Introduction
Bladder cancer (BC) is a common malignancy
worldwide. Approximately 70% of BC patients receive a preliminary
diagnosis of non-muscle invasive BC (NMIBC). Of these, 50–70% of
patients exhibit recurrence, and 10–20% progress to muscle-invasive
BC (MIBC) (1). Over the last
decade, significant progress has been made regarding the
mechanisms, diagnosis and therapy of BC (2). Nonetheless, a high rate of recurrence
of non-invasive BC and poor survival of patients with invasive BC
have not been thoroughly solved. Therefore, there is an unmet need
in understanding the molecular mechanisms of BC development,
improving the effective treatment and early detection of disease
recurrence.
MicroRNAs (miRNAs) are a group of non-coding small
RNAs (22 nucleotides in length) that negatively regulate gene
expression by binding to the 3′ untranslated region (3′-UTR) of
target mRNAs (3,4). There is strong evidence supporting
that the dysregulation of the expression of miRNAs is implicated in
human cancer development and progression (5,6). As
one of the three members of the miR-148/152 family, miR-152 has
been identified to accelerate growth of certain tumor types when
its expression is downregulated (7). Recent studies reported that the
aberrant expression of miR-152 plays an important role in the
pathogenesis of BC (8,9). However, the molecular mechanism
underlying the effect of aberrant expression of miR-152 on the
development of BC remains poorly understood.
As other protein-coding genes, miRNA expression is
regulated by the same mechanisms including epigenetic regulation
(10,11). As one of the most important
epigenetic regulators, DNA methylation is involved in various
biological processes including cancer (12–15).
DNA methylation plays an important role in regulating gene
expression, especially when it occurs in CpG island regions of gene
promoters (16). Previous studies
have indicated that several miRNAs are regulated by DNA methylation
in various types of cancers and metabolic diseases (17–19).
Notably, miRNAs control the chromatin structure by regulating
histone modifiers and DNA methyltransferases such as DNA
methyltransferases (DNMTs), ten-eleven translocations (TETs),
histone deacetylases (HDACs) and polycomb genes, indicating that
miRNAs can also indirectly modulate gene expression by controlling
epigenetic modifications (20–22).
In our previous study, we identified a functional
crosstalk between miR-152 and DNMT1 involved in Nis-induced
malignant transformation (23). In
the present study, we found that the expression of miR-152 in BC
cells and tissues was relatively lower than in normal bladder cells
and adjacent tissues of BC (ATBC), respectively. In addition, we
identified a higher expression of DNMT1 in BC cells and tissues
compared to normal bladder cells and adjacent tissues of BC
(24). Additionally,
methylation-specific PCR (MSP) revealed that miR-152 was methylated
in BC cells and tissues, whereas unmethlylated miR-152 was present
in normal bladder cells and adjacent tissues of BC. Since DNA
methylation can be regulated by miRNAs by targeting DNMTs (25), we aimed to determine the presence of
a regulatory circuit of miR-152 and DNMT1 in human BC. Furthermore,
we also determined whether miR-152 contributed to tumor cell
growth.
Materials and methods
Cell culture, treatment and
transfection
The immortalized human bladder epithelial cell line
SV-HUC-1 and the human urinary bladder transitional carcinoma cell
line T24 were both purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA). The human BC cell line
UM-UC-3 was purchased from the Cell Bank of the Chinese Academy of
Sciences (Shanghai, China). SV-HUC-1 cells were cultured in F12K,
T24 cells were cultured in RPMI-1640 medium, and UM-UC-3 cells were
cultured in MEM. All media were supplemented with 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), in a humidified air atmosphere of 5% CO2 at 37°C.
Trypsin (0.25% with 1 mM EDTA) (Invitrogen, Carlsbad, CA, USA) was
also used to harvest the cells for further experiments.
One day prior to transfection, the cells were plated
in growth medium without antibiotics at a density of 50–60%.
miR-152 mimic and mimic-control were transfected into UM-UC-3 cells
using Lipofectamine 2000 (Invitrogen) according to the
manufacturer's protocol. miR-152 inhibitor and inhibitor-control
were transfected into SV-HUC-1 cells as previously described
(23). Cells were harvested at 24,
48, 72 and 96 h post-transfection. For combined treatment of the
miR-152 inhibitor and the DNMT inhibitor, 5-aza-2-deoxycytidine
(DAC), SV-HUC-1 cells transfected with miR-152 inhibitor were
further divided into two groups, in which one group was treated
with 12.5 µM DAC (Sigma-Aldrich; Merck KGaA) for 72 h, and the
other group was used as a negative control.
Patients and tissue samples
A set of 24 bladder tumor specimens was obtained
from patients who underwent BC surgery between January 2012 and May
2015 at the First Affiliated Hospital of Guangzhou Medical
University (Guangzhou, Guangdong, China). None of the patients
received antitumor treatment prior to tumor sampling. A total of 24
adjacent tissues of bladder tumors from matched patients was
collected as the control group. Details of the characteristics of
the patients were described in our previous study (24). In order to further validate the
relationship between miR-152 and DNMT1 mRNA expression, public data
from both primary invasive and papillary BC tissues from ‘The
Cancer Genome Atlas’ (TCGA) data portal (https://tcga-data.nci.nih.gov) were used, and explored
through the cBio Cancer Genomics Portal (http://cbioportal.org). The data from 404 patients
from Illumina HiSeq gene expression platforms (Illumina, Inc., San
Diego, CA, USA) were extracted.
