Introduction
Erythropoietin-producing hepatocellular carcinoma
cell surface type-A receptor 3 (EPHA3) is a member of the Eph
subfamily of receptor tyrosine kinases, and is structurally
classified into cytoplasmic and extracellular regions. The
cytoplasmic region is composed of a regulatory juxtamembrane domain
with two tyrosine residues, a tyrosine kinase domain, a
sterile-α-motif interaction domain and a PSD95/Dlg/ZO-1 binding
motif. The extracellular region contains a cysteine-rich domain
comprised of a sushi and epidermal growth factor-like domain, two
fibronectin III repeats, and an N-terminal globular ligand-binding
domain (LBD) that can interact with cell membrane-bound ligands.
These ligands mainly include ephrinA3, ephrinA5, or ephrinB2, which
trigger bidirectional signaling of tyrosine kinase-dependent and
independent pathways involved in cell communication. EPHA3 is
highly expressed during the embryonic development of the brain and
spinal cord, lungs, kidneys and heart, and then drops to a low
level in most normal adult tissues (1). However, its expression is also
elevated in a wide range of malignancies, including gastric cancer
(2,3), melanoma (4–6),
hepatocellular carcinoma (7) and
glioblastoma (8), and is correlated
with tumorigenicity, tumor angiogenesis and cancer progression
(7–9). It was found that blocking the
activation of the EPHA3 receptor with soluble EPHA3-Fc inhibited
tumor growth in the 4T1 model of metastatic mammary adenocarcinoma
(10). The first-in-class
monoclonal antibody KB004 targeting the overexpressed receptor
tyrosine kinase EPHA3 was shown to be clinically active against
refractory hematological malignancies in humans (11). The conjugate cytotoxin of
ephrinA5-Fc and PE38QQR potently and specifically killed
glioblastoma tumor cells (12). The
EPHA3-specific monoclonal antibody IIIA4 was also found to have
antitumor effects in EPHA3-expressing leukemic xenografts (13). Accordingly, EPHA3 has been paid more
attention as one of the promising targets for the treatment of
several cancers (14,15). Conversely, there are also
contradictory reports concerning EphA3 expression in tumors and its
effect on the regulation of cancer progression. EPHA3 expression is
more commonly downregulated and does not play a major role in
colorectal cancer (16–18). EPHA3 is not highly expressed in the
primary tumor but in lymph node metastases, and its expression also
varies with disease stage in breast cancer (19). EPHA3 was found to suppress cell
adhesion and migration when EPHA3 phosphorylation was increased by
ephrinA5 stimulation in EPHA3-expressing TE671 and RD
rhabdomyosarcoma (RMS) cells, or when EPHA3 was ectopically
expressed in the EPHA3-negative CRL2061 RMS cell line (20), but EPHA3 and ephrinA1 upregulated by
the transcription factor PAX7 in RMS cell lines was associated with
cell migration and invasiveness (21). In addition, EPHA3 overexpression in
human lung cancer cell lines increased apoptosis by suppressing AKT
activation in vitro and inhibited the growth of tumor
xenografts (22). It also decreased
chemoresistance of small cell lung cancer via the PI3K/BMX/STAT3
signaling pathway (23).
Furthermore, the EphA3 level was found to be lower in primary lung
adenocarcinomas than in normal clinical specimens (22). Given the above-mentioned findings,
the role of EPHA3 either in promoting or suppressing oncogenesis is
quite complex and paradoxical in a variety of cancers.
Noteworthy, production of higher amounts of EPHA3 is
reported in the normal prostate compared with other benign human
tissues, such as in the uterus and bladder (24). However, although EPHA3 is also found
to be expressed in prostate tumor vasculature and stroma, there is
a great disparity in the EPHA3 levels, either elevated or reduced,
in various prostate cancer (PCa) cell lines compared with normal
prostate epithelium cells (25,26).
In addition, a positive correlation between the levels of EPHA3 and
the Gleason grade of PCa has also been found in clinical PCa
specimens (26). In addition, EphA3
enhanced the proliferation and survival of PCa LNCaP cells and
tumor development in nude mice subcutaneously implanted with
EPHA3-overexpressing LNCaP cells, and EPHA3 inhibition suppressed
the survival of the LNCaP-derived subline, C4-2B cells (27). The treatment of mice with an
agonistic α-EPHA3 antibody was found to inhibit tumor growth by
activation of EPHA3(+)/CD90(+)/Sca1(+) mesenchymal/stromal cells
(25). All of these findings
highlight the importance of further investigating EPHA3 expression
and the related mechanism in PCa. PCa is the second most common
carcinoma and the fifth leading cause of cancer-related deaths in
men worldwide (28), in which the
androgen receptor (AR) plays a pivotal role in the initiation and
progression of PCa (29–31). The regulation of EPHA3 in PCa
remains mostly unclear, and particularly the association between AR
and EPHA3 regulation has been less studied. In the present study,
we aimed to reveal the effects of AR signaling on EPHA3 expression
and the molecular mechanism of AR-regulated EPHA3 expression in
androgen-dependent PCa cells, and identified the DNA binding sites
within the EPHA3 promoter region that contribute to the regulation
of the expression of EPHA3 in response to androgen hormone.
Materials and methods
Cell culture and chemical
treatments
The media, fetal bovine serums (FBS) and
charcoal/dextran stripped FBS (SFBS) for cell culture were all
purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Human prostate adenocarcinoma cell lines PC-3, DU145, LNCaP
and 22Rv1 were obtained from the American Type Culture Collection
(ATCC; Manassas, VA, USA) and maintained in RPMI-1640 medium
containing 10% FBS, 100 IU/ml penicillin and 0.1 mg/ml streptomycin
at 37°C in 5% CO2. Human immortalized prostatic
myofibroblast stromal cell line WPMY-1 was obtained from Xiangf Bio
Technology Co., Ltd. (Shanghai, China) and maintained in Dulbecco's
modified Eagle's medium (DMEM) with 10% FBS. The androgens DHT and
Mib were obtained from Toronto Research Chemicals (North York, ON,
Canada) and J&K Scientific (Beijing, China), respectively, and
then dissolved in ethanol at a stock concentration of
10−2 mol/l. Mithramycin A (MTM) was obtained from Bio
Basic (Markham, ON, Canada) and dissolved in dimethyl sulfoxide
(DMSO) to prepare a 1 mM stock solution. For androgen stimulation,
cells maintained in phenol-free media with 10% FBS for 3–5 days
were plated in 6-well dishes and allowed to grow to near
confluence. Subsequently, the confluent cells were treated with
androgen at different concentrations for various time periods as
noted in the Figure legends. For MTM treatment, cells close to
confluence were treated in detail as noted in the Figure
legends.
Reverse transcription-polymerase chain
reaction (RT-PCR) and quantitative fluorescence real-time PCR
(qPCR)
Total RNA from cells was prepared with
TRIzol® reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. RT reaction was
performed with the HiScript Q RT SuperMix, PCR was carried out with
a 2X Taq Master Mix, and real-time monitoring PCR was conducted
with the AceQ qPCR SYBR-Green Master Mix (all from Vazyme Biotech,
Nanjing, China) using the Applied Biosystems StepOnePlus Real-Time
PCR System (Thermo Fisher Scientific, Inc.). Primers specific for
AR (32), PSA (33), EPHA3 (2), SP1 (34) and β-actin (forward,
5′-GAGCTACGAGCTGCCTGACG-3′ and reverse, 5′-CCTAGAAGCATTTGCGGTGG-3′)
were synthesized by Shanghai Generay Biotech Co., Ltd. (Shanghai,
China). Reaction mixtures were applied to gel electrophoresis
immediately after PCR amplification. In the RT-PCR analysis, the
expression of the target genes was normalized to β-actin levels in
the respective samples as an internal control, and in the qPCR, the
cycle threshold values were used to calculate the normalized
expression of the target genes against β-actin using the Q-Gene
software (http://www.gene-quantification.de/download.html#qgene)
(35).
Antibodies and western blotting
Antibodies against AR (dilution 1:1,000; cat. no.
sc-816), EPHA3 (dilution 1:1,000; cat. no. sc-919), or SP1
(dilution 1:1,000; cat. no. sc-14027) and the horseradish
peroxidase-conjugated secondary antibodies for rabbit (dilution
1:5,000; cat. no. sc-2357) was purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Antibody against PSA
(dilution 1:2,000; cat. no. DAK-A056201) was obtained from Dako
(Glostrup, Denmark) and against β-actin (dilution 1:10,000; cat.
no. AP0060) was obtained from Bioworld Technology (St. Louis Park,
MN, USA). The cells were lysed in ice-cold RIPA buffer (Beyotime
Institute of Biotechnology, Haimen, China) supplemented with 1 mM
phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) and protease inhibitor cocktail (Roche Applied
Science; Roche Diagnostics Corporation, Indianapolis, IN, USA) for
quantification with the DC Protein Assay kit purchased from Bio-Rad
Laboratories (Hercules, CA, USA). Equal amounts of whole cell
extracts were boiled for 5 min in sample loading buffer, separated
by SDS-PAGE and transferred onto polyvinylidene difluoride
membranes, which were probed with the indicated primary antibody
followed by incubation with the appropriate secondary antibody.
