Introduction
Reactive oxygen species (ROS) are highly unstable
and reactive molecules containing oxygen moieties. They include
hydrogen peroxide (H2O2), superoxide anion
(O2●−) and hydroxyl radical (●OH).
Although ROS are recognized as cytotoxic agents, they can serve as
second messengers to control many cellular events of gene
expression, differentiation, cell proliferation and cell death
(1,2). ROS are continuously generated by
endogenous aerobic metabolism within cells in the form of the
O2●− and/or are purposely made by oxidases,
such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase
and xanthine oxidase (3).
O2●− is converted to
H2O2 by the enzyme superoxide dismutase
(4). H2O2 is
further processed into O2 and H2O by catalase
or glutathione peroxidases (5).
Compared with other members of ROS, non-radical
H2O2 is able to freely penetrate cell
membranes, and it interacts with ferrous iron (Fenton chemistry),
which produces the highly destructive and short-lived
●OH. The production of different forms of ROS at various
levels can be either useful or harmful to cells and tissues.
Particularly, excessive amounts of ROS may be the outcome of either
their overproduction and/or downregulation of antioxidants.
Increased levels of ROS can result in damage to DNA, proteins and
lipids in cells, implicating them in the etiology of several human
diseases, including cancer (6–9).
Apoptosis is programmed cell death and occurs via
two different pathways: The mitochondrial intrinsic pathway and the
receptor mediated extrinsic pathway (10). The key step in the
mitochondrial-mediated apoptosis is the translocation of cytochrome
c from mitochondria to cytosol and its subsequent
interaction with Apaf-1 and caspase-9 to form a complex
(apoptosome). The apoptosome further activates executive caspase-3,
−6 and −7 (11). Conversely, the
extrinsic pathway begins with the binding of specific ligands, such
as TNF-α, TRAIL and Fas to the respective cell death receptors,
which stimulate the activities of caspase-8 and −3 (12). Caspase-8 cleaves BID, a
pro-apoptotic cytosolic protein of the Bcl-2 family, to generate a
truncated product, tBID that enters into the mitochondria and
decreases the mitochondrial membrane potential (MMP;
ΔΨm), causing the release of cytochrome c. The
translocation of another apoptotic protein, Bax from the cytosol to
the mitochondria also triggers similar loss of MMP
(ΔΨm). Caspase-3 is the key executive caspase; its
activation can systematically disassemble the integrity of cells
through cleaving several key proteins, such as poly(ADP-ribose)
polymerase (PARP) and RhoGDI.
Lungs are susceptible to a variety of airborne and
bloodborne injuries that may consequently cause lung fibrosis and
cancer (13). The carcinogenesis of
lung cancer is considered to be tightly linked to
H2O2-mediated tissue inflammation. During
inflammation, tissue concentrations of H2O2
are expected to achieve nearly millimolar levels, whereas low
levels of H2O2 produced by NADPH oxidases
under normal conditions are hypothesized not to have a higher
affect than the plasma membrane microenvironment, such as lipid
rafts (14,15). Nonetheless, in both cases,
H2O2 may modulate vital cellular functions of
cell proliferation, death and differentiation by changing signaling
cascades and gene expression and its higher levels may lead to
apoptosis and/or necrosis. Exogenous H2O2 is
frequently used as the representative ROS to simulate oxidative
stress in cells and tissues. H2O2 is
relatively non-toxic to the normal cells of human umbilical vein
endothelial cells and human pulmonary artery smooth muscle cells
(16,17). H2O2-triggered
cell death in lung cancer cells may have cytotoxicological research
interest.
In the present study, the molecular effects of
exogenous H2O2 on Calu-6 and A549 lung cancer
cells were evaluated with respect to cell growth and death, as well
as the anti-apoptotic effects of various caspase inhibitors were
investigated in H2O2-treated lung cancer
cells.
Materials and methods
Cell culture
The human lung cancer Calu-6 and A549 cell lines
were purchased from the Korean Cell Line Bank (Seoul, Korea) and
were cultivated in RPMI-1640 medium supplemented with 10% fetal
bovine serum (FBS; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
and 1% penicillin-streptomycin (Gibco-BRL; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). These cells were regularly
cultured in 100-mm plastic tissue culture dishes (Nunc, Roskilde,
Denmark) and harvested with a trypsin-EDTA solution (Gibco-BRL;
Thermo Fisher Scientific, Inc.).
Reagents
H2O2 was obtained from Merck
KGaA. Pan-caspase inhibitor (Z-VAD-FMK;
benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone), caspase-3
inhibitor (Z-DEVD-FMK;
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone), caspase-9
inhibitor (Z-LEHD-FMK;
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethyl ketone) and
caspase-8 inhibitor (Z-IETD-FMK;
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethyl ketone) were
purchased from R&D Systems, Inc. (Minneapolis, MN, USA) and
were dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck
KGaA). Cells were pre-incubated with each caspase inhibitor for 1 h
before the H2O2 treatment as previously
described (18).
Cell growth and cell number
assays
Cell growth changes were evaluated by assessing
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich; Merck KGaA) dye absorbance as previously described
(19). Viable and dead cell numbers
were determined by trypan blue cell staining method (20). Cells were exposed to the designated
amounts of H2O2 with or without 15 µM of each
caspase inhibitor for 24 h.
Cell cycle and sub-G1 cell
analysis
Cell cycle and sub-G1 cell analysis were performed
by propidium iodide (PI; Sigma-Aldrich; Merck KGaA) staining as
previously described (20). Cells
were exposed to the designated amounts of
H2O2 with or without 15 µM of each caspase
inhibitor for 24 h. Cell cycle distributions were analyzed with a
FACStar flow cytometer (Becton-Dickinson and Company, Franklin
Lakes, NJ, USA).
Annexin V-FITC staining for cell death
detection
Apoptotic cell death was verified by measuring cells
stained with Annexin V-fluorescein isothiocyanate (FITC;
Invitrogen; Thermo Fisher Scientific, Inc.) as previously described
(20). Cells were exposed to the
designated amounts of H2O2 with or without 15
µM of each caspase inhibitor for 24 h. Annexin V-FITC staining was
analyzed with a FACStar flow cytometer (Becton-Dickinson and
Company).
