Introduction
Gastric cancer (GC) is a common malignancy, which
has been suggested to be the third most common cause of
cancer-associated mortality worldwide (1). In general, GC is diagnosed at advanced
stages and prognosis is poor. Several drugs are available for the
treatment of GC; however, the prognosis for metastatic disease
remains unsatisfactory (2,3). The estimated overall survival rate
(OS) of patients with metastatic GC that receive conventional
chemotherapeutic agents has been reported to be as low as 10% at 5
years (4). However, several
innovative, biological therapies have been introduced for the
treatment of GC. The trastuzumab for GC (TOGA) trial was the first
randomized phase III trial, which demonstrated an advantage in
terms of progression-free survival (PFS) and OS rates for patients
with positive human epidermal receptor 2 (HER-2) GC tumors
(5). Unfortunately, only a small
percentage of patients (~20%) are ideal candidates for
HER-2-targeted therapy (5–7). In addition, ramucirumab, a human
monoclonal antibody directed against vascular endothelial growth
factor receptor (VEGFR)-2, which was initially developed for the
treatment of human tumors, has shown significant survival benefits
as a second line treatment option for patients with metastatic GC
who progressed on fluoropyrimidine- or platinum-based first-line
chemotherapy (8,9). Other antiangiogenic agents, including
bevacizumab, sunitinib and sorafenib, have failed to demonstrate
any survival benefit (10).
Previous studies have supported the inhibition of angiogenesis in
GC (11). Apatinib (Apa), a novel
receptor tyrosine kinase inhibitor that selectively targets the
intracellular ATP binding site of VEGFR-2 (12,13),
has shown promising results in preclinical and clinical trials
involving patients with GC. Apa has shown a suitable safety and
tolerance profile, and a sufficient treatment efficacy in phase
I/II trials. In a phase III trial, Apa prolonged the median OS
rates of patients with chemotherapy-refractory metastatic GC by 55
days and the median PFS rate by 25 days, when compared with
placebo-treated patients (14).
Dual-specificity phosphatase-1 (DUSP1) was initially
identified in cultured murine cells (15,16).
DUSP1 has two domains, including an amino terminal non-catalytic
domain and a C-terminal catalytic domain, the latter containing the
ATPase active site consensus sequence (17,18).
Previous studies have shown that DUSP1 regulates MAPK signaling and
is involved in cell proliferation, differentiation, transformation,
stress responses, inflammation, cycle arrest and apoptosis
(19–21). In addition, in vivo studies
have demonstrated that DUSP1 inactivates extracellular
signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and
p38 by a dephosphorylation processes (22–25).
In several human epithelial tumors, elevated levels of DUSP1 have
been reported, including in prostate, colon and bladder cancer
(26–28). However, the expression of DUSP1 in
tumors progressively decreased with a higher histological grade,
indicating that the function and mechanism of DUSP1 in tumors may
vary and is complex. In several studies, it has been reported that
tumor cell resistance was closely associated with DUSP1, including
lung cancer, ovarian cancer, osteosarcoma, breast cancer, hilar
cholangiocarcinoma, acute lymphoid system leukemia, prostate cancer
and glioma cancer cells (29–38).
Upon the expression of DUSP1, the chemotherapeutic resistance of
tumor cells is enhanced (31).
However, if DUSP1 activity is decreased, the chemotherapeutic
resistance of tumor cells reduces, resulting in tumor cells with
higher sensitivity (29).
Triptolide, a bioactive ingredient extracted from
Tripterygium wilfordii, which is a Chinese medicinal plant,
has been reported to exhibit antitumor effects in several types of
human cancer (39–41). Triptolide has been used in clinical
trials (42) and it is hypothesized
is that its antitumor effects are mediated by the suppression of
oncoproteins or growth factors, including DUSP1 (43), nuclear factor (NF)-κB (44), and heat shock proteins (HSPs)
(45). However, the antitumor
effects of triptolide in GC remain to be elucidated.
In the present study, the function and mechanism of
DUSP1 in Apa resistance were investigated. It was demonstrated that
combined treatment of Apa and triptolide, as protein inhibitors of
DUSP1, were able to overcome Apa resistance in GC cells.
Materials and methods
Cell lines
The human MGC803 and MKN45 GC cell lines were
purchased from the Cell Bank of Chinese Academy of Science
(Beijing, China). High-dose Apa resistant MGC803 cells (MGC803-AR)
and MKN45 cells (MKN-45AR) were derived from the MGC803 and MKN45
cells, respectively. In brief, the MGC803 and MKN45 cells were
treated with gradually increasing concentrations of Apa (5–80 µM)
for 2 months, and were maintained in RPMI medium (GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA), containing 10% fetal bovine
serum (FBS; GE Healthcare Bio-Sciences) and 80 µM Apa for 6 months
at 37°C in 5% CO2. Prior to experiments, the cells were
cultured in RPMI medium containing 10% FBS for 1 week.
Cell Counting Kit-8 (CCK-8) assay
A CCK-8 assay was used to determine the cell
proliferation ability of the MGC803, MGC803-AR, MKN45 and MKN45-AR
cells. In brief, the cells were seeded into 96-well cell culture
plates (Nest Biotechnology Co., Ltd., Wuxi, China) at a density of
5×103 cells/well in 100 µl RPMI medium containing 10%
FBS. Following culturing in RPMI medium for 24 h at 37°C, apatinib
and triptolide were administrated for 24 h. Subsequently, 10 µl of
CCK-8 reagent (Dojindo, Inc., Tokyo, Japan) was added to each well
and the cells were incubated for 2 h at 37°C according to the
manufacturer's protocol. Subsequently, the absorbance was read at a
wavelength of 450 nm using an automated plate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The experiments were
performed in triplicate.
cDNA synthesis and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Monolayers of MGC803, MGC803-AR, MKN45 and MKN45-AR
cells were harvested, cells were lysed, and total RNA was extracted
using the TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) method according to the manufacturer's protocol.
The total RNA (100 ng) was reverse transcribed into cDNA using a
Takara PrimeScript First Strand cDNA Synthesis kit (Takara Bio,
Inc., Shiga, Japan). RT-qPCR analysis was performed using the
Takara SYBR Premix Ex Taq kit (Takara Bio, Inc.). The sequences of
DUSP1 primers were as follows: Forward 5′-ACCACCACCGTGTTCAACTT-3′
and reverse 5′-CTCAAGGAGCATGGAGTCCC-3′. The sequences of ACTB
primers were as follows: Forward 5′-CGCCGCCAGCTCACC-3′ and reverse
5′-GACCCATGCCCACCATCAC-3′. Experiments were carried out in
triplicate, and the comparative CT was calculated to analyze the
expression levels of DUSP1 in GC or ARGC cells (46). ACTB were analyzed as a reference
gene for mRNA.
DUSP1 silencing with small interfering
(si)RNA
The MGC803-AR and MKN45-AR cells were transiently
transfected with DUSP1 siRNA or scrambled siRNA in 6-well plates
using TurboFect reagent according to the manufacturer's protocol.
The cells were used for experiments at 48 h post-transfection.
Cells transfected with scrambled siRNA were used as controls. The
siRNA sequences were as follows: DUSP1 siRNA-1, forward
5′-CUAAUCGAGUCAAGCUGGA-3′ and reverse 5′-UCCAGCUUGACUCGAUUAG-3′;
DUSP1 siRNA-2, forward 5′-GCUUACCUUAUGAGGACUA-3′ and reverse
5′-UAGUCCUCAUAAGGUAAGC-3′; DUSP1 siRNA-3, forward
5′-CGACGACACAUAUACAUAU-3′ and reverse
5′-AUAUGUAUAUGUGUCGUCG-3′.
Western blot analysis
For western blot analysis, the MGC803, MGC803-AR,
MKN45 and MKN45-AR cells were washed in PBS and lysed in RIPA
buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5%
deoxycholate, 0.1% SDS) containing SigmaFAST™ Protease Inhibitor
Cocktail (Sigma-Aldrich; Merck KGaA) for 15 min at 4°C. Cells were
removed by scraping and centrifuged at 8,000 × g rotations per
minute for 20 min at 4°C. Protein concentration was measured using
Bio-Rad DC Protein Assay kit (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). Equal quantities of total protein (40 µg) were loaded
onto 10% SDS-PAGE gels and separated. The proteins were the
transferred onto polyvinylidene fluoride membranes, and the
membranes were blocked in TBS-Tween (0.1%)/5% milk for 1 h at room
temperature. The membranes were then incubated overnight with
primary antibodies directed against DUSP1 (cat. no. Ab61201) from
Abcam (Cambridge, UK) and phosphorylated (p)-DUSP1 (cat. no. 2857),
JNK (cat. no. 9252), p-JNK (cat. no. 4668), ERK (cat. no. 469),
p-ERK (cat. no. 4370), P38 (cat. no. 8690), p-P38 (cat. no. 4511),
poly(ADP-ribose) polymerase (PARP) (cat. no. 9664), β-actin (ACTB)
(cat. no. 3700) from Cell Signaling Technology, Inc. (Danvers, MA,
USA) at 4°C. The primary antibodies were diluted 1:1,000 with
universal antibody dilution buffer (Sigma; EMD Millipore,
Billerica, MA, USA). The membranes were washed three times for 10
min with TBS-Tween (0.1%) at room temperature, and then incubated
with goat anti-rabbit IgG (H+L) secondary antibodies (dilution
1:3,000; cat. no. 70-GAR007; MultiSciences, Co., Ltd., Hangzhou,
China) for DUSP1, p-DUSP1, JNK, p-JNK, ERK, p-ERK, P38, p-P38 and
PARP, and goat anti-mouse IgG (H+L) secondary antibodies (dilution
1:3,000; cat. no. 70-GAM007; Hangzhou MultiSciences) for ACTB for 1
h at room temperature. The membranes were washed three times for 10
min with TBS-Tween (0.1%) at room temperature. The protein bands
were visualized using enhanced chemiluminescence reagents (EMD
Millipore). Images were captured and protein levels were quantified
using a ChemiDoc™ MP system (Bio-Rad Laboratories).
Cell cycle analyses
The MGC803 and MGC803-AR cells were seeded into
6-well plates at a density of 3×105 cells/well.
Following overnight incubation at 37°C, the cells were treated with
Apa at concentrations of 5, 10 and 20 µM for 24 h. For the
detection of apoptosis, the cells were harvested and washed with
PBS, and then stained with Annexin V/PI according to the
manufacturer's protocol. The stained cells were analyzed by flow
cytometry (FACSCalibur; BD Biosciences, San Jose, CA, USA). For the
analysis of cell cycle distribution, the supernatant was discarded,
and the attached cells were harvested and fixed overnight in cold
70% ethanol at −20°C. Cell cycle distribution was evaluated using a
flow cytometer provided with the PI staining kit according to the
manufacturer's protocol.