After washing, proteins were detected by a chemiluminescent
detection system (Tanon 5200; Tanon Science and Technology, Co.,
Ltd., Shanghai, China). The software of ImageJ 1.42 (NIH, Bethesda,
NY, USA) was used to perform densitometry analysis for protein
semi-quantification.
Construction of plasmids and transient
transfection of luciferase reporter and small interfering RNA
(siRNA)
The firefly basic luciferase reporter vector
pGL3-Basic and pEGFP-N1 plasmid were kindly provided by Dr Chuanjun
Wen (Nanjing Normal University, Nanjing, China). The EPHA3 core
promoter region (36) was analyzed
using GeneCards (Weizmann Institute of Science, Rehovot, Israel)
and the TRANSFAC databases (http://gene-regulation.com/, Gottingen, Germany) to
search transcription factor binding sites for AR and SP1. All the
primers designed for the construction of the reporter plasmids are
summarized in Table I. The
EPHA3-Luc-789 plasmid was generated by inserting a core promoter
sequence of the EPHA3 gene spanning from −789 to +146 bp into the
pGL3-Basic luciferase reporter vector. Construct EPHA3-Luc-317 is
composed of a PCR fragment spanning from −317 to +146 bp of the
EPHA3 promoter, and EPHA3-Luc-237 has core promoter sequence from
−237 to +146 bp instead. The mutant EPHA3-Luc-789ΔSp1 was
constructed mostly in the same way as EPHA3-Luc-789 except that the
small fragment of the SP1 binding region was replaced by
multi-point mutations using PCR amplification with the combinations
of specifically designed longer oligonucleotide ΔSp1-F and the
reverse primer for the EPHA3-Luc-789 construct, and the forward
primer for EPHA3-Luc-789 and ΔSP1-R. The AR expression plasmid
(pEGFP-AR) was constructed by inserting the full-length cDNA of the
AR from LNCaP cells amplified by PCR using the forward primer
attached with an EcoRI site
(5′-CCGGAATTCATGGAAGTGCAGTTAGGGCTG-3′) and the reverse primer with
a BamHI site
(5′-CGCGGATCCTCACTGGGTGCGCGGATCCTCACTGGGTGTGGAAATAGATGG-3′) added
into the multiple cloning site of pEGFP-N1. PCa cells in 24-well
plates were transiently transfected with 0.4 µg of the indicated
reporter plasmid, 0.4 µg of pEGFP-AR or 50 nM of siRNA AR (siAR)
(37), and 0.01 µg pRL-TK as an
internal control using Lipofectamine 2000 transfection reagent
(Thermo Fisher Scientific, Inc.). After 48 h transfection, cell
lysates were prepared according to the Dual-Luciferase Reporter
Assay system (Promega Corporation, Madison, WI, USA) protocol for
the luciferase assay in a Luminoskan Ascent (Thermo Fisher
Scientific, Inc.), and firefly luciferase activities were corrected
by the corresponding Renilla luciferase activities. The siAR
and siRNA SP1 (siSp1) (20) were
synthesized separately by Thermo Fisher Scientific, Inc., and
transfected as indicated in the Figure legends.
 | Table I.Primers used for the construction of
luciferase reporter plasmids. |
Table I.
Primers used for the construction of
luciferase reporter plasmids.
| Name |
Primersa |
|---|
| −789-EphA3-P-F |
CCGCTCGAGCTCCCTCCGTAAGATGA |
|
−789/-317/-237-EphA3-P-R |
CCCAAGCTTCTTTGAGAGCGTGAGCC |
| −317-EphA3-P-F |
CCGCTCGAGCTTCTTGTTTCCTGTGG |
| −237-EphA3-P-F |
CCGCTCGAGTGCCATCCCGCTCTGCTT |
| ΔSp1-F | AACCCACTAACACTCGGTCTACTTCACGAGCAGCTCTGGGCTGCAGAGCC |
| ΔSp1-R | TGAAGTAGACCGAGTGTTAGTGGGTTCGTCGCCCAGGGACAGGAAACAC |
Co-IP and ChIP assay
The 22Rv1 cells were maintained in SFBS medium for
3–5 days and grown to confluence in 100-mm dishes. Then, the cells
were lysed in a modified RIPA buffer [50 mM Tris-HCl (pH 7.4), 1%
NP-40, 150 mM NaCl] containing 1 mM PMSF and 1X protease inhibitor
cocktail after stimulation with 10 nM Mib for 24 h, and centrifuged
at 13,523 × g, and 4°C for 15 min. One aliquot of the supernatant
was saved as the input control, the remainder was incubated with
1.5 µg of anti-AR antibody, anti-SP1 antibody and negative control
IgG antibody (cat. no. A7016; Beyotime Institute of Biotechnology),
separately, followed by pull-down with 15 µl Protein A/G
PLUS-Agarose beads (Santa Cruz Biotechnology, Inc.). The beads were
then collected by centrifugation at 845 × g for 5 min and washed
three times with cold PBS. The immunoprecipitates were eluted by
boiling in reducing sample buffer for 5 min and subjected to
western blot analysis along with input sample as described
above.
Concurrently, 8×106 cells starved in
phenol-free RPMI-1640 medium with 10% SFBS medium were stimulated
with 10 nM DHT for 24 h, cross-linked by adding formaldehyde up to
a final concentration of 1% at room temperature for 10 min and
mixed with 0.125 M glycine for 5 min to quench cross-linking. The
cultures were then harvested by centrifugation and the pellets were
washed twice with ice-cold PBS and lysed in 300 µl of lysis buffer
[50 mM Tris-HCl (pH 8.0), 10 mM EDTA and 1% SDS] for shearing
chromatin on ice in 5 sec on/15 sec off pulses at 10% output for 10
min in a sonicator (Ningbo Scientz Biotechnology Co., Ltd., Ningbo,
China). Subsequenlty, the lysates were centrifuged to separate cell
debris from the supernatant, of which one tenth of the volume was
saved as input sample and the remainder was diluted 10-fold with
ChIP dilution buffer [16.7 mM Tris-HCl (pH 8.1), 1.2 mM EDTA, 167
mM NaCl, 1.1% Triton X-100 and 0.01% SDS] including protease
inhibitor. Subsequently, 500 µl aliquots were incubated with 2 µg
of the appropriate antibodies or IgG as negative control at 4°C
overnight, and 15 µl Protein A/G PLUS-Agarose beads (cat. no.
sc-2003; Santa Cruz Biotechnology, Inc.) were added for another 2-h
incubation at room temperature with gentle rotation to
immunoprecipitate protein-bound chromatin fragments. The beads were
collected and washed orderly with low salt, high salt and LiCl
buffers following the protocol of the ChIP assay kit (Upstate
Biotechnology Inc., Lake Placid, NY, USA), and eluted in 100 µl TE
buffer (20 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0). Cross-linking for
both the eluate and the input samples were reversed by incubation
in 0.2 M NaCl at 65°C for 2 h, and qPCR analysis was performed as
described above. The primer pair for the EPHA3 promoter surrounding
the SP1 binding sites is FW1 (5′-CACTCCCACCCGGTCTCCTT-3′) and RV1
(5′-GGGTCCTAGTGCCGTCTT-3′).
Image quantification and statistical
analysis
The results are representative of at least three
independent experiments. Images of the agarose gel electrophoresis
and western blotting membranes were analyzed with Quantity One
Software (Bio-Rad Laboratories) to quantify the mRNA and protein
expression. The data are expressed as the mean with standard
deviations for all groups, which were calculated using the GraphPad
Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). Comparisons
between two groups were performed using Student's t-test, and
comparisons among multiple groups were conducted by one-way
analysis of variance (ANOVA) using Bonferroni's post hoc test.
Differences were considered to be statistically significant if
P-value was <0.05.
