Introduction
Lung cancer is one of the most common and fatal
malignant tumor types worldwide (1). Non-small cell lung cancer (NSCLC)
accounts for 80–85% of all lung cancer cases (2). Since 2003, three generations of TKI
drugs have received FDA approval for the clinical treatment of
NSCLC (3). However, the increasing
rate of drug resistance in patients treated with molecular targeted
therapy is becoming a severe challenge for treatment. Moreover,
even patients who exhibit sensitivity to targeted drugs at the
start of treatment can become resistant within a few months
(4,5). The molecular mechanisms of drug
resistance, regarding both mutational and non-mutational pathways,
have only recently begun to be fully explored (6); however, at present, accumulating
evidence indicates that epithelial-mesenchymal transition (EMT)
plays an important role in the resistance to targeted therapy in a
non-mutational manner (7).
EMT is a process by which epithelial cells are
converted into mesenchymal cells. This involves a phenotypic switch
of cellular characteristics, whereby cells lose their adhesive
properties and cell-cell contacts while acquiring migratory
properties, which can promote tumor progression. The process of EMT
is mediated by a change in the expression of cell surface proteins
and the induction of cytoskeletal rearrangement, among other
phenotypic alterations (8,9). Diverse signaling molecules can trigger
EMT, including hepatocyte growth factor (HGF), epidermal growth
factor (EGF), fibroblast growth factor (FGF) (10,11).
In particular, transforming growth factor-β1 (TGF-β1) is considered
to be a key regulator in the EMT pathway (12). TGF-β1 belongs to a family of related
polypeptide factors with shared structural motifs, which serve
vital roles in processes such as cell growth, differentiation and
oxidative stress (13).
TGF-β-mediated EMT has been identified in many different tumor
types, including lung cancer (14).
TGF-β promotes EMT-related gene transcription in a Smad3-dependent
manner through the β-integrin signal transduction pathway or the
mitogen-activated protein kinase (MAPK) signaling pathway, in order
to influence gene expression and ultimately transform epithelial
cells from an epithelial phenotype to a stromal cell phenotype,
thus promoting the occurrence of EMT (15–17).
Programmed death-1 (PD-1) is so named due to its
association with apoptosis, and is mainly expressed in CD4+ and
CD8+ T lymphocytes, B lymphocytes, NK (natural killer) cells and
regulatory T cells (Tregs) (18,19).
The two ligands of PD-1 are PD-L1 (also known as CD274 or B7-H1)
and PD-L2 (also known as CD273 or B7-DC) (19). Cytokines, such as IL-4 and IL-10,
activate the PD-1/PD-L1 pathway, which affects T cells with high
expression of PD-1 molecules on their surface or tumor cells with
high surface-expression of PD-L1 (20,21).
As a result, T cell function is inhibited and the immune system is
suppressed in its recognition and attack of tumor cells. Recent
studies have indicated that the EMT in tumor cells is associated
with immune escape, which results in insensitivity to targeted
drugs (22,23)
In this study, we first speculated on the effect of
the EMT process induced by TGF-β1 and FGF2 on the expression of
PD-L1 in the NSCLC cell lines A549 (wild-type), H1650 (E746_A750
deletion) and H1975 (harboring the mutation of T790 in EGFR). We
then investigated the molecular mechanisms underlying the
regulation of PD-L1 expression during the EMT process, by treating
the three NSCLC cell lines with the AKT pathway inhibitor Ly294002,
the ERK pathway inhibitor PD98059 and the TAK1 pathway inhibitor
5Z-7. Furthermore, we assessed the expression of PD-L1 in the NSCLC
cells following treatment with EGFR-TKI (gefitinib). Collectively,
these tests aimed to provide a theoretical basis for the
development of new methods to reverse drug resistance in NSCLC.
Materials and methods
Reagents
Antibodies against E-cadherin (cat. no. 14472),
N-cadherin (cat. no. 14215), vimentin (cat. no. 5741), mTOR (cat.
no. 2983), phospho-mTOR (Ser 2448) (cat. no. 5536), Akt (cat. no.
4685), phospho-Akt (Ser473) (cat. no. 4060), p44/42 MAPK (Erk1/2)
(cat. no. 4695), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (cat.
no. 4370), TAK1 (cat. no. 4505), p65 (cat. no. 8242), GADPH (cat.
no. 5174) and human Basic Fibroblast Growth Factor (hFGF
basic/FGF2) (cat. no. 8910) were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). PD-L1 (cat. no. 205921) was
purchased from Abcam (Cambridge, MA, USA). Gefitinib (cat. no.
S1025), LY294002 (cat. no. S1105) and PD98059 (cat. no. S1177) were
purchased from Selleck Chemicals (Houston, TX, USA) and stock
solution was prepared in dimethyl sulfoxide (DMSO) at 10 mM.
5Z-7-oxozeaenol (5Z-7) was obtained from Sigma-Aldrich (Rehovot,
Israel) and dissolved in DMSO. Recombinant human TGF-β1 was
purchased from PeproTech (cat. no. 100-21C; Rocky Hill, NJ, USA).
Rhodamine phalloidin and DAPI were obtained from Sigma-Aldrich.
PE-conjugated anti-Human PD-L1 (cat. no. 12-5983-41) was purchased
from eBioscience/Thermo Fisher Scientific (San Diego, CA, USA).
NE-PER™ Nuclear and Cytoplasmic Extraction Reagents were obtained
from Thermo Fisher Scientific™ (Rockford, IL, USA).
Cell lines
Non-small cell lung cancer cell lines A549,
NCI-H1975 and NCI-H1650 were purchased from the China
Infrastructure of Cell Line Resources (Beijing, China). The cells
were grown in McCoy's medium (Gibco; Invitrogen, Carlsbad, Calif)
or RPMI-1640 modified medium (HyClone; GE Healthcare Life Sciences,
Logan, UT, USA) supplemented with 10% fetal bovine serum (Gibco;
Invitrogen, Carlsbad, Calif), 100 U/ml penicillin and 100 µg/ml
streptomycin. The cells were maintained in humidified air
containing 5% CO2 at 37°C in a monolayer culture.