Assessement of MMP
(ΔΨm)
MMP (ΔΨm) was evaluated by a rhodamine
123 fluorescent dye (Sigma-Aldrich; Merck KGaA) as previously
described (21). Cells were exposed
to the designated amounts of H2O2 with or
without 15 µM of each caspase inhibitor for 24 h. Rhodamine 123
staining intensity was analyzed by a FACStar flow cytometer
(Becton-Dickinson and Company). The absence of rhodamine 123 from
the cells indicated the loss of MMP (ΔΨm) in lung cancer
cells. MMP (ΔΨm) levels in cells not including MMP
(ΔΨm)-loss cells were expressed as the mean fluorescence
intensity, which was estimated by CellQuest software (version 5.1;
Becton-Dickinson and Company).
Western blot analysis
The changes in Bcl-2, caspase-3 and PARP in
H2O2-treated cells were analyzed by western
blotting. Briefly, 1×106 cells in 60-mm culture dish
(Nunc) were incubated with the designated amounts of
H2O2 for 24 h. Samples containing 20 µg total
protein were separated by 8 or 12.5% SDS-PAGE gel, transferred to
Immobilon-P PVDF membranes (EMD Millipore, Billerica, MA, USA) by
electroblotting and then probed with anti-Bcl-2, anti-caspase-3,
anti-PARP and anti-β-actin antibodies (dilution 1:5,000; Santa Cruz
Biotechnology, Santa Cruz, CA, USA). Membranes were treated with
horseradish peroxidase-conjugated secondary antibodies (dilution
1:5,000; Cell signaling Technology, Inc.). Blots were developed by
means of an ECL kit (Amersham Life Science, Arlington Heights, IL,
USA).
Quantification of caspase-3 and −8
activities
The activities of caspase-3 and −8 were evaluated by
caspase-3 and −8 colorimetric assay kits (R&D Systems, Inc.) as
previously described (20). In
brief, 1×106 cells in 60-mm culture dish (Nunc) were