Results
Androgen induces mRNA and protein
expression of EphA3 in PCa 22Rv1 cells in a dose- and
time-dependent manner
To determine whether EPHA3 expression is induced by
androgen in PCa cells, androgen responsive 22Rv1 cells were
stimulated with various doses of DHT for different time periods for
RT-PCR and western blot analysis. The data presented in Fig. 1A reveals that with the increase of
the DHT concentration, EPHA3, AR and PSA mRNA levels were gradually
increased compared with the untreated control. Similarly, EPHA3, AR
and PSA protein levels were also induced by DHT, as displayed in
Fig. 1B, where the two bands for
the AR protein in 22Rv1 cells correspond to one full-length and the
other to a truncated AR lacking the COOH-terminal domain (CTD). The
mRNA and protein expression of EPHA3 was continually increased in a
dose-dependent manner by treatment with DHT from 0.1 to 10 nM, in
which EPHA3 expression was significantly increased at 1 nM and
achieved a more than 2-fold increase at 5 nM (Fig. 1A and B, right pattern). Similar
time-dependent trends were observed for mRNA and protein expression
of EPHA3, AR and PSA in 22Rv1 cells treated with 10 nM DHT from
time 0 to 72 h (Fig. 1C and D). In
particular, EphA3 expression was significantly elevated after 8 h
of DHT treatment, and up to more than 2-fold after 24 h of DHT
treatment (Fig. 1C and D, right
pattern). In addition, another androgen responsive PCa cell line,
LNCaP, was also stimulated by both DHT and Mib at different
concentrations (Fig. 2), where it
is shown that the EPHA3 protein level was gradually elevated along
with the increases in AR and PSA levels.
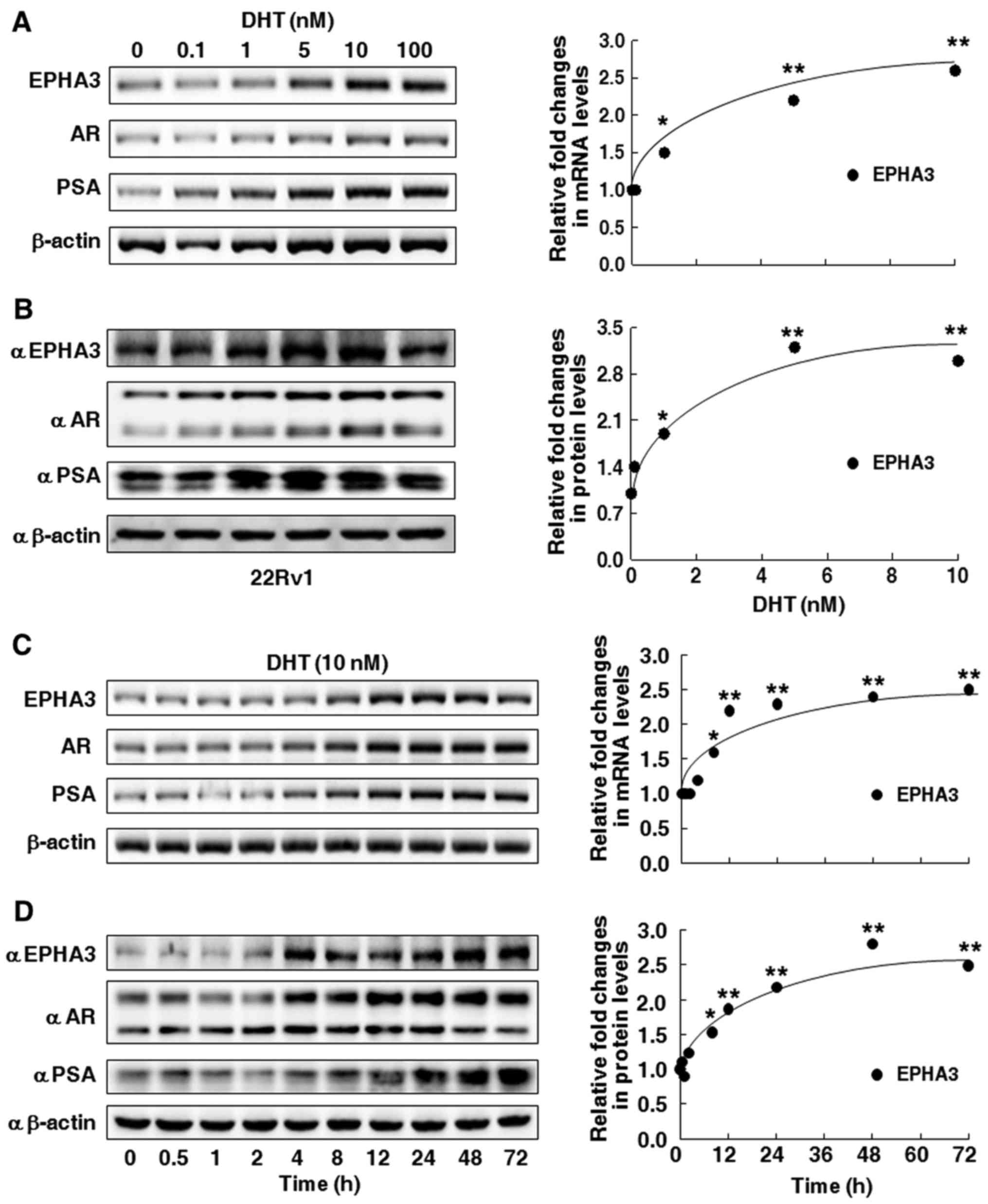 | Figure 1.Dose-response and time course effect
of DHT treatment on EPHA3 expression in prostate cancer 22Rv1
cells. (A and B) Following androgen starvation, 22Rv1 cells were
treated with DHT at final concentrations of 0.1, 1, 5, 10 and 100
nM for 24 h, or (C and D) treated with 10 nM DHT for 0–72 h. (A and
C) RT-PCR or (B and D) western blotting was performed to measure
mRNA and protein expression, respectively, for AR, PSA and EPHA3
levels as described in the text. β-actin levels were used as
loading controls, and images of the agarose gel electrophoresis and
western blotting membranes were analyzed by densitometry for
quantification of the mRNA and protein expressions of AR, PSA and
EPHA3, and the results are shown as relative fold-change,
normalized to β-actin and relative to the untreated cells (A-D:
