Introduction
Colorectal cancer (CRC) is the third most common
cancer worldwidewith about 1.4 million new cases diagnosed in 2012
(1). It is predicted that by 2035
the number of diagnosed CRC cases will reach 2.4 million annually
worldwide (1). Therefore,
developing novel chemotherapeutic agents for the treatment of CRC
is an urgent need. Mammalian target of rapamycin (mTOR) is a
serine/threonine protein kinase which is a member of the
PI3K-related kinase (PIKK) family. In mammalian cells mTOR is
composed of two large distinct multi-protein complexes, mTORC1 and
mTORC2. mTORC1 consists of mTOR and five accessory proteins whereas
mTORC2 consists of mTOR and six accessory proteins. Both complexes
share mTOR as the catalytic subunit of the complex and share mLST8,
DEPTOR and Tti1/Tel2 in the complex (2). The unique components of mTORC1 are
RAPTOR and PRAS40 whereas RICTOR, mSIN1 and PROTOR are specific to
mTORC2 (3). mTORC1 promotes protein
synthesis and lipid biogenesis which favours cell growth and
proliferation (3,4). mTORC1 plays a central role in
tumourigenesis via controlling the mRNA translation and elongation
by phosphorylation of its two downstream targets, eukaryotic
initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and the p70
ribosomal S6 kinase 1 (S6K1) (2).
The resulting activation of S6K1 results in the phosphorylation and
inhibition of insulin receptor substrate 1 (IRS1), an upstream
regulator of phosphoinositide 3-kinase (PI3K) and AKT kinases.
4E-BP1 regulates the function of eukaryotic translation initiation
factor 4E (eIF4E) and when it is phosphorylated by mTORC1 it
results in the separation of 4E (eIF4E) from 4E-BP1 and initiates
translation by recruiting the translation initiation factor eIF4G
to the 5′ end of mRNAs (3). mTORC2
plays a part in tumourigenesis by phosphorylating AKT on the Ser473
residue (5). AKT promotes
proliferation, apoptosis and survival through the phosphorylation
of several effectors within the mTOR pathway.
Inhibiting mTORC1 and mTORC2 have become attractive
therapeutic strategies for cancer including CRC due to their
central roles in carcinogenesis. The first-generation mTOR
inhibitor rapamycin and its analogues (rapalogs) demonstrated
limited efficacy in cancer treatment. These compounds partially
suppressed the mTORC1 activity due to incomplete phosphorylation of
4E-BP1 (6). Secondly, they enhanced
PI3K/Akt signalling activities via the negative feedback loop that
is suppressed upon mTORC1 inhibition resulting in the promotion of
cancer cell survival (7). Recently,
second generation mTOR inhibitors have emerged including
ATP-competitive mTOR kinase and dual mTOR/PI3K inhibitors. They
have been used in clinical trials as anticancer agents (8). ATP-competitive mTOR kinase inhibitors
(e.g. PP424) target the kinase domain of mTOR and inhibit the
kinase activity of both mTORC1 and mTORC2 complexes, there by
suppressing the feedback activation of Akt (9). Dual mTOR/PI3K inhibitors (e.g.
NVP-BEZ235) inhibit mTORC1, mTORC2 and PI3K by targeting
ATP-binding sites of mTOR complexes and the p110α, β, and γ
isoforms of PI3K leading to complete blockage of the PI3K/Akt
pathway (10).
A growing body of evidence indicates that mTORC1 and
mTORC2 are overexpressed in human CRCs and the inhibition of these
components inhibits cell proliferation and induces apoptosis in
in vitro and in vivo models (11,12).
The aim of thepresent study was to compare the antitumor activity
of ATP competitive mTOR kinase inhibitor PP424 with dual PI3K/mTOR
inhibitor NVP-BEZ235 in human CRC cell lines. Our findings provided
evidence supporting the notion that targeting mTOR pathway with
PP424 and NVP-BEZ235 could be useful as a potential therapeutic
strategy for anticancer therapy in CRC.
Materials and methods
Cell culture and reagents
NVP-BEZ235 and PP242 were purchased from ChemdeaLLC
(Ridgewood, NJ, USA) and used at a concentration of 1,000 nM in all
experiments. Rapamycin (RAD001) was obtained from LC Laboratories
(Woburn, MA, USA) and used at a concentration of 1,000 nM. Each
inhibitor was dissolved in dimethyl sulfoxide (DMSO) and stored at
−20°C until use. All drugs were diluted in culture medium
immediately before use. The human CRC cell lines HT29, HCT116,
SW620 and SW480 were purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA). CSC480 cell line was
purchased from BioMedicure (San Diego, CA, USA) (13). All cell lines were cultured in
Dulbecco's modified Eagle's medium (DMEM), 4.5 g/l D-glucose,
L-glutamine and 110 mg/l sodium pyruvate (all from Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10%
fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific,
Inc.) at 37°C in a humidified atmosphere consisting of 5%
CO2 and 95% air. Cells were starved 24 h in serum-free
medium and stimulated with insulin (200 nM) for 10 min. All primary
antibodies against, pan 4E-BP1 (1:1,000; cat. no. 9644), pho-4E-BP1
(Thr37/46) (1:1,000; cat. no. 9459), pan AKT (1:1,000; cat. no.
4691) and pho-AKT (Ser473) (1:2,000; cat. no. 4060) were purchased
and validated by the manufacturer (Cell Signalling Technology,
Inc., Danvers, MA, USA).
Cell viability analysis
Determination of cell viability was performed by the
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT)
assay under both normoxic and hypoxic conditions. CRC cells were
seeded into 96-well plates in triplicate at a density of
1×104 cells/well. After 24 h, the cells were exposed to
different concentrations of RAD001, PP242 and NVP-BEZ235
(0.1–10,000 nmol/l) in 100 µl of medium. After incubation for the
indicated time-points, the MTT reagent (Life Technologies; Thermo
Fisher Scientific, Inc.) was added to each well, following the
manufacturer's instructions and incubated (at 37°C with 5%
CO2) for another 2 h and the medium was removed. For the
hypoxic treatment, the cells were incubated in an anaerobic chamber
(Forma Anaerobic System; Thermo Fisher Scientific, Inc.). DMSO (100
µl) was added to each well to dissolve the dark purple crystal
formed, and the plate was shaken gently for 5 min at room
temperature. Subsequently, absorbance was assessed at 540 nm using
an absorbance plate reader (POLARstar Omega reader; BMG Labtech,
Cary, NC, USA). For examining the effect of removing NVP-BEZ235,
after three days the cells were cultured with drug-free DMEM for
another three days.
Cell proliferation assay
The cells were seeded in 100-mm plates at
1×106 cells in 10 ml of media. After 24 h, cells were
treated with RAD001, PP242 and NVP-BEZ235 (1 µM). After incubation
at 37°C with the drugs for 1, 2, 3, 4 or 5 days, the cells were
extensively rinsed in phosphate-buffered saline (PBS) to remove any
loosely attached or floating cells. The untreated control and
treated cells were harvested by trypsinization and cell numbers
were determined using a hemocytometer.