Real-time quantitative PCR
Total RNA from tissues or cell lines was extracted
using TRIzol reagent (Invitrogen) according to the manufacturer's
instructions. The quantity and quality of the RNA were determined
by UV spectrophotometer. Bulge-loop™ miRNA qRT-PCR Primer sets (one
RT primer and a pair of qPCR primers for each set) specific for
miR-152 were designed by Guangzhou RiboBio Co., Ltd. (Guangzhou,
China). The relative quantification value of the target, normalized
to the control, was calculated by the the ΔΔCq method (26). PCR was carried out as follows: 95°C
for 20 sec, 40 cycles of 10 sec at 95°C, 20 sec at 58°C and 30 sec
at 72°C. Samples were analyzed in triplicate.
Western blot analysis
Protein was extracted from T24, UM-UC-3 and SV-HUC-1
cells for western blotting. Cultured cells were collected and
washed three times with PBS. Following incubation with RIPA buffer
(Sigma-Aldrich; Merck KGaA) containing 5 mM EDTA, PMSF, cocktail
inhibitor and phosphatase inhibitor cocktail, cells were collected
in a centrifuge tube. Cell lysates were centrifuged at 13,000 × g
for 15 min at 4°C and insoluble debris was discarded. The proteins
were determined by BCA quantitative method. Soluble proteins (30
µg) were subjected to 8% SDS-PAGE and transferred to a Hybond-P
polyvinylidene difluoride (PVDF) membrane. Membranes were
blocked in TPBS (PBS with 0.05% Tween-20) containing 5% (w/v)
non-fat dry milk for 1 h at room temperature, washed in TPBS and
then incubated with primary antibody. Immunoblottings were
performed with 1:1,000 diluted anti-DNMT1 antibody (cat. no. D4692;
Sigma-Aldrich; Merck KGaA) and 1:1,000 diluted anti-GAPDH antibody
(cat. no. sc-166545; Santa Cruz Biotechnology, Santa Cruz, CA,
USA). Blots were visualized using the Immobilon solutions (EMD
Millipore, Billerica, MA, USA) under a chemiluminescence detection
system, the ChemiDoc XRS (Bio-Rad Laboratories). Band area
intensity was analyzed using Quantity One software (Bio-Rad
Laboratories).
Methylation-specific PCR (MSP)
Genomic DNA of tissues and cells was extracted using
standard phenol/chloroform purification and ethanol precipitation.
Bisulfite modification of the genomic DNA was performed using EZ
DNA Methylation-Gold kit (Zymo Research Corp., Irvine, CA, USA).
Methylation of each sample was evaluated in triplicate using MSP.
PCR products were electrophoresed on a 2% agarose gel for analysis.
Primer sequences for miR-152 are listed in Table I.
 | Table I.Primers used in this study. |
Table I.
Primers used in this study.
|
| Primer sequence
(5′-3′) |
|---|
|
|
|
|---|
| Primer name | F | R |
|---|
| MSP |
| miR-152
(methylated) |
TTTAGAATTCGCGCGTTC |
CCGACGAACTCAAAACAAA |
| miR-152 (non
methylated) |
GGAGGAAAAGTTTGTTTTAGTGTT |
ACCCACCAATAATAAAAACCAAA |
| ChIP |
| miR-152
promoter |
AGAGGAGGCCTGTCCTGAGT |
CGGGTAGACTCCAGAAGCAT |
Chromatin immunoprecipitation
(ChIP)
Chromatin immunoprecipitation was performed using
EZ-Magna ChIP A/G kit (EMD Millipore) according to the
manufacturer's instructions. Briefly, cells were grown on 100-mm
plates to 85% confluence; formaldehyde was added to a final
concentration of 1%, and the plates were incubated for 10 min at
37°C; the cross-linking reaction was stopped by the addition of
0.125 M glycine for 5 min at room temperature. Cells were sonicated
and sheared to yield fragments between 200 and 1,000 bp. Anti-DNMT1
(5 µg; Abcam, Cambridge, UK) was added to the sonicated samples and
incubated at 4°C overnight with rotation. Immune complexes were
collected with Protein A/G agarose beads and washed with buffer to
remove nonspecific binding. Protein/DNA complex was reverse
crosslinked and DNA was purified using spin columns. DNA was
detected with quantitative PCR. Primers for ChIP-qPCR are listed in
Table I.
Cell proliferation analysis. The miR-152 mimic was
transfected into the T24 and UM-UC-3 cells and the miR-152
inhibitor was transfected into the SV-HUC-1 cells lasting 6 h.
Subsequently, the cells were plated into a new dish. Cells
(2×103) were plated in triplicate and measured at the
indicated time-points: 24, 48, 72 and 96 h. The number of cells was
determined using the CellTiter 96® AQueous One Solution
assay (Promega Corp., Madison, WI, USA). Triplicate plates were
counted for each cell line.