treated with 75 µM H2O2 for 24 h. Samples
containing 50 µg total protein were used to assess caspase-3 and −8
activities.
Statistical analysis
Data representing at least two independent
experiments (mean ± SD) were analyzed through InStat software
(GraphPad Prism4; GraphPad Software, Inc., San Diego, CA, USA). The
Student's t-test or one-way analysis of variance (ANOVA) with post
hoc analysis using Tukey's multiple comparison test was used for
parametric data. P<0.05 was considered to indicate a
statistically significant difference.
Results
H2O2 affects the
cell growth and cycle distribution in lung cancer cells
The cellular effects of H2O2
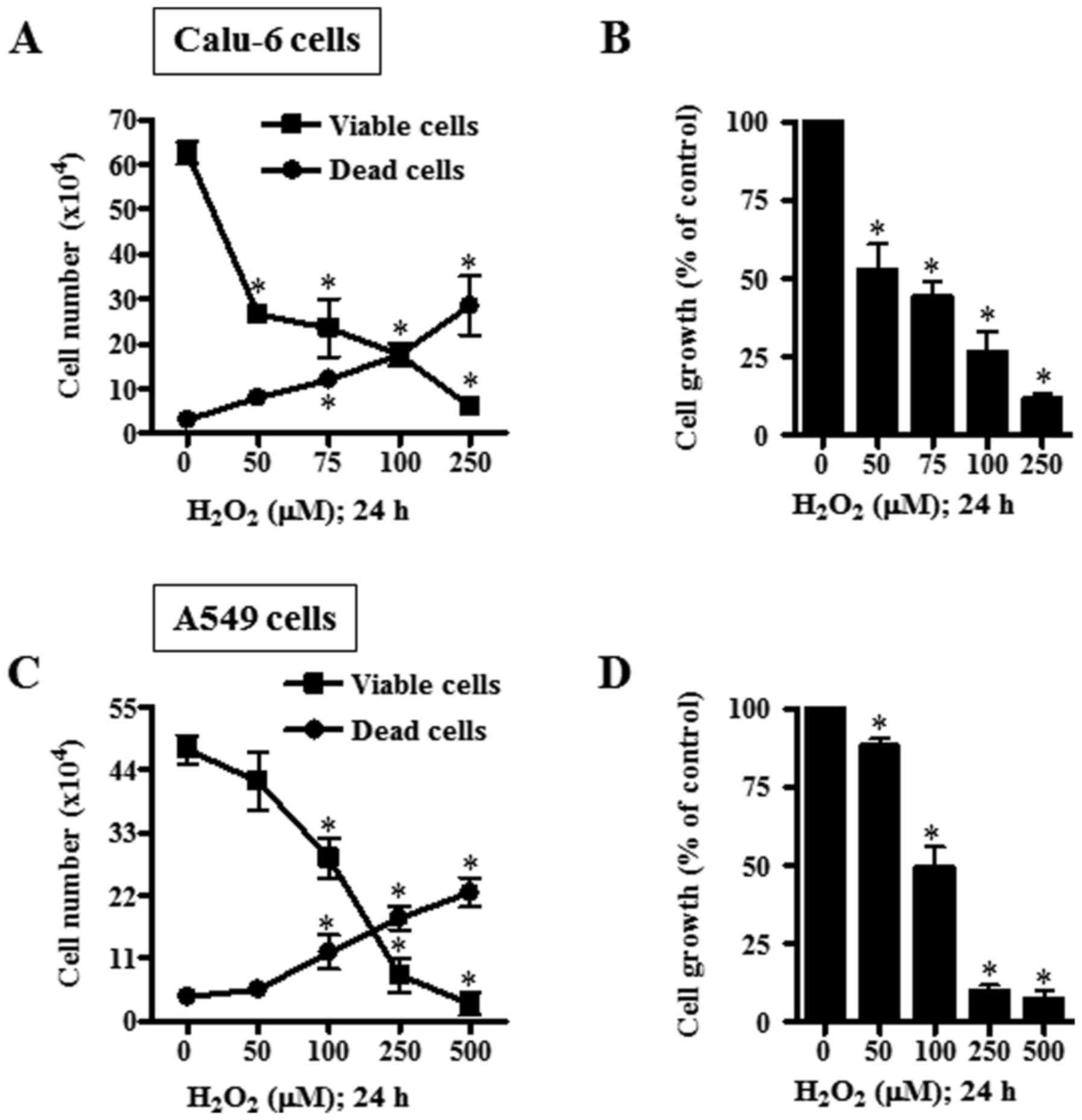
on the growth of lung cancer cells were examined at 24 h. Treatment
with 50–250 µM H2O2 significantly reduced
viable (trypan blue-negative) and increased dead (trypan
blue-positive) Calu-6 cells in a dose-dependent manner (Fig. 1A). Based on MTT assays, 50–250 µM
H2O2 significantly attenuated the growth of
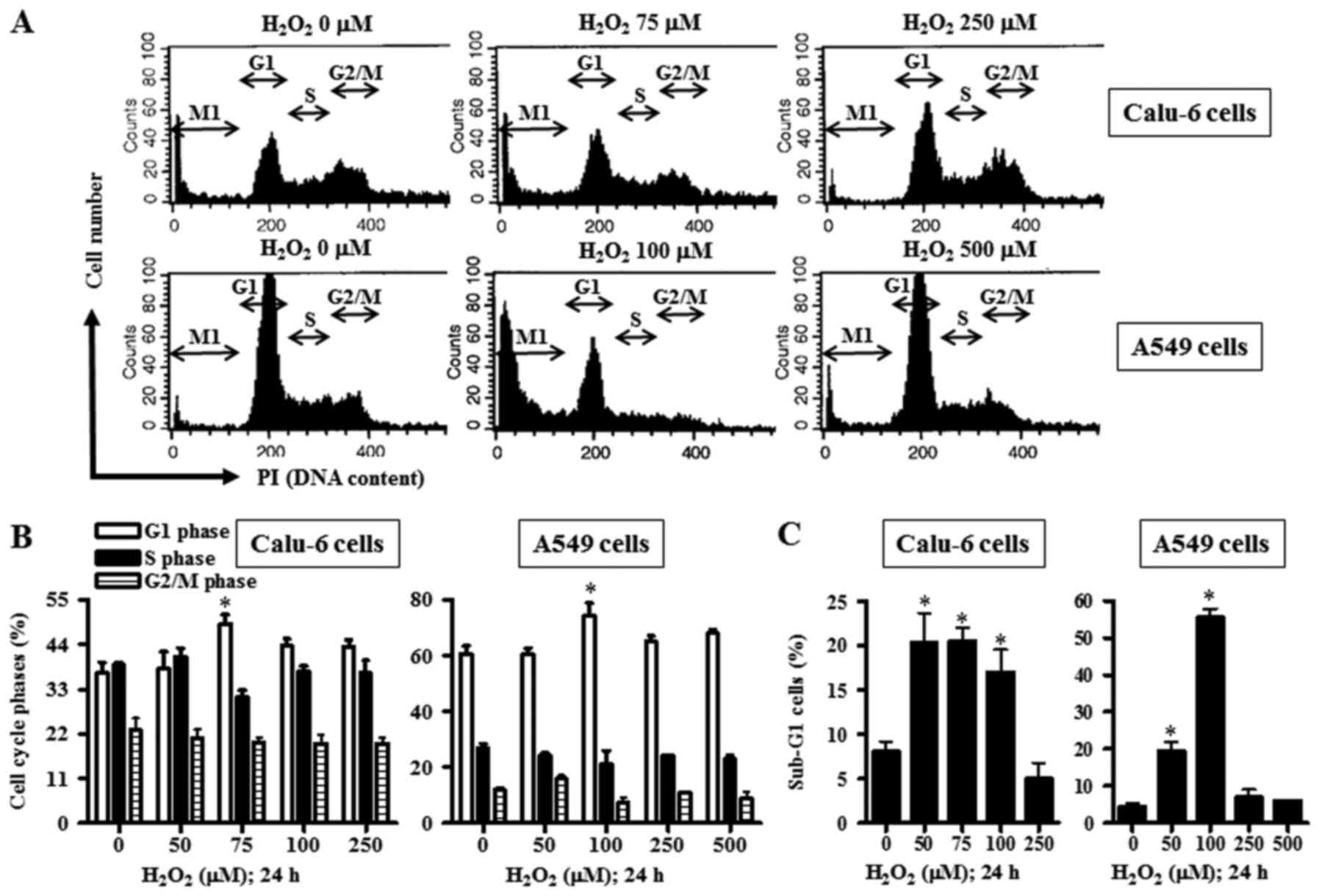
Calu-6 cells with an IC50 of ~50 µM (Fig. 1B). When the cell cycle distribution
in H2O2-treated Calu-6 cells was examined,
Calu-6 cells treated with 75-µM H2O2
demonstrated a significant G1-phase arrest of the cell cycle
compared with the control cells (Fig.
2A and B). As in Calu-6, upon H2O2
treatment, the number of A549 viable cells decreased and dead cells
increased significantly in a dose-dependent manner (Fig. 1C). In addition,
H2O2 dose-dependently reduced the growth of
A549 cells with an IC50 of ~100 µM (Fig. 1D). Treatment with 100 µM
H2O2 also significantly induced a G1-phase
arrest in A549 cells compared with the control cells (Fig. 2A and B).