Hoechst 33342 staining assay
The MGC803-AR and MKN45-AR cells were seeded into
96-well plates at a density of 1×106 cells/well. The
cells were then transfected with siRNA and/or treated with 10 µM
Apa or 1 µM triptolide. Following incubation in fresh RPMI medium
containing 10% FBS for 6 h, the cells were incubated in 5 µl of
Hoechst 33342 (Beyotime Institute of Biotechnology, Shanghai,
China) solution per well at 37°C for 10 min, and evaluated using a
fluorescence microscope. The nuclei of apoptotic cells exhibited a
high level fluorescence, whereas the fluorescence observed in
non-apoptotic cells was weak. Images were captured and
quantification of apoptotic cells was performed by counting at
least 200 cells in four randomly selected fields per well.
Colony formation assays
For the colony formation assay, the cells
(3×104 cells/well) were seeded into 6-well plates and
were treated with the indicated agents for 5 days. The medium was
then replaced with drug-free medium and the cells were incubated
for another 10 days. Subsequently, the cells were cultured with 4%
paraformaldehyde for 15 min then were stained with crystal violet
solution for 30 min before being washed with PBS.
Immunohistochemical (IHC)
staining
A total of 101 tumors collected at various
departments between January 2009 and March 2011 in Zhejiang
Province Tumor Hospital, from patients with newly diagnosed gastric
adenocarcinoma, were included in this study. There are 71 male and
30 female patients in this study and the age is from 31 to 80 years
with the average age 61 years. All available clinical factors were
evaluated. Informed consent was obtained from all patients before
testing. The study was approved by the Medical Ethics Committee of
Zhejiang Province Tumor Hospital (Hangzhou, China). Tumor specimens
were collected to analyze the expression of DUSP1 using IHC
staining. These tissues were fixed in 10% formaldehyde and embedded
in paraffin. Sections (3–5 µm) were continuously sliced. After
dewaxing by xylene, the tissues were dehydrated in 100, 100, 85 and
75% gradient alcohol. Hydrogen peroxide (3%) was applied to repair
the antigen. The non-specific staining was blocked by 10% goat
serum at room temperature for 30 min. Immunostaining of
histological sections was performed using monoclonal antibodies
against DUSP1 (dilution 1:1,000; cat. no. Ab61201; Abcam) overnight
at 4°C followed by a 30-min incubation with goat anti-rabbit IgG
(H&L) Biotin secondary antibody (dilution 1:1,000; cat. no
AB97049; BioVision, Inc., Milpitas, CA, USA) and visualization with
3,3′-diaminobenzidine (DAB) for 3 min. Harris hematoxylin was used
to stain the nuclei. The sections were left to dehydrate, and
sealed with neutral gel. Categorization of the scanned images was
performed using density quant software in the Quant Center
(3DHistech, Ltd., Budapest, Hungary). A staining intensity score
between 0 and 3 was assigned for the intensity of tumor cells (0,
no staining; 1, weak staining; 2, intermediate staining; 3, strong
staining). A score was assigned by the estimated proportion of
positively-stained tumor cells. To assess the average degree of
staining within a tumor sample, multiple regions were analyzed, and
at least 100 tumor cells were assessed. The cytoplasmic expression
was assessed using the H-score system (22). The formula for the H-score was as
follows: H-score = ∑ (I × Pi), where I represents the staining
intensity and Pi represents the percentage of positively-stained
tumor cells, producing a cytoplasmic score ranging between 0 and
300 (47). Scoring was performed
independently by two assessors who were not informed of the
clinical outcomes.
Statistical analysis
For data analysis, SPSS version 18.0 (SPSS, Inc.,
Chicago, IL, USA) was used. Data are expressed as the mean ±
standard deviation. The unpaired t-test was used for comparing data
between two groups. Multiple group data and multiple comparisons
were analyzed by one-way analysis of variance and the LSD test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Expression of DUSP1 is upregulated in
AR gastric cancer cell lines
In the present study, two high-dose Apa-resistant GC
cell lines were established, MGC803-AR and MKN45-AR, respectively.
In brief, the MGC803 and MKN45 cells were treated with 80 µM of Apa
for 6 months. To verify the resistance of the MGC803-AR and
MKN45-AR cells against Apa, parental cells and AR cells were
treated with Apa or culture medium alone (control) for 24 h at
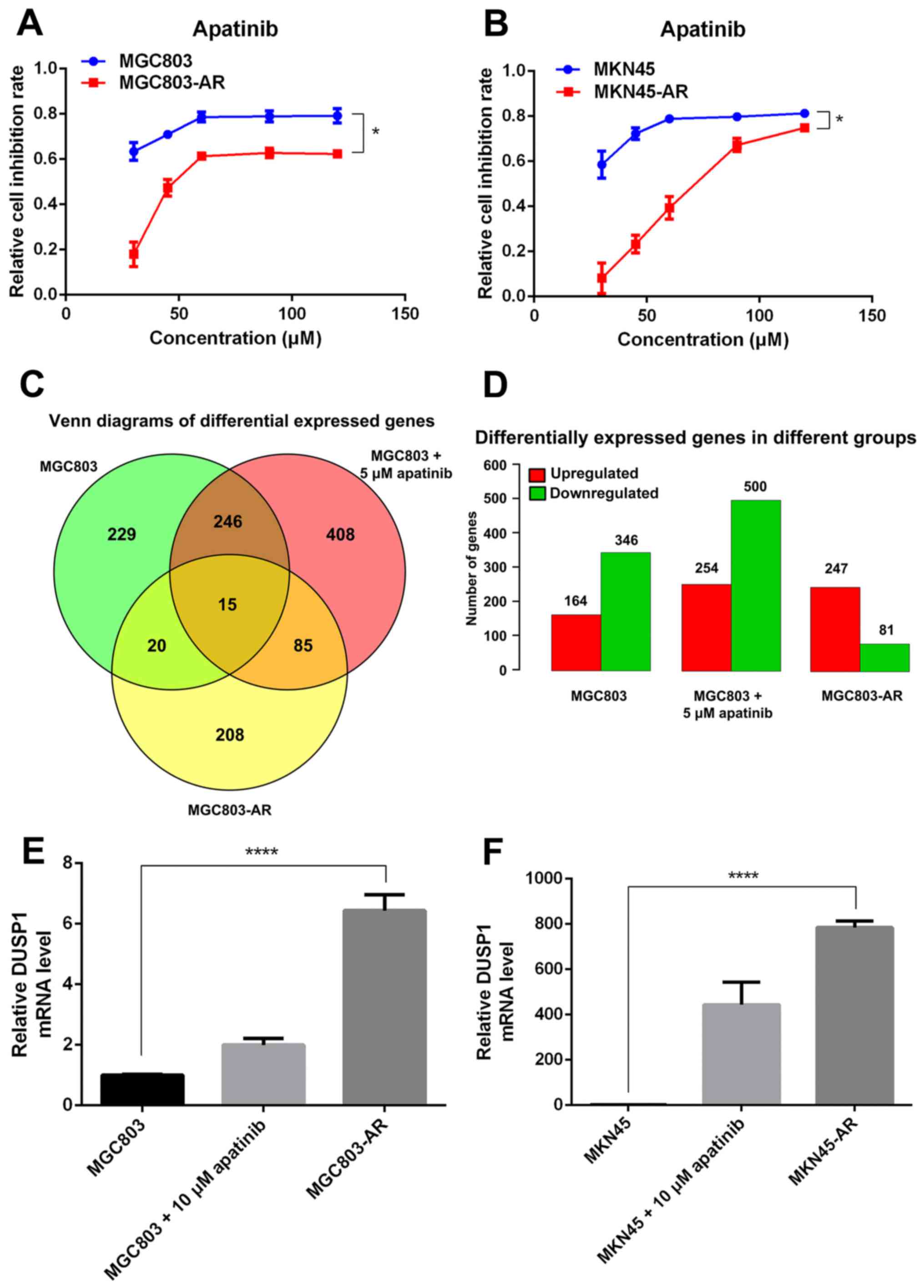
37°C. Cell proliferation was then determined using an CCK-8 assay.
As expected, treatment with Apa markedly inhibited the viability of
the parental MGC803 and MKN45 cells with IC50 values of
10.30 and 16.23 µM, respectively (Fig.
1A and B). By contrast, the MGC803-AR and MKN45-AR cells were
resistant to Apa inhibition with IC50 values of 60.83
and 72.36 µM, respectively (Fig. 1A and
B).
The Apa-treated cells were divided into three
groups, comprising MGC803 cells, MGC803 cells treated with 5 µM Apa
for 24 h, and MGC803-AR cells. Venn diagrams indicated that,
between the MGC803 and MGC803-AR cells, 20 genes were
differentially expressed. The number of differentially expressed
genes between the low-dose Apa-treated MGC803 cells and MGC803-AR
cells was 85. In addition, the number of genes differentially
expressed between these groups was 15 (Fig. 1C and D). As experimental subjects,
six of the 15 differentially expressed genes were selected, which
included DUSP1, HSPA1A, HSPA1B, serum response factor, tubulin β
(TUBB)2A and TUBB4B. RT-qPCR analysis demonstrated that, among
these six genes, DUSP1 showed the most marked significant
difference; therefore, DUSP1 was selected for further analysis.
To investigate the possible role of DUSP1 in Apa
resistance, the mRNA levels of DUSP1 in the MGC803-AR, MKN45-AR,
MGC803 and MKN45 cells were evaluated using RT-qPCR analysis. The
data indicated that the mRNA expression of DUSP1 was higher in the
MGC803-AR and MKN45-AR cells than in the MGC803 and MKN45 cells
(Fig. 1E and F).