Right graphs, *P<0.05, **P<0.01). DHT, dihydrotestosterone;
AR, androgen receptor; PSA, prostate-specific antigen. |
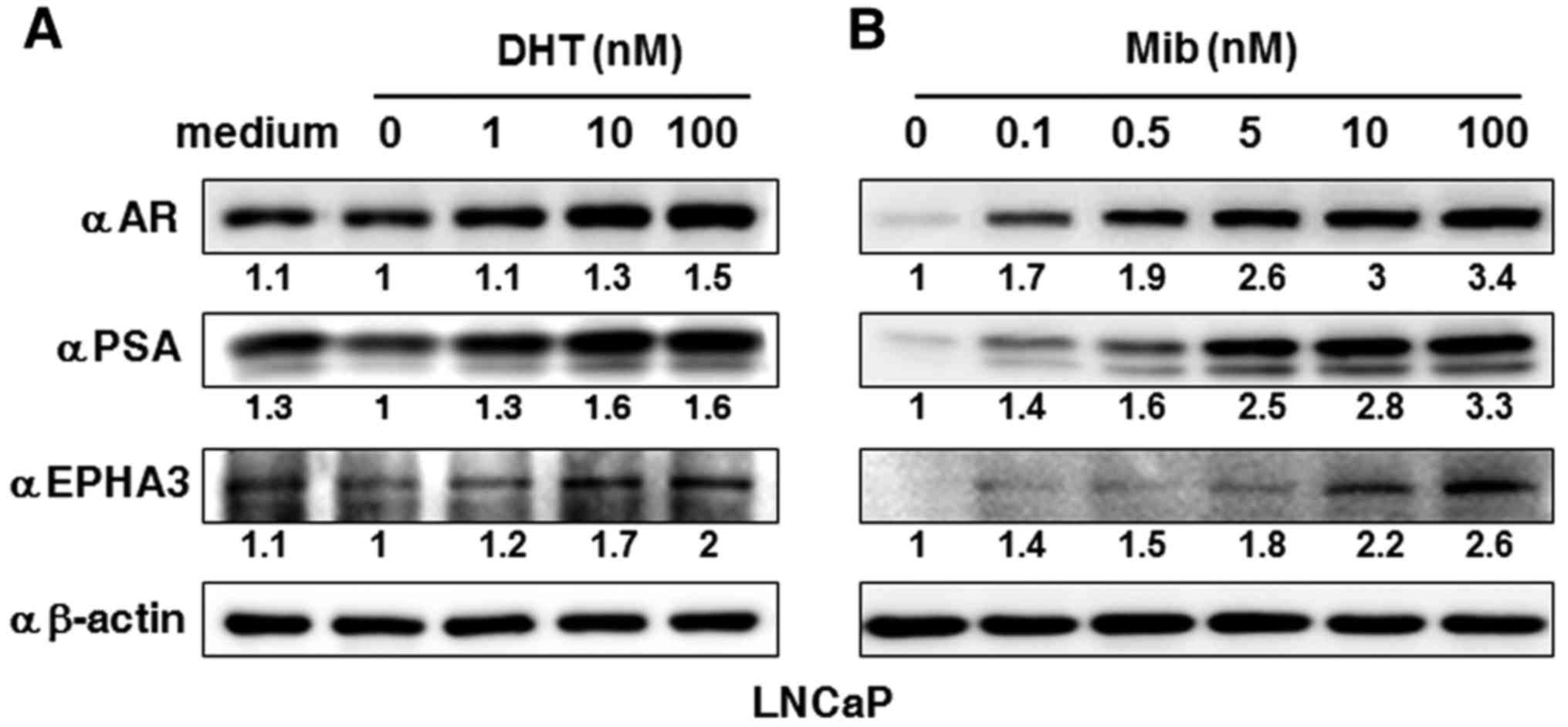 | Figure 2.Dose effects of androgen treatment on
EPHA3 expression in prostate cancer LNCaP cells. (A) After androgen
starvation, LNCaP cells were treated with DHT at final
concentrations of 1, 10 and 100 nM for 24 h, or (B) Mib at
concentrations of 0, 0.1, 0.5, 5, 10 and 100 nM. Western blotting
was performed to determine protein expression for AR, PSA and EPHA3
levels as described in the text, and β-actin levels were used as
loading controls. Images were analyzed by densitometry for relative
protein expression of AR, PSA and EPHA3. The results are shown as
relative fold-change, normalized to β-actin and relative to
untreated cells. Mib, mibolerone; DHT, dihydrotestosterone; AR,
androgen receptor; PSA, prostate-specific antigen. |
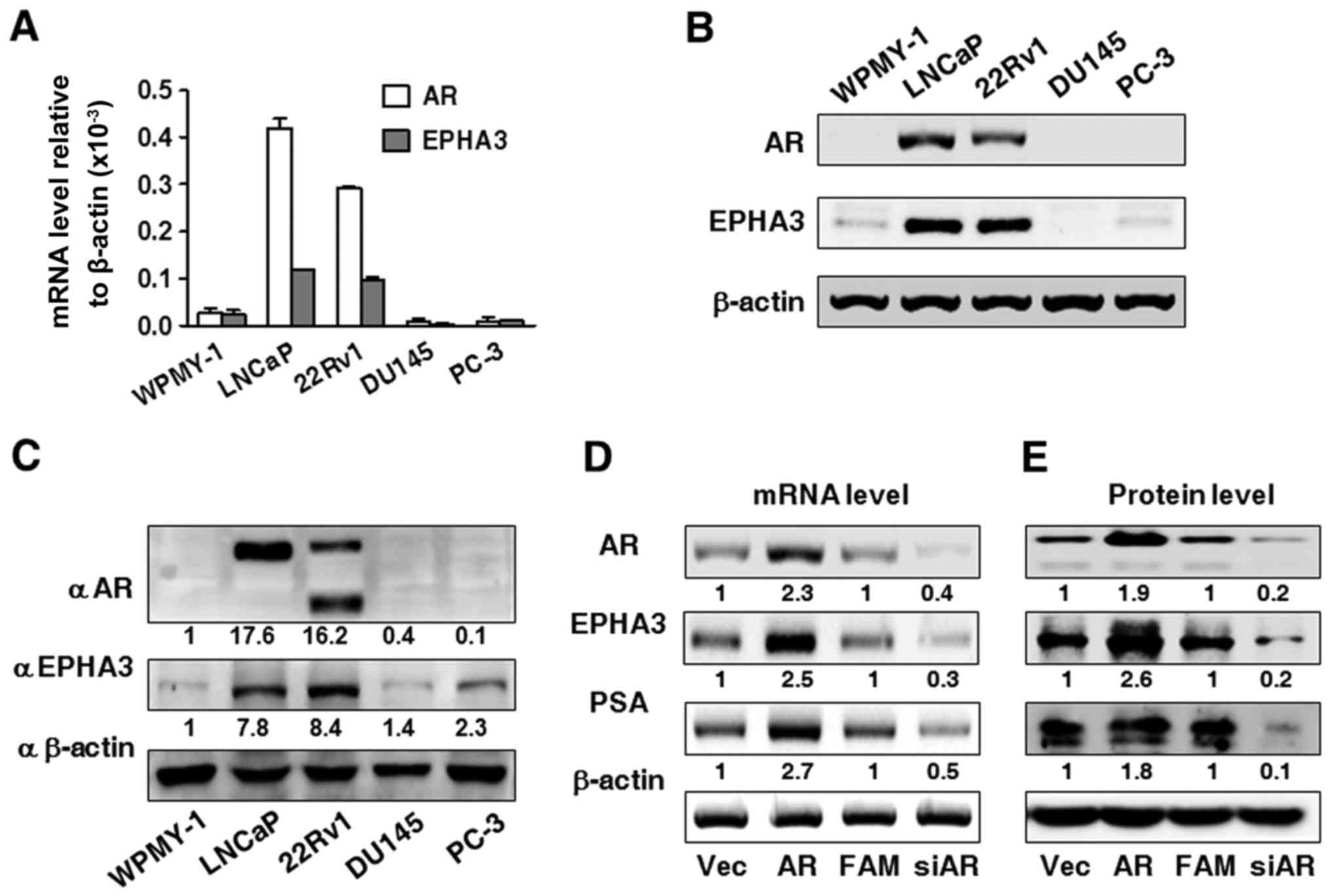
AR expression level affects EPHA3 mRNA
and protein expression in PCa 22Rv1 cells
To investigate the association between AR and EPHA3
expression, qPCR analysis and western blotting were performed to
measure AR and EPHA3 expression in the prostate stromal cell WPMY-1
and PCa cell lines LNCaP, 22Rv1, PC-3 and DU145. The results shown
in Fig. 3A and B revealed that the
mRNA levels of EPHA3 and AR were higher in LNCaP and 22Rv1 cells,
and almost undetectable in WPMY-1, PC-3, while DU145 cells hardly
expressed AR, which was confirmed at the protein level, as
displayed in Fig. 3C. To evaluate
whether AR can regulate EPHA3 expression, AR was overexpressed or
silenced to determine EPHA3 and PSA levels in 22Rv1 cells. The
results showed that overexpression of AR markedly increased PSA and
EPHA3 expression both at the mRNA (Fig.
3D) and protein levels (Fig.
3E). In contrast, AR-specific inhibition of siAR markedly
decreased both the mRNA and protein expression of PSA and EPHA3
(Fig. 3D and E).
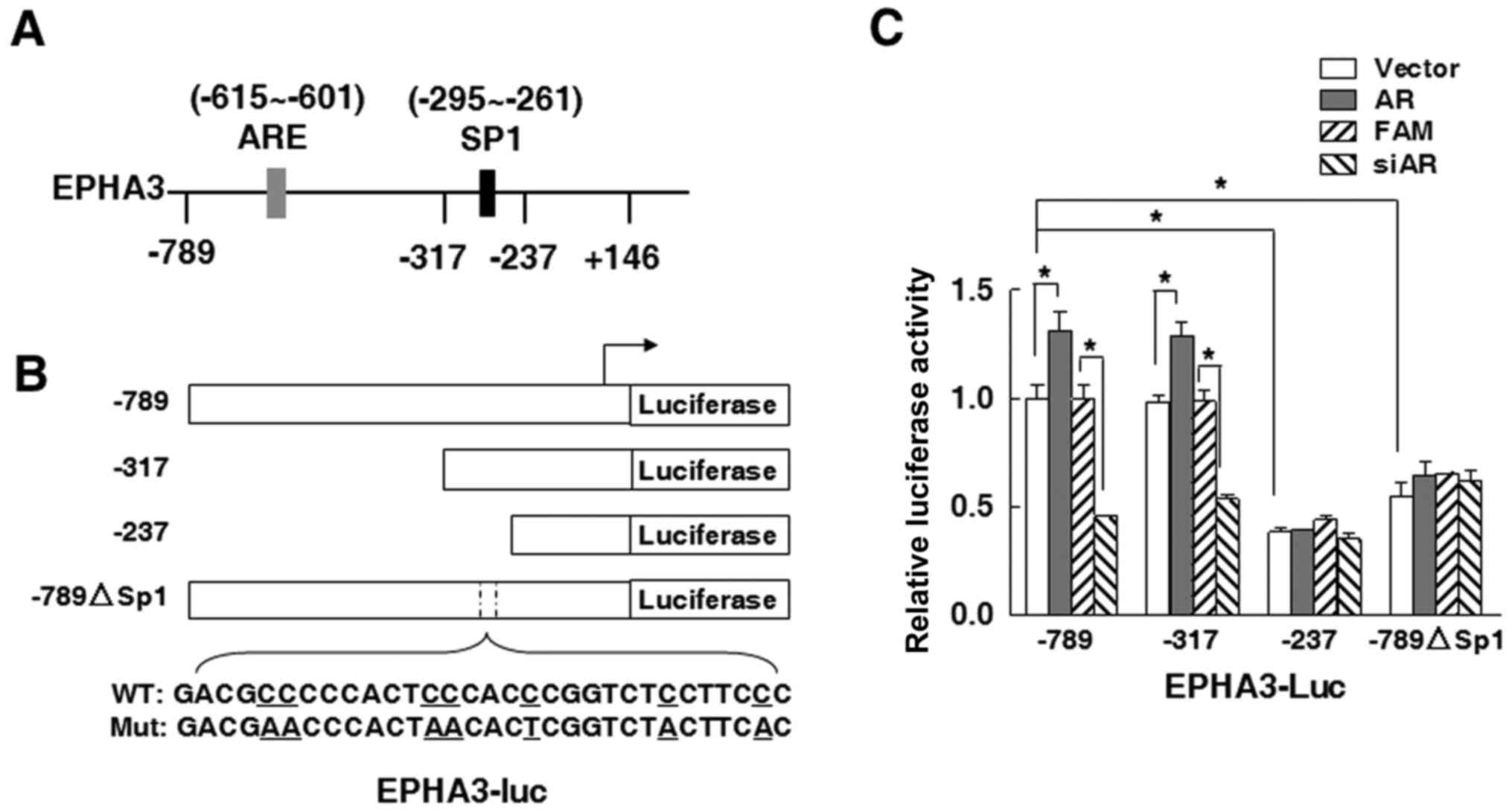
SP1 binding sites are required to
regulate EPHA3 expression in response to AR signaling
To further investigate whether AR binds the EPHA3
promoter region and directly regulates EPHA3 transcription, two
important transcription factor binding sites in the EPHA3 core
promoter region were inferred, one of which was an
androgen-responsive element (ARE) for AR binding located between
−615 and −601 bp, and the other one ranging from −295 to −261 bp
for Sp1 binding, as shown in Fig.
