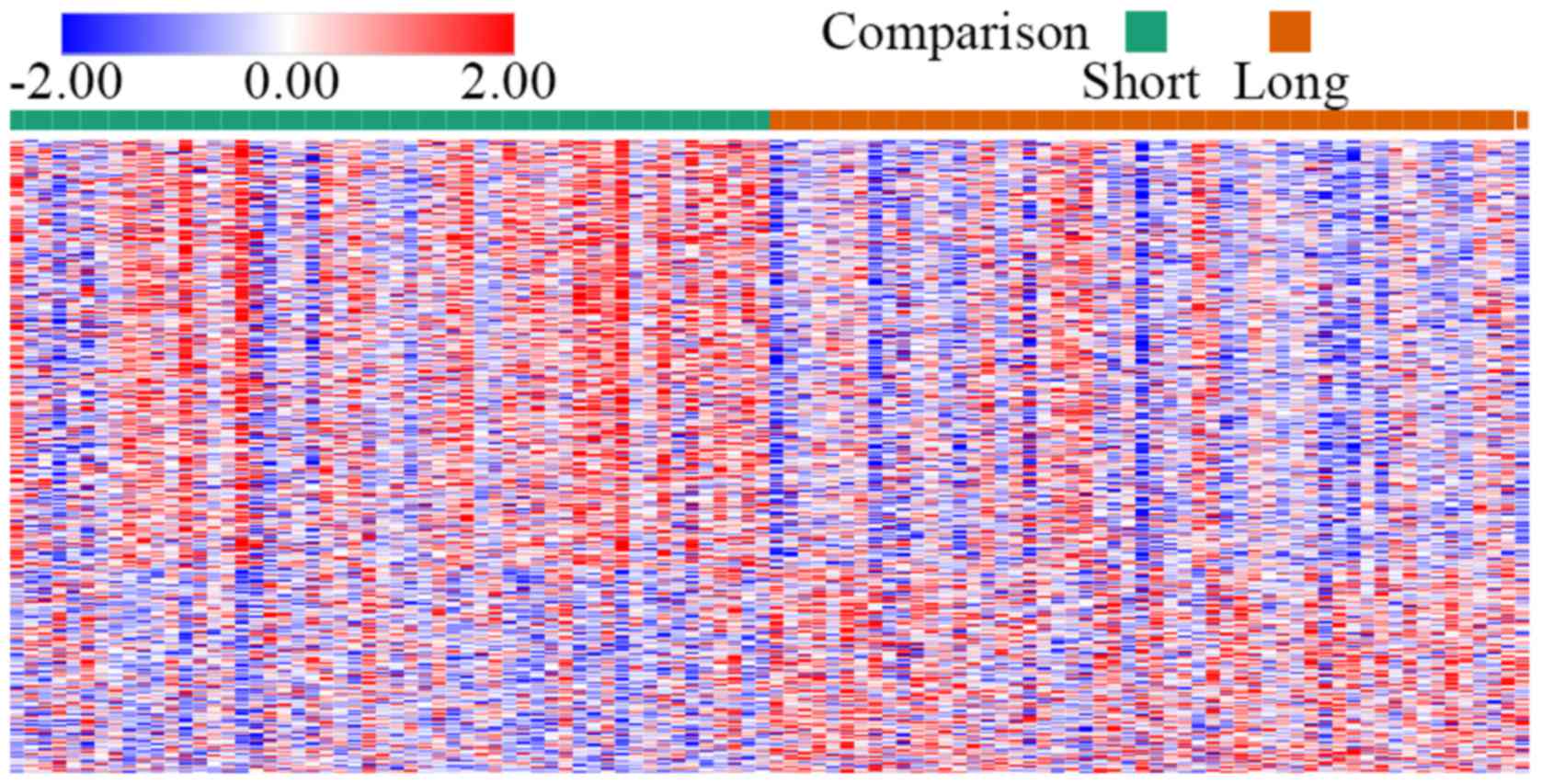
identified after analysis of the two groups. A DEG expression heat
map is shown in Fig. 1.
GO term enrichment analysis and KEGG
pathway analysis
All DEGs were uploaded to the online software DAVID
(https://david.ncifcrf.gov/home.jsp)
to identify overrepresented GO categories and KEGG pathways.
According to the reverse order of P-values, the top two terms are
shown in Tables II and III.
| Table II.Gene Ontology analysis of the
differentially expressed genes correlated with the survival of the
GBM patients. |
Table II.
Gene Ontology analysis of the
differentially expressed genes correlated with the survival of the
GBM patients.
| Expression | Category | Term | Count | % | P-value |
|---|
| Upregulated | BP |
GO:0001525-angiogenesis | 13 | 5.306122 | 7.73E-05 |
|
| BP | GO:0008285-negative
regulation of cell proliferation | 16 | 6.530612 | 5.01E-04 |
|
| MF |
GO:0004553-activity, hydrolyzing
O-glycosyl compounds | 6 | 2.44898 | 5.59E-05 |
|
| MF | GO:0005178-integrin
binding | 9 | 3.673469 | 1.16E-04 |
|
| CC |
GO:0070062-extracellular exosome | 92 | 37.55102 | 1.54E-17 |
|
| CC |
GO:0005615-extracellular space | 43 | 17.55102 | 1.75E-07 |
| Downregulated | BP | GO:0000122-negative
regulation of transcription from RNA polymerase II promoter | 14 | 12.61261 | 3.39E-04 |
|
| BP | GO:0007268-chemical
synaptic transmission | 8 | 7.207207 | 5.78E-04 |
|
| MF |
GO:0003682-chromatin binding | 9 | 8.108108 | 0.002705 |
|
| MF | GO:0046982-protein
heterodimerization activity | 8 | 7.207207 | 0.023714 |
|
| CC |
GO:0005634-nucleus | 50 | 45.04505 | 1.32E-04 |
|
| CC |
GO:0014069-postsynaptic density | 7 | 6.306306 | 6.53E-04 |
| Table III.KEGG pathway analysis of
differentially expressed genes associated with the survival of the
GBM patients. |
Table III.