Luciferase reporter assay
The cells (1×105 cells/well) were seeded
in 24-well plates. When cells reached 70–80% confluence, 0.5 µg of
reporter plasmids alone or co-transfected with or without microRNA
mimics were transiently transfected using Lipofectamine 2000
according to the manufacturer's instructions (Invitrogen; Thermo
Fisher Scientific, Inc.). Luciferase activity was determined as
previously reported (23).
Statistical analysis
All analyses were performed with the SPSS 13
statistical software (SPSS, Inc., Chicago, IL, USA). Statistical
significance was determined by Student's t-test and Mann-Whitney U
test. The relationship between the expression level of miR-152 and
clinicopathological parameters was analyzed using the Mann-Whitney
U test. The correlations were analyzed using Spearman's correlation
rank test. Differences in probability values (P-values) of <0.05
were considered to indicate a statistically significant
difference.
Results
miR-152 is downregulated in BC cells
and tumor tissues
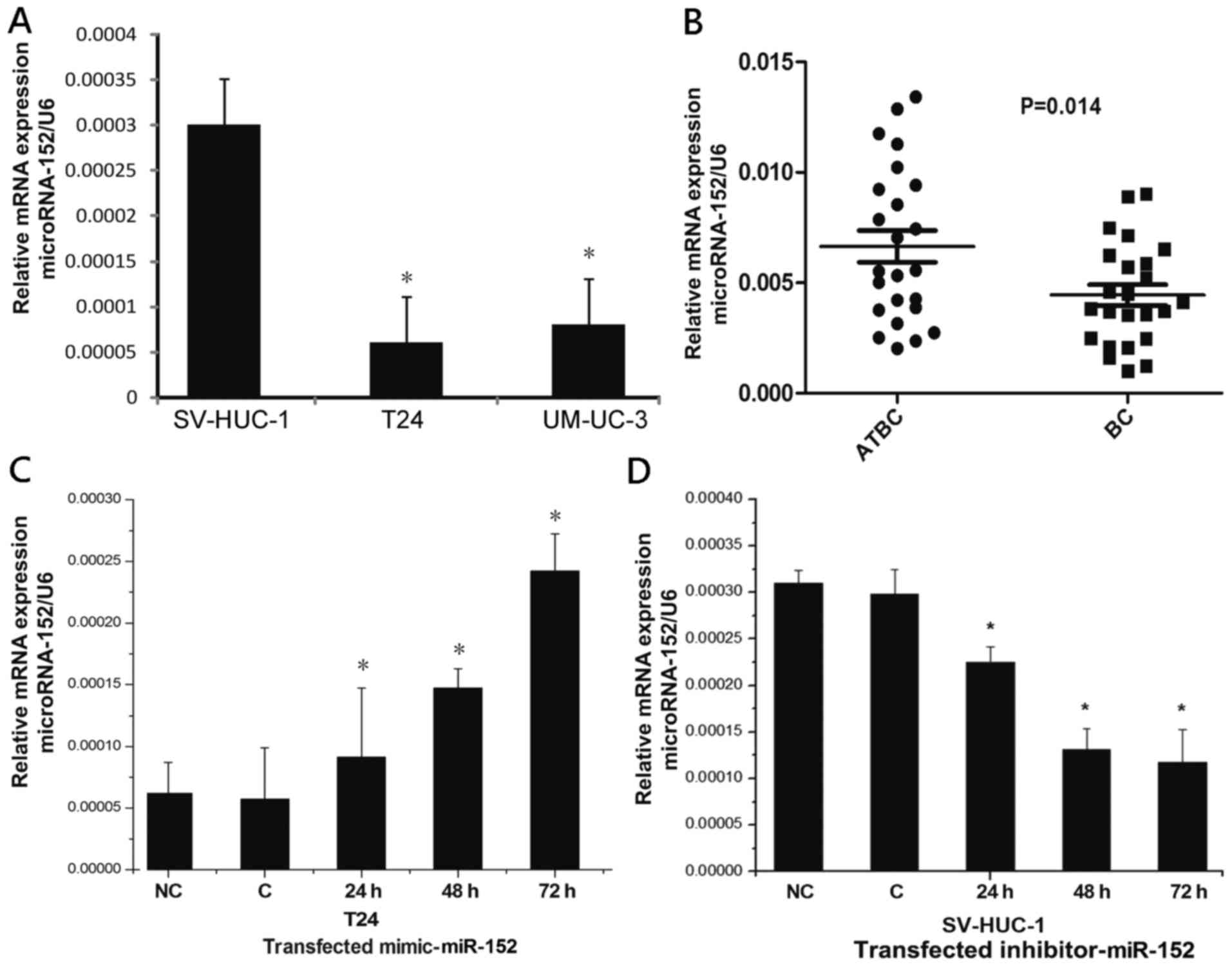
To determine the expression of miR-152 in BC,
real-time PCR was used to compare the expression levels of miR-152
in T24 cells, UM-UC-3 cells and SV-HUC-1 cells. A lower level of
miR-152 was observed in T24 cells and UM-UC-3 cells compared with
SV-HUC-1 cells (Fig. 1A). We
further compared the expression of miR-152 between 24 BC tissue
specimens and the matched adjacent normal tissues. Consistent with
our observations in cells, compared to the matched adjacent tissues
of BC, miR-152 had a relatively lower expression in BC tissues
(Fig. 1B). Additionally, after
transfection with the miR-152 mimic, the expression of miR-152 in
T24 cells had an increased level in a time-dependent manner
(Fig. 1C). Conversely, treatment
with miR-152 inhibitor in SV-HUC-1 cells resulted in a reduction of
miR-152 expression in a time-dependent manner (Fig. 1D). These results indicated that
miR-152 was downregulated in BC cell and tumor tissues. Notably,
the analysis of the clinicopathological features of BC patients
revealed that a lower miR-152 expression level was significantly
correlated with the stage and grade of BC patients, which was
independent of patient sex and age (Table II).