Mitochondrial membrane potential
assay
Mitochondrial membrane potential was determined by
staining cells with
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetra-ethylbenzimidazole
carbocyanide iodide (JC-1) fluorescence dye (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany). JC-1 fluorescence emanating from the
mitochondria was analysed by fluorescence microscopy (14). Cells were incubated with JC-1 (2.5
µmol/l) in 96-well plates at 37°C and 5% CO2 for 30 min.
Drugs (1 µmol/l) and valinomycin (1 µmol/l) were added 30 min
before JC-1. Red fluorescence (excitation 550 nm, emission 600 nm)
and green fluorescence intensity (excitation 485 nm and emission
535 nm) were detected using a POLARstar Omega microplate reader
(BMG Labtech).
Phospho-specific protein microarray
analysis
The mTORsignalling phospho-specific antibody array
was conducted (FullMoon BioSystems, Inc., Sunnyvale, CA, USA)
according to the mancufacturer's protocol. The array consisted of
138 phospho-specific antibodies against phosphorylated and
unphosphorylated proteins. All antibodies were related to proteins
involved in the mTOR pathway. Briefly, SW620 cells were
serum-starved for 24 h and treated with vehicle or NVP-BEZ235 (10
µM) for 2 h. Untreated cells served as controls. Cells were lysed
using protein extraction buffer provided by Full Moon BioSystems
Inc. Proteins were labelled using biotin dissolved in
N,N-dimethylformamide (DMF) and hybridised to the (76×25 mm)
microarray slides. Biotin-labelled proteins conjugated to
antibodies were detected using streptavidin-Cy3 (Full Moon
BioSystems). Slides were scanned using GenePix 4100 microarray
scanner (Molecular Devices, San Jose, CA, USA). Phosphorylated
protein expression was normalized to its corresponding total
protein. Fold change in intensities from six replicates between the
phosphorylation of SW620-treated and untreated cells was
calculated. Ratios of phosphorylated to unphosphorylated proteins
were calculated as follows: Average signal intensity of
phospho-site-specific antibody/average signal intensity of
site-specific antibody. The ratio change between samples was
calculated as follows: treated/control.
Protein extraction and immunoblot
analysis
HT29, HCT116, SW620, SW480 and CSC480 cells were
serum starved for 24 h. Subsequently, the cells were pre-treated
with drugs (1 µmol/l) or DMSO as a control for 2 h in the presence
of insulin (200 nmol/l). For protein lysates, cells were washed
extensively with ice-cold PBS. Cells were subsequently harvested in
RIPA lysis buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl,
1% Nonidet P-40, 0.5% sodium deoxycholate, 1% SDS, 5 mM EDTA, 1 mM
phenylmethylsulfonyl fluoride, 10 µg/ml leupeptine and 1 mM sodium
orthovanadate. RIPA lysis buffer was supplemented with Protease and
Phosphatase Inhibitor Cocktail (cat. no. 78440; Thermo Fisher
Scientific, Inc.). Lysates were used immediately or stored at −80°C
for later use. Protein concentrations were assessed using POLARstar
Omega (BMG Labtech). For immunoblotting, equal amounts of lysates
(20 µg) were diluted in reducing 4XSDS buffer, boiled and then
separated on 4–15% polyacrylamide gel (Mini-PROTEAN TGX; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Subsequently, the resolved
proteins were transferred onto polyvinylidene difluoride (PVDF)
membrane (Trans-Blot Turbo Transfer 0.2 µm PVDF; Bio-Rad
Laboratories). Membranes were blocked with Tris buffered saline
(TBS) in 1% BSA pH 8.0 (Sigma-Aldrich). Membranes were washed and
incubated with primary antibodies overnight at 4°C. After extensive
washing with TBS plus 0.1% Tween-20 (TBST), the membranes were
incubated with appropriate anti-rabbit IgG, HRP-linked secondary
antibody (1:1,000; cat. no. 7074) (Cell Signaling Technology,
Inc.). Following 1-h incubation, the membranes were washed with
TBST and the proteins of interest were detected by using Super
Signal West Pico substrate (Thermo Fisher Scientific, Inc.). Bands
from immunoreactive proteins were visualised by VersaDoc imaging
system (Bio-Rad Laboratories).
Statistical analysis
Curve fitting was conducted using GraphPad Prism
ver. 6 (GraphPad Software, Inc., La Jolla, CA, USA). Data are
presented as the mean of at least three replicates with standard
error of the mean. Student's t-test was used to analyse the data
and a significance level of P<0.05 was used to indicate
statistically significant differences. In the Student's t-test each
drug group was individually compared to the control group (no drug)
and the multiple group graphs represented significance in relation
to the no drug control.
Results
mTOR inhibitors suppress CRC cell
proliferation in vitro
We evaluated the effects of mTOR inhibitors on CRC
cells in vitro using the cell viability and proliferation
assays under both normoxic and hypoxic conditions. To explore the
effect of mTOR inhibitors on CRC cells, serial dilutions of RAD001,
PP242 and NVP-BEZ235 ranging from 0.1 to 10,000 nM were added to
different CRC cell lines, including HT29, HCT116, SW620 and SW480,
as well as CSC480 cells. For MTT assay, a comparison was made among
RAD001, PP242 and NVP-BEZ235 in normoxia and hypoxia.