Wound healing assay
Cells (5×105) in 6-well plates were
allowed to grow until reaching 80% confluency. After starvation
overnight, wounds were scratched on the cell surface using a 100 µl
micropipette tip. The cells were treated with 10 ng/ml TGF-β1, 10
ng/ml FGF2 or 10 ng/ml TGF-β1 plus 10 ng/ml FGF2 for 48 h. The
changes of the wound area were captured by Nikon camera.
Immunofluorescence staining
Cells 5×105 were seeded on 6-well plates
coated with coverslips in McCoy's medium or RPMI-1640 medium.
Following starvation overnight in serum-free medium, the cells were
treated with 10 ng/ml TGF-β1 plus 10 ng/ml FGF2 for 48 h. The cells
were fixed for 20 min in paraformaldehyde and permeabilized in 0.2%
Triton X-100 for 5 min. Then, F-actin was stained with Rhodamine
phalloidin and the nuclei with DAPI. The images were captured using
a Leica SP5 spectral confocal microscope.
Western blot analysis
The cells in 6-well plates were washed with cold
phosphate-buffered saline (PBS) twice and lysed with 80 µl RIPA
solution containing 1 mM PMSF in each well. Lysates were incubated
on ice and vortexed every 5 min, in total 30 min. Then the
supernatant was collected after centrifugation (15,000 × g for 5
min at 4°C). The concentration of the total protein was assessed
using the BCA Protein Assay Kit (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China). Total protein (35 µg) was
subjected to 10–12% SDS-PAGE and blotted on PVDF membranes (EMD
Millipore, Bedford, MA, USA). Then the membranes were incubated
with primary antibodies mentioned above (1:1,000 dilution)
overnight at 4°C. The results were detected using the Odyssey
detection system (LI-COR Biosciences, Lincoln, NE, USA).
Flow cytometric analysis
Cells were plated in 6-well plates
(1–5×105) and treated with 0.05% trypsin. Then, the
cells were harvested by centrifugation (800 × g, 5 min). Flow
cytometric cells were stained with the PE-conjugated anti-human
PD-L1 clone M1H1. The cells were assessed using Accuri™ C6 (BD
Biosciences, Franklin Lakes, NJ, USA). The data was analyzed using
FlowJo software (TreeStar, Inc., Ashland, OR, USA) and the PD-L1
levels were determined by calculating the Median Fluorescence
Intensity (MFI).
Nuclear translocation analysis
Cells were harvested with trypsin-EDTA, and then
centrifuged at 500 × g for 5 min. The cells were then transferred
to a 1.5 ml microcentrifuge tube and pelleted by centrifugation at
500 × g for 2–3 min. A pipette was used to carefully remove and
discard the supernatant, leaving the cell pellet as dry as
possible. Ice-cold CER I was added to the cell pellet. The tube was
then vortexed vigorously on the highest setting for 15 sec to fully
suspend the cell pellet and subsequently the tube was incubated on
ice for 10 min. Ice-cold CER II was added to the tube and then the
tube was vortexed for 5 sec on the highest setting. Incubation
followed on ice for 1 min and then the tube was centrifuged for 5
min at a maximum speed (~16,000 × g). The supernatant was then
immediately transferred to a clean pre-chilled tube and vortexed on
the highest setting for 15 sec in ice-cold NER. Subsequently, the
sample was placed on ice and vortexing continued for 15 sec every
10 min, for a total of 40 min. The tube was then centrifuged at
maximum speed (~16,000 × g) in a microcentrifuge for 10 min.
Finally, the supernatant (nuclear extract) fraction was immediately
transferred to a clean pre-chilled tube and the extracts were
stored at −80°C until use.
Animals and tumor xenograft
experiments
As mentioned in our previous study (24), total 12 five- to six-week-old female
BALB/c nude mice were raised under specific pathogen-free
conditions. Approximately 2×106 H1975 cells were
injected subcutaneously into the right flank region of nude mice.
The tumor volume was assessed every three days. Mice bearing tumors
~50 mm3 in volume were randomized into six mice per
group (n=6). The two group were administrated every day for 21 days
by oral gavage as followed: (a) saline; (b) gefitinib. At harvest,
the mice were sacrificed and the tumor tissues were fixed in
formalin for immunohistochemistry.
Immunohistochemistry
A series of 3-µm sections were obtained from each
paraffin block. After heat immobilization, deparaffinization and
rehydration, the sections were treated with 10 mM sodium citrate
buffer at pH 6.0 in boiling temperature for antigen retrieval. Then
the tissue sections were incubated with the primary antibodies
PD-L1 (1:100) overnight at 4°C and incubated with the secondary
antibody biotin (cat. no. 926-32211; (LI-COR Biosciences) for 20
min at room temperature. Targeted proteins PD-L1 were visualized
using peroxidase substrate diaminobenzidine (DAB). Staining
intensities were estimated in five random fields per section by
three independent observers individually using microscope equipped
with camera.
Statistics
Data were computed using Student's t-test
(two-tailed) or one-way ANOVA analysis. All values were presented
as the mean ± SD. P<0.05 was considered to indicate a
statistically significant difference.