KEGG pathway analysis of
differentially expressed genes associated with the survival of the
GBM patients.
| Expression | Term | Count | % | P-value | Genes |
|---|
| Upregulated | hsa04142:
Lysosome | 14 | 5.714286 | 7.71E-07 | GNS, SLC11A1, NPC1,
TPP1, PSAP, ATP6AP1, HEXA, HEXB, CTSD, CTSA, CTSB, M6PR, MANBA,
GLB1 |
|
| hsa04512:
ECM-receptor interaction | 8 | 3.265306 | 0.001753 | SDC1, LAMB3, LAMA3,
ITGA5, COL6A2, ITGA3, LAMB1, COL5A1 |
| Downregulated | hsa04015: Rap1
signaling pathway | 6 | 5.405405 | 0.010241 | P2RY1, SIPA1L3,
RAPGEF4, PRKCG, MAPK11, FGF21 |
|
| hsa04550: Signaling
pathways regulating pluripotency of stem cells | 5 | 4.504505 | 0.011701 | NANOG, WNT3, RIF1,
ID4, MAPK11 |
GO analysis results (Table II) showed that the upregulated DEGs
were significantly enriched in the biological processes (BP) of
angiogenesis and negative regulation of cell proliferation. For
molecular function (MF), upregulated DEGs were enriched in
hydrolase activity, hydrolysing O-glycosyl compounds and
integrin binding. Moreover, GO cell component (CC) analysis also
showed that the upregulated DEGs were significantly enriched in
extracellular exosome and extracellular space. Downregulated DEGs
were involved in the negative regulation of transcription from RNA
polymerase II promoter and chemical synaptic transmission for BP.
For MF, downregulated DEGs were enriched in chromatin binding,
protein heterodimerization activity. For CC, they were enriched in
nucleus and postsynaptic density.
KEGG analysis results (Table III) illustrated that the
upregulated DEGs were significantly enriched in the pathways of
lysosome and extracellular matrix (ECM)-receptor interaction.
Downregulated DEGs were enriched in the Rap1 signalling pathway and
signalling pathways that regulate the pluripotency of stem
cells.