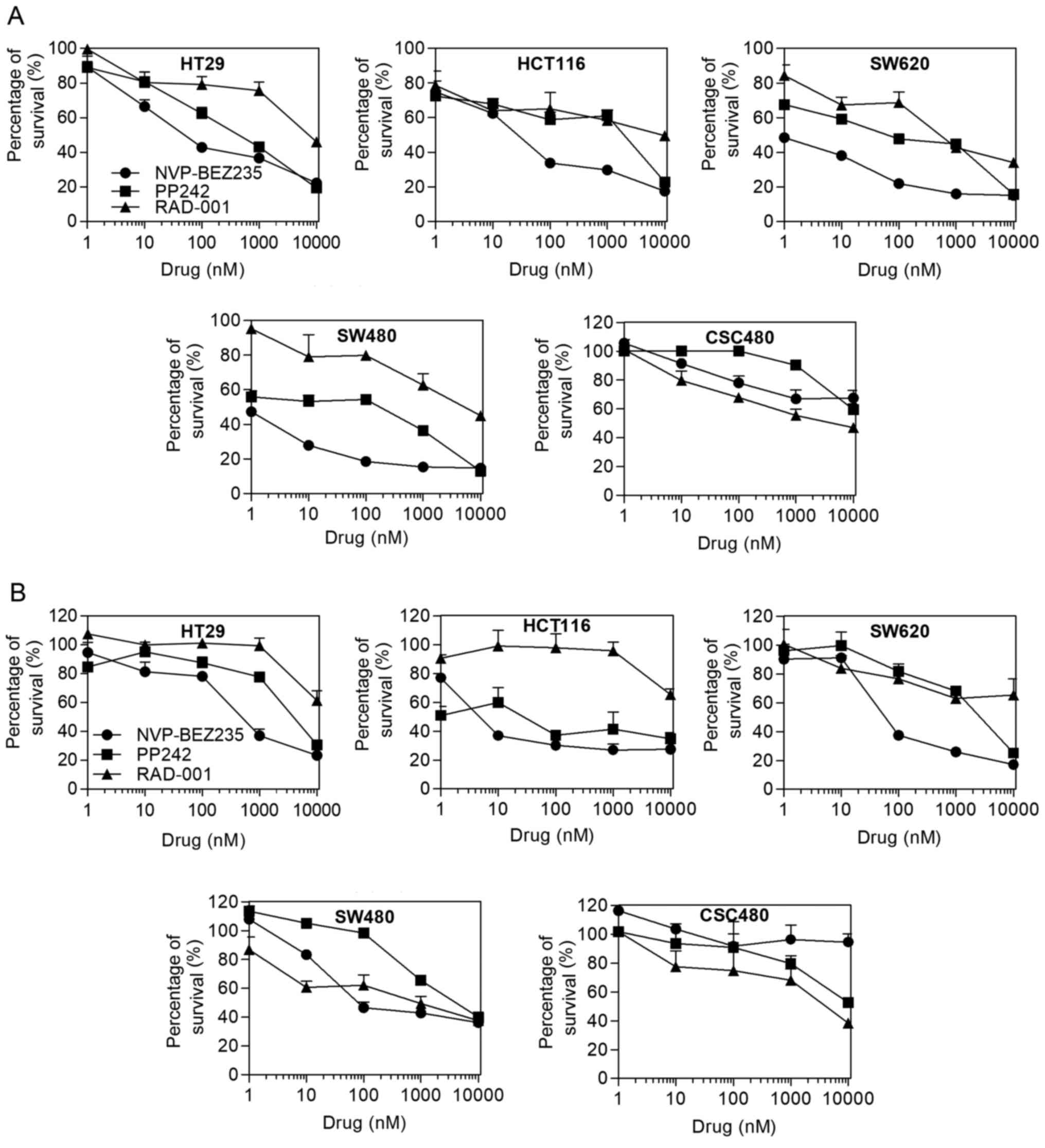
IC50 values were evaluated using the MTT assay. Table I displays the IC50 values
obtained after the cell lines were treated with the mTOR
inhibitors. In normoxia, a progressive decrease of the viable cell
numbers was observed on different CRC cell lines by treating the
cells with increasing concentrations of the drugs (Fig. 1A). The capacity of RAD001 and PP242
to reduce cell proliferation was limited, except for PP242 having
an increased inhibitory effect on SW620 cells (IC50, 62
nmol/l). Most cell lines displayed an IC50 for
NVP-BEZ235 ranging from 0.47 to 50 nmol/l, whereas the
IC50 for CSC480 cells was 282 nmol/l. The CRC cell lines
SW480 and SW620 had the highest sensitivity to NVP-BEZ235
(IC50, 0.47 and 0.5 nmol/l, respectively). In hypoxia,
the cell lines demonstrated more resistance to the drugs except for
HCT116 cells. PP242 (IC50, 4.4 nmol/l) and NVP-BEZ235
(IC50, 3.25 nmol/l) had significant effects on HCT116
cell viability (Fig. 1B). Notably,
in both normoxic and hypoxic conditions, RAD001 exhibited better
effect than NVP-BEZ235 in SW480 and CSC480.
 | Table I.IC50 values obtained after
the CRC cell lines were treated with mTOR inhibitors. |
Table I.
IC50 values obtained after
the CRC cell lines were treated with mTOR inhibitors.
|
| Normoxia | Hypoxia |
|---|
|
|
|
|
|---|
| Cell line | RAD001 | PP242 | NVP-BEZ235 | RAD001 | PP242 | NVP-BEZ235 |
|---|
| HT29 | 7731.60 | 445.13 | 50.14 | 10699.68 | 4598.27 | 480.36 |