Results
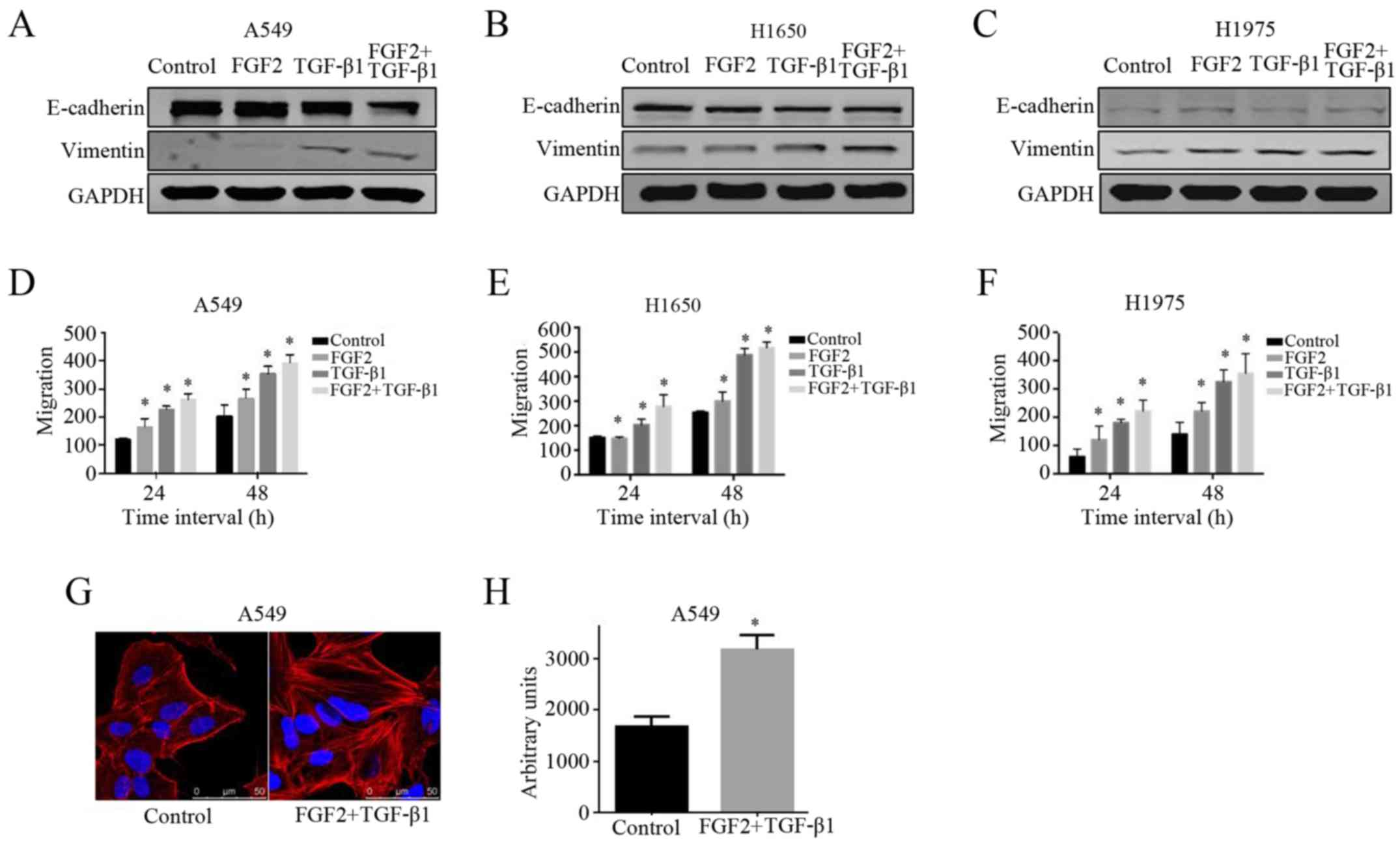
FGF2 plus TGF-β1 induced EMT in NSCLC
cell lines
In this study, we explored the effects of FGF2 plus
TGF-β1 on promoting the EMT process in three NSCLC cell lines: A549
(wild-type EGFR), H1650 (E746_A750 deletion) and H1975 (T790M). We
stimulated the cells with TGF-β1, FGF2 or FGF2 plus TGF-β1 for 48 h
in serum-free medium. The EMT phenotype was confirmed in the FGF2
plus TGF-β1 group when the expression of E-cadherin (an epithelial
marker) was decreased and the expression of vimentin and N-cadherin
(mesenchymal markers) were markedly increased compared with the
other groups (Fig. 1A-C).
Subsequently, cell migration capacity was assessed by a wound
healing assay. We observed that the migration ability of cells
treated with FGF2 plus TGF-β1 was enhanced in comparison with the
other groups at both 24 and 48 h (Fig.
1D-F). These results indicated that treatment with FGF2 plus
TGF-β1 downregulated E-cadherin, upregulated vimentin and
N-cadherin and increased migration ability in all three NSCLC cell
lines.
In addition to affecting cell migratory ability and
EMT-related protein expression, morphological changes further
confirmed the process of EMT. An increase in cell size mediated by
FGF2 plus TGF-β1 was one of the predominant characterizations of