4A. Based on this, different truncated promoters were used for
construction of luciferase reporter plasmids to analyze the EPHA3
promoter activity (Fig. 4B). The
results in Fig. 4C showed that, in
22Rv1 cells, the promoter luciferase activity of the EPHA3-Luc-789
reporter plasmid with both AR and SP1 binding sites was similarly
as high as that of the EPHA3-Luc-317 reporter plasmid with the SP1
binding site only. In addition, AR overexpression also remarkably
increased the EPHA3 promoter activity in both the −789 and −317
truncation plasmids, whereas the siRNA-mediated knockdown of AR
markedly decreased the promoter activity in 22Rv1 cells (Fig. 4C). The EPHA3 promoter activity was
the lowest in 22Rv1 cells transfected with the EPHA3-Luc-237
plasmid including neither the ARE nor SP1 binding sites, and was
not affected by either AR overexpression or siAR interference
(Fig. 4C). Compared to the
EPHA3-Luc-789 plasmid transfected group, the EPHA3 promoter
activity was downregulated by about half in 22Rv1 cells after
transfection with the EPHA3-Luc-789ΔSP1 plasmid containing modified
SP1 binding sites in the GC-rich region, and scarcely influenced by
co-transfection with AR or siAR (Fig.
4C).
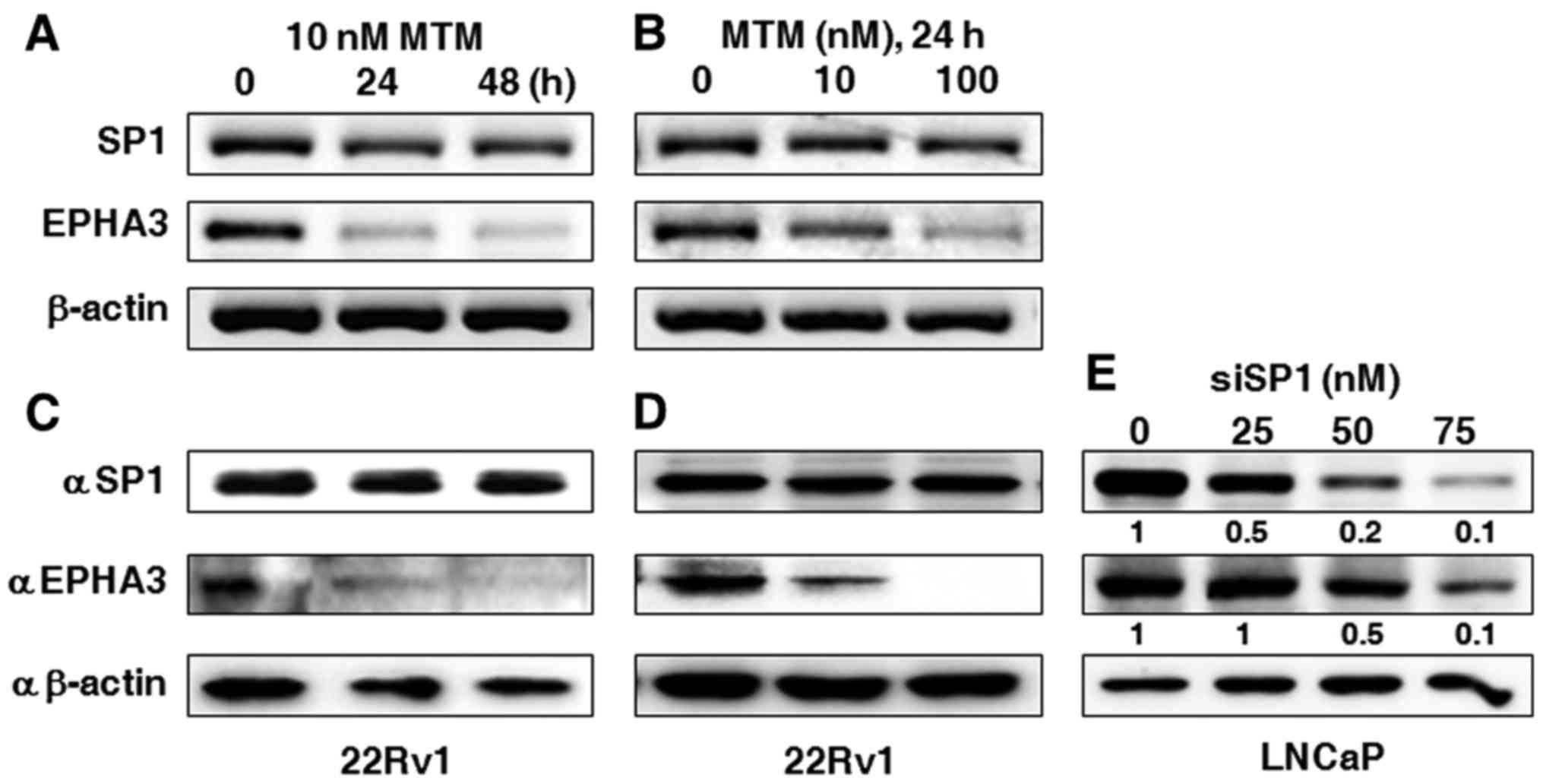
Transcription factor SP1 still
regulates EPHA3 expression even when AR is not exogeneously
altered
In order to further characterize the involvement of
SP1 in the regulation of EPHA3 expression when AR is not
artificially inhibited or overexpressed, MTM, as an inhibitor of
SP1 transcription activity, was added to 22Rv1 cells to determine
the effect of SP1 transcriptional activation on EPHA3 expression.
The data displayed in Fig. 5A shows
that the EPHA3 mRNA level was severely reduced in the presence of
10 nM MTM from 24 to 48 h, or by the treatment with 10 and 100 nM
MTM for 24 h, although the SP1 mRNA level was rarely affected
(Fig. 5B). Accordingly, the EPHA3
protein level was also markedly reduced under the same conditions,
while the SP1 protein level was barely changed, as shown in
Fig. 5C and D. In addition,
specific inhibition of SP1 in LNCaP cells using siSP1 at an
increasing gradient mode also gradually abated EPHA3 protein
expression (Fig. 5E).