Hub gene screening from the PPI
network
With the information from the STRING database, the
top 11 hub genes (the last two genes had the same nodes) with the
most nodes were screened. These hub genes included the following:
serpin family E member 1 (SERPINE1), cathepsin B
(CTSB), plasminogen activator, urokinase receptor
(PLAUR), galactosidase β1 (GLB1), FOS such as 1, AP-1
transcription factor subunit (FOSL1), ferritin heavy chain 1
(FTH1), glutamate metabotropic receptor 1 (GRM1),
hexosaminidase subunit α (HEXA), heat shock protein family B
member 1 (HSPB1), DNA damage inducible transcript 3
(DDIT3), and cation dependent mannose-6-phosphate receptor
(M6PR).
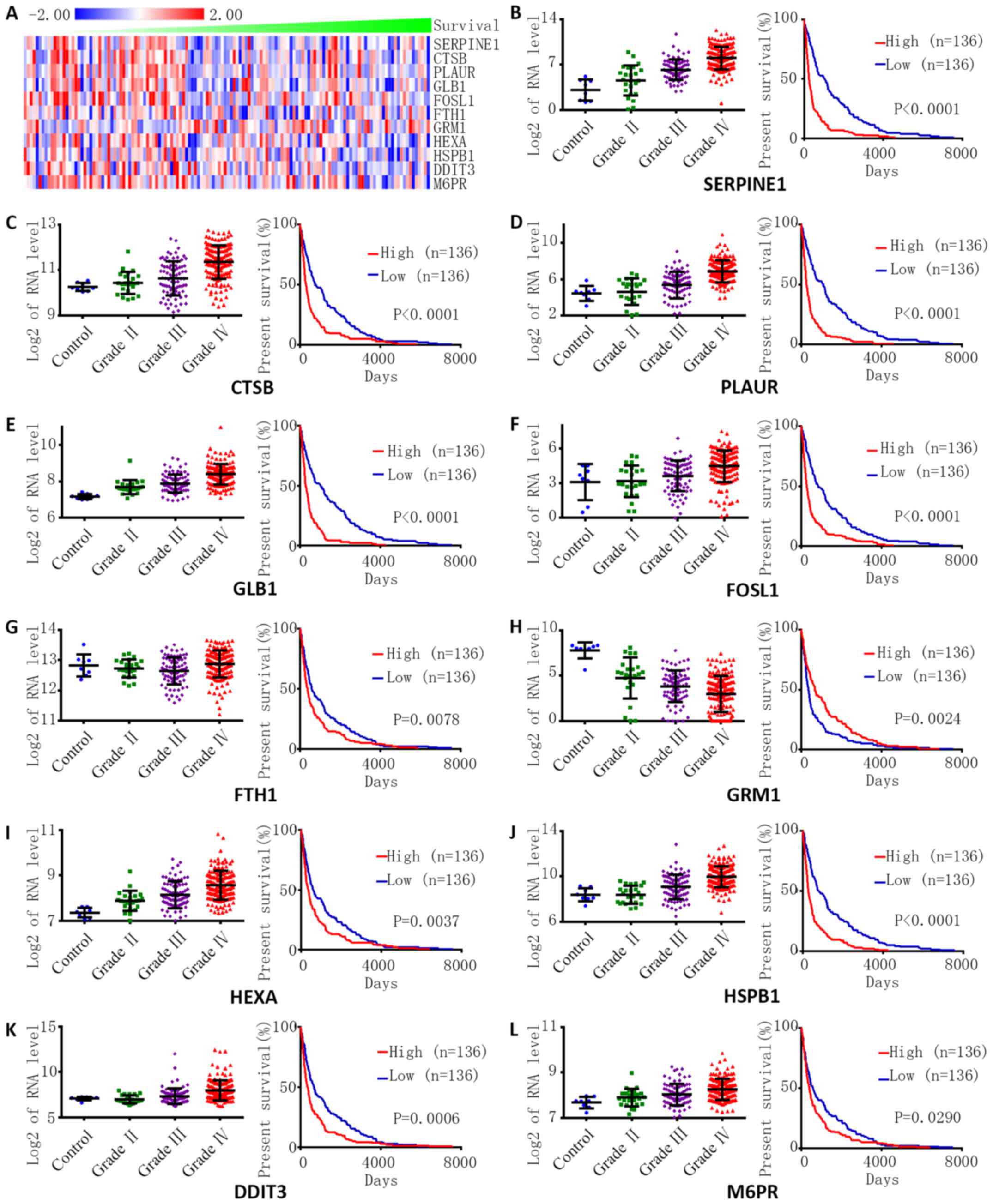
The levels of hub gene expression were closely
related to the survival time of 136 eligible GBM patients who had
undertaken standard therapy (Fig.
2A). The relationships between hub gene expression and
histology and survival were also explored with another dataset
(Fig. 2B-L). The expression of
FOSL1 was negatively correlated with the grade and survival of
glioma (Fig. 2F). The expression of
FTH was not significantly related to the grade but positively
correlated with survival of glioma (Fig. 2G), and the expression of other genes
was positively correlated with the grade and survival of
glioma.
Module screening from the PPI
network
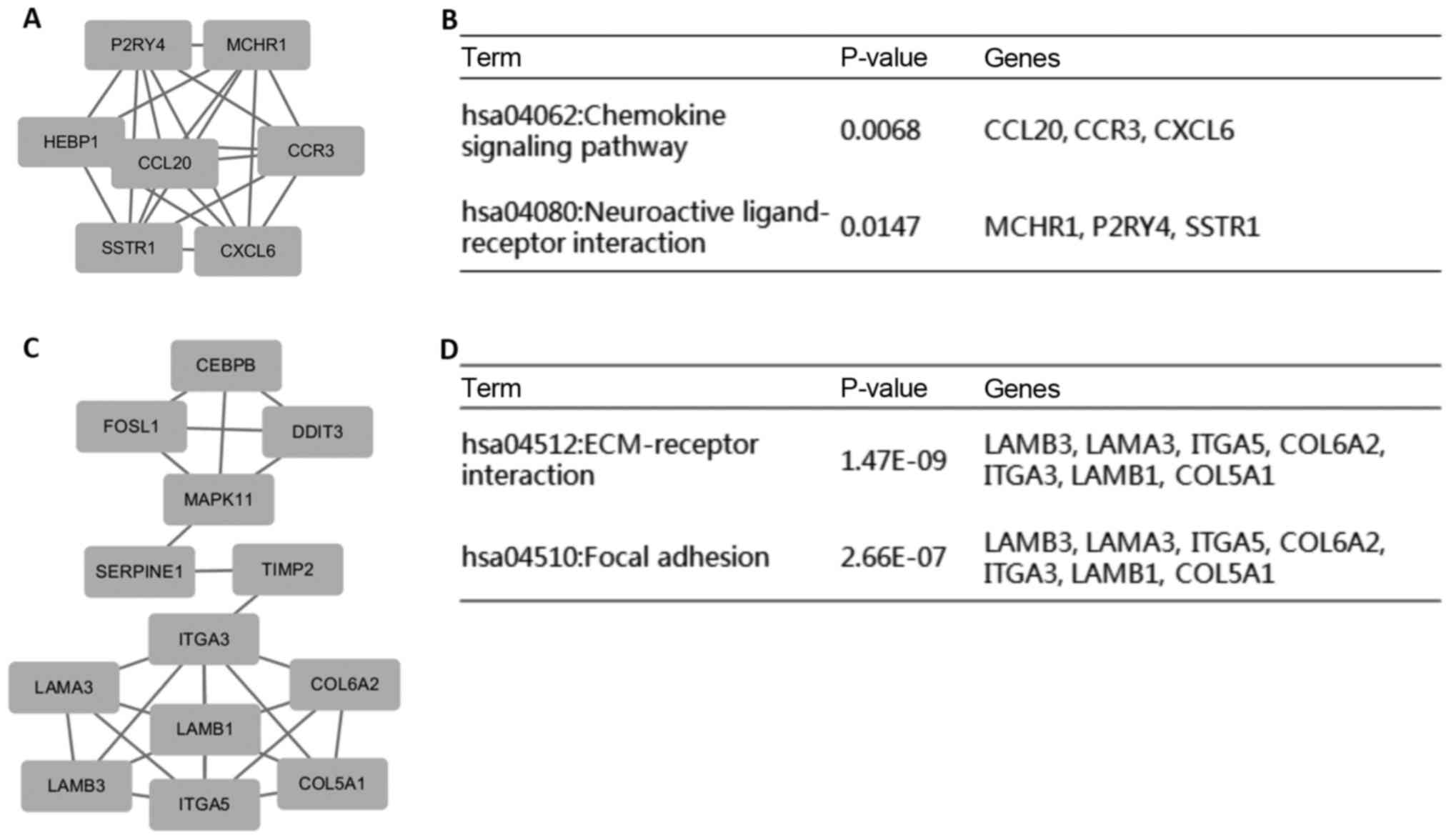
In addition, a total of 265 nodes and 568 edges were
analysed with the Cytoscape plug-in MCODE. The top 2 most
significant modules were selected, and the pathway enrichment of
the genes involved in these modules was analysed (Fig. 3). Enrichment analysis indicated that
the genes in these modules were mainly associated with the
chemokine signalling pathway, neuroactive ligand-receptor
interactions, ECM-receptor interactions and focal adhesion.