| Table II.Clinicopathological features of BC
patients and the levels of miR-152 expression in cancer
tissues. |
Table II.
Clinicopathological features of BC
patients and the levels of miR-152 expression in cancer
tissues.
|
|
| Molecular
expression level |
|---|
|
|
|
|
|---|
|
Characteristics | No. of patients
(%) |
miR-152c | P-value |
|---|
| All patients | 24 (100) |
|
|
| Sexa |
|
| 0.725 |
|
Females | 10 (41.6) | 0.0046
(0.0016–0.0088) |
|
|
Males | 14 (58.4) | 0.0042
(0.001–0.0089) |
|
| Age at
diagnosisa, years |
|
| 0.184 |
|
≤70 | 12 (50.0) | 0.0037
(0.001–0.0074) |
|
|
>70 | 12 (50.0) | 0.0051
(0.0012–0.0089) |
|
| Pathological
gradea |
|
| 0.03d |
|
Low | 8 (33.3) | 0.0064
(0.0035–0.0089) |
|
|
High | 16 (66.7) | 0.0034
(0.0010–0.0062) |
|
| Pathological
stageb |
|
| 0.04d |
|
pTa | 5 (20.8) | 0.0061
(0.0045–0.0088) |
|
|
pT1 | 13 (54.2) | 0.0045
(0.0012–0.0089) |
|
|
≥pT2 | 6 (25.0) | 0.0027
(0.001–0.0038) |
|
Downregulation of miR-152 is due to
DNA hypermethylation via DNMT1 in BC
The reduction or absence of gene expression mediated
by DNA hypermethylation plays an important role in cancer
development (27). We hypothesized
that downregulation of miR-152 in BC was due to DNA methylation. To
verify our hypothesis, we used an MSP method to determine the
methylation status of miR-152 promoter in T24 and SV-HUC-1 cells.
We observed that miR-152 was methylated in T24 cells, but not in
SV-HUC-1 cells (Fig. 2A).
Similarly, 17 out of 24 (70.8%) samples had CpG island methylation
of miR-152 promoter in BC tissues, while only 9 out of 24 (37.5%)
samples had CpG island methylation of miR-152 promoter in the
matched normal tissues adjacent to BC (Fig. 2B and C). Conversely, our previous
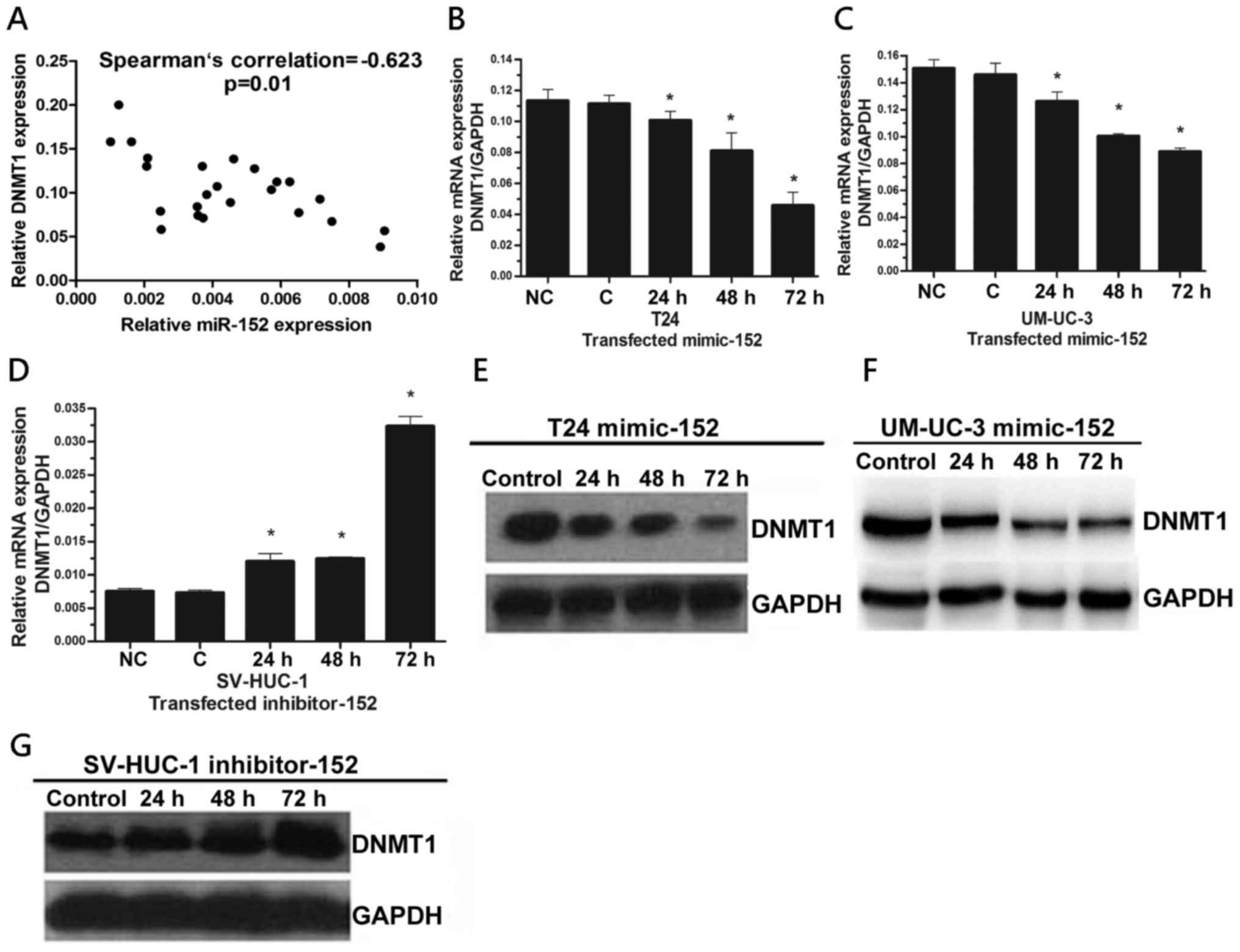
study demonstrated that the expression level of DNMT1 was
significantly higher in BC cells and tissues (24). To explore the potential regulatory
circuit of DNMT1 and miR-152, we transfected miR-152 mimics in T24