H2O2 influences
cell death and MMP (ΔΨm) in
H2O2-treated lung cancer cells
Subsequently, the role of H2O2
in lung cancer cell death was further investigated to gain more
understanding. While 50–100 µM H2O2
significantly augmented the percentages of sub-G1 cells in Calu-6
cells, 250 µM H2O2 did not increase the
percentage of sub-G1 cells in these cells (Fig. 2A and C). However, treatment with
50–250 µM H2O2 dose-dependently increased the
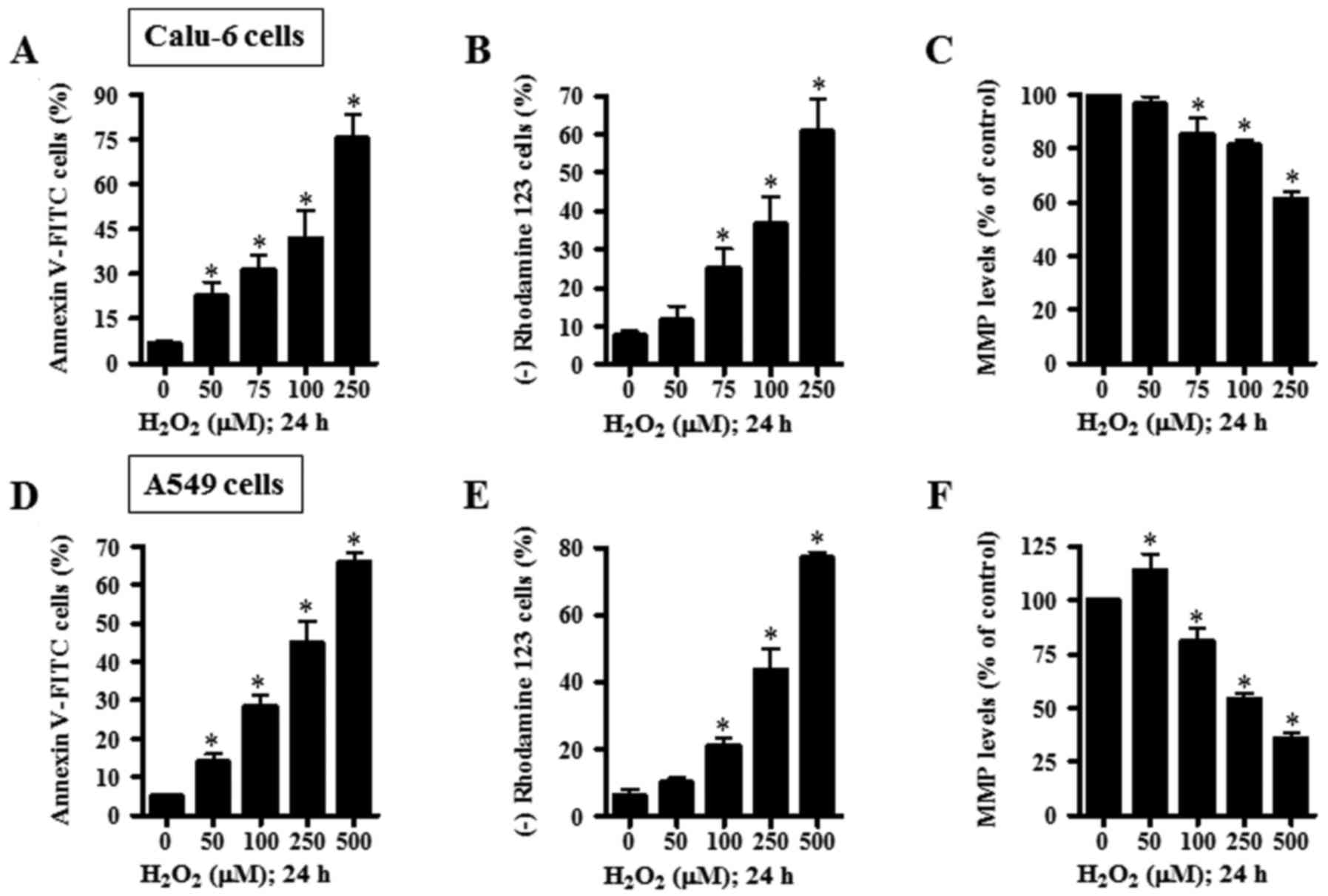
numbers of Annexin V-FITC-stained cells in Calu-6 cells (Fig. 3A). When the effect of
H2O2 on MMP (ΔΨm) in Calu-6 cells
was assessed using rhodamine 123, H2O2
provoked the loss of MMP (ΔΨm) in a dose-dependent
manner (Fig. 3B). With regard to
MMP (ΔΨm) level in Calu-6 cells excluding negative
rhodamine 123 staining cells, H2O2 decreased
the MMP (ΔΨm) level in Calu-6 cells in a dose-dependent
manner (Fig. 3C). In A549 cells,
treatment of 50 and 100 µM H2O2 significantly
increased the percentages of sub-G1 cells, but treatment with 250
and 500 µM H2O2 did not show this effect
(Fig. 2A and C).
H2O2 dose-dependently enhanced the numbers of
Annexin V-FITC-stained A549 cells (Fig.
3D). Additionally, H2O2 dose-dependently
induced the loss of MMP (ΔΨm) in A549 cells (Fig. 3E). While 50 µM
H2O2 increased MMP (ΔΨm) level in
A549 cells, 100–250 µM H2O2 significantly
decreased MMP (ΔΨm) levels in these cells (Fig. 3F).
H2O2 influences
apoptosis-related proteins and caspases in
H2O2-treated lung cancer cells
Assessment of apoptosis-related proteins during
H2O2-induced lung cell death revealed that
Bcl-2, an anti-apoptotic protein, decreased upon
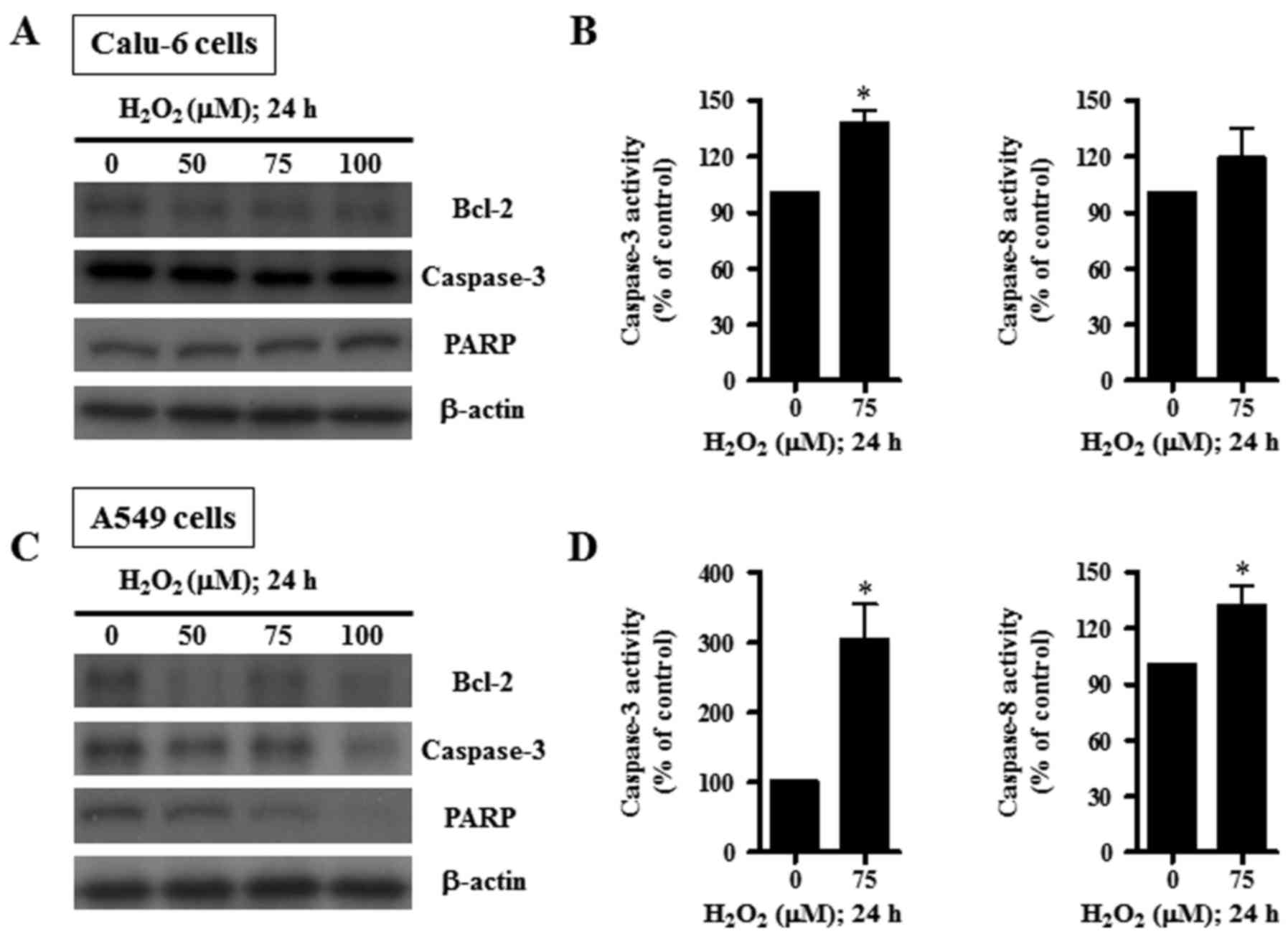