Discussion
In the present study, the pivotal genes and pathways
related to the survival of GBM patients who had undergone standard
therapy were explored. Surgical resection of the tumours followed
by TMZ chemoradiation and long-term therapy with TMZ were required
for eligible samples. To eliminate interference from confounding
factors, treated tumours, recurrent tumours, normal tissues, and
patients with a Karnofsky Performance Status (KPS) <60 were
excluded. Additionally, the exact survival time was vital for group
assignment and gene expression files were essential for further
analysis. Finally, 136 eligible patients were included. Their
median survival was 448 days (95% CI, 421–475 days), which was a
little longer than the 14.6 months (438 days) reported in a study
by Stupp et al (6). This is
most likely since all these eligible patients had undergone
surgical resection, whereas only 84% of patients had undergone
debulking surgery in the previous study.
Regarding the results of GO pathway analysis, it was
significant that the negative regulation of cell proliferation was
correlated with the short survival of GBM patients, although
unlimited proliferation is one of the essential characteristics of
cancers. The process involving growth restraints exerted by ectopic
tissue that leads to reversible mitotic arrest is called tumour
dormancy (7). The literature
published in recent years has revealed a growing focus on tumour
cell dormancy, which is probably one of the main reasons for
refractory to targeted or conventional therapies (7–10). In
glioma, the ‘peri-necrotic niche’ harbouring HIF-1α+
quiescent stem-like cells have been proposed as candidates for
long-term tumourigenic cells (11).
For the enrichment in angiogenesis of upregulated DEGs, it is
widely recognized that the extreme proliferation of new blood
vessels in GBM is structurally and functionally abnormal and
contributes to a hostile microenvironment (low oxygen tension and
high interstitial fluid pressure) that selects for a more malignant
phenotype with increasing morbidity and mortality (12–14).
In addition, the extracellular space is closely involved with the
invasion and migration of glioma cells. It has been reported that a
pore space of appropriate size can promote the amoeboid movement of
glioma cells (15). Clinically, we
also observed that glioma cells spread along the structure of the
white matter fibre bundle.
Furthermore, the KEGG pathway analysis showed that
the downregulated DEGs were enriched in signalling pathways
regulating pluripotency of stem cells. It has been reported that
the anti-differentiation strategies of quiescent cells are co-opted
by cancer cells (16). Sosa et
al reported that downregulated pluripotency is part of tumour
cell quiescence and contributes to fuel incurable local or distal
recurrences (17). Regarding the
enrichment of upregulated DEGs in the lysosome, it is known that
the lysosome mediates the process of autophagy and plays multiple
context-dependent roles in tumourigenesis and treatment resistance.
Previous studies have also shown that the endolysosome is critical
for the organization and turnover of epidermal growth factor
receptor (EGFR) to maintain tumour growth and invasion (18). For the enrichment of upregulated
DEGs in ECM-receptor interaction, glioma cells produce their own
ECM environment and use that of the host cells as a bed for
migration (19). Integrin directly
binds the components of the ECM and provides the traction necessary
for cell motility and invasion (20). Regarding the enrichment of
downregulated DEGs in the Rap1 signalling pathway, the Ras family
of small GTPases is highly expressed in the normal brain, and the
Rap1 signalling pathway is necessary to regulate neurite growth and
arborization in mammalian neurons (21, 22). However, it has been
reported that Rap1 signalling also regulates glioma cell motility
(23).
PPI networks of DEGs were constructed, and the
top-degree hub genes were SERPINE1, CTSB, PLAUR, GLB1, FOSL1, FTH1,
GRM1, HEXA, HSPB1, DDIT3 and M6PR. The relationships between RNA
expression and histology/survival were also explored with another
glioma dataset (Fig. 2). SERPINE1
(also known as PAI-1) encodes a member of the serine proteinase
inhibitor (serpin) superfamily. Previous publications have
demonstrated that the overexpression of PAI-1 in GBM is
significantly correlated with a shorter survival rate (24). Recent research has revealed that the
glioma-derived plasminogen activator inhibitor-1 (PAI-1) affects
the tumour microenvironment by regulating the recruitment of
LRP1-positive mast cells (25).