cells and detected the expression of DNMT1 and the methylation
level of miR-152 CpG island promoter. We determined that both DNMT1
mRNA expression and the DNA methylation of miR-152 promoter were
decreased in T24 cells transfected with miR-152 mimics (Figs. 3B and 2D). Notably, the binding of DNMT1 to the
miR-152 CpG island promoter was significantly decreased in T24
cells transfected with miR-152 mimics (Fig. 2E). In addition, DNMT1 binding to the
miR-152 CpG island promoter was significantly increased in SV-HUC-1
cells transfected with the miR-152 inhibitor compared to the
control group (Fig. 2F). To further
determine the relationship between the expression of miR-152 and
DNMT1, we performed Spearman's rank correlation analysis and found
a significantly negative correlation between the expression levels
of miR-152 and DNMT1 in BC specimens (r=−0.623, P=0.01) (Fig. 3A). In order to further confirm these
findings, we determined DNMT1 and miR-152 expression in The Cancer
Genome Atlas (TCGA) dataset which includes a total of 404 BC
samples. Notably, we also identified a negative correlation
(r=−0.145) in BC samples of stage IV, although there was no
correlation of DNMT1 with miR-152 mRNA expression among BC samples
of all stages. In addition, miR-152 levels may affect disease-free
survival (P=0.026) (data not shown). Collectively, our results
indicated that hypermethylation via DNMT1 is responsible for the
silencing of miR-152 expression in BC.
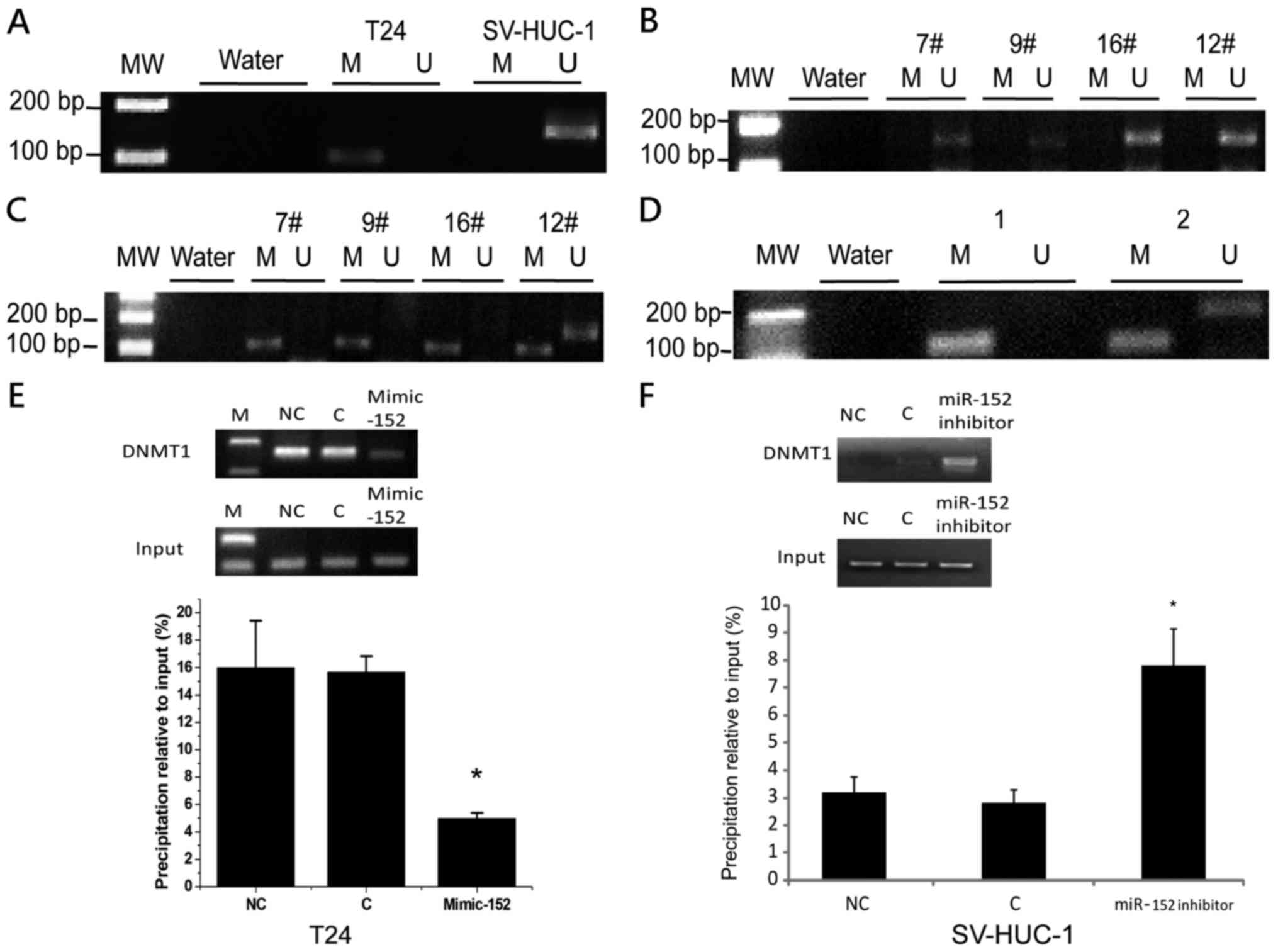 | Figure 2.The expression of miR-152 was
regulated by DNA methylation via DNMT1. (A) MSP analysis of miR-152
gene promoter in T24 and SV-HUC-1 cells. MSP, methylation-specific
PCR; MW, molecular weight; U, unmethylated status; M, methylated
status. Water was used as a control for PCR. (B) MSP analysis of
miR-152 gene promoter in BC tumors. MW, molecular weight;
7#, 9#, 16#, 12#,