EMT. As shown in Fig. 1G and H, the
A549 cells treated with FGF2 plus TGF-β1 for 48 h were markedly
enlarged compared with the control cells. Collectively, these
results indicated that FGF2 plus TGF-β1 treatment is an effective
way of promoting the induction of a physiological EMT
phenotype.
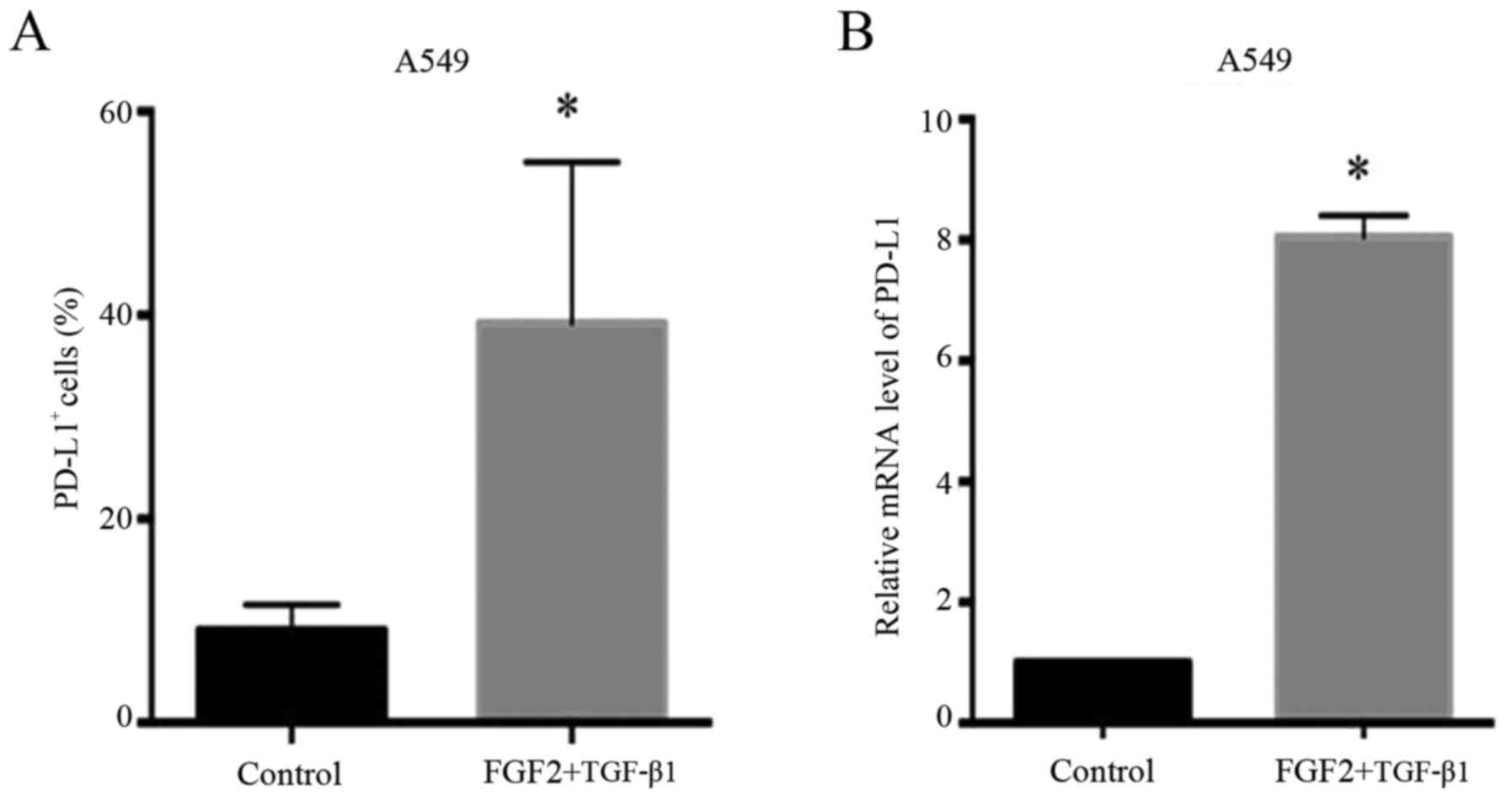
EMT promotes the expression of PD-L1
in the wild-type EGFR NSCLC cell line
The expression of PD-L1 in A549 cells was markedly
increased during the EMT process induced by FGF2 plus TGF-β1
treatment. As shown in Fig. 2A, the
PD-L1-positive staining on the surface of A549 cells was
significantly increased after treatment with FGF2 plus TGF-β1 as
determined by flow cytometric analysis (P=0.0029). Moreover, this
result was further confirmed by RT-PCR analysis. The mRNA level of
PD-L1 during the EMT process was higher than that in the control
cells (P=0.009) (Fig. 2B). These
results indicated that the EMT process markedly promoted the
expression of PD-L1 in A549 cells.
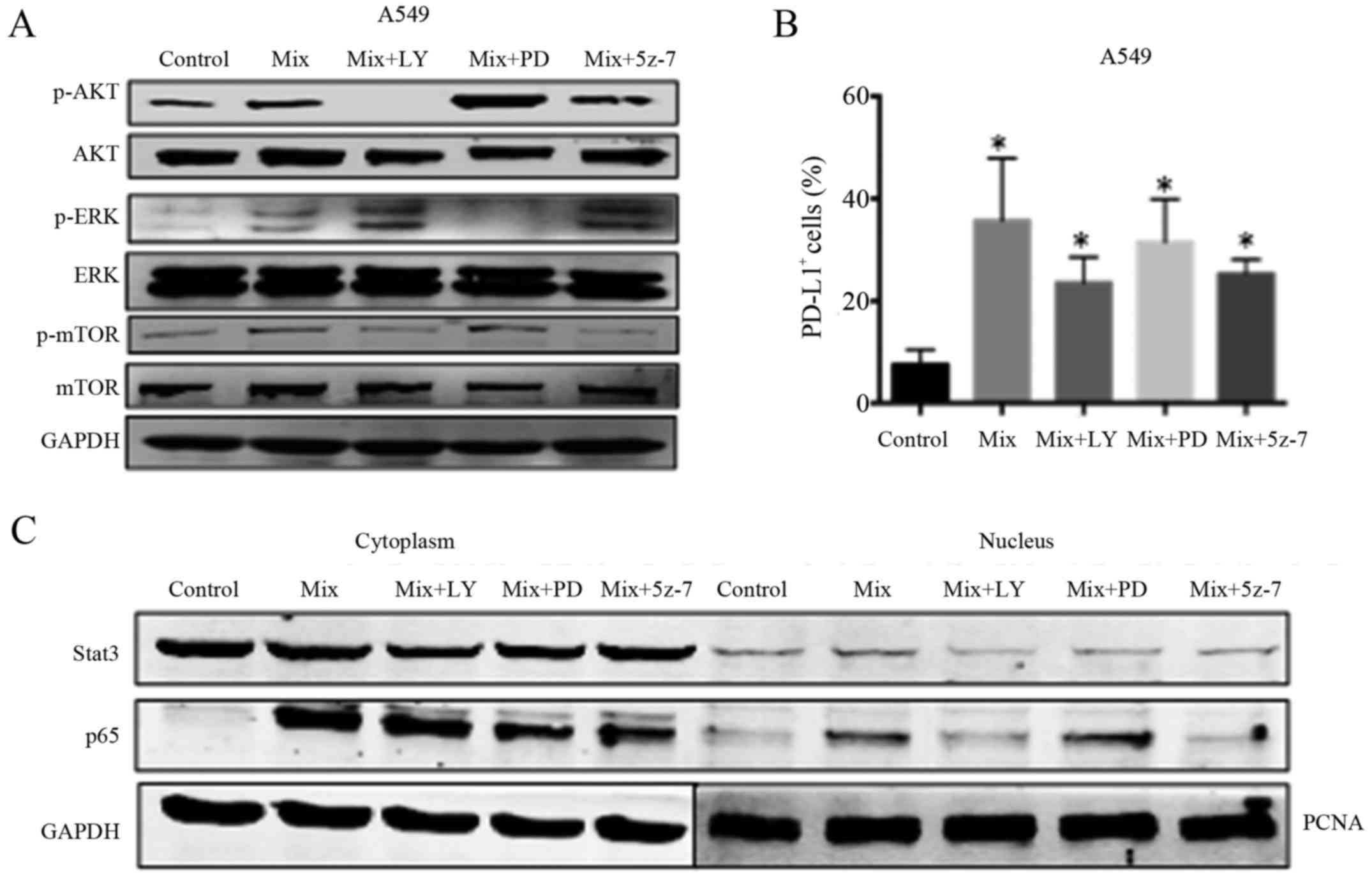
Related signaling pathways during the
EMT process in wild-type EGFR A549 cells
To further explore the molecular mechanisms of
EMT-induced PD-L1 expression, we assessed several EMT-related
signaling pathways. As shown in Fig.