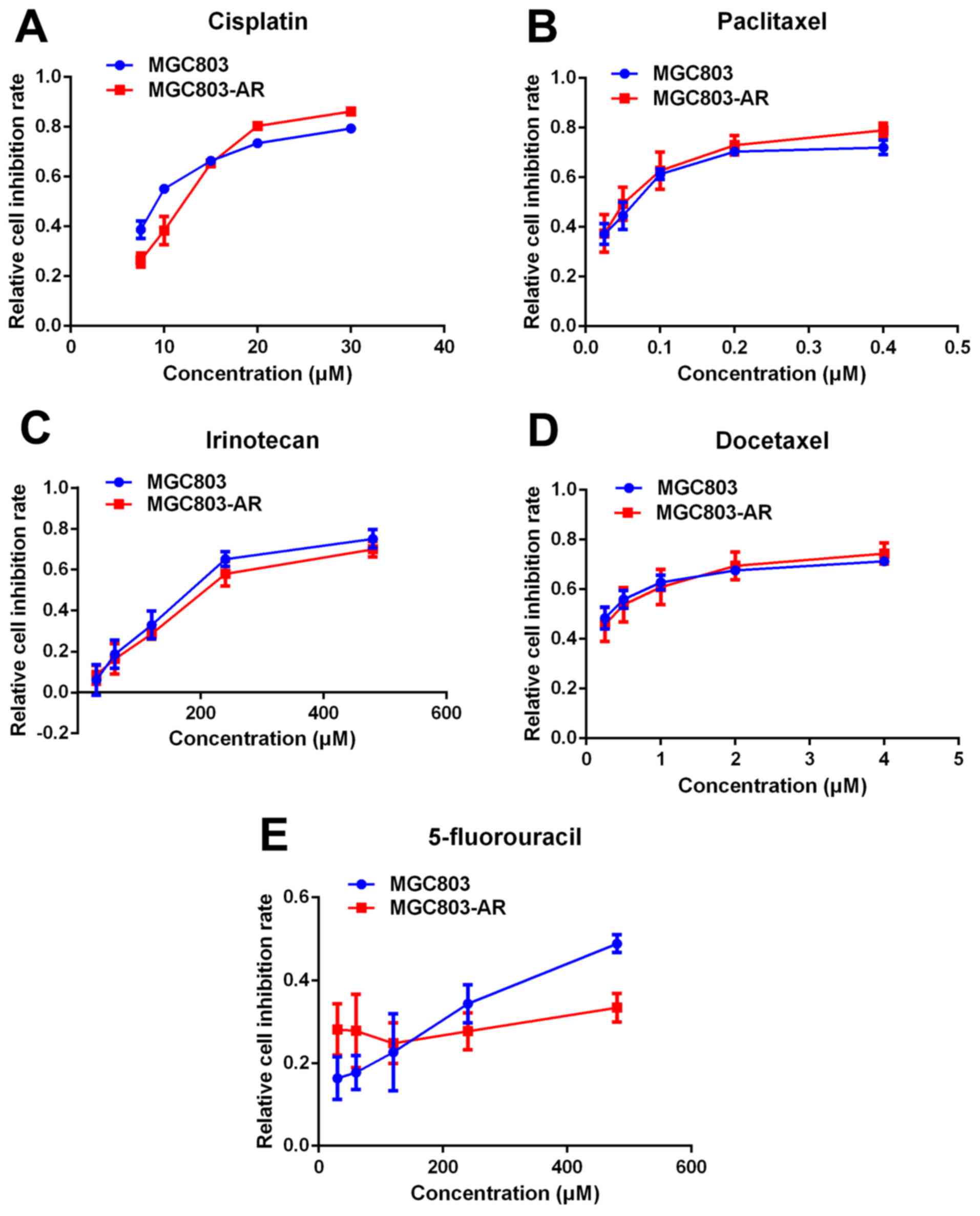
Apatinib resistance does not cause
multi-drug resistance of chemotherapy
Previous studies indicated that elevated levels of
DUSP1 may be involved in cellular responses to chemotherapy
(48). Therefore, it was
hypothesized that AR GC cells may be resistant to other drugs used
in chemotherapy. In the present study, the effect of five widely
used chemotherapeutic agents, cisplatin, paclitaxel, docetaxel,
irinotecan and 5-fluorouracil (5-FU), were analyzed in MGC803 and
MGC803-AR cells using a CCK-8 assay. The results showed no
significant difference in the effects of the five chemotherapeutic
agents between the MGC803 and MGC803-AR cells (Fig. 2A-E). Therefore, these findings
suggested that Apa resistance in GC cells did not cause multi-drug
resistance.
Apatinib resistance in GC cells
affects cell cycle, apoptosis and the MAPK signaling pathway
DUSP1 is located at a key position in the MAPK
signaling pathway, and directly interacts with downstream proteins,
including JNK, ERK and P38 (49).
Western blot analysis was performed to investigate whether changes
in the mRNA expression of DUSP1 in AR cells affected the expression
of these four proteins (Fig. 3A and
B).
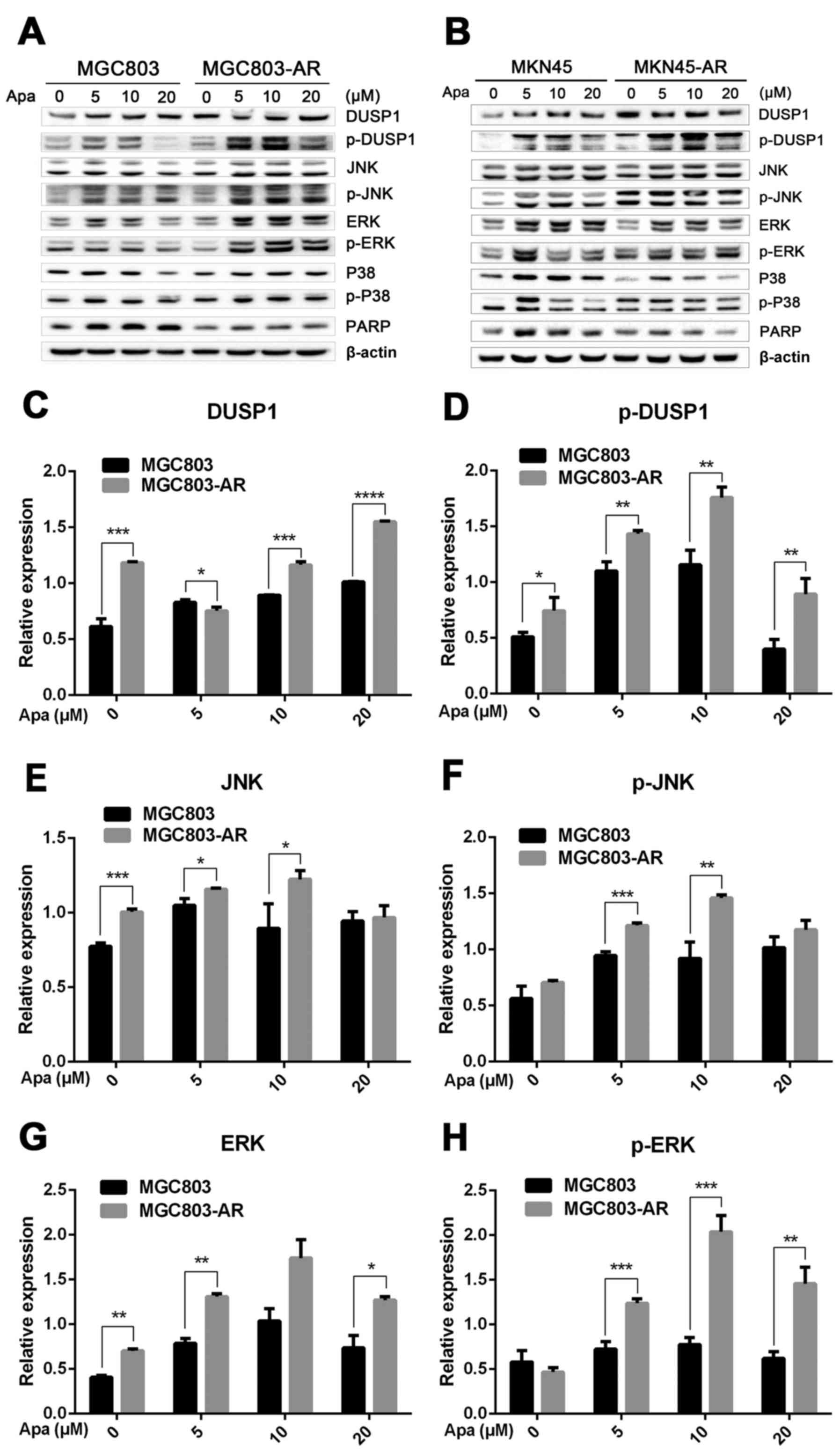 | Figure 3.Apa resistance in gastric cancer
cells affect cell cycle, apoptosis and MAPK signaling pathways. (A)
MGC803, MGC803-AR, (B) MKN45 and MKN45-AR cells were treated for 24
h with Apa at the indicated concentrations. Total cell lysates were
prepared and analyzed by western blot analysis using antibodies
directed against MAPK signaling molecules (C) DUSP, (D) p-DUSP, (E)
JNK, (F) p-JNK, (G) ERK, (H) p-ERK.. (I) P38, (J) p-P38, and (K)
PARP in MGC803, MGC803-AR cells, and (L) DUSP, (M) p-DUSP (N) JNK,
(O) p-JNK. (P) ERK, (Q) p-ERK, (R) P38, (S) p-P38, and (T) PARP in
MKN45 and MKN45-AR cells. β-actin served as a loading control.
Histograms representing the relative quantitative comparison of
proteins between parental cells and resistant cells. (U) MGC803 and
MGC803-AR cells were treated with Apa for 24 h at indicated
concentrations, and cell cycle distribution was analyzed by flow
cytometry. Histograms representing the relative cell cycle
distribution between sensitive cells and resistant cells at (V) 0,
(W) 5, (X) 10 and (Y) 20 µM Apa. Data are presented as the mean ±
standard deviation (n=3; Student's t-test; *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001). Apa, apatinib; AR, Apa-resistant;
MAPK, mitogen-activated protein kinase; DUSP, dual-specificity
phosphatase-1; JNK, c-Jun N-terminal kinase; ERK, extracellular
signal-regulated kinase; p-, phosphorylated; PARP, poly(ADP-ribose)
polymerase. |
The results showed that, compared with MGC803 cells,
the expression of DUSP1 was significantly increased in MGC803-AR
cells (P<0.001). In addition, p-DUSP1 was significantly
increased in MGC803-AR cells compared with that in MGC803 cells
(P<0.05). The levels of JNK and p-JNK, ERK and p-ERK were all
expressed at higher levels in the AR cells (P<0.05), regardless
of the concentration of Apa used. In addition, PARP was
significantly decreased in the MGC803-AR cells compared with the
MGC803 cells (P<0.05; Fig.
3C-K).