Number of tumor samples. (C) MSP analysis of miR-152 gene promoter
in the matched adjacent tissues of BC. MW, molecular weight;
7#, 9#, 16#, 12#,
Number of adjacent tissues samples. (D) MSP analysis of miR-152
gene promoter in T24 cells transfected with miR-152 mimics and
control. MW, molecular weight; 1, Control; 2, T24 mimic-152. (E)
The ChIP analysis of the binding of DNMT1 in miR-152 CpG island
promoter in T24 cells transfected with miR-152 mimics and control.
Quantitative-ChIP analysis was conducted to assess the binding of
DNMT1 in miR-152 CpG island promoter with specific antibodies of
DNMT1 with normalization by total input DNA. Results represent the
mean ± SD from three independent experiments. *P<0.05, compared
with the negative control group. Right images are representative
results from quantitative PCR after ChIP assay. M, marker; NC,
negative control; C, control cells; ChIP, Chromatin
immunoprecipitation. (F) The ChIP analysis of the binding of DNMT1
in miR-152 CpG island promoter in SV-HUC-1 cells transfected with
miR-152 inhibitor and control. *P<0.05, compared with the
negative control group. miR-152, microRNA-152; DNMT1, DNA
methyltransferase 1; BC, bladder cancer. |
Expression of DNMT1 is regulated by
miR-152 by targeting the 3′-UTR in BC cells
DNMT1 is the key enzyme in DNA methylation which has
been identified as one of the high-scoring candidate genes of
miR-152 targets (28). To further
determine whether the expression of DNMT1 is regulated by miR-152,
we first determined the levels of DNMT1 mRNA in T24 and UM-UC-3
cells transfected miR-152 mimics by real-time PCR. We found that
DNMT1 expression was decreased in both of T24 and UM-UC-3 cells
transfected with the miR-152 mimic in a time-dependent manner
(Fig. 3B and C). Conversely, the
level of DNMT1 mRNA in SV-HUC-1 cells transfected the miR-152
inhibitor was increased in a time-dependent manner (Fig. 3D). These results were ascertained by
western blot analysis (Fig. 3E-G).