CTSB encodes cathepsin B, a lysosomal cysteine cathepsin. This is
consistent with the fact that cathepsin B is a strong predictor of
survival in GBM (24). PLAUR (also
known as uPAR) encodes the receptor for urokinase plasminogen
activator. A correlation between uPAR and invasion of GBM has been
repeatedly demonstrated in recent years (26, 27). Cathepsin B and
uPAR regulate self-renewal of glioma-initiating cells (28). Inhibition of cathepsin B and uPAR
inhibits cell invasion in glioma (29) and enhanced radiation-induced
apoptosis in glioma-initiating cells (30). GLB1 (also known as EBP) encodes
galactosidase β1. When bound to elastin-derived peptides, EBP
allows GBM cells to adhere to the newly synthesized matrix and
increases their aggressiveness (31). Generally, the molecular mechanisms
underlying GLB1 in GBM have not been well clarified. FOSL1 (also
known as FRA1) encodes the AP-1 transcription factor Fra-1. Only a
few literature reports have proposed that Fra-1 takes part in
migratory behaviour of GBM cells (32). However, FRA1 expression is
associated with the migration of breast cancers (33), and the depletion of FRA1 results in
a mesenchymal-epithelial transition (34). FTH1 encodes ferritin heavy chain 1,
the major intracellular iron storage protein in cells. Ferritin
protects DNA from iron-induced oxidative damage (35), and silencing the ferritin heavy
chain can effectively sensitize tumours to chemotherapy in glioma
(36). However, the ferritin heavy
chain has also been reported to be a negative regulator of ovarian
cancer stem cell expansion and epithelial-to-mesenchymal transition
(37). GRM1 encodes glutamate
metabotropic receptor 1. Ligand binding to this protein activates a
phosphatidylinositol-calcium second messenger system. The Human
Protein Atlas (http://www.proteinatlas.org) shows that GRM1 is highly
expressed in normal brain tissue and less frequently expressed in
glioma tissue and cells. This is consistent with the negative
relationship between GRM1 expression and glioma grade (Fig. 2). However, it has also been
demonstrated that mGluR1 inhibition induces cell cycle arrest,
caspase-dependent apoptosis, and prevents invasion and migration in
glioma (38). HEXA encodes a
preproprotein that is proteolytically processed to generate the α
subunit of the lysosomal enzyme β-hexosaminidase. In the last
century, β-hexosaminidase was found to be overexpressed in colonic
carcinoma (39), ovarian
adenocarcinoma (40) and lung
cancer (41), but no further
research has been conducted to clarify its underlying mechanisms,
which need to be further verified. HSPB1 (also known as HP27)
encodes heat shock protein family B member 1, which functions as a
molecular chaperone, probably maintaining denatured proteins in a
folding-competent state. Recently, HSPB1 was proposed to be a
discriminating short protein that is a long survival factor in GBM
(42). HSP27 mediates SPARC-induced
changes in glioma morphology and invasion (43), and inhibition of HSP27 alone or in
combination with pAKT inhibition may be an effective therapeutic
approach (44). DDIT3 (also known
as CHOP) encodes DNA damage inducible transcript 3, which functions
as an inhibitor by forming heterodimers with other C/EBP members.
In addition, it blocks their DNA binding activity. CHOP facilitates
autophagy and contributes to resistance to treatment in glioma
(45,46). Overexpression of CHOP enables immune
inhibitory activity of tumour-infiltrating myeloid-derived
suppressor cells (MDSCs), which promotes cancer development
(47). M6PR encodes
mannose-6-phosphate receptor, which is related to transportation of
phosphorylated lysosomal enzymes. There have been few research
efforts involved in determining the influence of M6PR on cancers.
Selective M6PR downregulation has a critical role in
CD8+ T cell survival, which provides protection against
cancer (48). M6PR mediates TMEPAI
transport from the Golgi directly into the endo-lysosomal pathway
and then indirectly boosts tumourigenesis in lung cancer (49).
Module analysis of the PPI network revealed that the
DEGs related to survival were mainly associated with cell
migration. GBM secretes chemokines that can promote tumour growth
and progression or induce stromal cells to provide a hotbed for
tumour growth (50). Neuroactive
ligand-receptor interactions have been reported as having a
negative effect (51, 52) on glioma cell proliferation. It was a
foregone conclusion that ECM-receptor interaction and focal
adhesion contribute to a poor prognosis, as both are closely
related to cancer cell invasion.
It must be acknowledged that microarray analysis has
limitations. First, it can only report the level of mRNA
expression, usually comparing tumour tissues with non-tumour
tissues, and the genes that are highly expressed in tumour tissue
are determined by statistical analysis. However, this method cannot
analyse gene modification or detect protein function. For example,
NLGN3 is a functional protein expressed in normal brain tissue, but
its associated normal functional activity promotes cancer growth
(53). On the other hand, due to
statistical analysis, bioinformatic analysis is not applicable to
all cases and samples. For example, the trend in gene expression
profiles in each sample is not consistent in Fig. 1, but the trend in most samples is
consistent. This is due to small probability events that are almost
impossible in a single experiment but inevitable in repeated tests.