3A, the cells were treated with the AKT inhibitor LY294002, the
ERK inhibitor PD98059 and the TAK1 inhibitor 5Z-7 after the
induction of EMT. Following treatment, we observed that the
expression of phosphorylated AKT, phosphorylated mTOR,
phosphorylated ERK was inhibited compared to FGF2 plus TGF-β1
treatment. Furthermore, the expression of PD-L1 was decreased
following treatment with the inhibitors (Fig. 3B). According to previous research,
phosphorylated proteins mediate downstream transcription factors in
their signaling pathways. Our study revealed that the inhibitors of
AKT, ERK and TAK1 reduced the expression of Stat3 in the nucleus to
a similar extent. Additionally, treatment with AKT inhibitor
(LY294002) and TAK1 inhibitor (5Z-7), but not ERK inhibitor
(PD98059), elicited decreased nuclear expression of p65 (Fig. 3C). The results confirmed our
hypothesis that inhibition of AKT, ERK and TAK1 phosphorylation
could inhibit the import of Stat3 transcription factor into the
nucleus, and thus inhibit the expression. Furthermore, our results
confirmed that inhibition of AKT and TAK1 phosphorylation could
further inhibit the nuclear import of the p65 subunit of NF-κB,
thus indicating its synergistic role in influencing the expression
of PD-L1.
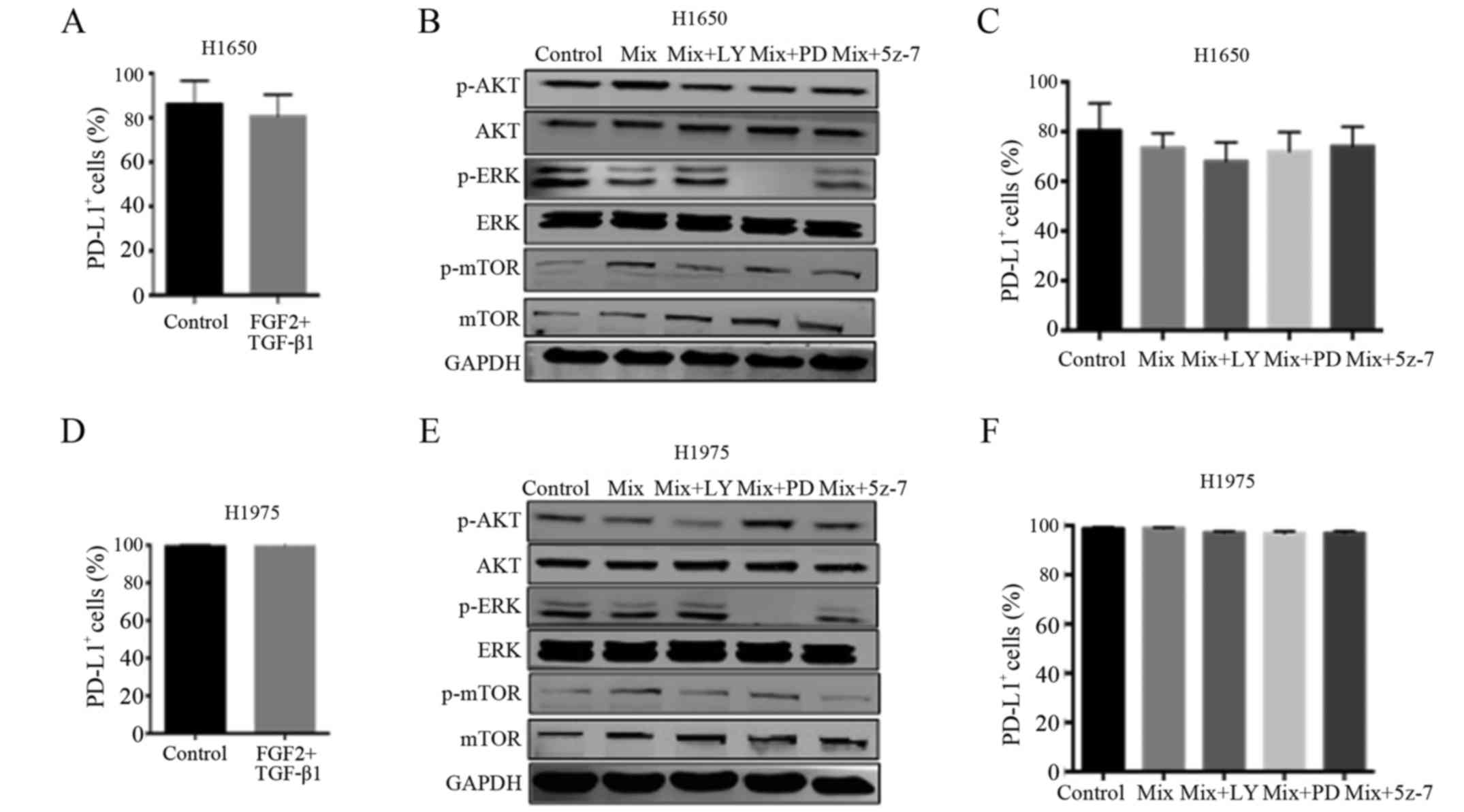
Related signaling pathways regulate
PD-L1 expression in H1650 cells while having no influence on H1975
cells during the EMT process
In this study, we further explored the regulatory
mechanism underlying PD-L1 expression in the H1650 cell line. As
shown in Fig. 4A, the expression of
PD-L1 during the EMT process underwent no significant change in the
H1650 cells. And the expression of relative proteins was reduced
after the treatment of inhibitors (Fig.