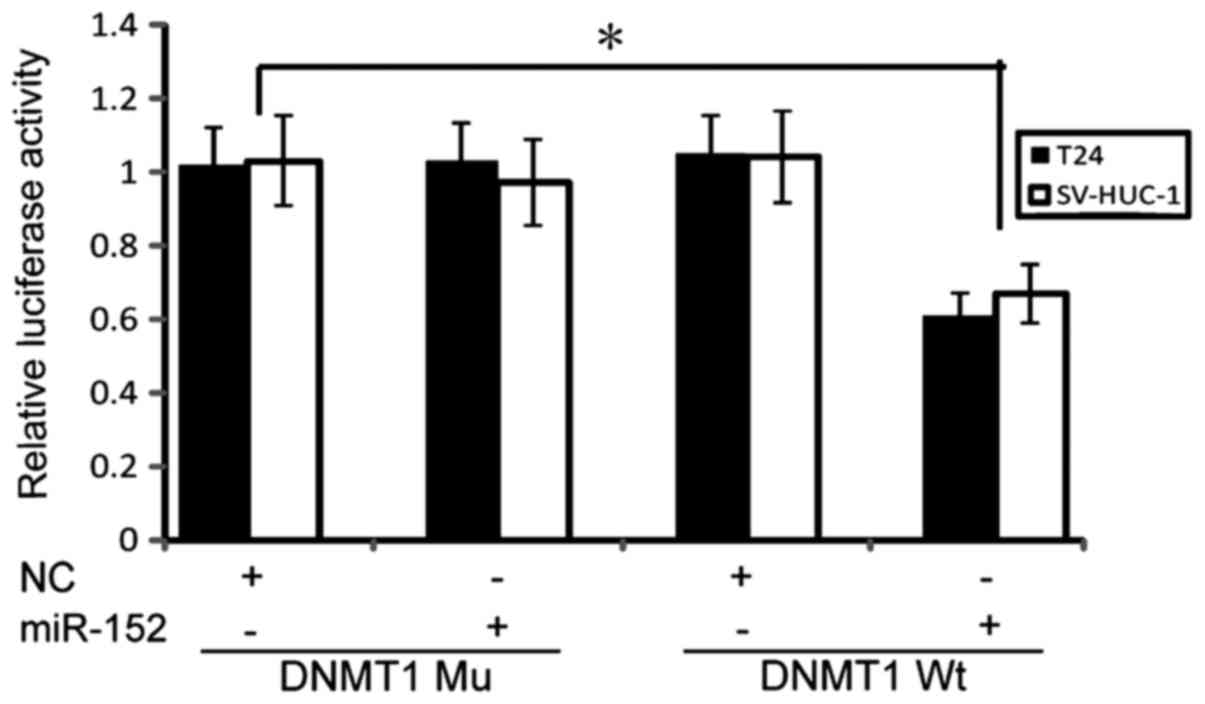
Notably, we revealed that miR-152 could directly target DNMT1
3′-UTR and suppress its expression in NiS-transformed cells
(23). To further validate our
results in T24 cells and SV-HUC-1 cells, a luciferase reporter
assay was performed, in which miR-152 significantly reduced DNMT1
wild-type 3′-UTR luciferase activity, but not DNMT1 Mu 3′-UTR
activity (Fig. 4), indicating that
miR-152 directly bound to the DNMT1 3′-UTR and inhibited its
expression in BC cells.
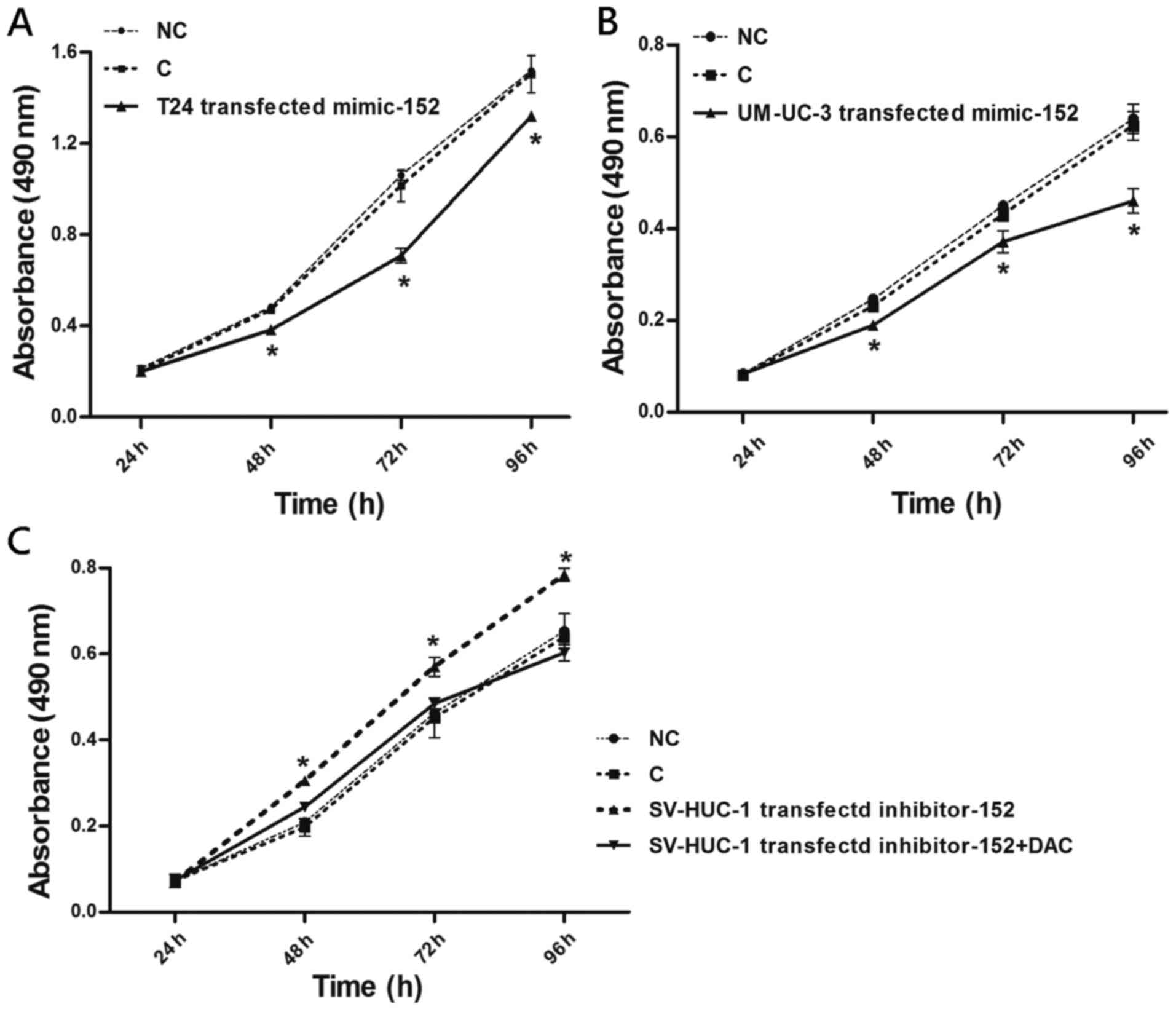
miR-152 inhibits BC cell growth
In order to clarify whether the expression of
miR-152 plays a significant role in BC malignant phenotypes, we
explored the effects of ectopically expressed miR-152 on the
proliferation of T24, UM-UC-3 and SV-HUC-1 cells. Both T24 and
UM-UC-3 cells transfected with miR-152 mimics exhibited
significantly decreased cell proliferation at 48, 72 and 96 h,
compared to the miR-control (Fig. 5A
and B). Conversely, SV-HUC-1 cells transfected with miR-152
inhibitor had increased cell proliferation at 48, 72, and 96 h,
compared to the miR-control (Fig.
5C). Notably, the combined treatment of the miR-152 inhibitor
and the DNMT inhibitor, 5-aza-2-deoxycytidine (DAC), led to
decreased cell proliferation compared to suppression of the miR-152
inhibitor alone, whereas the combined treatment resulted in a
slight elevation of cell proliferation compared to that in control
SV-HUC-1 cells at 48 and 72 h post treatment. These findings
highlighted a reciprocal regulatory loop between miR-152 and
DNMT1.
Discussion
In recent years, there has been an increasing
interest in the role of epigenetic regulation in human diseases
(29). For instance, numerous
studies have revealed that epigenetic dysregulation of miRNA
expression plays a key role in cancer development (30,31).
In the present study, we demonstrated that upregulation of DNMT1
was responsible for the hypermethylation of miR-152, which resulted
in the downregulation of miR-152. miR-152 mediated DNA methylation
by directly targeting DNMT1 3′-UTR, indicative of a novel
regulatory circuit between DNMT1 and miR-152 in human BC.
MiRNAs are a group of post-transcriptional