| HCT116 | 8640.46 | 1966.92 | 27.58 | <> | 4.40 | 3.25 |
| SW620 | 523.39 | 62.24 | 0.50 | 11696.43 | 2711.80 | 58.35 |
| SW480 | 3484.61 | 169.11 | 0.47 | 860.41 | 3932.78 | 81.00 |
| CSC480 | 3983.25 | 6500.46 | 281.63 | 11769.11 | 9548.71 | <> |
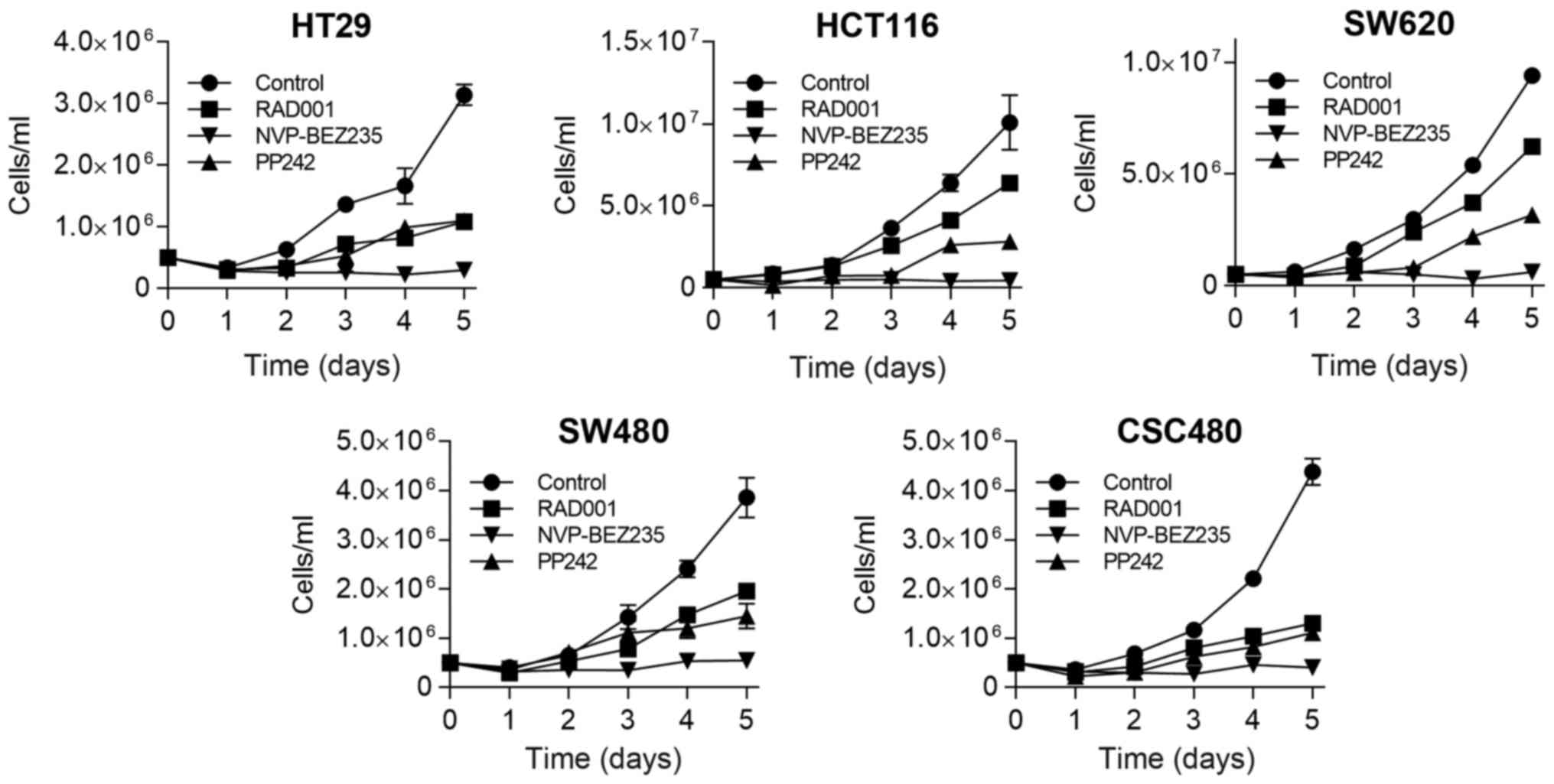
Subsequently, we evaluated the capacity of mTOR
inhibitors to reduce cell growth at different time-points by
proliferation assay. Using these cell lines, in vitro
growth-inhibitory effect of mTOR inhibitors was examined in 1 µM
concentration of the drugs. Although some cell lines displayed
sensitivity to RAD001 and PP242 treatment at days 2 and 3, the
cells started to proliferate again thereafter, resulting in a 2 to
3-fold increase in the number of cells (Fig. 2). In contrast, when all cell lines
were treated by NVP-BEZ235, it caused persistent inhibition of cell
growth, which remained at a low level thereafter.
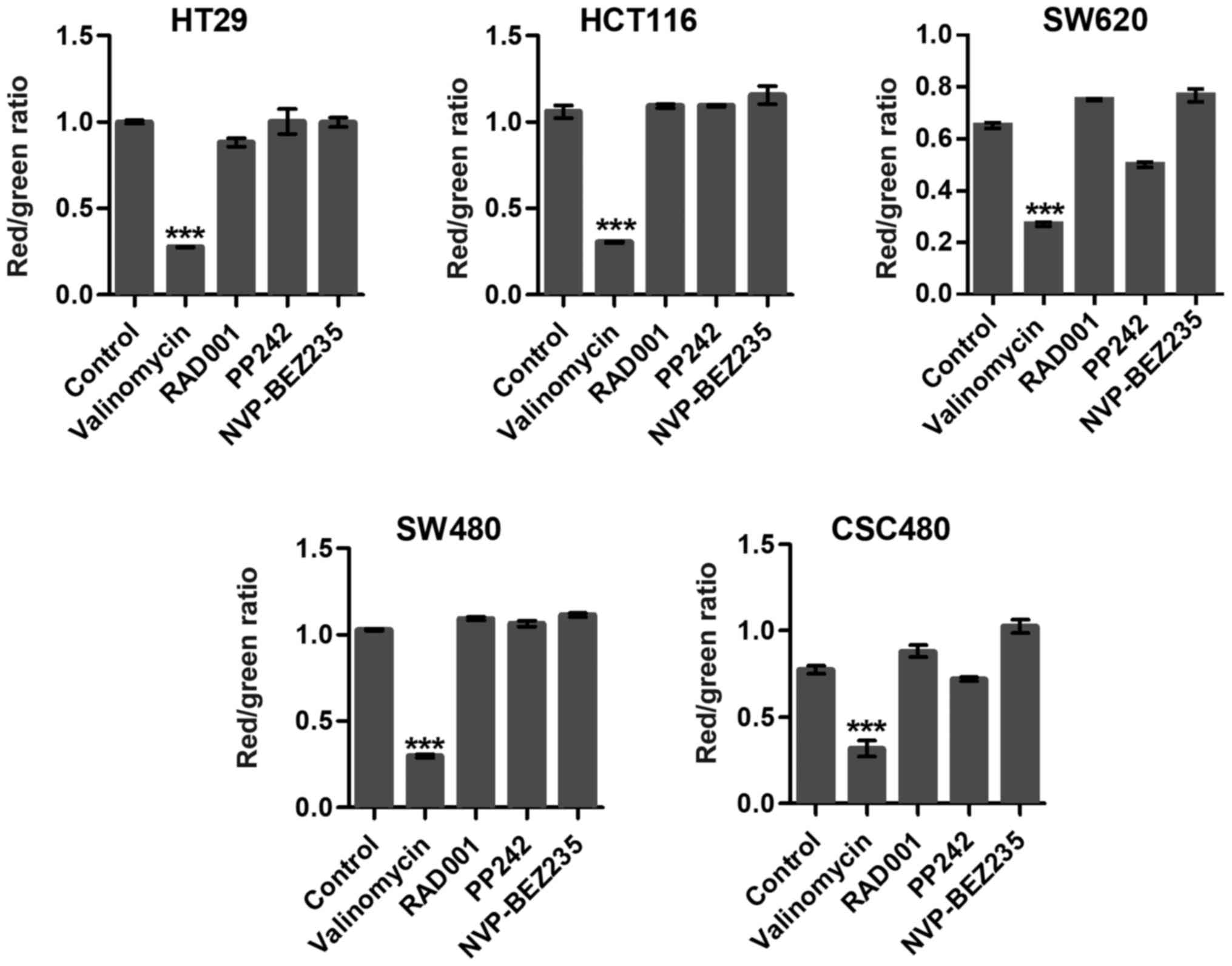
Mitochondrial membrane potentials
To examine whether targeting mTOR induced collapse
of the mitochondrial membrane potential (ΔΨm), we used JC-1 dye,
which concentrates in the mitochondrial matrix of healthy cells
forming red fluorescent aggregates at high-membrane potential. In
apoptotic cells, the dye is dispersed throughout the entire cell
leading to a shift from red to green fluorescence. RAD001 and PP242
slightly induced mitochondrial dysfunction and activated cell
apoptosis in SW620 cells (Fig. 3).