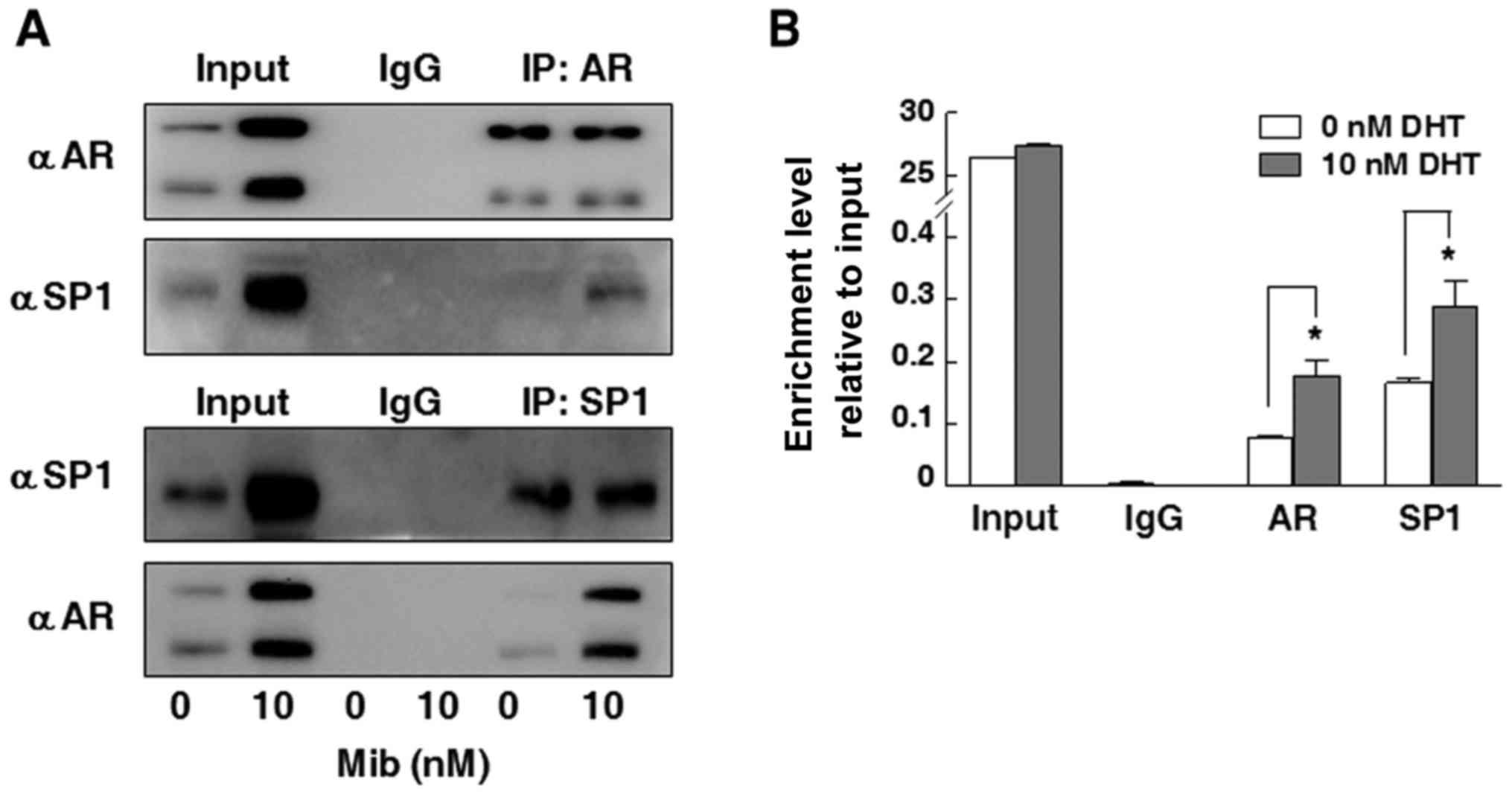
Interaction between AR and SP1
contributes to the hormone response of the EPHA3 promoter
Co-IP assay was performed to evaluate whether AR can
bind to the Sp1 transcription factor. As shown in Fig. 6A, the AR protein was detected in
22Rv1 cells after immunoprecipitation with antibodies not only to
AR but also to SP1 in the presence of Mib. In addition, the SP1
protein was also immunoprecipitated by the SP1 or AR antibody in
these cells after Mib stimulation (Fig.
6A). To determine whether androgen-induced EPHA3 expression is
regulated through SP1 binding sites in the EPHA3 core promoter
region, ChIP assay was carried out to analyze the association of
the SP1 protein with specific core promoter regions of EPHA3 in the
case of DHT stimulation. As shown in Fig. 6B, both SP1 and AR were found to be
bound to the SP1 binding sites in the EPHA3 core promoter region
even in the androgen-starvation state. In addition, the SP1 binding
capacity was around 2-fold higher than that of AR, and their
binding capacities to this GC-rich region were increased almost 2
folds in the presence of DHT.
Discussion
Although EPHA3 plays an important role in the
progression of various invasive and metastatic cancers (14,15,27,14), the mechanisms involved in the
regulation of EPHA3 are largely unknown. In the current study, we
demonstrated that EPHA3 expression was induced by DHT hormone, in
PCa cells 22Rv1, at both the mRNA and protein levels in a time- and
dose-dependent manner, while the expression of AR and the
AR-targeted gene, PSA revealed the increasing trend along with DHT
stimulation. Similarly, EPHA3 was also markedly induced in PCa
LNCaP cells treated with DHT or Mib. The results indicated that
EPHA3 expression is responsive to androgen hormone, which is
possibly due to AR regulation since most biological effects of
androgens are mediated through the action of nuclear AR as a master
regulator of downstream androgen-dependent signaling pathway
networks (40). We also found that
the EPHA3 mRNA expression level was rather high in the
androgen-dependent PCa LNCaP and 22Rv1 cells with higher AR
expressions compared with the AR non-expressing prostate stromal
cell WPMY-1 and PCa cell lines PC-3 and DU145. This finding is
consistent with a previous report that mRNA level of EPHA3 was
highly expressed in LNCaP cells (26), and, additionally, so was the EPHA3
protein expression in these cells, i.e., EPHA3 appeared to exhibit
a similar expression pattern as the AR at both the mRNA and protein
levels. When the AR was overexpressed in 22Rv1 cells, the EPHA3
mRNA and protein levels were significantly increased with the rise
of the PSA expression. In contrast, mRNA and protein expressions of
PSA and EPHA3 were markedly decreased after knocking down the AR,
which clearly suggested that AR activity can affect EPHA3
expression in androgen-dependent PCa cells. A reported study showed
that prostate androgen induces the prostate leucine zipper gene
promoted EPHA3 expression (27),
suggesting from another point of view that the AR is probably
associated with EPHA3 regulation.
AR is composed of four domains, the N terminal
transactivation domain, the DNA binding domain, the flexible hinge
region and the LBD. Usually, the inactivated AR in the cytoplasm
can be translocated into the nucleus upon binding with ligands,
such as androgen hormone, and then attached to specific DNA binding
sites of ARE loci in the promoter region of the target gene to
start the AR-mediated transcription activity (33). In the present study, we assumed that
the potential ARE site was between −615 to −601 bp and the SP1
binding sites of highly GC-rich region was between −295 and −261 bp
in the EPHA3 promoter region. We found that the SP1 binding sites
were indeed required for the AR-mediated EPHA3 promoter activity,
while the elimination of the ARE site hardly affected the EPHA3
reporter promoter activity. Specifically, AR regulation of EPHA3
expression is not due to direct binding of the AR with the EPHA3
core promoter region, but AR may regulate EPHA3 through a
SP1-dependent pathway whereby AR can regulate downstream gene
expression not only as a transcription factor binding to ARE sites
in the promoter of target genes, but also as an auxiliary
transcription factor interacting with the transcription
complex.
SP1 is a well-known member of the Sp transcription
factor family including SP2, SP3 and SP4, which are implicated in
varieties of biological processes (41). SP1 activates gene transcription by
binding to specific CG-rich SP-binding sites within gene promoters,
and has been considered as a therapeutic target for human cancers,
including PCa (41–44). In this study, we demonstrated that
EPHA3 expression was markedly downregulated when 22Rv1 cells were
treated with MTM, which inhibited SP1 binding to GC rich promoter
region. Similarly, it was also downregulated when LNCaP cells were
treated with siSP1 to knock down SP1 expression, which suggested
that SP1 may regulate EPHA3 expression as a transcription factor.
The co-IP assay further showed that AR forms a complex with SP1.
Additionally, the ChIP assay confirmed that SP1 mediated androgen
induction of EPHA3 core promoter activity involved with DNA
binding. These findings suggested that androgen-initiated AR
signaling transduction is achieved through the interaction of AR
and SP1 to mediate EPHA3 expression, whereby SP1 is able to bind
the EPHA3 core promoter in chromatin responding to AR signaling.
This is in accordance with the functional mechanism of AR and the
transcription factor SP1 complex to mediate vascular endothelial
growth factor (VEGF) expression in PCa cells in response to
androgen induction, which is also dependent upon a critical SP1
binding site within the VEGF core promoter (45). Similarly, AR and SP1 induced cyclin
dependent kinase inhibitor gene p21 transcription in LNCaP cells
through binding to the ARE, as well as SP1 binding site in the p21
promoter after androgen stimulation (46). Our results showed that the binding
between SP1 and the EPHA3 core promoter region to induce EPHA3
expression occurred no matter whether androgen exists or not, which
may result from constitutively active AR mutants expressed only in
22Rv1 cells (47–49) and/or incomplete androgen-starvation
of 22Rv1 cells used in our experiments.