H2O2 treatment in Calu-6 cells (Fig. 4A). The level of pro-caspase-3 was
reduced by 75 µM H2O2 (Fig. 4A). The unbroken form of 116 kDa PARP
was not altered by H2O2 (Fig. 4A). The activity of caspase-3 was
found to be increased in H2O2-treated Calu-6
cells, while that of caspase-8 was not significantly changed
(Fig. 4B). Treatment with 50–100 µM
H2O2 appeared to decrease Bcl-2,
pro-caspase-3, and PARP protein levels in A549 cells (Fig. 4C). Specifically, 100 µM
H2O2 showed a marked decrease in the levels
of these proteins. Treatment with 75 µM H2O2
significantly augmented the activity of caspase-3 in A549 cells and
significantly increased the activity of caspase-8 (Fig. 4D).
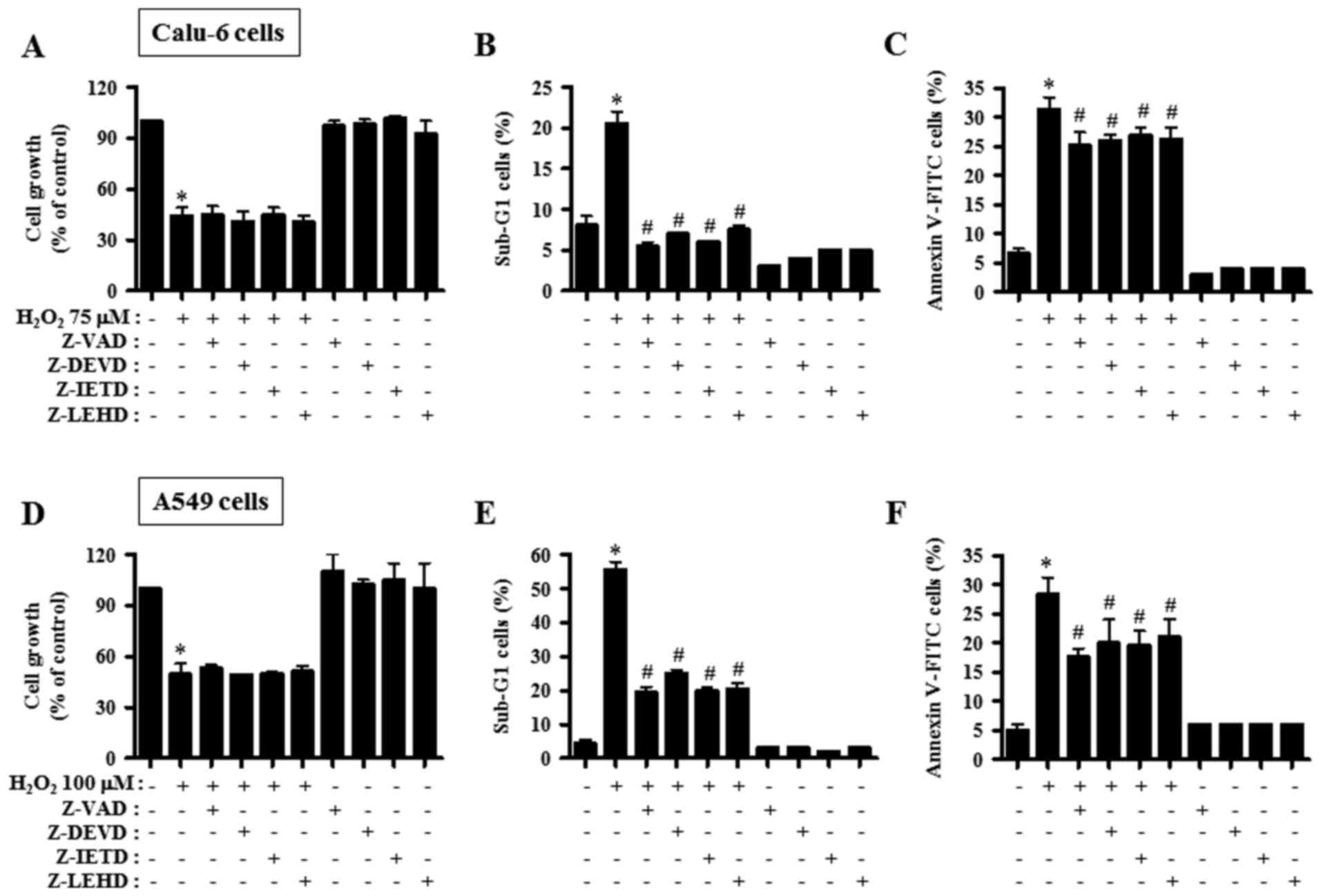
Caspase inhibitors affect cell growth
and death in H2O2-treated lung cancer
cells
Subsequently we sought to decipher the role of
individual caspases in H2O2-induced cell
death at 24 h in lung carcinoma cell lines. Calu-6 and A549 cells
were pre-incubated with 15 µM caspase inhibitor for 1 h before
treatment with 75 or 100 µM H2O2. None of the
tested caspase inhibitors influenced the growth inhibition induced
by H2O2 in both Calu-6 and A549 cell lines
(Fig. 5A and D). However, all the
caspase inhibitors tested in H2O2-treated
Calu-6 decreased the percentages of sub-G1 cells to the level of
the control cells (Fig. 5B). In
addition, treatment with all the tested caspase inhibitors
significantly reduced the number of Annexin V-FITC-stained cells in
H2O2-treated Calu-6 cells, but the decreased
effect was weaker compared with the decrease in sub-G1 cells
(Fig. 5C). All the caspase
inhibitors markedly rescued A549 cells from
H2O2-promoted cell death, as assessed by the
population of sub-G1 cells (Fig.