When the concentration of Apa increased, the
expression of DUSP1 gradually increased in MKN45 cells, whereas
that in MKN45-AR cells gradually decreased. In addition, the
expression of p-DUSP1 in AR cells was higher than that in parental
cells. This was the case at each concentration assessed in the
present study. The levels of p-ERK in the MKN45-AR cells gradually
increased, whereas levels decreased in MKN45 cells when higher
concentrations of Apa were used. At an Apa concentration of 5 µM,
the levels of p-ERK in MKN45 cells were significantly higher than
those in MKN-45AR cells (P<0.05). However, at 20 µM of Apa, the
levels of p-ERK in MKN45-AR cells was significantly higher compared
with those in MKN45 cells (P<0.05). At each concentration of Apa
examined, the levels of JNK were significantly higher in the
MKN45-AR cells compared with those in the MKN45 cells (P<0.01),
whereas the levels of PARP were higher in the MKN45 cells than in
the MKN45-AR cells (P<0.05; Fig.
3L-T).
As Apa affected the expression of PARP, the cell
cycle of MGC803 and MGC803-AR cells were analyzed following
treatment with different concentrations of Apa by flow cytometry.
As shown in Fig. 3U-Y, the cell
populations in the G1 and S phases were decreased, whereas the
population of cells in the G2 phase was increased in MGC803-AR
cells when compared with that of control cells. In particular, the
proportion of MGC803-AR cells in the G2 phase was significantly
higher when compared with that of MGC803 cells (P=0.04).
The protein expression of DUSP1 was high in ARGC
cells. Due to the overall physiological changes in resistant cells,
downstream proteins, including JNK, ERK and P38 were activated
(Fig. 4A-H). The protein expression
levels of JNK and ERK were higher in AR cells, compared with levels
in their corresponding parental cell lines, which may further
enhance cellular resistance to Apa. Following Apa treatment, the
protein expression of apoptotic PARP in AR cells was lower than
that in sensitive cells (P<0.05), indicating that Apa led to a
decrease of apoptosis in AR GC cells.
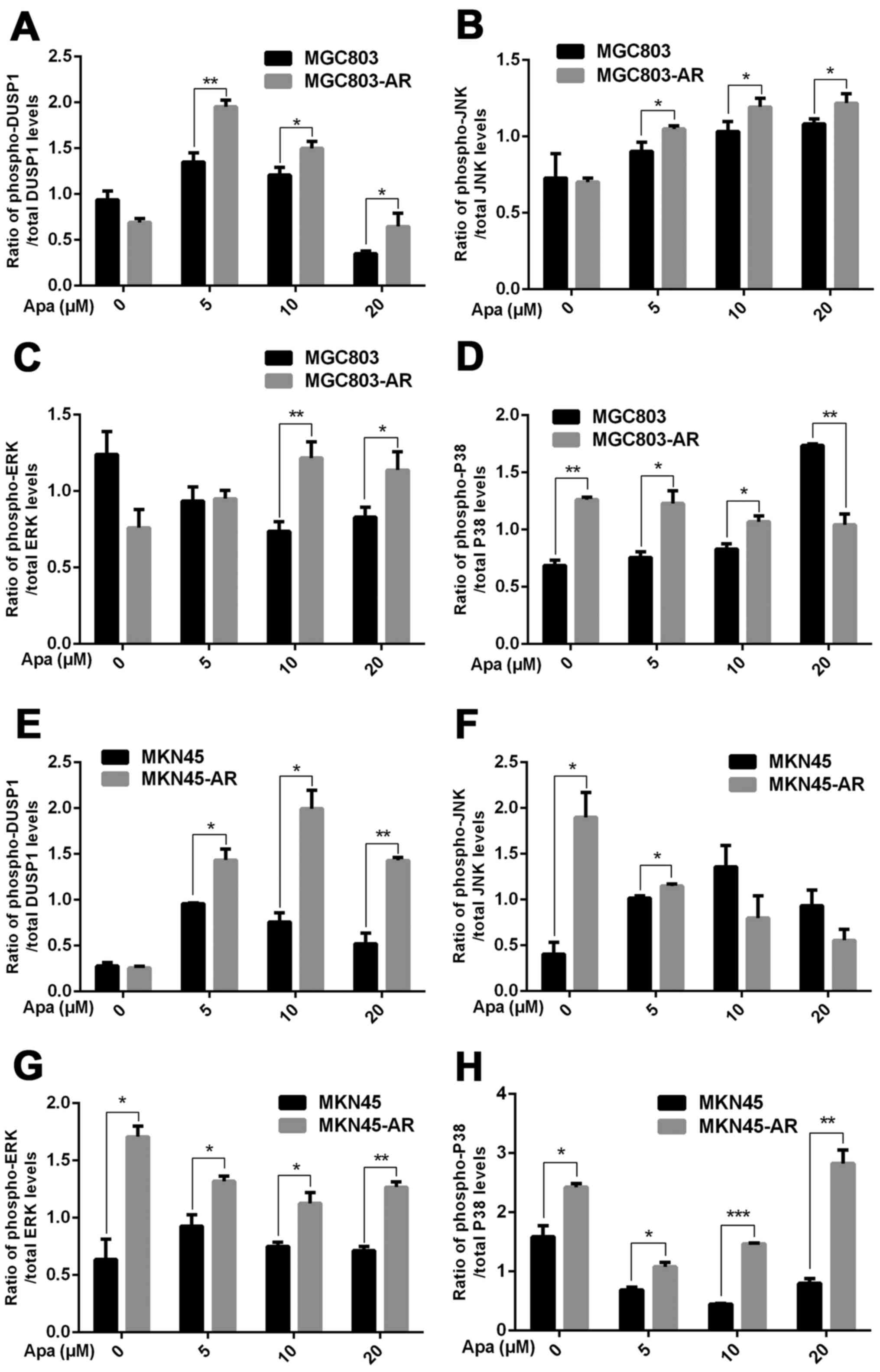 | Figure 4.Ratios of proteins involved in the
mitogen-activated protein kinase pathway between AR-resistant and
non-resistant gastric cancer cells. Ratios of (A) p-DUSP1/total
DUSP1 (B) p-JNK/total JNK, (C) p-ERK/total ERK and (D) p-P38/total
P38 in MGC803 and MGC803-AR cells, respectively. Ratios of (E)
p-DUSP1/total DUSP1 (F) p-JNK/total JNK, (G) p-ERK/total ERK and
(H) p-P38/total P38 in MKN45 and MKN45-AR cells, respectively (n=3;
Student's t-test; *P<0.05, **P<0.01, ***P<0.001). AR,
apatinib-resistant; p-/phospho, phosphorylated DUSP,
dual-specificity phosphatase-1; JNK, c-Jun N-terminal kinase; ERK,
extracellular signal-regulated kinase; p-, phosphorylated; PARP,
poly(ADP-ribose) polymerase. |
Knockdown of DUSP1 can overcome
apatinib resistance
In the present study, it was found that the mRNA and
protein levels of DUSP1 were elevated in MGC803-AR and MKN45-AR
cells. In previous studies, DUSP1 has been shown to be associated
with the resistance of several molecular targeting agents (50,51).
Therefore, transient transfection of siRNA targeting DUSP1 was
performed in the present study to silence the expression of DUSP1,
and changes indicating drug resistance were observed. As shown in
Fig. 5A-D, siRNA-2 had the most
marked inhibitory effect on DUSP1 protein, therefore, siRNA-2 and
the scramble siRNA sequence were selected for further
investigation. Following transfection of the corresponding oligos
into MGC803-AR and MKN45-AR cells, the inhibition rates of cells
treated with Apa were determined using a CCK-8 assay. The knockdown
of DUSP1 by siRNA treatment decreased the IC50 values of
the MGC803-AR and MKN45-AR cells to 32.19 and 25.18 µM,
respectively, indicating that DUSP1 was involved in Apa resistance
(Fig. 5E and F). In addition, a
colony formation assay showed that the knockdown of DUSP1 decreased
the colony-forming ability of cells exposed to Apa (Fig. 5G and H).
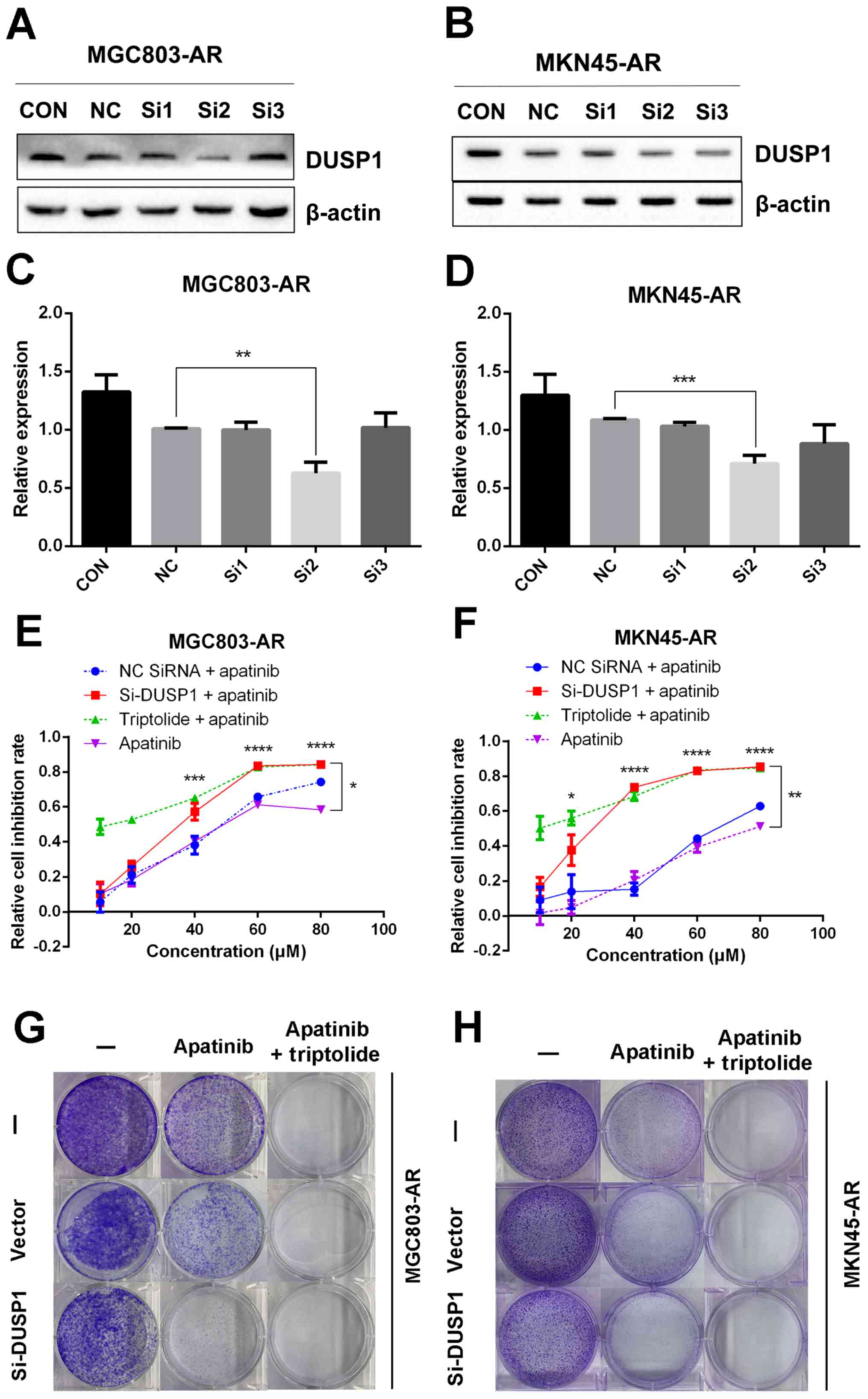 | Figure 5.Knockdown of DUSP1 overcomes Apa
resistance. (A) MGC803-AR and (B) MKN45-AR cells were treated with
siRNA targeting DUSP1 (clones Si1, Si2 and Si3), and expression of
DUSP1 was evaluated by western blot analysis. β-actin served as a
loading control. Histograms represent the relative quantitative
expression in (C) MGC803-AR and (D) MKN45-AR cells. Data are
presented as the mean ± standard deviation (n=3; Student's t-test;
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001). (E)
MGC803-AR and (F) MKN45-AR cells were treated with siRNA targeting
DUSP1, siRNA control, or triptolide, respectively, and exposed to
Apa for 24 h at different concentrations. Cell viability was
determined using a Cell Counting Kit-8 assay. The statistical
differences between Apa + triptolide and Apa only groups are above
the curve; statistical differences between the scramble siRNA NC +
Apa and si-DUSP1 + Apa groups are shown to the right of the curve.