4B). In addition, the inhibitors of AKT (LY294002), ERK
(PD98059) and TAK1 (5Z-7) decreased the expression of PD-L1
(Fig. 4C).
In addition, we explored the influence of EMT on the
T790M EGFR mutant cell line H1975. As shown in Fig. 4D, the EMT process had no influence
on the expression of PD-L1 (P=0.3739). Even following the
inhibition of relative proteins (Fig.
4E), the expression of PD-L1 had no changes (Fig. 4F). These results revealed that the
signaling pathways of AKT, ERK and TAK1 served an important role in
promoting the expression of PD-L1 during the EMT process in H1650
cells, but not in H1975 cells.
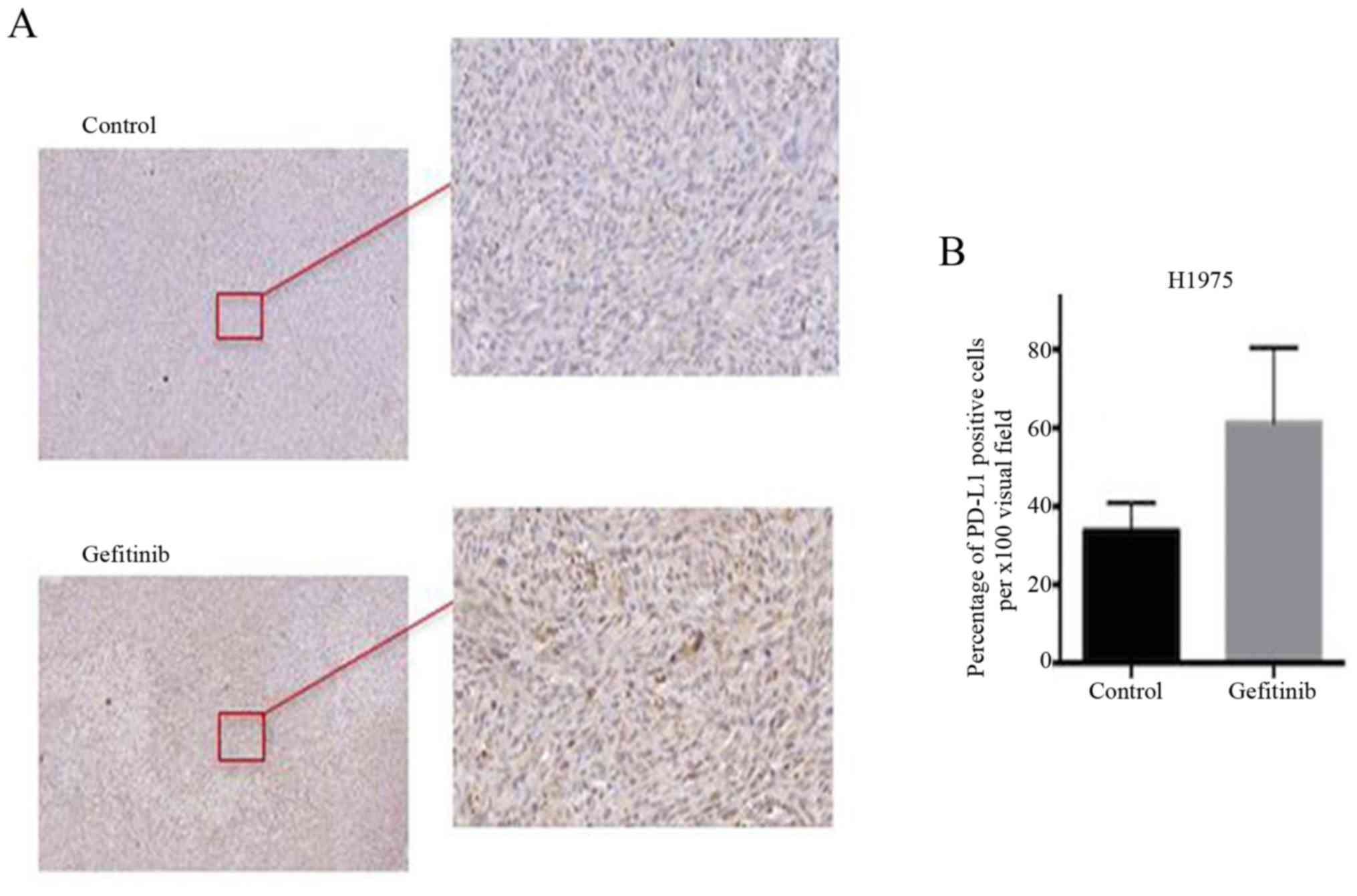
Gefitinib promotes the expression of
PD-L1 in H1975 xenograft nude mice
In the present study, we also assessed the
expression of PD-L1 in the tumor tissue of an H1975 xenograft nude
mouse model. As shown in Fig. 5,
the expression of PD-L1 in the gefitinib treatment group was
markedly higher compared with that in the control group. This
result revealed that gefitinib promoted the expression of PD-L1 in
the H1975 xenograft nude mice.
Discussion
Currently, researchers are becoming increasingly
aware that the occurrence and development of tumors are processes
involving mutual antagonism between the tumor and hosT cells. The
interaction between the tumor and host serves an important role in
tumor progression and metastasis. Furthermore, the host immune
system and the immune microenvironment are considered to be crucial
determinants of tumor progression and metastasis. Under normal
conditions, the immune system of the host body should be able to
recognize and kill the malignanT cells to remove or control the
growth of tumor tissue. However, the immune system of cancer
patients is typically inhibited. One of the most obvious
characteristics of this inhibition is the clustering of immune
suppressor cells around the tumor mass in its microenvironment.
PD-1/PD-L1 is one of the main pathways involved in tumor immune
evasion. PD-L1 mRNA is widely expressed in normal human tissues and
organs, while the PD-L1 protein is rarely expressed in normal human
tissues, being limited to embryonic tonsil tissue, macrophage-like
cells, lung cells and liver cells at a low level of expression
(25). This phenomenon reveals that
the expression of PD-L1 receives post-transcriptional regulation
under normal physiological conditions. However, PD-L1 is abundantly
expressed on the cell membranes of various tumor cells. Moreover,
PD-L1 is expressed in tumor lesions, but not in metastatic cells in
most tumors (25). These findings
indicate that PD-L1 is selectively expressed in tumor tissues, and
that its expression is closely related to the tumor
microenvironment.