5E). Furthermore, these inhibitors significantly reduced the
number of Annexin V-FITC-stained cells in
H2O2-treated A549 cells (Fig. 5F). Each caspase inhibitor had very
similar anti-death effects in the
H2O2-treated lung cancer cells.
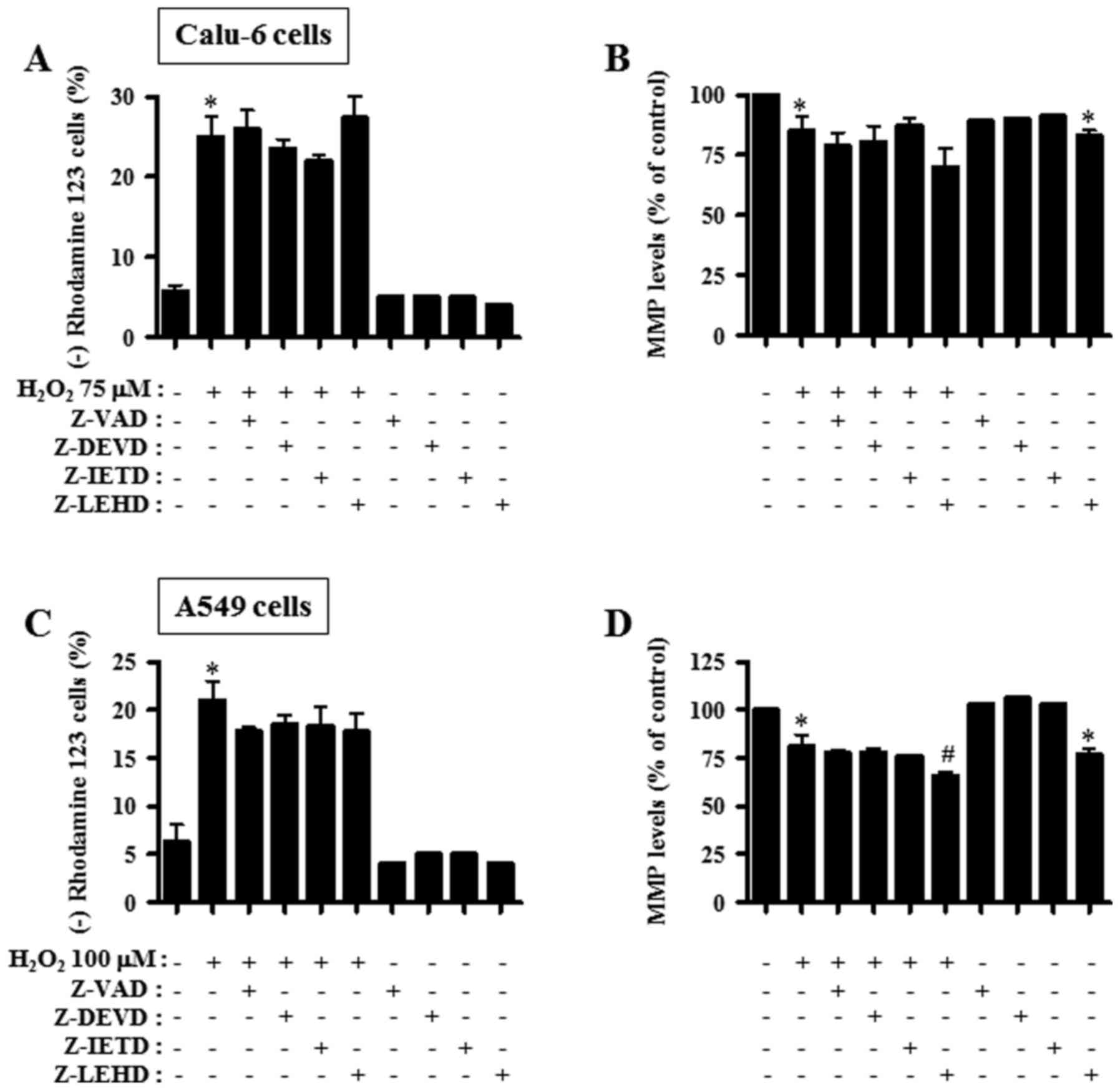
Caspase inhibitors affect MMP
(ΔΨm) in H2O2-treated lung cancer
cells
Cell death is strongly associated with the collapse
of MMP (ΔΨm) (22).
Thus, MMP (ΔΨm) in 75 or 100 µM
H2O2-treated lung cancer cells was determined
with or without each caspase inhibitors at 24 h. However, all the
caspase inhibitors did not significantly reduce the loss of MMP
(ΔΨm) in H2O2-treated Calu-6 cells
(Fig. 6A). Additionally, most of
these inhibitors did not influence the MMP (ΔΨm) level
in H2O2-treated Calu-6 cells. However,
caspase-9 inhibitor (Z-LEHD) appeared to enhance the decrease of
the level in these cells (Fig. 6B).
In A549 cells, all the caspase inhibitors partially prevented the
loss of MMP (ΔΨm) by H2O2
(Fig. 6C). In
H2O2-treated A549 cells, caspase-9 inhibitor
selectively further enhanced the decrease in MMP (ΔΨm)
level (Fig. 6D). This inhibitor
alone significantly reduced MMP (ΔΨm) levels in Calu-6
and A549 control cells (Fig. 6B and
D).
Discussion
Lung cancer represents one of the main causes of
cancer-related mortality worldwide and is related to the malicious
activity of ROS. In the present study, exogenous
H2O2 was used for generating oxidative stress
in lung cancer cells. This study focused on defining the molecular
mechanisms of cell growth inhibition and cell death in
H2O2-treated Calu-6 and A549 lung cancer
cells. Based on MTT assays, after 24-h exposure, the
IC50 values for H2O2 were ~50 and
100 µM in Calu-6 and A549 cells, respectively.
H2O2 dose-dependently increased the number of
dead and Annexin V-FITC-stained Calu-6 and A549 cells, indicating
that H2O2-induced lung cancer cell death
occurred through apoptosis. Evidently, H2O2
decreased the levels of Bcl-2 and pro-caspase-3 in both cell types.