Data are presented as the mean ± standard deviation (n=5; Student's
t-test; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001).
(G) MGC803-AR and (H) MKN45-AR cells were transiently transfected
with a scramble siRNA or DUSP1 siRNA, then treated with 10 µM Apa
or 10 µM Apa + 1 µM triptolide for 5 days. Cells were stained with
crystal violet and analyzed. Apa, apatinib; AR, Apa-resistant DUSP,
dual-specificity phosphatase-1; si, small interfering RNA; CON,
control; NC, negative control. |
Triptolide combined with apatinib
overcomes apatinib resistance by inhibiting MAPK signaling and
inducing apoptosis
Subsequently, the present study examined the
specific inhibition of triptolide on DUSP1 protein. Triptolide, a
specific inhibitor of the DUSP1 protein, can arrest the cell cycle
without causing cell apoptosis (43,52,53).
Therefore, a triptolide concentration gradient was established to
identify the optimal concentration of triptolide that inhibits
DUSP1 in GC cell lines. In the MGC803-AR cells, 1 µM of triptolide
inhibited the expression of DUSP1. In addition, 0.25 µM of
triptolide significantly decreased the levels of DUSP1 in MKN45-AR
cells. Considering all factors, a concentration of 1 µM triptolide
appeared optimal (Fig. 6A-D). To
verify whether triptolide treatment combined with Apa can overcome
Apa resistance, the cell inhibition rate of triptolide alone or
triptolide combined with Apa was determined using a CCK-8 assay
(Fig. 5E and F). The results
demonstrated that, when 1 µM of triptolide and Apa were
administered, the IC50 of the MGC803-AR and MKN45-AR
cells was reduced to 13.61 and 12.06 µM, respectively. When Apa was
used alone, the IC50 values obtained were 60.83 µM in
the MGC803-AR cells and 72.36 µM in the MKN45-AR cells.
Subsequently, Hoechst staining was performed to detect cells
undergoing apoptosis (Fig. 6E). To
identify differences between AR GC cells exposed to Apa following
treatment with siRNA-2 or control siRNA, the cells were stained
with Hoechst. Within similar-sized fields, and following exposure
to 10 µM of Apa, a higher number of apoptotic cells were present in
the cells with DUSP1 knockdown, compared with cells in the scramble
control group. Together, these findings suggested that the
knockdown of DUSP1 resulted in the apoptosis of AR GC cells and
reversed Apa resistance. The MGC803-AR cells and MKN45-AR cells
showed a weak fluorescence intensity for Hoechst when treated with
triptolide alone. These findings were consistent with the findings
presented in previous studies in which triptolide affected the cell
cycle, but did not affect apoptosis (43,52,53).
However, when triptolide and Apa were combined, a significant
increase in Hoechst-related fluorescence intensity was found. This
observation indicated that triptolide not only inhibited the
expression of DUSP1, but also enhanced the effect of Apa on
apoptosis. Furthermore, the present study examined the effect of
triptolide and Apa treatment on the MAPK signaling pathway
(Fig. 6F and G). Compared with Apa
treatment alone, the combination of Apa and triptolide
significantly inhibited MAPKs. The combined treatment effects
include promotion of cell proliferation and survival via the ERK
pathway, inhibition of cell apoptosis via the JNK pathway, and
regulation of the cell cycle via the P38 pathway (54–60).
The levels of DUSP1 were significantly inhibited by combination
treatment (Fig. 6F-Y), which may
explain why triptolide combined with Apa reversed Apa
resistance.
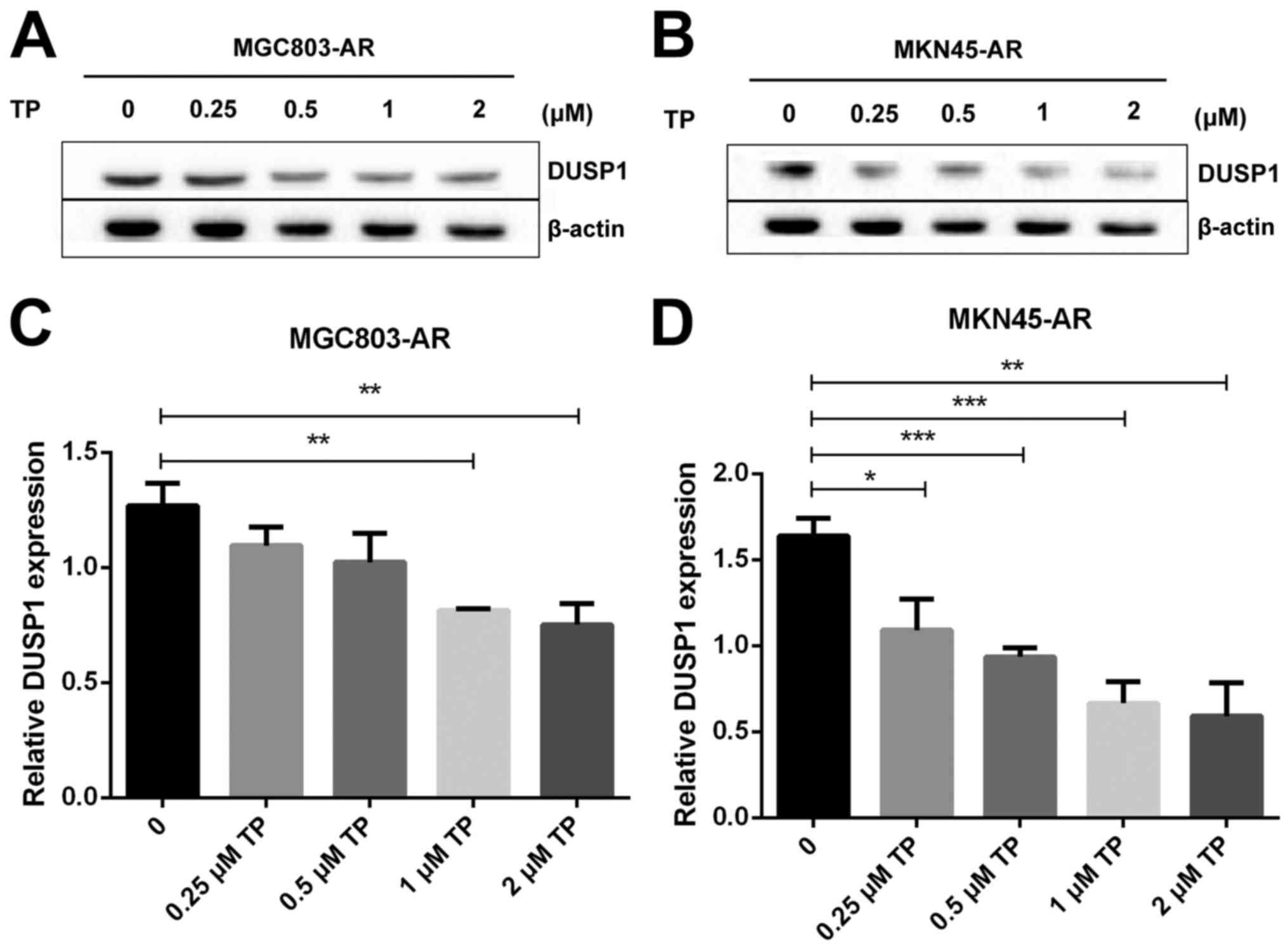 | Figure 6.Triptolide combined with Apa
overcomes Apa resistance by inhibiting MAPK signaling and inducing
apoptosis. (A) MGC803-AR and (B) MKN45-AR cells were treated with a
concentration gradient of triptolide, and expression of DUSP1 was
evaluated by western blot analysis using β-actin as a loading
control. Histograms show the relative quantitative expression in
(C) MGC803-AR and (D) MKN45-AR cells. Data are presented as the
mean ± standard deviation (n=3; Student's t-test; *P<0.05,
**P<0.01, ***P<0.001, ****P<0.0001). (E) MGC803-AR and
MKN45-AR cells were treated with 10 µM Apa and cells with DUSP1
knockdown were treated with 10 µM Apa or 1 µM triptolide alone or
10 µM Apa + 1 µM triptolide for 6 h. Cells were stained with
Hoechst 33342 and images were captured using an Olympus BH-2
fluorescence microscope (magnification, ×40). (F) MGC803-AR and (G)
MKN45-AR cells were treated with Apa or Apa + 1 µM triptolide for
24 h at different Apa concentrations. Total cell lysates were
prepared and analyzed by western blot analysis using antibodies
directed against MAPK signaling molecules (H) DUSP1, (I) p-DUSP1,
(J) JNK and (K) p-JNK. (L) ERK, (M) p-ERK, (N) P38, (O) p-P38, (P)
PARP in MGC803-AR cells; and (Q) DUSP1, (R) p-DUSP1, (S) JNK. and
(T) p-JNK (U) ERK, (V) p-ERK, (W) P38, (X) p-P38, (Y) PARP in
MKN45-AR cells. β-actin was a loading control. Data are presented
as the mean ± standard deviation (n=3; Student's t-test;
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001). Apa/AP,
apatinib; AR, Apa resistant; MAPK, mitogen-activated protein
kinase; p-, phosphorylated DUSP, dual-specificity phosphatase-1;
JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated
kinase; p-, phosphorylated; PARP, poly(ADP-ribose) polymerase. |
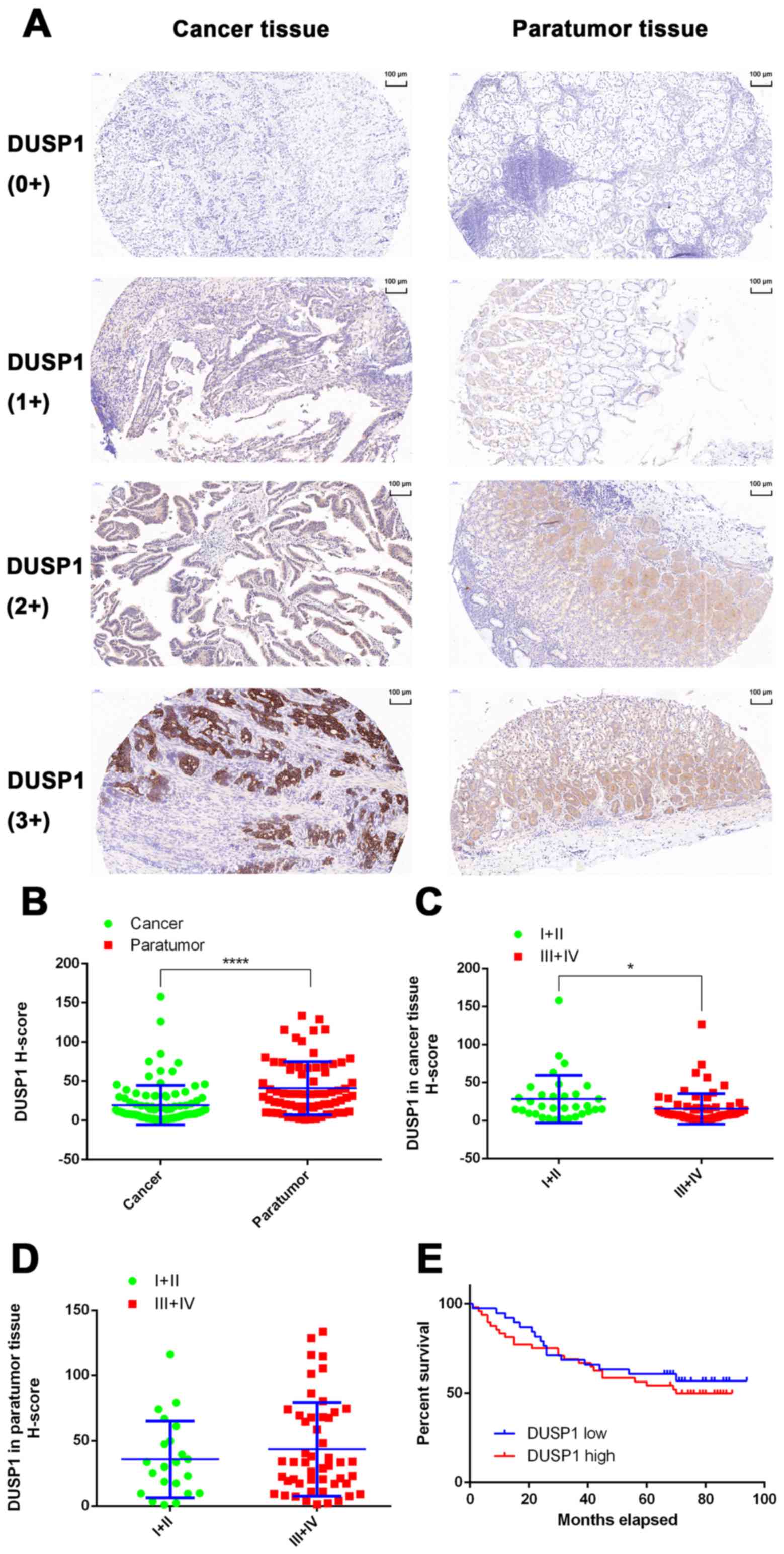
Expression of DUSP1 in tissues of
patients with GC and the effect of DUSP1 on the prognosis of
GC
In the present study, the expression of DUSP1 was