This indicates that bioinformatic analysis is only an analytical
method. Scientific research cannot be entirely dependent on such a
method, and the hypotheses yielded by bioinformatic approaches must
be demonstrated by various experimental methods and finally proven
in clinical practice.
The present study identified key pathways and genes
correlated with survival of GBM patients who had undertaken
standard therapy. As expected, the angiogenesis and migration of
GBM cells contributed to a poor prognosis. Notably, the upregulated
genes were enriched in negative regulation of cell proliferation,
and the downregulated genes were enriched in signalling pathways
regulating pluripotency of stem cells. A total of 3 hub genes
(FTH1, GRM1 and DDIT3) had negative effects on cancer
cell proliferation, and the module genes of the PPI network were
enriched in neuroactive ligand-receptor interaction, which has been
reported to have a negative effect on glioma cell proliferation.
All the above results highlight that cell dormancy is an essential
contributor to the shorter survival of GBM patients who have
undertaken standard therapy. The change in the dormant state is
probably determined through a process of epigenetic regulation
(54). Presumably, the expression
and functional level of the dormant genes would be reduced in
recurrent tumours. However, as little research has been published
concerning this issue, further experimental validation is required
to confirm the pathogenesis and mechanisms. Modulating persistent
cell dormancy or preventing tumour cells from translating into
quiescence during radiochemotherapy should be helpful to prolong
the survival of GBM patients under standard treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (no. 81472352) and the Tianjin
Research Program of Application Foundation and Advanced Technology
(no. 15JCZDJC36200).
Availability of data and materials
The datasets used during the present study are
freely available from the corresponding websites.
Authors' contributions
LT and XY conceived and designed the study. LY, PL,
LH and TL downloaded the datasets, selected eligible samples,
assigned groups and screened differentially expressed genes (DEGs).
They also analyzed the differences of age, sex and subtypes of the
groups. ZT and HM underwent Gene Ontology and pathway enrichment
analysis of DEGs. YX, YH and SY conducted PPI analysis and selected
hub genes. In addition, they analyzed the relationships of hub
genes between expression and glioma histology or survival with
another glioma dataset. JL and FY conducted module analysis. LT, LY
and PL reviewed many relevant articles and analyzed the research
progress of related pathways and genes. LT, XY and IRA wrote the
paper. LT, LY, PL and IRA reviewed and edited the manuscript. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
No procedures performed in the study involved human
participants.
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Ostrom QT, Gittleman H, Fulop J, Liu M,
Blanda R, Kromer C, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS Statistical Report: Primary Brain and Central Nervous System
Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 17
Suppl 4:iv1–iv62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nabors LB, Portnow J, Ammirati M, Baehring
J, Brem H, Brown P, Butowski N, Chamberlain MC, Fenstermaker RA,
Friedman A, et al: Central Nervous System Cancers, Version 1.2015.
J Natl Compr Canc Netw. 13:1191–1202. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Venteicher AS, Tirosh I, Hebert C, Yizhak
K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman
C, et al: Decoupling genetics, lineages, and microenvironment in
IDH-mutant gliomas by single-cell RNA-seq. Science. 355:3552017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brennan CW, Verhaak RG, McKenna A, Campos
B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ,
Berman SH, et al: TCGA Research Network: The somatic genomic
landscape of glioblastoma. Cell. 155:462–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Verhaak RG, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al:
Cancer Genome Atlas Research Network: Integrated genomic analysis
identifies clinically relevant subtypes of glioblastoma
characterized by abnormalities in PDGFRA, IDH1, EGFR, and
NF1. Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: European Organisation for Research and Treatment of
Cancer Brain Tumor and Radiotherapy Groups; National Cancer
Institute of Canada Clinical Trials Group: Radiotherapy plus
concomitant and adjuvant temozolomide for glioblastoma. N Engl J
Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Klein CA: Framework models of tumor
dormancy from patient-derived observations. Curr Opin Genet Dev.
21:42–49. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goss PE and Chambers AF: Does tumour
dormancy offer a therapeutic target? Nat Rev Cancer. 10:871–877.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aguirre-Ghiso JA: Models, mechanisms and
clinical evidence for cancer dormancy. Nat Rev Cancer. 7:834–846.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sosa MS, Bragado P and Aguirre-Ghiso JA:
Mechanisms of disseminated cancer cell dormancy: An awakening
field. Nat Rev Cancer. 14:611–622. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishii A, Kimura T, Sadahiro H, Kawano H,
Takubo K, Suzuki M and Ikeda E: Histological characterization of
the tumorigenic ‘peri-necrotic niche’ harboring quiescent stem-like
tumor cells in glioblastoma. PLoS One. 11:e01473662016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jain RK, di Tomaso E, Duda DG, Loeffler
JS, Sorensen AG and Batchelor TT: Angiogenesis in brain tumours.