Lung cancer is one of the most common and fatal
malignant tumor types worldwide. Non-small cell lung cancer (NSCLC)
accounts for 80–85% of all lung cancer cases (26). Since 2003, three generations of TKI
drugs have received FDA approval for the clinical treatment of
NSCLC (27). However, the
increasing rate of drug resistance in patients treated with
chemotherapy increasingly becomes a serious challenge; even
patients who are sensitive to chemotherapeutic drugs at the start
of treatment develop resistance within a few months (28). The molecular mechanisms of drug
resistance, including both mutational and non-mutational, have only
recently begun to be explored (29–32).
At present, accumulating evidence has indicated that the
epithelial-mesenchymal transition (EMT) plays an important role in
chemo-resistance in a non-mutational way. In the present study, we
selected three NSCLC cell lines, A549 (wild-type EGFR), NCI-H1975
(L858R and T790M mutation; EGFR-TKI-resistant) and NCI-H1650
(E746_A750 deletion mutation; EGFR-TKI-sensitive), and explored the
relationship between the expression of PD-L1 and the EMT
process.
Numerous primary studies have demonstrated that EMT
is strongly associated with cancer metastasis (33–34).
Cadherin switching is a main characteristic alteration in several
types of cancer, including NSCLC. The inactivation of the
epithelial marker E-cadherin is the pivotal event of EMT in tumor
progression (9). Furthermore,
activation of certain mesenchymal markers, such as vimentin and
N-cadherin, may play a positive role in the process of metastasis
since the normal cell-cell and cell-matrix contacts are disrupted
(35). The aforementioned changes
promote cell migration and contribute to loss of cell adhesion. A
recent study suggested a relationship between PD-L1 expression and
the EMT process in promoting the occurrence of immune evasion and
metastasis progression (22). In
this study, we created an effective way of promoting the induction
of a physiological-like EMT phenotype in A549 cells. Additionally,
our data elucidated the key role of EMT in triggering the
expression of PD-L1 in the A549 cell line. We further explored the
mechanisms of EMT in promoting the expression of PD-L1. The results
indicated that the AKT, ERK and TAK1 pathways play synergistic
roles in regulating the expression of PD-L1 during the EMT process,
by mediating nuclear import of the transcription factor Stat3 and
the p65 subunit of NF-κB in A549 cells. These regulatory mechanisms
may be applied to the EGFR-TKI sensitive cell line H1650, but not
the EGFR-TKI resistant cell line H1975. Furthermore, gefitinib, an
oral EGFR-TKI, increased the expression of PD-L1 in an H1975
xenograft tumor. Collectively, the aforementioned results revealed
that EMT could mediate PD-L1 expression via multiple pathways, and
thereby may serve an important role in immune evasion in the tumor
microenvironment. However, our research is in the primary stage and
further study is warranted to confirm the present conclusions.
In the clinical treatment of NSCLC, certain patients
exhibit primary drug resistance to EGFR-TKI without having gene
mutations. Considering the results of previous studies together
with our findings leads us to make the assumption that EMT plays an
important role in promoting the expression of PD-L1 in NSCLC cases
expressing either wild-type EGFR or activated EGFR mutant by
mediating the AKT-mTOR, ERK and TAK1-p65 pathways, but not in cases
with EGFR-TKI resistance. Therefore, EMT may play a crucial role in
primary drug resistance to EGFR-TKI in patients without mutation by
promoting immune evasion, while having little effect on NSCLC with
EGFR-TKI-resistant gene mutation (T790M). Although the process of
tumor metastasis is regulated by various key factors, such as the
circulatory system (26), the
phosphorylation of Smad2 and Smad3 (37), and diverse growth factors in the
tumor microenvironment (38), EMT
may be one of the key cross-linking components mediating all of the
necessary elements during the process of metastasis. Therefore,
inhibition of EMT induction could be applied as a novel therapeutic
method for the treatment of NSCLC harboring wild-type EGFR or
EGFR-TKI-sensitizing mutation.
Acknowledgements
Not applicable.
Funding
This present study was supported by a grant from
Peking University Third Hospital of China (no. BYSY2014011).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
LF and ZTJ conceived and designed the study. LF, YY
and XZ performed the experiments. LL and WJD reviewed and edited
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Institutional Review Board of the Department of Laboratory Animal
Science of Peking university Health Science Center (Beijing,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Siegel RL and Jemal A: Lung
cancer statistics. Adv Exp Med Biol. 893:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi L, Tang J, Tong L and Liu Z: Risk of
interstitial lung disease with gefitinib and erlotinib in advanced
non-small cell lung cancer: A systematic review and meta-analysis
of clinical trials. Lung Cancer. 83:231–239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li H, Schmid-Bindert G, Wang D, Zhao Y,
Yang X, Su B and Zhou C: Blocking the PI3K/AKT and MEK/ERK
signaling pathways can overcome gefitinib-resistance in non-small
cell lung cancer cell lines. Adv Med Sci. 56:275–284. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rolff J, Becker M, Merk J, Hoffmann J and
Fichtner I: Preclinical study of a combination of erlotinib and
bevacizumab in early stages of unselected non-small cell lung
cancer patient-derived xenografts. Target Oncol. 11:507–514. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin L and Bivona TG: Mechanisms of
resistance to epidermal growth factor receptor inhibitors and novel
therapeutic strategies to overcome resistance in NSCLC patients.
Chemother Res Pract. 2012:8172972012.PubMed/NCBI
|
|
7
|
Sato M, Shames DS and Hasegawa Y: Emerging
evidence of epithelial-to-mesenchymal transition in lung
carcinogenesis. Respirology. 17:1048–1059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Voena C, Varesio LM, Zhang L, Menotti M,
Poggio T, Panizza E, Wang Q, Minero VG, Fagoonee S, Compagno M, et
al: Oncogenic ALK regulates EMT in non-small cell lung carcinoma
through repression of the epithelial splicing regulatory protein 1.
Oncotarget. 7:33316–33330. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu F, Li J, Jang C, Wang J and Xiong J:
The role of Axl in drug resistance and epithelial-to-mesenchymal
transition of non-small cell lung carcinoma. Int J Clin Exp Pathol.