Furthermore, it has been previously reported that
EPHA3 is significantly increased during the conversion of LNCaP
cells from androgen-dependent (LNCaP-C33) to androgen-independent
(LNCaP-C81) phenotypes using Affymetrix GeneChip array analysis
(50). Nevertheless, this does not
imply that the increase of EPHA3 expression is independent from
androgen induction, or that AR is not associated with EPHA3
regulation in androgen-independent prostate cells as such. Indeed,
compared to androgen-dependent LNCaP cells, the AR level is also
increased in androgen-independent LNCaP sublines, and two AR target
genes, namely UGT2B15 and UGT2B17, which are not expressed in
AR-negative PCa cells, were both positively correlated with
upregulated AR in androgen-independent LNCaP subline, though PSA as
one of the AR main targets, was markedly decreased (51,52).
This suggested that upregulation of AR targets in
androgen-dependent PCa cells may occur in AR positive
androgen-independent state. On the other hand, PSA would also be
downregulated under androgen-independent condition, e.g., increased
microR-100 in androgen-independent PCa cells suppresses PSA
expression even if AR transcription activity initiated by ligands
is blocked, mostly due to androgen deprivation (53). Other factors, such as the NF-κB
level also mediates PSA expression in androgen-independent PCa
cells (54). Thus, there may be
additional molecules and other pathways besides AR signaling
involved in regulating EPHA3 in AR-positive castration-resistant
PCa and various stages of PCa progression, which remains to be
further examined.
The present study found that EPHA3 is increased at
the transcript and protein expression levels in 22Rv1 cells in a
time- and dose-dependent manner under treatment with DHT, and the
EPHA3 expression pattern is similar to that of AR in the prostate
(cancer) cell lines. AR overexpression or AR inhibition markedly
affected the EPHA3 levels, due to the interaction of AR and SP1 as
a transcription factor to bind SP1 binding sites in the core
promoter region of EPHA3. These findings indicated the association
among EPHA3, AR and SP1, which could be useful to gain further
insight into the importance of EPHA3 in PCa development and
progression and will additionally facilitate our understanding of
AR and SP1 as targets for the treatment of PCa.
Acknowledgements
We are grateful to Chuanjun Wen (Nanjing Normal
University) for providing us the plasmids.
Funding
The present study was supported by funding from the
National Natural Science Foundation of China (nos. 81172007,
81472415 and 81272850).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SL, XD, XC and PL conceived and designed the
experiments; XD, XC, YP, YZ and FW performed experiments; SL, XD,
XC, PL, YG, XW and SY analyzed the data; PL, YG, XW, SY and SL
provided reagents, materials and tools; SL, YG, SY and XW wrote and
reviewed the manuscript. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EPHA3
|
erythropoietin-producing
hepatocellular carcinoma cell surface type-A receptor 3
|
|
PCa
|
prostate cancer
|
|
AR
|
androgen receptor
|
|
RT-PCR
|
reverse transcription-polymerase chain
reaction
|
|
qPCR
|
quantitative fluorescence real-time
PCR
|
|
PSA
|
prostate-specific antigen
|
|
ARE
|
androgen response element
|
|
Sp
|
specific protein
|
|
siRNA
|
small interfering RNA
|
|
Co-IP
|
co-immunoprecipitation
|
|
ChIP
|
chromatin IP
|
|
RMS
|
rhabdomyosarcoma
|
|
FBS
|
fetal bovine serum
|
|
SFBS
|
charcoal/dextran-stripped FBS
|
|
DHT
|
dihydrotestosterone
|
|
Mib
|
mibolerone
|
|
MTM
|
mithramycin A
|
|
PMSF
|
phenylmethylsulfonyl fluoride
|
|
LBD
|
ligand binding domain
|
|
VEGF
|
vascular endothelial growth factor
|
References
|
1
|
Hood G, Laufer-Amorim R, Fonseca-Alves CE
and Palmieri C: Overexpression of ephrin A3 receptor in canine
prostatic carcinoma. J Comp Pathol. 154:180–185. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xi HQ, Wu XS, Wei B and Chen L: Aberrant
expression of EphA3 in gastric carcinoma: Correlation with tumor
angiogenesis and survival. J Gastroenterol. 47:785–794. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nasri B, Inokuchi M, Ishikawa T, Uetake H,
Takagi Y, Otsuki S, Kojima K and Kawano T: High expression of EphA3
(erythropoietin-producing hepatocellular A3) in gastric cancer is
associated with metastasis and poor survival. BMC Clin Pathol.
17:82017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tímár J, Mészáros L, Ladányi A, Puskás LG
and Rásó E: Melanoma genomics reveals signatures of sensitivity to
bio- and targeted therapies. Cell Immunol. 244:154–157. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Caivano A, La Rocca F, Laurenzana I,
Annese T, Tamma R, Famigliari U, Simeon V, Trino S, De Luca L,
Villani O, et al: Epha3 acts as proangiogenic factor in multiple
myeloma. Oncotarget. 8:34298–34309. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
La Rocca F, Airoldi I, Di Carlo E, Marotta
P, Falco G, Simeon V, Laurenzana I, Trino S, De Luca L, Todoerti K,
et al: EphA3 targeting reduces in vitro adhesion and invasion and
in vivo growth and angiogenesis of multiple myeloma cells. Cell
Oncol. 40:483–496. 2017. View Article : Google Scholar
|
|
7
|
Lu CY, Yang ZX, Zhou L, Huang ZZ, Zhang
HT, Li J, Tao KS and Xie BZ: High levels of EphA3 expression are
associated with high invasive capacity and poor overall survival in
hepatocellular carcinoma. Oncol Rep. 30:2179–2186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Day BW, Stringer BW, Al-Ejeh F, Ting MJ,
Wilson J, Ensbey KS, Jamieson PR, Bruce ZC, Lim YC, Offenhäuser C,
et al: EphA3 maintains tumorigenicity and is a therapeutic target
in glioblastoma multiforme. Cancer Cell. 23:238–248. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li M, Yang C, Liu X, Yuan L, Zhang F, Wang
M, Miao D, Gu X, Jiang S, Cui B, et al: EphA3 promotes malignant
transformation of colorectal epithelial cells by upregulating
oncogenic pathways. Cancer Lett. 383:195–203. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brantley DM, Cheng N, Thompson EJ, Lin Q,
Brekken RA, Thorpe PE, Muraoka RS, Cerretti DP, Pozzi A, Jackson D,
et al: Soluble Eph A receptors inhibit tumor angiogenesis and
progression in vivo. Oncogene. 21:7011–7026. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Swords RT, Greenberg PL, Wei AH, Durrant
S, Advani AS, Hertzberg MS, Jonas BA, Lewis ID, Rivera G,
Gratzinger D, et al: KB004, a first in class monoclonal antibody
targeting the receptor tyrosine kinase EphA3, in patients with
advanced hematologic malignancies: Results from a phase 1 study.
Leuk Res. 50:123–131. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferluga S, Tomé CM, Herpai DM, D'Agostino
R and Debinski W: Simultaneous targeting of Eph receptors in
glioblastoma. Oncotarget. 7:59860–59876. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Charmsaz S, Al-Ejeh F, Yeadon TM, Miller
KJ, Smith FM, Stringer BW, Moore AS, Lee FT, Cooper LT, Stylianou
C, et al: EphA3 as a target for antibody immunotherapy in acute
lymphoblastic leukemia. Leukemia. 31:1779–1787. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Janes PW, Slape CI, Farnsworth RH,
Atapattu L, Scott AM and Vail ME: EphA3 biology and cancer. Growth
Factors. 32:176–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lodola A, Giorgio C, Incerti M, Zanotti I
and Tognolini M: Targeting Eph/ephrin system in cancer therapy. Eur
J Med Chem. 142:152–162. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Herath NI, Spanevello MD, Doecke JD, Smith
FM, Pouponnot C and Boyd AW: Complex expression patterns of Eph
receptor tyrosine kinases and their ephrin ligands in colorectal
carcinogenesis. Eur J Cancer. 48:753–762. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xi HQ and Zhao P: Clinicopathological
significance and prognostic value of EphA3 and CD133 expression in
colorectal carcinoma. J Clin Pathol. 64:498–503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Andretta E, Cartón-García F,
Martínez-Barriocanal Á, de Marcondes PG, Jimenez-Flores LM, Macaya
I, Bazzocco S, Bilic J, Rodrigues P, Nieto R, et al: Investigation
of the role of tyrosine kinase receptor EPHA3 in colorectal cancer.