Nat Rev Neurosci. 8:610–622. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tate MC and Aghi MK: Biology of
angiogenesis and invasion in glioma. Neurotherapeutics. 6:447–457.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jhaveri N, Chen TC and Hofman FM: Tumor
vasculature and glioma stem cells: Contributions to glioma
progression. Cancer Lett. 380:545–551. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang Y, Tong L, Yi L, Zhang C, Hai L, Li
T, Yu S, Wang W, Tao Z, Ma H, et al: Three-dimensional hydrogel is
suitable for targeted investigation of amoeboid migration of glioma
cells. Mol Med Rep. 17:250–256. 2018.PubMed/NCBI
|
|
16
|
Sang L, Roberts JM and Coller HA:
Hijacking HES1: How tumors co-opt the anti-differentiation
strategies of quiescent cells. Trends Mol Med. 16:17–26. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sosa MS, Parikh F, Maia AG, Estrada Y,
Bosch A, Bragado P, Ekpin E, George A, Zheng Y, Lam HM, et al:
NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven
quiescence programmes. Nat Commun. 6:61702015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kondapalli KC, Llongueras JP,
Capilla-González V, Prasad H, Hack A, Smith C, Guerrero-Cázares H,
Quiñones-Hinojosa A and Rao R: A leak pathway for luminal protons
in endosomes drives oncogenic signalling in glioblastoma. Nat
Commun. 6:62892015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Noreen R, Moenner M, Hwu Y and Petibois C:
FTIR spectro-imaging of collagens for characterization and grading
of gliomas. Biotechnol Adv. 30:1432–1446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Desgrosellier JS and Cheresh DA: Integrins
in cancer: Biological implications and therapeutic opportunities.
Nat Rev Cancer. 10:9–22. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kawabe H, Neeb A, Dimova K, Young SM Jr,
Takeda M, Katsurabayashi S, Mitkovski M, Malakhova OA, Zhang DE,
Umikawa M, et al: Regulation of Rap2A by the ubiquitin ligase
Nedd4-1 controls neurite development. Neuron. 65:358–372. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schwamborn JC and Püschel AW: The
sequential activity of the GTPases Rap1B and Cdc42 determines
neuronal polarity. Nat Neurosci. 7:923–929. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barrett A, Evans IM, Frolov A, Britton G,
Pellet-Many C, Yamaji M, Mehta V, Bandopadhyay R, Li N, Brandner S,
et al: A crucial role for DOK1 in PDGF-BB-stimulated glioma cell
invasion through p130Cas and Rap1 signalling. J Cell Sci.
127:2647–2658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Colin C, Voutsinos-Porche B, Nanni I, Fina
F, Metellus P, Intagliata D, Baeza N, Bouvier C, Delfino C, Loundou
A, et al: High expression of cathepsin B and plasminogen activator
inhibitor type-1 are strong predictors of survival in
glioblastomas. Acta Neuropathol. 118:745–754. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Roy A, Coum A, Marinescu VD, Põlajeva J,
Smits A, Nelander S, Uhrbom L, Westermark B, Forsberg-Nilsson K,
Pontén F, et al: Glioma-derived plasminogen activator inhibitor-1
(PAI-1) regulates the recruitment of LRP1 positive mast cells.
Oncotarget. 6:23647–23661. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu J, Muller KA, Furnari FB, Cavenee WK,
VandenBerg SR and Gonias SL: Neutralizing the EGF receptor in
glioblastoma cells stimulates cell migration by activating
uPAR-initiated cell signaling. Oncogene. 34:4078–4088. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Raghu H, Gondi CS, Dinh DH, Gujrati M and
Rao JS: Specific knockdown of uPA/uPAR attenuates invasion in
glioblastoma cells and xenografts by inhibition of cleavage and
trafficking of Notch-1 receptor. Mol Cancer. 10:1302011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gopinath S, Malla R, Alapati K, Gorantla
B, Gujrati M, Dinh DH and Rao JS: Cathepsin B and uPAR regulate
self-renewal of glioma-initiating cells through GLI-regulated Sox2
and Bmi1 expression. Carcinogenesis. 34:550–559. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Malla Rao R, Gopinath S, Alapati K,
Gorantla B, Gondi CS and Rao JS: Knockdown of cathepsin B and uPAR
inhibits CD151 and α3β1 integrin-mediated cell adhesion and
invasion in glioma. Mol Carcinog. 52:777–790. 2013.PubMed/NCBI
|
|
30
|
Malla RR, Gopinath S, Alapati K, Gorantla
B, Gondi CS and Rao JS: uPAR and cathepsin B inhibition enhanced
radiation-induced apoptosis in gliomainitiating cells. Neuro Oncol.
14:745–760. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Coquerel B, Poyer F, Torossian F, Dulong
V, Bellon G, Dubus I, Reber A and Vannier JP: Elastin-derived
peptides: Matrikines critical for glioblastoma cell aggressiveness
in a 3-D system. Glia. 57:1716–1726. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Debinski W and Gibo DM: Fos-related
antigen 1 (Fra-1) pairing with and transactivation of JunB in GBM
cells. Cancer Biol Ther. 11:254–262. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Belguise K, Cherradi S, Sarr A, Boissière
F, Boulle N, Simony-Lafontaine J, Choesmel-Cadamuro V, Wang X and
Chalbos D: PKCθ-induced phosphorylations control the ability of
Fra-1 to stimulate gene expression and cancer cell migration.
Cancer Lett. 385:97–107. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E,
Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, et al: Protein
kinase C α is a central signaling node and therapeutic target for
breast cancer stem cells. Cancer Cell. 24:347–364. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thompson KJ, Fried MG, Ye Z, Boyer P and
Connor JR: Regulation, mechanisms and proposed function of ferritin
translocation to cell nuclei. J Cell Sci. 115:2165–2177.