evaluated in clinical tissue samples from 101 patients with GC.
None of the patients had undergone chemotherapy or targeted-drug
therapy prior to surgery. In 72 of the 101 patients, both cancerous
and paracancerous tissues were collected. In the remaining 29
patients, only cancerous tissue was obtained (Table I). A total of 101 GC tissues and 72
paracancerous tissues were used for IHC experiments and tissues
were scanned using the Pannoramic MIDI Tissue Chip Scanner
(3DHistech, Ltd.). The GC cells were quantified in tumor tissues,
whereas fundic gland cells were quantified in non-tumor tissues.
The system automatically identified all dark-brown colored areas in
the tissue section as strongly positive, brown-yellow as moderately
positive, light-yellow as weak positive, and blue nuclei as
negative (Fig. 7A). For statistical
analysis, an H-scoring system was used. The results showed that the
expression level of DUSP1 in GC tissues was significantly lower
than that in adjacent tissues (P<0.0001; Fig. 7B). In addition, it was found that
the expression of DUSP1 in cancerous tissue derived from stage I
and II patients was higher than that of patients with a GC stage of
III and IV (P<0.05; Fig. 7C). In
paracancerous tissue, no significant differences were observed
between the expression of DUSP1 and tumor stage (Fig. 7D). Finally, the tissue samples were
grouped based on the H-Score. A score of >10 points was assigned
for high levels of DUSP1, whereas a score of <10 points was
assigned for low levels of DUSP1. A total of 39 patients were
grouped in the high expression group, whereas the low expression
group contained 48 patients. According to the patients' follow-up
data, a survival curve was plotted and no statistically significant
differences were observed (Fig.
7E). Therefore, it was hypothesized that, in patients who did
not receive drugs or surgical treatment, the expression of DUSP1 in
cancerous tissue had minimal effect on survival rate or
prognosis.
 | Table I.Information on patient tissues used
for immunohistochemical analysis. |
Table I.
Information on patient tissues used
for immunohistochemical analysis.
|
| Tissue samples
(n) |
|---|
|
|
|
|---|
| Factor | Cancer | Paratumor |
|---|
| Patients | 101 | 72 |
| Tumor stage |
|
I+II | 33 | 22 |
|
III+IV | 68 | 50 |
| Follow-up DUSP1
H-score |
| High
(≥10) | 39 | – |
| Low
(<10) | 48 | – |
Discussion
VEGFR2-targeted therapy not only represents a novel
treatment regimen but also provides novel biologic insight into GC,
in which Apa leads to increased PFS and significantly prolonged OS
rates in patients with GC (61).
However, the emergence of acquired resistance is inevitable and
remains a major obstacle. Resistance to Apa suggests the
involvement of signaling pathways additional to VEGFR2, including
c-Met amplification (62), PI3K
catalytic subunit α mutations (63), or BRAF mutations (64). In the present study, two AR GC cell
lines were generated, and the role of DUSP1 in the resistance of GC
cells to Apa was investigated.
In previous studies, DUSP1 has been reported to be
associated with the drug resistance of tumor cells (31,50,51,65–67).
In the present study, it was shown that the mRNA expression of
DUSP1 was significantly increased in AR GC cells. To further
demonstrate whether the overexpression of DUSP1 in AR GC cells
resulted in resistance to chemotherapeutic agents, five commonly
used chemotherapeutic agents were used to determine the sensitivity
of AR GC cells. No significant differences were observed in the
sensitivity of these five agents between parental and AR GC cells,
suggesting that the resistance of GC cells to Apa does not cause
multi-drug resistance to chemotherapy.
To overcome the resistance of GC cells to Apa, two
approaches were examined to reverse drug resistance. siRNA
transfection technology was used to interfere with the protein
expression of DUSP1 in the AR cells, or a specific protein
inhibitor of DUSP1, triptolide, was used to inhibit the protein
synthesis of DUSP1 in AR cells. The data indicated that, on
knocking down the expression of DUSP1 by siRNA, the IC50
of Apa in MGC803-AR cells decreased from 60.83 to 32.19 µM. In
addition, in the MKN45-AR cells, the IC50 decreased from
72.34 to 25.18 µM. When the AR GC cells were treated with Apa in
combination with triptolide, the expression of DUSP1 decreased
further, and the IC50 of Apa in MGC803-AR cells
decreased to 13.61 µM. In the MKN45-AR cells, the IC50
decreased to 12.06 µM. Neither were significantly different to the
respective parental GC cells. Taken together, a high expression of
DUSP1 was required for Apa-resistance in GC cells.
Protein kinase and phosphatase can maintain
homeostasis of cellular signaling, including the MAPK signaling
pathway. DUSP1, which acts as a phosphatase, can deactivate the
MAPKs, ERKs, P38 MAPKs and JNKs by dephosphorylating threonine and
tyrosine, which are involved in cellular proliferation,
differentiation, and apoptosis, and the progression of tumor
carcinogenesis (68). It has been
demonstrated that the JNK inhibitor SP-600125 may have antitumor
activity in GC cells by inhibiting cell proliferation, promoting
apoptosis, or causing cell cycle arrest (54). DUSP1 is known to dephosphorylate ERK
(55,69). Activation of the ERK1/2 pathway has
been shown to promote cell proliferation (56–60)
and result in malignant transformation (70,71).
The P38 protein prolongs phosphorylation and induces apoptosis,
whereas transient phosphorylation contributes to cell survival
(55).