7:6653–6661. 2014.PubMed/NCBI
|
|
10
|
Jiao D, Wang J, Lu W, Tang X, Chen J, Mou
H and Chen QY: Curcumin inhibited HGF-induced EMT and angiogenesis
through regulating c-Met dependent PI3K/Akt/mTOR signaling pathways
in lung cancer. Mol Ther Oncolytics. 3:160182016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu ZC, Chen XH, Song HX, Wang HS, Zhang
G, Wang H, Chen DY, Fang R, Liu H, Cai SH and Du J: Snail regulated
by PKC/GSK-3β pathway is crucial for EGF-induced
epithelial-mesenchymal transition (EMT) of cancer cells. Cell
Tissue Res. 358:491–502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bi WR, Yang CQ and Shi Q: Transforming
growth factor-β1 induced epithelial-mesenchymal transition in
hepatic fibrosis. Hepatogastroenterology. 59:1960–1963.
2012.PubMed/NCBI
|
|
13
|
Kaminska B, Wesolowska A and Danilkiewicz
M: TGF beta signalling and its role in tumour pathogenesis. Acta
Biochim Pol. 52:329–337. 2005.PubMed/NCBI
|
|
14
|
Shintani Y, Okimura A, Sato K, Nakagiri T,
Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, et
al: Epithelial to mesenchymal transition is a determinant of
sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann
Thorac Surg. 92:1794–1804; discussion 1804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen XF, Zhang HJ, Wang HB, Zhu J, Zhou
WY, Zhang H, Zhao MC, Su JM, Gao W, Zhang L, et al: Transforming
growth factor-β1 induces epithelial-to-mesenchymal transition in
human lung cancer cells via PI3K/Akt and MEK/Erk1/2 signaling
pathways. Mol Biol Rep. 39:3549–3556. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang HJ, Wang HY, Zhang HT, Su JM, Zhu J,
Wang HB, Zhou WY, Zhang H, Zhao MC, Zhang L and Chen XF:
Transforming growth factor-β1 promotes lung adenocarcinoma invasion
and metastasis by epithelial-to-mesenchymal transition. Mol Cell
Biochem. 355:309–314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cervantes-Arias A, Pang LY and Argyle DJ:
Epithelial-mesenchymal transition as a fundamental mechanism
underlying the cancer phenotype. Vet Comp Oncol. 11:169–184. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Keir ME, Butte MJ, Freeman GJ and Sharpe
AH: PD-1 and its ligands in tolerance and immunity. Annu Rev
Immunol. 26:677–704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dai S, Jia R, Zhang X, Fang Q and Huang L:
The PD-1/PD-Ls pathway and autoimmune diseases. Cell Immunol.
290:72–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bachy E and Coiffier B: Anti-PD1 antibody:
A new approach to treatment of lymphomas. Lancet Oncol. 15:7–8.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gunturi A and McDermott DF: Potential of
new therapies like anti-PD1 in kidney cancer. Curr Treat Options
Oncol. 15:137–146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during Snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia
XF, Sun X, Li GG, Hu QD, Fu QH and Liang TB: Hypoxia-induced
epithelial-to-mesenchymal transition in hepatocellular carcinoma
induces an immunosuppressive tumor microenvironment to promote
metastasis. Cancer Res. 76:818–830. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li F, Zhu T, Cao B, Wang J and Liang L:
Apatinib enchance antitumor activity of EGFR-TKIs in non-small cell
lung cancer with EGFR-TKI resistance. Eur J Cancer. 84:184–192.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hellmann M, Rizvi N, Wolchok JD and Chan
TA: Genomic profile, smoking, and response to anti-PD-1 therapy in
non-small cell lung carcinoma. Mol Cell Oncol. 3:e10489292015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Riihimäki M, Hemminki A, Fallah M, Thomsen
H, Sundquist K, Sundquist J and Hemminki K: Metastatic sites and
survival in lung cancer. Lung Cancer. 86:78–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Singh D, Attri BK, Gill RK and Bariwal J:
Review on EGFR Inhibitors: Critical updates. Mini Rev Med Chem.
16:1134–1166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nurwidya F, Takahashi F, Murakami A,
Kobayashi I, Kato M, Shukuya T, Tajima K, Shimada N and Takahashi
K: Acquired resistance of non-small cell lung cancer to epidermal
growth factor receptor tyrosine kinase inhibitors. Respir Investig.
52:82–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Z, Lee JC, Lin L, Olivas V, Au V,
LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al:
Activation of the AXL kinase causes resistance to EGFR-targeted
therapy in lung cancer. Nat Genet. 44:852–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nurwidya F, Takahashi F, Murakami A and
Takahashi K: Epithelial mesenchymal transition in drug resistance
and metastasis of lung cancer. Cancer Res Treat. 44:151–156. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saijo N: Present status and problems on
molecular targeted therapy of cancer. Cancer Res Treat. 44:1–10.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesemchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Diepenbruck M and Christofori G:
Epithelial to mesemchymal transition (EMT) and metastais: Yes, no,
maybe? Curr Opin Cell Biol. 43:7–13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Strippoli R, Benedicto I, Perez Lozano ML,
Pellinen T, Sandoval P, Lopez-Cabrera M and del Pozo MA: Inhibition
of transforming growth factor-activated kinase 1 (TAK1) blocks and
reverses epithelial to mesenchymal transition of mesothelial cells.
PLoS One. 7:e314922012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu W, Kovacevic Z, Peng Z, Jin R, Wang P,
Yue F, Zheng M, Huang ML, Jansson PJ, Richardson V, et al: The
molecular effect of metastasis suppressors on Src signaling and
tumorigenesis: New therapeutic targets. Oncotarget. 6:35522–35541.
2015.PubMed/NCBI
|
|
37
|
Sequist LV, Bell DW, Lynch TJ and Haber
DA: Molecular predictors of response to epidermal growth factor
receptor antagonists in non-small-cell lung cancer. J Clin Oncol.
25:587–595. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Said NA and Williams ED: Growth factors in
induction of epithelial-mesenchymal transition and metastasis.
Cells Tissues Organs. 193:85–97. 2011. View Article : Google Scholar : PubMed/NCBI
|