PARP was reduced in the H2O2-treated A549
cells. Furthermore, the activities of caspase-3 and −8 were
increased in both H2O2-treated cell types.
Apoptosis is strongly related to the collapse of MMP
(ΔΨm) (22).
H2O2 triggered the loss of MMP
(ΔΨm) in Calu-6 and A549 cells in a dose-dependent
manner, indicating that lung cancer cell death by
H2O2 was closely related with the collapse of
MMP (ΔΨm). In addition, H2O2
decreased the MMP (ΔΨm) level in lung cancer cells
containing the rhodamine 123 dye.
Although 50–100 µM H2O2
significantly increased the percentages of sub-G1 Calu-6 and A549
cells, 250 or 500 µM H2O2 did not demonstrate
a similar effect, indicating that the higher doses of
H2O2 fixed these lung cancer cells in a
similar way to ethanol or methanol. Thus,
H2O2 appeared to induce lung cancer cell
death simultaneously via necrosis and apoptosis, depending on its
concentration. In particular, 75 and 100 µM
H2O2 appeared to concurrently trigger both
apoptosis and necrosis in Calu-6 cells, since these doses of
H2O2 did not increase the percentages of
sub-G1 cells compared with 50 µM H2O2-treated
cells, as well as there was no change in the levels of the intact
form of PARP protein. It is required to evaluate the activity of
the extracellular lactate dehydrogenase in lung cancer cells
treated with 50- 500 µM H2O2 for the
detection of necrotic cell death. Previous studies revealed a role
of H2O2 in cell-cycle phase arrest and
progression by adjusting cell cycle-related proteins (23,24).
In line with this, treatment with 75 or 100 µM
H2O2 among the tested doses significantly
showed a G1 phase arrest in Calu-6 and A549 cells. Thus, the G1
phase arrest together with induction of cell death is the potential
mechanism behind the attenuation of cell growth upon
H2O2 treatment. However,
H2O2 did not make any specific phase arrests
of the cell cycle in HeLa cells (20). These results indicated that
H2O2-induced oxidative stress manifested its
effects on cell cycle progression depending on the cell type and
H2O2 dose.
Caspase inhibitors used in this experiment failed
to attenuate the growth inhibition in
H2O2-treated Calu-6 and A549 cancer cells,
whereas these inhibitors considerably prevented
H2O2-induced cell death in these cells.
Although H2O2 to some extent augmented the
activity of caspase-8 in both lung cancer cells, caspase-8
inhibitor significantly attenuated cell death triggered by
H2O2. Thus, a slight alteration in the
activity of caspase-8 appeared to have strong impact on the
pro-apoptotic pathway in H2O2-treated lung
cancer cells. These results also indicated that both mitochondrial
and cell death receptor pathways were mutually required for the
entire induction of apoptosis in H2O2-treated
lung cancer cells. It would be important to ascertain how
H2O2 affects the cell death receptor pathway
to induce apoptosis in lung cancer cells. Concerning MMP
(ΔΨm), caspase inhibitors did not have any significant
effect on the loss of MMP (ΔΨm) in
H2O2-treated Calu-6 and A549 cells. In
addition, these inhibitors did not restore the decreased MMP
(ΔΨm) levels in H2O2-treated lung
cancer cells. Instead, the caspase-9 inhibitor enhanced the
decreased levels in these cells. It is plausible that the loss of
MMP (ΔΨm) following treatment with
H2O2 activated various caspases related to
mitochondrial and cell death receptor pathways, consequently
inducing apoptosis, and the activation of caspases by
H2O2 could not positively enhance the MMP
(ΔΨm) loss. In addition, the loss of MMP
(ΔΨm) induced by H2O2 may not be
enough to completely provoke apoptosis in Calu-6 and A549 cells
under the downregulation of caspase activity.
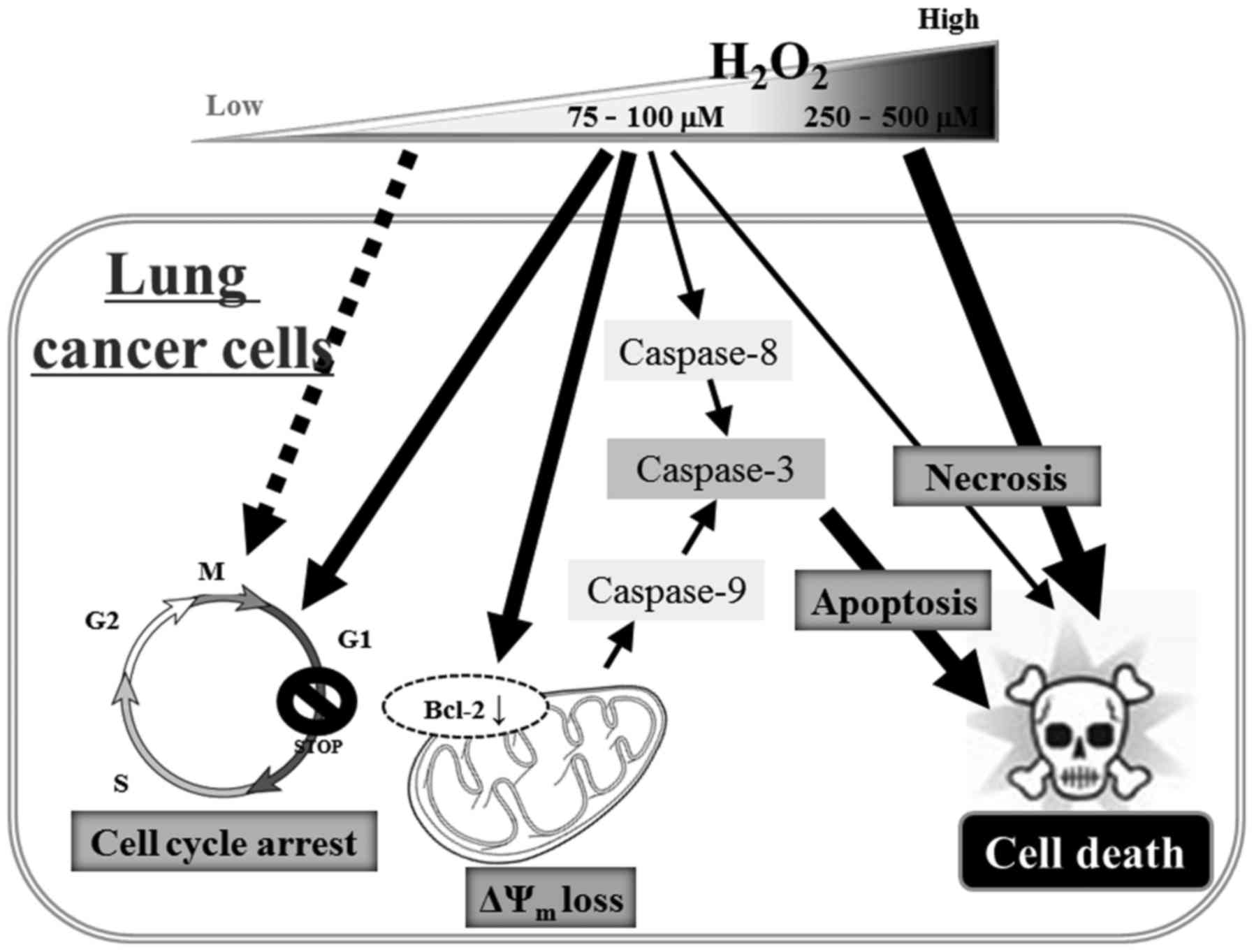
In conclusion, H2O2 inhibited
the growth of lung cancer cells through cell death and G1-phase
arrest of the cell cycle. Calu-6 and A549 cell death caused by
H2O2 resulted from necrosis, as well as
caspase-dependent apoptosis (Fig.