DUSP1 has been reported as a negative regulator of
the MAPK signaling pathway, however, in the present study, JNK,
ERK, P38 and their corresponding phosphorylated proteins were not
significantly decreased due to the high expression and
phosphorylation of DUSP1. This may be associated with other
regulatory factors of the JNK, ERK and P38 signaling pathways. In
addition, the specific MAPK pathway regulated by DUSP1 in
conferring drug resistance appears to be dependent on the
cell/tissue type in addition to the chemotherapeutic agent used
(49). The AR GC cells showed
significant phosphorylation of JNK, ERK and P38. A marked
anti-apoptotic effect was observed for p-JNK, whereas p-ERK was
closely associated with cell proliferation and survival and p-P38
affected the cell cycle. Activation of the JNK, ERK and P38
proteins is likely to further enhance drug resistance. When AR GC
cells were treated with a combination of Apa and triptolide, the
expression of DUSP1 was downregulated, and phosphorylation of the
JNK, ERK and P38 proteins in the MAPK pathway were significantly
inhibited, particularly in the MGC803-AR cells, which conferred its
proapoptotic and in vivo antitumor activities (72). In the present study, it was
demonstrated that DUSP1 was associated with drug resistance. Even
when the single factor of DUSP1 in the MAPK pathway was an
inhibitor, the overall physiological changes in resistant cells
were more marked in changes of the MAPK pathway. This may explain
why Apa combined with triptolide reversed drug resistance from the
perspective of MAPK signaling pathways. Therefore, the results of
the present study confirmed that downregulation of the expression
of DUSP1 with triptolide may be a useful strategy to overcome
Apa-acquired resistance.
In clinical GC specimens from patients who had not
received chemotherapy or targeted drugs, the protein levels of
DUSP1 were significantly higher in paracarcinoma tissues than in
carcinoma tissues (P<0.0001). In addition, an increase in the
expression of DUSP1 was associated with cancer progression, drug
resistance and poor prognosis.
In conclusion, DUSP1 may serve as a predictive
biomarker for Apa treatment and its increase may be one possible
reason for Apa-acquired resistance. Targeting DUSP1 may overcome
the impaired efficacy caused by drug resistance and thereby
significantly improve the effectiveness of current antitumor drugs.
The present study not only demonstrated a novel mechanism for
acquired resistance in GC, but also provided an effective
combinatorial approach to overcome Apa-acquired resistance.
Acknowledgements
I would like to express my sincere thanks to
Professor Juqian Guo for the English language revisions of this
manuscript.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81573953), the
Program of Zhejiang Provincial TCM Sci-tech Plan (grant no.
2016ZZ012), the Zhejiang Provincial Science and Technology Projects
(grant no. 2013C03044-4), the Natural Science Foundation of
Zhejiang Province (grant nos. LY16H280011 and LY13H160027) and the
Zhejiang Provincial Medical and Healthy Science and Technology
Projects (grant nos. WKJ-ZJ-1728, 2016KYB220 and 2017PY009).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FT was the senior author of the study. He
participated in every step of the design project and the specific
experiment, and was also the writer of this manuscript. ZX also
participated in the overall design of this topic and proposed many
feasible solutions. JC cultured apatinib-resistant gastric cancer
cells. GuowZ participated in the collection of case data. GuodZ
participated in IHC-related experiments. HL gave many practical
advice and guidance. YW modified the language of the manuscript. LW
participated in the analysis and interpretation of IHC results. The
XC gave the overall idea of the study and controlled the quality of
all the work throughout the entire process. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All procedures performed involving human
participants were in accordance with the ethical standards of the
Institutional and/or National Research Committee, and with the 1964
Helsinki declaration and its later amendments or comparable ethical
standards. Informed consent was obtained from all participants
enrolled in the study. From all participants, tissue samples were
included in the sample pool and informed consent was collected from
all participants prior to storage of the sample. An ethical review
of the sample library has been submitted and the review contains
the informed consent of all participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wagner AD, Unverzagt S, Grothe W, Kleber
G, Grothey A, Haerting J and Fleig WE: Chemotherapy for advanced
gastric cancer. Cochrane Database Syst Rev: CD004064. 2010.
View Article : Google Scholar
|
|
3
|
Petrioli R, Francini E, Roviello F,
Marrelli D, Fiaschi AI, Laera L, Rossi G, Bianco V, Brozzetti S and
Roviello G: Sequential treatment with epirubicin, oxaliplatin and
5FU (EOF) followed by docetaxel, oxaliplatin and 5FU (DOF) in
patients with advanced gastric or gastroesophageal cancer: A
single-institution experience. Cancer Chemother Pharmacol.
75:941–947. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lordick F, Allum W, Carneiro F, Mitry E,
Tabernero J, Tan P, Van Cutsem E, van de Velde C and Cervantes A:
Unmet needs and challenges in gastric cancer: The way forward.
Cancer Treat Rev. 40:692–700. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meulendijks D, Beerepoot LV, Boot H, de
Groot JW, Los M, Boers JE, Vanhoutvin SA, Polee MB, Beeker A,
Portielje JE, et al: Trastuzumab and bevacizumab combined with
docetaxel, oxaliplatin and capecitabine as first-line treatment of
advanced HER2-positive gastric cancer: A multicenter phase II
study. Invest New Drugs. 34:119–128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
de Mello RA, Marques AM and Araujo A: HER2
therapies and gastric cancer: A step forward. World J
Gastroenterol. 19:6165–6169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Janjigian YY, Werner D, Pauligk C,
Steinmetz K, Kelsen DP, Jäger E, Altmannsberger HM, Robinson E,
Tafe LJ, Tang LH, et al: Prognosis of metastatic gastric and
gastroesophageal junction cancer by HER2 status: A European and USA
international collaborative analysis. Ann Oncol. 23:2656–2662.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fuchs CS, Tomasek J, Yong CJ, Dumitru F,
Passalacqua R, Goswami C, Safran H, Dos Santos LV, Aprile G, Ferry
DR, et al: Ramucirumab monotherapy for previously treated advanced
gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An
international, randomised, multicentre, placebo-controlled, phase 3
trial. Lancet. 383:31–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wilke H, Muro K, Van Cutsem E, Oh SC,
Bodoky G, Shimada Y, Hironaka S, Sugimoto N, Lipatov O, Kim TY, et
al: Ramucirumab plus paclitaxel versus placebo plus paclitaxel in
patients with previously treated advanced gastric or
gastro-oesophageal junction adenocarcinoma (RAINBOW): A
double-blind, randomised phase 3 trial. Lancet Oncol. 15:1224–1235.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roviello G, Petrioli R, Marano L, Polom K,
Marrelli D, Perrella A and Roviello F: Angiogenesis inhibitors in
gastric and gastroesophageal junction cancer. Gastric Cancer.
19:31–41. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cancer Genome Atlas Research Network, :
Comprehensive molecular characterization of gastric adenocarcinoma.
Nature. 513:202–209. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tian S, Quan H, Xie C, Guo H, Lü F, Xu Y,
Li J and Lou L: YN968D1 is a novel and selective inhibitor of
vascular endothelial growth factor receptor-2 tyrosine kinase with
potent activity in vitro and in vivo. Cancer Sci. 102:1374–1380.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43–9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Geng R and Li J: Apatinib for the
treatment of gastric cancer. Expert Opin Pharmacother. 16:117–122.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lau LF and Nathans D: Identification of a
set of genes expressed during the G0/G1 transition of cultured
mouse cells. EMBO J. 4:3145–3151. 1985.PubMed/NCBI
|
|
16
|
Farooq A and Zhou MM: Structure and
regulation of MAPK phosphatases. Cell Signal. 16:769–779. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kwak SP, Hakes DJ, Martell KJ and Dixon
JE: Isolation and characterization of a human dual specificity
protein-tyrosine phosphatase gene. J Biol Chem. 269:3596–3604.
1994.PubMed/NCBI
|
|
18
|
Tanoue T, Adachi M, Moriguchi T and
Nishida E: A conserved docking motif in MAP kinases common to
substrates, activators and regulators. Nat Cell Biol. 2:110–116.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Theodosiou A and Ashworth A: MAP kinase
phosphatases. Genome Biol. 3:Reviews30092002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guan KL, Broyles SS and Dixon JE: A
Tyr/Ser protein phosphatase encoded by vaccinia virus. Nature.
350:359–362. 1991. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alessi DR, Smythe C and Keyse SM: The
human CL100 gene encodes a Tyr/Thr-protein phosphatase which
potently and specifically inactivates MAP kinase and suppresses its
activation by oncogenic ras in Xenopus oocyte extracts. Oncogene.
8:2015–2020. 1993.PubMed/NCBI
|
|
22
|
Camps M, Nichols A and Arkinstall S: Dual
specificity phosphatases: A gene family for control of MAP kinase
function. FASEB J. 14:6–16. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Duff JL, Monia BP and Berk BC:
Mitogen-activated protein (MAP) kinase is regulated by the MAP
kinase phosphatase (MKP-1) in vascular smooth muscle cells. Effect
of actinomycin D and antisense oligonucleotides. J Biol Chem.
270:7161–7166. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chu Y, Solski PA, Khosravi-Far R, Der CJ
and Kelly K: The mitogen-activated protein kinase phosphatases
PAC1, MKP-1, and MKP-2 have unique substrate specificities and
reduced activity in vivo toward the ERK2 sevenmaker mutation. J
Biol Chem. 271:6497–6501. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Slack DN, Seternes OM, Gabrielsen M and
Keyse SM: Distinct binding determinants for ERK2/p38alpha and JNK
map kinases mediate catalytic activation and substrate selectivity
of map kinase phosphatase-1. J Biol Chem. 276:16491–16500. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bang YJ, Kwon JH, Kang SH, Kim JW and Yang
YC: Increased MAPK activity and MKP-1 overexpression in human
gastric adenocarcinoma. Biochem Biophys Res Commun. 250:43–47.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Loda M, Capodieci P, Mishra R, Yao H,
Corless C, Grigioni W, Wang Y, Magi-Galluzzi C and Stork PJ:
Expression of mitogen-activated protein kinase phosphatase-1 in the
early phases of human epithelial carcinogenesis. Am J Pathol.