2002.PubMed/NCBI
|
|
36
|
Liu X, Madhankumar AB, Slagle-Webb B,
Sheehan JM, Surguladze N and Connor JR: Heavy chain ferritin siRNA
delivered by cationic liposomes increases sensitivity of cancer
cells to chemotherapeutic agents. Cancer Res. 71:2240–2249. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lobello N, Biamonte F, Pisanu ME, Faniello
MC, Jakopin Ž, Chiarella E, Giovannone ED, Mancini R, Ciliberto G,
Cuda G, et al: Ferritin heavy chain is a negative regulator of
ovarian cancer stem cell expansion and epithelial to mesenchymal
transition. Oncotarget. 7:62019–62033. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang C, Yuan XR, Li HY, Zhao ZJ, Liao YW,
Wang XY, Su J, Sang SS and Liu Q: Anti-cancer effect of
metabotropic glutamate receptor 1 inhibition in human glioma U87
cells: Involvement of PI3K/Akt/mTOR pathway. Cell Physiol Biochem.
35:419–432. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brattain MG, Green C, Kimball PM, Marks M
and Khaled M: Isoenzymes of beta-hexosaminidase from normal rat
colon and colonic carcinoma. Cancer Res. 39:4083–4090.
1979.PubMed/NCBI
|
|
40
|
Chatterjee SK, Chowdhury K, Bhattacharya M
and Barlow JJ: Beta-hexosaminidase activities and isoenzymes in
normal human ovary and ovarian adenocarcinoma. Cancer. 49:128–135.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Narita M, Taniguchi N, Makita A, Kodama T,
Araki E and Oikawa K: Elevated activity of beta-hexosaminidase and
sulfhydryl modification in the B-variant of human lung cancer.
Cancer Res. 43:5037–5042. 1983.PubMed/NCBI
|
|
42
|
Gimenez M, Marie SK, Oba-Shinjo S, Uno M,
Izumi C, Oliveira JB and Rosa JC: Quantitative proteomic analysis
shows differentially expressed HSPB1 in glioblastoma as a
discriminating short from long survival factor and NOVA1 as a
differentiation factor between low-grade astrocytoma and
oligodendroglioma. BMC Cancer. 15:4812015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Golembieski WA, Thomas SL, Schultz CR,
Yunker CK, McClung HM, Lemke N, Cazacu S, Barker T, Sage EH, Brodie
C, et al: HSP27 mediates SPARC-induced changes in glioma
morphology, migration, and invasion. Glia. 56:1061–1075. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schultz CR, Golembieski WA, King DA, Brown
SL, Brodie C and Rempel SA: Inhibition of HSP27 alone or in
combination with pAKT inhibition as therapeutic approaches to
target SPARC-induced glioma cell survival. Mol Cancer. 11:202012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rouschop KM, van den Beucken T, Dubois L,
Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W,
Voncken JW, et al: The unfolded protein response protects human
tumor cells during hypoxia through regulation of the autophagy
genes MAP1LC3B and ATG5. J Clin Invest. 120:127–141. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu WT, Huang CY, Lu IC and Gean PW:
Inhibition of glioma growth by minocycline is mediated through
endoplasmic reticulum stress-induced apoptosis and autophagic cell
death. Neuro Oncol. 15:1127–1141. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Thevenot PT, Sierra RA, Raber PL, Al-Khami
AA, Trillo-Tinoco J, Zarreii P, Ochoa AC, Cui Y, Del Valle L and
Rodriguez PC: The stress-response sensor chop regulates the
function and accumulation of myeloid-derived suppressor cells in
tumors. Immunity. 41:389–401. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ahmed KA, Wang L, Griebel P, Mousseau DD
and Xiang J: Differential expression of mannose-6-phosphate
receptor regulates T cell contraction. J Leukoc Biol. 98:313–318.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Luo S, Jing L, Zhao T, Li Y, Liu Z and
Diao A: Ubiquitination and dynactin regulate TMEPAI lysosomal
trafficking. Sci Rep. 7:426682017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Somasundaram R and Herlyn D: Chemokines
and the microenvironment in neuroectodermal tumor-host interaction.
Semin Cancer Biol. 19:92–96. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Barbieri F, Pattarozzi A, Gatti M, Aiello
C, Quintero A, Lunardi G, Bajetto A, Ferrari A, Culler MD and
Florio T: Differential efficacy of SSTR1, −2, and −5 agonists in
the inhibition of C6 glioma growth in nude mice. Am J Physiol
Endocrinol Metab. 297:E1078–E1088. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Barbieri F, Pattarozzi A, Gatti M, Porcile
C, Bajetto A, Ferrari A, Culler MD and Florio T: Somatostatin
receptors 1, 2, and 5 cooperate in the somatostatin inhibition of
C6 glioma cell proliferation in vitro via a phosphotyrosine
phosphatase-eta-dependent inhibition of extracellularly regulated
kinase-1/2. Endocrinology. 149:4736–4746. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Venkatesh HS, Johung TB, Caretti V, Noll
A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS,
et al: Neuronal Activity Promotes Glioma Growth through
Neuroligin-3 Secretion. Cell. 161:803–816. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lyu T, Jia N, Wang J, Yan X, Yu Y, Lu Z,
Bast RC Jr, Hua K and Feng W: Expression and epigenetic regulation
of angiogenesis-related factors during dormancy and recurrent
growth of ovarian carcinoma. Epigenetics. 8:1330–1346. 2013.
View Article : Google Scholar : PubMed/NCBI
|