Notably, in the present study, there was no significant difference
among other mTOR inhibitor-treated groups compared to the control
groups (Fig. 3). Valinomycin was
used as a positive control which collapsed ΔΨm and indicated cell
apoptosis or necrosis.
mTOR phospho-antibody microarray
reveals effect of mTOR inhibitors
To achieve a comprehensive understanding of the
behaviour of key proteins within the mTOR pathway under inhibitory
conditions, SW620 cells were treated with the dual specificity
inhibitor NVP-BEZ235 and the phosphorylation status of the proteins
was analysed using anmTOR specific antibody phospho array. The
array contained 138 highly specific and well-characterized
phospho-specific antibodies for upstream and downstream proteins in
the mTOR pathway. To allow determination of the relative level of
phosphorylation, pairs of antibodies (phosphorylated and
unphosphorylated) for the same target sites were included in the
array. The antibody array data revealed that NVP-BEZ235 decreased
phosphorylation of AKT on Ser473 by 0.6-fold. In addition,
phosphorylation of AKT on the other site Thr308 decreased by
0.7-fold. Furthermore, the data demonstrateddifferential regulation
of many proteins related to the mTOR pathway (Table II). For example, phosphorylation of
mTOR at Ser2481 was decreased by about 0.4-fold. In response to
NVP-BEZ235 treatment, a negative regulator of mTORC1, TSC2 protein
tuberin decreased by 0.35-fold at Thr1462 and 3-fold at Ser939.
| Table II.Fold change in phosphorylated
proteins in response to drugs. |
Table II.
Fold change in phosphorylated
proteins in response to drugs.
|
|
| SW620 Non | SW620 NVP | SW620 NVP/SW620
Non |
|---|
|
|
|
|
|---|
| Phosphorylation
site | Swiss-Prot No. | Fold change |
|---|
| GSK3 beta
(Phospho-Ser9) | P49841 | 1.65 | 3.50 | 2.12 |
| PIP5K
(Phospho-Ser307) | Q9Y2I7 | 0.68 | 1.14 | 1.68 |
|
6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase
2 (PFKFB2) (Phospho-Ser483) | O60825 | 0.81 | 1.36 | 1.68 |
| PTEN
(Phospho-Ser370) | P60484 | 1.76 | 2.85 | 1.61 |
| P90RSK
(Phospho-Thr573) | Q15418 | 0.13 | 0.17 | 1.38 |
| eIF4G
(Phospho-Ser1108) | Q04637 | 0.46 | 0.60 | 1.31 |
| PTEN
(Phospho-Ser380/Thr382/Thr383) | P60484 | 2.75 | 3.57 | 1.30 |
| BAD
(Phospho-Ser136) | Q92934 | 1.82 | 2.33 | 1.28 |
| RSK1/2/3/4
(Phospho-Ser221/227/218/232) |
Q15418/P51812/Q15349/Q9UK32 | 1.38 | 1.69 | 1.22 |
| P70S6K
(Phospho-Thr229) | P23443 | 0.85 | 1.00 | 1.18 |
| mTOR
(Phospho-Ser2448) | P42345 | 1.02 | 1.17 | 1.16 |
| AKT1
(Phospho-Thr450) | P31749 | 0.87 | 0.95 | 1.09 |
| P90RSK
(Phospho-Thr359/Ser363) | Q15418 | 0.18 | 0.19 | 1.03 |
| P70S6K
(Phospho-Thr421) | P23443 | 1.46 | 1.48 | 1.01 |
| AKT1S1
(Phospho-Thr246) | Q96B36 | 0.30 | 0.30 | 1.00 |
| BAD
(Phospho-Ser91/128) | Q92934 | 5.67 | 5.12 | 0.90 |
| AMPKbeta1
(Phospho-Ser182) | Q9Y478 | 0.85 | 0.76 | 0.89 |
| 4E-BP1
(Phospho-Ser65) | Q13541 | 0.34 | 0.30 | 0.89 |
| PTEN
(Phospho-Ser380) | P60484 | 1.10 | 0.95 | 0.86 |
| PDK1
(Phospho-Ser241) | O15530 | 0.76 | 0.62 | 0.82 |
| AMPK1/AMPK2
(Phospho-Ser485/491) | Q13131/P54646 | 3.25 | 2.59 | 0.80 |
| BAD
(Phospho-Ser134) | Q92934 | 0.72 | 0.57 | 0.79 |
| PKC alpha/beta II
(Phospho-Thr638) | P17252 | 1.30 | 0.95 | 0.73 |
| P70S6k-beta
(Phospho-Ser423) | Q9UBS0 | 6.21 | 4.32 | 0.70 |
| PI3-kinase
p85-subunit alpha/gamma | P27986/ | 2.05 | 1.40 | 0.68 |
|
(Phospho-Tyr467/Tyr199) | Q92569 |
|
|
|
| AKT1
(Phospho-Ser246) | P31749 | 1.54 | 1.05 | 0.68 |
| AKT
(Phospho-Thr308) | P31749 | 2.04 | 1.37 | 0.67 |
| AKT1
(Phospho-Ser124) | P31749 | 0.83 | 0.53 | 0.64 |
| PPAR-b
(Phospho-Thr1457) | Q15648 | 1.52 | 0.96 | 0.63 |
| Mnk1
(Phospho-Thr385) | Q9BUB5 | 1.57 | 0.95 | 0.60 |
| AKT
(Phospho-Ser473) | P31749 | 2.11 | 1.27 | 0.60 |
| Rho/Rac guanine
nucleotide exchange factor 2 (Phospho-Ser885) | Q8TDA3 | 2.14 | 1.28 | 0.60 |
| ERK3
(Phospho-Ser189) | Q16659 | 3.00 | 1.77 | 0.59 |
| BAD
(Phospho-Ser112) | Q92934 | 2.31 | 1.33 | 0.58 |
| GSK3 alpha
(Phospho-Ser21) | P49840 | 2.79 | 1.56 | 0.56 |