Sci Rep. 7:415762017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vecchi M, Confalonieri S, Nuciforo P,
Viganò MA, Capra M, Bianchi M, Nicosia D, Bianchi F, Galimberti V,
Viale G, et al: Breast cancer metastases are molecularly distinct
from their primary tumors. Oncogene. 27:2148–2158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clifford N, Smith LM, Powell J,
Gattenlöhner S, Marx A and O'Connor R: The EphA3 receptor is
expressed in a subset of rhabdomyosarcoma cell lines and suppresses
cell adhesion and migration. J Cell Biochem. 105:1250–1259. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chiappalupi S, Riuzzi F, Fulle S, Donato R
and Sorci G: Defective RAGE activity in embryonal rhabdomyosarcoma
cells results in high PAX7 levels that sustain migration and
invasiveness. Carcinogenesis. 35:2382–2392. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhuang G, Song W, Amato K, Hwang Y, Lee K,
Boothby M, Ye F, Guo Y, Shyr Y, Lin L, et al: Effects of
cancer-associated EPHA3 mutations on lung cancer. J Natl Cancer
Inst. 104:1182–1197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peng J, Wang Q, Liu H, Ye M, Wu X and Guo
L: EPHA3 regulates the multidrug resistance of small cell lung
cancer via the PI3K/BMX/STAT3 signaling pathway. Tumour Biol.
37:11959–11971. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hafner C, Schmitz G, Meyer S, Bataille F,
Hau P, Langmann T, Dietmaier W, Landthaler M and Vogt T:
Differential gene expression of Eph receptors and ephrins in benign
human tissues and cancers. Clin Chem. 50:490–499. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vail ME, Murone C, Tan A, Hii L, Abebe D,
Janes PW, Lee FT, Baer M, Palath V, Bebbington C, et al: Targeting
EphA3 inhibits cancer growth by disrupting the tumor stromal
microenvironment. Cancer Res. 74:4470–4481. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fox BP, Tabone CJ and Kandpal RP:
Potential clinical relevance of Eph receptors and ephrin ligands
expressed in prostate carcinoma cell lines. Biochem Biophys Res
Commun. 342:1263–1272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu R, Wang H, Wang J, Wang P, Huang F, Xie
B, Zhao Y, Li S and Zhou J: EphA3, induced by PC-1/PrLZ,
contributes to the malignant progression of prostate cancer. Oncol
Rep. 32:2657–2665. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
World Health Organization: Global battle
against cancer won't be won with treatment alone-effective
prevention measures urgently needed to prevent cancer crisis. Cent
Eur J Public Health. 22:23–28. 2014.PubMed/NCBI
|
|
29
|
Roviello G, Sigala S, Sandhu S, Bonetta A,
Cappelletti MR, Zanotti L, Bottini A, Sternberg CN, Fox SB and
Generali D: Role of the novel generation of androgen receptor
pathway targeted agents in the management of castration-resistant
prostate cancer: A literature based meta-analysis of randomized
trials. Eur J Cancer. 61:111–121. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Komura K, Jeong SH, Hinohara K, Qu F, Wang
X, Hiraki M, Azuma H, Lee GS, Kantoff PW and Sweeney CJ: Resistance
to docetaxel in prostate cancer is associated with androgen
receptor activation and loss of KDM5D expression. Proc Natl Acad
Sci USA. 113:6259–6264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Geynisman DM, Plimack ER and Zibelman M:
Second-generation androgen receptor-targeted therapies in
nonmetastatic castration-resistant prostate cancer: Effective early
intervention or intervening too early? Eur Urol. 70:971–973. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Juang HH, Hsieh ML and Tsui KH:
Testosterone modulates mitochondrial aconitase in the full-length
human androgen receptor-transfected PC-3 prostatic carcinoma cells.
J Mol Endocrinol. 33:121–132. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao H, Zhu C, Qin C, Tao T, Li J, Cheng
G, Li P, Cao Q, Meng X, Ju X, et al: Fenofibrate down-regulates the
expressions of androgen receptor (AR) and AR target genes and
induces oxidative stress in the prostate cancer cell line LNCaP.
Biochem Biophys Res Commun. 432:320–325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee WS, Kwon J, Yun DH, Lee YN, Woo EY,
Park MJ, Lee JS, Han YH and Bae IH: Specificity protein 1
expression contributes to Bcl-w-induced aggressiveness in
glioblastoma multiforme. Mol Cells. 37:17–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Simon P: Q-Gene: Processing quantitative
real-time RT-PCR data. Bioinformatics. 19:1439–1440. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dottori M, Down M, Hüttmann A, Fitzpatrick
DR and Boyd AW: Cloning and characterization of EphA3 (Hek) gene
promoter: DNA methylation regulates expression in hematopoietic
tumor cells. Blood. 94:2477–2486. 1999.PubMed/NCBI
|
|
37
|
Liao X, Tang S, Thrasher JB, Griebling TL
and Li B: Small-interfering RNA-induced androgen receptor silencing
leads to apoptotic cell death in prostate cancer. Mol Cancer Ther.
4:505–515. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Boyd AW, Ward LD, Wicks IP, Simpson RJ,
Salvaris E, Wilks A, Welch K, Loudovaris M, Rockman S and Busmanis
I: Isolation and characterization of a novel receptor-type protein
tyrosine kinase (hek) from a human pre-B cell line. J Biol
Chem. 267:3262–3267. 1992.PubMed/NCBI
|
|
39
|
Pasquale EB: Eph receptors and ephrins in
cancer: Bidirectional signalling and beyond. Nat Rev Cancer.
10:165–180. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Matsumoto T, Sakari M, Okada M, Yokoyama
A, Takahashi S, Kouzmenko A and Kato S: The androgen receptor in
health and disease. Annu Rev Physiol. 75:201–224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vizcaíno C, Mansilla S and Portugal J: Sp1
transcription factor: A long-standing target in cancer
chemotherapy. Pharmacol Ther. 152:111–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Beishline K and Azizkhan-Clifford J: Sp1
and the ‘hallmarks of cancer’. FEBS J. 282:224–258. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Safe S, Imanirad P, Sreevalsan S, Nair V
and Jutooru I: Transcription factor Sp1, also known as specificity
protein 1 as a therapeutic target. Expert Opin Ther Targets.
18:759–769. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sankpal UT, Goodison S, Abdelrahim M and
Basha R: Targeting Sp1 transcription factors in prostate cancer
therapy. Med Chem. 7:518–525. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Eisermann K, Broderick CJ, Bazarov A,
Moazam MM and Fraizer GC: Androgen up-regulates vascular
endothelial growth factor expression in prostate cancer cells via
an Sp1 binding site. Mol Cancer. 12:72013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lu S, Jenster G and Epner DE: Androgen
induction of cyclin-dependent kinase inhibitor p21 gene: Role of
androgen receptor and transcription factor Sp1 complex. Mol
Endocrinol. 14:753–760. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia
L, Lee LF, Pretlow TG and Kung HJ: Characterization of a novel
androgen receptor mutation in a relapsed CWR22 prostate cancer
xenograft and cell line. Cancer Res. 62:6606–6614. 2002.PubMed/NCBI
|
|
48
|
Attardi BJ, Burgenson J, Hild SA and Reel
JR: Steroid hormonal regulation of growth, prostate specific
antigen secretion, and transcription mediated by the mutated
androgen receptor in CWR22Rv1 human prostate carcinoma cells. Mol
Cell Endocrinol. 222:121–132. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li Y, Hwang TH, Oseth LA, Hauge A,
Vessella RL, Schmechel SC, Hirsch B, Beckman KB, Silverstein KA and
Dehm SM: AR intragenic deletions linked to androgen receptor splice
variant expression and activity in models of prostate cancer
progression. Oncogene. 31:4759–4767. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Singh AP, Bafna S, Chaudhary K,
Venkatraman G, Smith L, Eudy JD, Johansson SL, Lin MF and Batra SK:
Genome-wide expression profiling reveals transcriptomic variation
and perturbed gene networks in androgen-dependent and
androgen-independent prostate cancer cells. Cancer Lett. 259:28–38.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yuan TC, Veeramani S and Lin MF:
Neuroendocrine-like prostate cancer cells: Neuroendocrine
transdifferentiation of prostate adenocarcinoma cells. Endocr Relat
Cancer. 14:531–547. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bao BY, Chuang BF, Wang Q, Sartor O, Balk
SP, Brown M, Kantoff PW and Lee GS: Androgen receptor mediates the
expression of UDP-glucuronosyltransferase 2 B15 and B17 genes.
Prostate. 68:839–848. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang Y, Wang Y, Liu Q, Xu G, Mao F, Qin T,
Teng H, Cai W, Yu P, Cai T, et al: Comparative RNA-seq analysis
reveals potential mechanisms mediating the conversion to androgen
independence in an LNCaP progression cell model. Cancer Lett.
342:130–138. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen CD and Sawyers CL: NF-kappa B
activates prostate-specific antigen expression and is upregulated
in androgen-independent prostate cancer. Mol Cell Biol.
22:2862–2870. 2002. View Article : Google Scholar : PubMed/NCBI
|