7). The present results provide useful information to
comprehend the cytotoxicological effect of exogenous
H2O2 on lung cancer cells in regards to cell
growth and death. In addition, novel strategies for the treatment
of lung cancer based on the use of H2O2 may
be helpful in reducing the mortality related to this
malignancy.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
National Research Foundation of Korea (NRF) funded by the Korean
government (MSIP; 2016R1A2B4007773) and supported by the ‘Research
Base Construction Fund Support Program’ funded by Chonbuk National
University in 2018.
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors' contributions
WHP was the sole contributor to the conception and
design, acquisition of data, analysis and interpretation of data
and writing of the manuscript. WHP is accountable for all aspects
of the work in ensuring that questions related to the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that he has no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
H2O2
|
hydrogen peroxide
|
|
ROS
|
reactive oxygen species
|
|
NADPH
|
nicotinamide adenine dinucleotide
phosphate
|
|
Z-VAD-FMK
|
benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone
|
|
Z-DEVD-FMK
|
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethyl ketone
|
|
Z-IETD-FMK
|
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethyl ketone
|
|
Z-LEHD-FMK
|
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethyl ketone
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
FITC
|
fluorescein isothiocyanate
|
|
PI
|
propidium iodide
|
References
|
1
|
Gonzalez C, Sanz-Alfayate G, Agapito MT,
Gomez-Niño A, Rocher A and Obeso A: Significance of ROS in oxygen
sensing in cell systems with sensitivity to physiological hypoxia.
Respir Physiol Neurobiol. 132:17–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: An update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: A comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wilcox CS: Reactive oxygen species: Roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen TJ, Jeng JY, Lin CW, Wu CY and Chen
YC: Quercetin inhibition of ROS-dependent and -independent
apoptosis in rat glioma C6 cells. Toxicology. 223:113–126. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dasmahapatra G, Rahmani M, Dent P and
Grant S: The tyrphostin adaphostin interacts synergistically with
proteasome inhibitors to induce apoptosis in human leukemia cells
through a reactive oxygen species (ROS)-dependent mechanism. Blood.
107:232–240. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wallach-Dayan SB, Izbicki G, Cohen PY,
Gerstl-Golan R, Fine A and Breuer R: Bleomycin initiates apoptosis
of lung epithelial cells by ROS but not by Fas/FasL pathway. Am J
Physiol Lung Cell Mol Physiol. 290:L790–L796. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sarsour EH, Kumar MG, Chaudhuri L, Kalen
AL and Goswami PC: Redox control of the cell cycle in health and
disease. Antioxid Redox Signal. 11:2985–3011. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Budihardjo I, Oliver H, Lutter M, Luo X
and Wang X: Biochemical pathways of caspase activation during
apoptosis. Annu Rev Cell Dev Biol. 15:269–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mehmet H: Caspases find a new place to
hide. Nature. 403:29–30. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hinz B, Phan SH, Thannickal VJ, Prunotto
M, Desmoulière A, Varga J, De Wever O, Mareel M and Gabbiani G:
Recent developments in myofibroblast biology: Paradigms for
connective tissue remodeling. Am J Pathol. 180:1340–1355.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rhee SG, Kang SW, Jeong W, Chang TS, Yang
KS and Woo HA: Intracellular messenger function of hydrogen
peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol.
17:183–189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vilhardt F and van Deurs B: The phagocyte
NADPH oxidase depends on cholesterol-enriched membrane microdomains
for assembly. EMBO J. 23:739–748. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park WH: The effects of exogenous H2O2 on
cell death, reactive oxygen species and glutathione levels in calf
pulmonary artery and human umbilical vein endothelial cells. Int J
Mol Med. 31:471–476. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park WH: Exogenous H2O2 induces growth
inhibition and cell death of human pulmonary artery smooth muscle
cells via glutathione depletion. Mol Med Rep. 14:936–942. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of lung cancer Calu-6 cells via
caspase-dependent apoptosis. Chem Biol Interact. 177:107–114. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park WH, Seol JG, Kim ES, Hyun JM, Jung
CW, Lee CC, Kim BK and Lee YY: Arsenic trioxide-mediated growth
inhibition in MC/CAR myeloma cells via cell cycle arrest in
association with induction of cyclin-dependent kinase inhibitor,
p21, and apoptosis. Cancer Res. 60:3065–3071. 2000.PubMed/NCBI
|
|
20
|
Park WH: Anti-apoptotic effect of caspase
inhibitors on H2O2-treated HeLa cells through early suppression of
its oxidative stress. Oncol Rep. 31:2413–2421. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
You BR, Kim SH and Park WH: Reactive
oxygen species, glutathione, and thioredoxin influence suberoyl
bishydroxamic acid-induced apoptosis in A549 lung cancer cells.
Tumour Biol. 36:3429–3439. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han YH, Kim SH, Kim SZ and Park WH:
Antimycin A as a mitochondria damage agent induces an S phase
arrest of the cell cycle in HeLa cells. Life Sci. 83:346–355. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of human lung cancer Calu-6 cells
via arresting the cell cycle arrest. Toxicol In Vitro.
22:1605–1609. 2008. View Article : Google Scholar : PubMed/NCBI
|