| PKC alpha
(Phospho-Tyr657) | P17252 | 4.13 | 2.29 | 0.55 |
| GSK3a-b
(Phospho-Tyr216/279) | P49840 | 4.49 | 2.49 | 0.55 |
| AKT2
(Phospho-Ser474) | P31751 | 2.63 | 1.44 | 0.55 |
| mTOR
(Phospho-Thr2446) | P42345 | 4.08 | 2.21 | 0.54 |
| PPAR-r
(Phospho-Ser112) | P37231 | 0.16 | 0.08 | 0.51 |
| AKT1
(Phospho-Tyr474) | P31749 | 0.64 | 0.32 | 0.51 |
| eIF2 alpha
(Phospho-Ser51) | P05198 | 2.33 | 1.14 | 0.49 |
| PP2A-a
(Phospho-Tyr307) | P67775 | 0.39 | 0.18 | 0.47 |
| eIF4E
(Phospho-Ser209) | P06730 | 2.10 | 0.96 | 0.46 |
| AKT1
(Phospho-Thr72) | P31749 | 0.34 | 0.15 | 0.45 |
| P70S6K
(Phospho-Ser418) | P23443 | 0.95 | 0.42 | 0.44 |
| P70S6K
(Phospho-Ser411) | P23443 | 2.90 | 1.18 | 0.41 |
| BAD
(Phospho-Ser155) | Q92934 | 3.39 | 1.34 | 0.39 |
| AKT
(Phospho-Tyr326) | P31749 | 1.73 | 0.64 | 0.37 |
| P70S6K
(Phospho-Ser371) | P23443 | 1.68 | 0.61 | 0.37 |
| mTOR
(Phospho-Ser2481) | P42345 | 2.17 | 0.80 | 0.37 |
| 4E-BP1
(Phospho-Thr70) | Q13541 | 2.55 | 0.91 | 0.36 |
| Tuberin/TSC2
(Phospho-Thr1462) | P49815 | 0.50 | 0.17 | 0.35 |
| AMPK1
(Phospho-Thr174) | Q13131 | 2.57 | 0.88 | 0.34 |
| P90RSK
(Phospho-Ser380) | Q15418 | 0.94 | 0.32 | 0.34 |
| ERK1-p44/42 MAP
Kinase (Phospho-Tyr204) | P27361/P28482 | 5.45 | 1.84 | 0.34 |
| ERK1-p44/42 MAP
Kinase (Phospho-Thr202) | P27361/P28482 | 5.98 | 1.92 | 0.32 |
| Tuberin/TSC2
(Phospho-Ser939) | P49815 | 0.64 | 0.18 | 0.28 |
| P70S6K
(Phospho-Ser424) | P23443 | 5.96 | 1.36 | 0.23 |
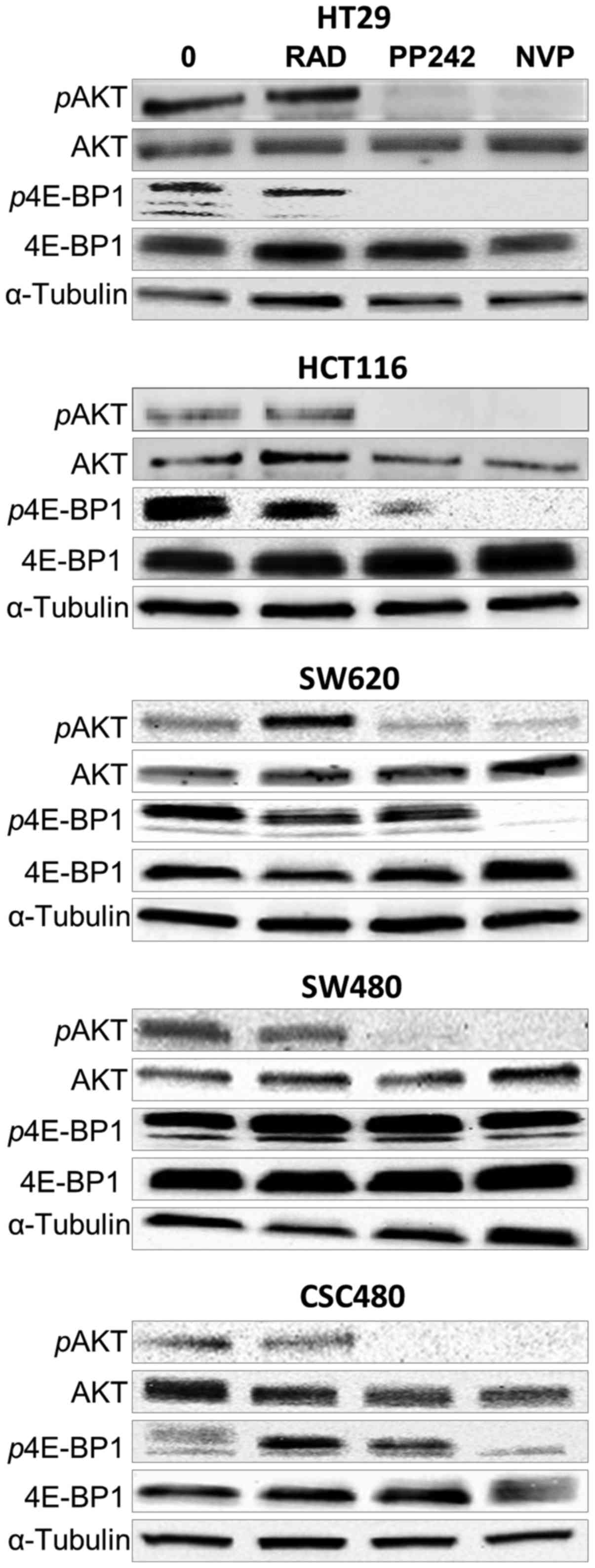
PP242 and NVP-BEZ235 treatment of
human colorectal cell lines results in sustained mTORC1 and mTORC2
inhibition
To examine the effects of RAD001, PP242 and
NVP-BEZ235 treatment on mTORC1 (p4E-BP1 T37/46) and mTORC2 (pAKT
S473) mTOR signalling, western blot analysis was performed. After
2-h treatment of HT29, HCT116, SW620, SW480 and CSC480 cells with
all agents at concentrations of 1 µmol/l (Fig. 4), the expression of
non-phosphorylated AKT and 4E-BP1 (functionally inactive) was
unchanged in all cell lines. However, the expression of
phosphorylated AKT (pAKT, functionally active) and phosphorylated
p4E-BP1 (p4E-BP1, functionally active) (Fig. 4) was differentiated in response to
different drugs. Concerning pAKT S473, PP242 and NVP-BEZ235 were
able to inhibit phosphorylation of S473 in all cell lines (Fig. 4). In the same cell lines, the
phosphorylation status of pAKT S473 was not affected by RAD001. In
contrast, the expression of p4E-BP1 T37/46 was not affected by
PP242 in three cell lines (SW620, SW480 and CSC480). However, the
expression decreased in HCT116 cells and showed full inhibition in
HT29 cells in response to PP242 treatment. NVP-BEZ235 inhibited
phosphorylation of T37/46 in HCT116, HT29, SW620 and CSC480.
Notably, NVP-BEZ235 had no effect on the expression of p4E-BP1
(T37/46) in SW480 cells. Collectivelly, these data indicated that
treatment by PP242 and NVP-BEZ235 resulted in sustained inhibition
of mTORC1 and mTORC2 signalling in vitro.
Discussion
Rapamycin and its analogues are inhibitors for the
mTOR signalling pathway. Previous research revealed that they have
antitumour activities through binding to its intracellular receptor
FKBP12 forming a rapamycin-FKBP12 complex which binds directly to
mTORC1 and indirectly to mTORC2 inhibiting their activities
(15). Other studies also indicated
that inhibiting the S6K1-IRS1 feedback loop by rapamycin leads
mTORC1 to hyper-activate the PI3K-Akt pathway (16). However, treatment of cancer with
rapamycin has limited efficacy in the mTOR pathway (17). Recent advances in the development of
next generation mTOR inhibitors PP242 (ATP-competitive mTOR
inhibitor) and NVP-BEZ235 (dual PI3K-mTOR inhibitor) have the
potential to overcome the development of drug resistance due to
targeting of the mTOR pathway at multiple levels (18). PP242 and NVP-BEZ235 target both mTOR
complexes and upstream activators and downstream targets of mTOR,
providing promising approaches for cancer therapy (10,19).