149:1553–1564. 1996.PubMed/NCBI
|
|
28
|
Manzano RG, Montuenga LM, Dayton M, Dent
P, Kinoshita I, Vicent S, Gardner GJ, Nguyen P, Choi YH, Trepel J,
et al: CL100 expression is down-regulated in advanced epithelial
ovarian cancer and its re-expression decreases its malignant
potential. Oncogene. 21:4435–4447. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Haagenson KK and Wu GS: The role of MAP
kinases and MAP kinase phosphatase-1 in resistance to breast cancer
treatment. Cancer Metastasis Rev. 29:143–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shi YY, Small GW and Orlowski RZ:
Proteasome inhibitors induce a p38 mitogen-activated protein kinase
(MAPK)-dependent anti-apoptotic program involving MAPK
phosphatase-1 and Akt in models of breast cancer. Breast Cancer Res
Treat. 100:33–47. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Z, Xu J, Zhou JY, Liu Y and Wu GS:
Mitogen-activated protein kinase phosphatase-1 is required for
cisplatin resistance. Cancer Res. 66:8870–8877. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chattopadhyay S, Machado-Pinilla R,
Manguan-Garcia C, Belda-Iniesta C, Moratilla C, Cejas P,
Fresno-Vara JA, de Castro-Carpeño J, Casado E, Nistal M, et al:
MKP1/CL100 controls tumor growth and sensitivity to cisplatin in
non-small-cell lung cancer. Oncogene. 25:3335–3345. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu JJ and Bennett AM: Essential role for
mitogen-activated protein (MAP) kinase phosphatase-1 in
stress-responsive MAP kinase and cell survival signaling. J Biol
Chem. 280:16461–16466. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abrams MT, Robertson NM, Litwack G and
Wickstrom E: Evaluation of glucocorticoid sensitivity in 697 pre-B
acute lymphoblastic leukemia cells after overexpression or
silencing of MAP kinase phosphatase-1. J Cancer Res Clin Oncol.
131:347–354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang J, Zhou JY and Wu GS: ERK-dependent
MKP-1-mediated cisplatin resistance in human ovarian cancer cells.
Cancer Res. 67:11933–11941. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang HY, Cheng Z and Malbon CC:
Overexpression of mitogen-activated protein kinase phosphatases
MKP1, MKP2 in human breast cancer. Cancer Lett. 191:229–237. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Small GW, Shi YY, Higgins LS and Orlowski
RZ: Mitogen-activated protein kinase phosphatase-1 is a mediator of
breast cancer chemoresistance. Cancer Res. 67:4459–4466. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Z, Zhou JY, Kanakapalli D, Buck S, Wu
GS and Ravindranath Y: High level of mitogen-activated protein
kinase phosphatase-1 expression is associated with cisplatin
resistance in osteosarcoma. Pediatr Blood Cancer. 51:754–759. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang M, Zhang H, Liu T, Tian D, Gu L and
Zhou M: Triptolide inhibits MDM2 and induces apoptosis in acute
lymphoblastic leukemia cells through a p53-independent pathway. Mol
Cancer Ther. 12:184–194. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang W, Yang S, Su Y, Xiao Z, Wang C, Li
X, Lin L, Fenton BM, Paoni SF, Ding I, et al: Enhanced antitumor
effect of combined triptolide and ionizing radiation. Clin Cancer
Res. 13:4891–4899. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mujumdar N, Mackenzie TN, Dudeja V, Chugh
R, Antonoff MB, Borja-Cacho D, Sangwan V, Dawra R, Vickers SM and
Saluja AK: Triptolide induces cell death in pancreatic cancer cells
by apoptotic and autophagic pathways. Gastroenterology.
139:598–608. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kitzen JJ, de Jonge MJ, Lamers CH, Eskens
FA, van der Biessen D, van Doorn L, Ter Steeg J, Brandely M, Puozzo
Ch and Verweij J: Phase I dose-escalation study of F60008, a novel
apoptosis inducer, in patients with advanced solid tumours. Eur J
Cancer. 45:1764–1772. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Koo HS, Kang SD, Lee JH, Kim NH, Chung HT
and Pae HO: Triptolide inhibits the proliferation of immortalized
ht22 hippocampal cells via persistent activation of extracellular
signal-regulated kinase-1/2 by down-regulating mitogen-activated
protein kinase phosphatase-1 expression. J Korean Neurosurg Soc.
46:389–396. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu W, Ou Y, Li Y, Xiao R, Shu M, Zhou Y,
Xie J, He S, Qiu P and Yan G: A small-molecule triptolide
suppresses angiogenesis and invasion of human anaplastic thyroid
carcinoma cells via down-regulation of the nuclear factor-kappa B
pathway. Mol Pharmacol. 75:812–819. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Phillips PA, Dudeja V, McCarroll JA,
Borja-Cacho D, Dawra RK, Grizzle WE, Vickers SM and Saluja AK:
Triptolide induces pancreatic cancer cell death via inhibition of
heat shock protein 70. Cancer Res. 67:9407–9416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yeo W, Chan SL, Mo FK, Chu CM, Hui JW,
Tong JH, Chan AW, Koh J, Hui EP, Loong H, et al: Phase I/II study
of temsirolimus for patients with unresectable hepatocellular
carcinoma (HCC)- a correlative study to explore potential
biomarkers for response. BMC Cancer. 15:3952015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Denkert C, Schmitt WD, Berger S, Reles A,
Pest S, Siegert A, Lichtenegger W, Dietel M and Hauptmann S:
Expression of mitogen-activated protein kinase phosphatase-1
(MKP-1) in primary human ovarian carcinoma. Int J Cancer.
102:507–513. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Low HB and Zhang Y: Regulatory roles of
MAPK phosphatases in cancer. Immune Netw. 16:85–98. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lin YC, Lin YC, Shih JY, Huang WJ, Chao
SW, Chang YL and Chen CC: DUSP1 expression induced by HDAC1
inhibition mediates gefitinib sensitivity in non-small cell lung
cancers. Clin Cancer Res. 21:428–438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ma G, Pan Y, Zhou C, Sun R, Bai J, Liu P,
Ren Y and He J: Mitogen-activated protein kinase phosphatase 1 is
involved in tamoxifen resistance in MCF7 cells. Oncol Rep.
34:2423–2430. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Donaubauer EM, Law NC and Hunzicker-Dunn
ME: Follicle-stimulating hormone (FSH)-dependent regulation of
extracellular regulated kinase (ERK) phosphorylation by the
mitogen-activated protein (MAP) kinase phosphatase MKP3. J Biol
Chem. 291:19701–19712. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Boulding T, Wu F, McCuaig R, Dunn J,
Sutton CR, Hardy K, Tu W, Bullman A, Yip D, Dahlstrom JE and Rao S:
Differential roles for DUSP family members in
epithelial-to-mesenchymal transition and cancer stem cell
regulation in breast cancer. PLoS One. 11:e01480652016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Xia HH, He H, De Wang J, Gu Q, Lin MC, Zou
B, Yu LF, Sun YW, Chan AO, Kung HF and Wong BC: Induction of
apoptosis and cell cycle arrest by a specific c-Jun NH2-terminal
kinase (JNK) inhibitor, SP-600125, in gastrointestinal cancers.
Cancer Lett. 241:268–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Owens DM and Keyse SM: Differential
regulation of MAP kinase signalling by dual-specificity protein
phosphatases. Oncogene. 26:3203–3213. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Krysan K, Reckamp KL, Dalwadi H, Sharma S,
Rozengurt E, Dohadwala M and Dubinett SM: Prostaglandin E2
activates mitogen-activated protein kinase/Erk pathway signaling
and cell proliferation in non-small cell lung cancer cells in an
epidermal growth factor receptor-independent manner. Cancer Res.
65:6275–6281. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Andradas C, Caffarel MM, Perez-Gomez E,
Salazar M, Lorente M, Velasco G, Guzmán M and Sánchez C: The orphan
G protein-coupled receptor GPR55 promotes cancer cell proliferation
via ERK. Oncogene. 30:245–252. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kanai M, Konda Y, Nakajima T, Izumi Y,
Kanda N, Nanakin A, Kubohara Y and Chiba T:
Differentiation-inducing factor-1 (DIF-1) inhibits STAT3 activity
involved in gastric cancer cell proliferation via MEK-ERK-dependent
pathway. Oncogene. 22:548–554. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tyagi N, Bhardwaj A, Singh AP, McClellan
S, Carter JE and Singh S: p-21 activated kinase 4 promotes
proliferation and survival of pancreatic cancer cells through AKT-
and ERK-dependent activation of NF-κB pathway. Oncotarget.
5:8778–8789. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Pulverer BJ, Kyriakis JM, Avruch J,
Nikolakaki E and Woodgett JR: Phosphorylation of c-jun mediated by
MAP kinases. Nature. 353:670–674. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Roviello G, Ravelli A, Fiaschi AI,
Cappelletti MR, Gobbi A, Senti C, Zanotti L, Polom K, Reynolds AR,
Fox SB, et al: Apatinib for the treatment of gastric cancer. Expert
Rev Gastroenterol Hepatol. 10:887–892. 2016.PubMed/NCBI
|
|
62
|
Yang Y, Wu N, Shen J, Teixido C, Sun X,
Lin Z, Qian X, Zou Z, Guan W, Yu L, et al: MET overexpression and
amplification define a distinct molecular subgroup for targeted
therapies in gastric cancer. Gastric Cancer. 19:778–788. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tran P, Nguyen C and Klempner SJ:
Targeting the phosphatidylinositol-3-kinase pathway in gastric
cancer: Can omics improve outcomes? Int Neurourol J. 20 Suppl
2:S131–S140. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lee HS, Kim WH, Kwak Y, Koh J, Bae JM, Kim
KM, Chang MS, Han HS, Kim JM, Kim HW, et al: Molecular testing for
gastrointestinal cancer. J Pathol Transl Med. 51:103–121. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Candas D and Li JJ: MKP1 mediates
resistance to therapy in HER2-positive breast tumors. Mol Cell
Oncol. 2:e9975182015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kang YS, Seok HJ, Jeong EJ, Kim Y, Yun SJ,
Min JK, Kim SJ and Kim JS: DUSP1 induces paclitaxel resistance
through the regulation of p-glycoprotein expression in human
ovarian cancer cells. Biochem Biophys Res Commun. 478:403–409.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liu F, Gore AJ, Wilson JL and Korc M:
DUSP1 is a novel target for enhancing pancreatic cancer cell
sensitivity to gemcitabine. PLoS One. 9:e849822014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Boutros T, Chevet E and Metrakos P:
Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase
regulation: Roles in cell growth, death, and cancer. Pharmacol Rev.
60:261–310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Keyse SM: Dual-specificity MAP kinase
phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 27:253–261.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sebolt-Leopold JS and Herrera R: Targeting
the mitogen-activated protein kinase cascade to treat cancer. Nat
Rev Cancer. 4:937–947. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Webb CP, Van Aelst L, Wigler MH and Vande
Woude GF: Signaling pathways in Ras-mediated tumorigenicity and
metastasis. Proc Natl Acad Sci USA. 95:8773–8778. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Xie CQ, Zhou P, Zuo J, Li X, Chen Y and
Chen JW: Triptolide exerts pro-apoptotic and cell cycle arrest
activity on drug-resistant human lung cancer A549/Taxol cells via
modulation of MAPK and PI3K/Akt signaling pathways. Oncol Lett.
12:3586–3590. 2016. View Article : Google Scholar : PubMed/NCBI
|