In the present study, we used multiple human CRC
cell lines (HT29, HCT116, SW620, SW480 and CSC480) to evaluate the
interaction of mTORC1 and mTORC2 with their substrate targets
4E-BP1 and AKT, respectively. Furthermore, we investigated the role
of mTORC1 and mTORC2 in human CRC using the new generation of mTOR
inhibitors, PP242 and NVP-BEZ235.
In cancer cells, mTOR signalling induces cell
proliferation in two ways, downstream of mTORC1 at the level of
4E-BP1 and by activating AKT by mTORC2 (5,20). The
present study demonstrated the antiproliferative effect of RAD001,
PP242 and NVP-BEZ235 on CRC cells using MTT and proliferation
assays. This data revealed that NVP-BEZ235 (1 µM) markedly reduced
the viability of all cell lines. Consistent with the
antiproliferative effects of NVP-BEZ235, current literature reveals
that NVP-BEZ235 inhibits cell proliferation in different cancer
cell lines (21,22). However, the present study revealed
that in SW480 and CSC480 cells, RAD001 had a slightly better
efficacy than PP242 and NVP-BEZ235. Furthermore, when the cells
were exposed to the different drugs under hypoxia, significant
resistance was observed in all cell lines except for HCT116 which
was significantly sensitive to PP242 and NVP-BEZ235. HCT116 is
wild-type for APC and has an heterozygous mutation in β-catenin
which acts in a dominant negative fashion increasing
β-catenin/TCF-4-mediated transcriptional activity (23). Newton et al (2010) observed
that downregulation of APC is important for survival under hypoxia
(24). APC is negative regulator of
β-catenin and mutations in APC are commonly associated with
increased β-catenin activity (23).
Recent evidence suggests crosstalk between hypoxia and the
upregulation of β-catenin activity leading to increased cell
proliferation migration and invasion (25). Therefore, the wild-type APC gene
could be contributing to the apparent sensitivity of HCT116 to
PP242 and NVP-BEZ235 through the suppression of β-catenin
activity.
To determine the effects of the mTOR inhibitors on
the induction of apoptosis via the mitochondrial pathway, the
mitochondrial potential of CRC cells was analysed in the presence
of the drugs. Induction of apoptosis by the mitochondria is due to
the release of cytochrome c in response to loss of
mitochondrial potential (26). Our
data revealed that the mTOR inhibitors did not demonstrate
significant disruption of the mitochondrial potential, with the
exception of PP242 having a slight effect on SW620 cells. The data
indicated that NVP-BEZ235 and RAD001 did not induce apoptosis via
the mitochondrial pathway. In agreement with our results, induction
of apoptosis in breast cancer cells by NVP-BEZ235 was found to be
independent of mitochondrial-induced caspases, but rather induced
via the extrinsic caspase-7 pathway (27,28).
Furthermore, NVP-BEZ235 was demonstrated not to induce the
activation of Bax, a key initiator of mitochondrial-induced
apoptosis in the human neuroblastoma cell line SH-EP (29).
To ascertain the effect of NVP-BEZ235 on the mTOR
pathway, we conducted a phospho-antibody microarray analysis of the
key proteins in the mTOR pathway using SW620 cells. Using an
mTOR-specific phospho array, changes in the expression levels of
phosphoproteins were identified as follows: Downstream target of
mTORC1, 4E-BP1 at both sites Thr37 and Thr46 and upstream effector
of mTORC2, AKT at site Ser473.
Comparing between 4E-BP1 and S6K as indicators of
the functional state of mTOR, a recent study revealed that the
phosphorylation status of 4E-BP1 is a better biomarker than S6K to
assess the efficacy ATP-competitive inhibitors (30). mTORC1 regulates protein synthesis
through the phosphorylation and inactivation of 4E-BP1 (15). When 4E-BP1 is not phosphorylated
(inactive), it binds tightly to eukaryotic translation initiation
factor 4E (eIF4E). However, when phosphorylated by mTORC1, 4E-BP1
is released from eIF4E resulting in inhibition of cap-dependent
translation and recruitment of the translation initiation factor
eIF4G and other cap-binding proteins (3). A previous study reported that RAD001
partially inhibited 4E-BP1 Thr37/46 phosphorylation (31). Our data revealed no effect of RAD001
on 4EBP1 Thr37/46 effector pathway downstream of mTORC1 in all
colorectal cell lines. Furthermore, when PP242 (1,000 nM) was used
to treat the cells it was found to have a weak inhibitory effect on
4E-BP1 Thr37/46. However, at the same dose NVP-BEZ235 effectively
inhibited 4E-BP1 phosphorylation (at Thr37/46). Conversely, we
found that in SW480 cells, mTORC1 inhibition by NVP-BEZ235 did not
affect the phosphorylation of 4E-BP1, leading us to suggest that
alternative mechanisms could also account for the increased
4E-BP1.
The mTORC2 complex phosphorylates protein kinase AKT
at a serine residue Ser473 (5). It
has been previously reported that NVP-BEZ235 did not inhibit p-AKT
in some cancer cell lines at low concentrations (1–10 nM) and that
the inhibitory effect lasted for 48 h at higher concentrations
(21). On the basis of this, we
chose to treat the cancer cells with mTOR inhibitors at the
concentrations of 1,000 nM for 2 h to conduct our experiments. We
observed that RAD001 increased AKT phosphorylation in SW620 cells.
This observation is consistent with previous studies (32,33).
In the present study however, PP242 and NVP-BEZ235 decreased p-AKT
levels in all cell lines. This is in agreement with previous
studies, in which both inhibitors were shown to decrease AKT
phosphorylation at high doses (21,34).
In conclusion, our preclinical findings demonstrated
that the new generation, ATP-competitive and dual mTOR/PI3K
inhibitors of mTOR, are potent in inhibiting CRC cells in
vitro. In respect to apoptosis, however, NVP-BEZ235 did not
affect the mitochondrial potential. Collectively, these results
demonstrated the potential of the new generation of dual mTOR
inhibitors in the treatment of CRC. However, further mechanistic,
in vivo studies and clinical studies are required to
ascertain their usefulness.
Acknowledgements
We would like to thank the other members of the Wei
Laboratory for their support and helpful comments.
Funding
The present study was supported by a PhD scholarship
awarded to Naif Alqurashi by Imam Abdulrahman Bin Faisal
University.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
NA conceived, designed and performed experiments.
SMH and FA performed experiments and contributed to the writing of
the manuscript. SMH, SI and MW reviewed and edited the manuscript.
All authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Ervik M,
Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and
Bray F: Cancer incidence and mortality worldwide: sources, methods
and major patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zoncu R, Efeyan A and Sabatini DM: mTOR:
From growth signal integration to cancer, diabetes and ageing. Nat
Rev Mol Cell Biol. 12:21–35. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Laplante M and Sabatini DM: mTOR signaling
at a glance. J Cell Sci. 122:3589–3594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Choo AY, Yoon SO, Kim SG, Roux PP and
Blenis J: Rapamycin differentially inhibits S6Ks and 4E-BP1 to
mediate cell-type-specific repression of mRNA translation. Proc
Natl Acad Sci USA. 105:17414–17419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guertin DA and Sabatini DM: The
pharmacology of mTOR inhibition. Sci Signal. 2:pe242009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang YJ, Duan Y and Zheng XF: Targeting
the mTOR kinase domain: The second generation of mTOR inhibitors.
Drug Discov Today. 16:325–331. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu K, Toral-Barza L, Shi C, Zhang WG,
Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, et al:
Biochemical, cellular, and in vivo activity of novel
ATP-competitive and selective inhibitors of the mammalian target of
rapamycin. Cancer Res. 69:6232–6240. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zaytseva YY, Valentino JD, Gulhati P and
Evers BM: mTOR inhibitors in cancer therapy. Cancer Lett. 319:1–7.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang YJ, Dai Q, Sun DF, Xiong H, Tian XQ,
Gao FH, Xu MH, Chen GQ, Han ZG and Fang JY: mTOR signaling pathway
is a target for the treatment of colorectal cancer. Ann Surg Oncol.
16:2617–2628. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gulhati P, Cai Q, Li J, Liu J, Rychahou
PG, Qiu S, Lee EY, Silva SR, Bowen KA, Gao T and Evers BM: Targeted
inhibition of mammalian target of rapamycin signaling inhibits
tumorigenesis of colorectal cancer. Clin Cancer Res. 15:7207–7216.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qian Y: Tumorigenic CancerStemCells,
Methods of Isolating and Using the Same. Google Patents,
US20110206735A1. 2011
|
|
14
|
Bedner E, Li X, Gorczyca W, Melamed MR and
Darzynkiewicz Z: Analysis of apoptosis by laser scanning cytometry.
Cytometry. 35:181–195. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma XM and Blenis J: Molecular mechanisms
of mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
O'Reilly KE, Rojo F, She QB, Solit D,
Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al:
mTOR inhibition induces upstream receptor tyrosine kinase signaling
and activates Akt. Cancer Res. 66:1500–1508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang S, Bjornsti MA and Houghton PJ:
Rapamycins: Mechanism of action and cellular resistance. Cancer
Biol Ther. 2:222–232. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gossage L and Eisen T: Targeting multiple
kinase pathways: A change in paradigm. Clin Cancer Res.
16:1973–1978. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alqurashi N, Hashimi SM and Wei MQ:
Chemical inhibitors and microRNAs (miRNA) targeting the mammalian
target of rapamycin (mTOR) pathway: Potential for novel anticancer
therapeutics. Int J Mol Sci. 14:3874–3900. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dowling RJ, Topisirovic I, Alain T,
Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj
A, Liu Y, et al: mTORC1-mediated cell proliferation, but not cell
growth, controlled by the 4E-BPs. Science. 328:1172–1176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Serra V, Markman B, Scaltriti M, Eichhorn
PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S,
et al: NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K
signaling and inhibits the growth of cancer cells with activating
PI3K mutations. Cancer Res. 68:8022–8030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roper J, Richardson MP, Wang WV, Richard
LG, Chen W, Coffee EM, Sinnamon MJ, Lee L, Chen PC, Bronson RT, et
al: The dual PI3K/mTOR inhibitor NVP-BEZ235 induces tumor
regression in a genetically engineered mouse model of PIK3CA
wild-type colorectal cancer. PLoS One. 6:e251322011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ilyas M, Tomlinson IP, Rowan A, Pignatelli
M and Bodmer WF: Beta-catenin mutations in cell lines established
from human colorectal cancers. Proc Natl Acad Sci USA.
94:10330–10334. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Newton IP, Kenneth NS, Appleton PL, Näthke
I and Rocha S: Adenomatous polyposis coli and hypoxia-inducible
factor-1{alpha} have an antagonistic connection. Mol Biol Cell.
21:3630–3638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu W, Zhou W, Cheng M, Wang J, Liu Z, He
S, Luo X, Huang W, Chen T, Yan W and Xiao J: Hypoxia activates
Wnt/β-catenin signaling by regulating the expression of BCL9 in
human hepatocellular carcinoma. Sci Rep. 7:404462017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Perry SW, Norman JP, Barbieri J, Brown EB
and Gelbard HA: Mitochondrial membrane potential probes and the
proton gradient: A practical usage guide. Biotechniques. 50:98–115.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brachmann SM, Hofmann I, Schnell C,
Fritsch C, Wee S, Lane H, Wang S, Garcia-Echeverria C and Maira SM:
Specific apoptosis induction by the dual PI3K/mTor inhibitor
NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells.
Proc Natl Acad Sci USA. 106:22299–22304. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cuda CM, Pope RM and Perlman H: The
inflammatory role of phagocyte apoptotic pathways in rheumatic
diseases. Nat Rev Rheumatol. 12:543–558. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Seitz C, Hugle M, Cristofanon S,
Tchoghandjian A and Fulda S: The dual PI3K/mTOR inhibitor
NVP-BEZ235 and chloroquine synergize to trigger apoptosis via
mitochondrial-lysosomal cross-talk. Int J Cancer. 132:2682–2693.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ducker GS, Atreya CE, Simko JP, Hom YK,
Matli MR, Benes CH, Hann B, Nakakura EK, Bergsland EK, Donner DB,
et al: Incomplete inhibition of phosphorylation of 4E-BP1 as a
mechanism of primary resistance to ATP-competitive mTOR inhibitors.
Oncogene. 33:1590–1600. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thoreen CC, Kang SA, Chang JW, Liu Q,
Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM and Gray NS: An
ATP-competitive mammalian target of rapamycin inhibitor reveals
rapamycin-resistant functions of mTORC1. J Biol Chem.
284:8023–8032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang X, Yue P, Kim YA, Fu H, Khuri FR and
Sun SY: Enhancing mammalian target of rapamycin (mTOR)-targeted
cancer therapy by preventing mTOR/raptor inhibition-initiated,
mTOR/rictor-independent Akt activation. Cancer Res. 68:7409–7418.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu CX, Li Y, Yue P, Owonikoko TK,
Ramalingam SS, Khuri FR and Sun SY: The combination of RAD001 and
NVP-BEZ235 exerts synergistic anticancer activity against non-small
cell lung cancer in vitro and in vivo. PLoS One. 6:e208992011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Blaser B, Waselle L, Dormond-Meuwly A,
Dufour M, Roulin D, Demartines N and Dormond O: Antitumor
activities of ATP-competitive inhibitors of mTOR in colon cancer
cells. BMC Cancer. 12:862012. View Article : Google Scholar : PubMed/NCBI
|