regulators which inhibit target gene expression by mRNA degradation
and translation inhibition (32,33).
Several studies have revealed that miRNAs are altered in many
cancers, and that they can initiate carcinogenesis or drive tumor
progression (11,34). miR-152 is located at 17q21.32, and a
typical CpG island is surrounded by miR-152 promoter. Huang et
al have suggested a tumor-suppressive role of miR-152 in the
epigenetic aberration of HBV-related hepatocellular carcinoma
(25). Stumpel et al
(35) reported that downregulation
of miR-152 was related to CpG hypermethylation. Further research
indicated that the hypermethylation of the miR-152 CpG island was
associated with poor clinical outcome in mixed-lineage leukemia
(MLL). In BC, Kohler et al (9) reported that a reduced expression of
miR-152 was related to increased DNA methylation. Consistent with
this study, our results demonstrated the downregulation of miR-152
expression in BC as well as the hypermethylation at the promoter
region of miR-152. Notably, we identified that the DNMT1 bound to
the miR-152 CpG island promoter was markedly decreased in T24 cells
transfected with miR-152 mimics, which was similar to a previous
study revealing that the overexpression of miR-152 decreased DNMT1
and MeCP2 binding to the CpG island promoter of miR-152 in an FLS
model (36). In addition, it was
revealed that miR-152 could act as a tumor suppressor by targeting
TGF, and inhibit prostate cancer cell migration and invasion
(37). Overexpression of miR-152
suppressed cell proliferation and colony formation in
non-small-cell lung cancer (NSCLC) cells by inhibiting FGF2
(38). Furthermore, miR-152 was
downregulated in human glioblastoma stem cells (GSCs). Restoring
the expression of miR-152 inhibited the cell proliferation of GSCs
by downregulating KLF4 (39).
Recently, miR-152 was identified as a regulator of lung metastasis
which increased metastasis rates, but failed to promote primary
tumor growth through a genome-wide in vivo CRISPR/Cas9
screening (40). Consistent with
these studies, we also found that the proliferation of T24 cells
and UM-UC-3 cells was significantly inhibited after treatment with
miR-152 mimics. Conversely, the proliferation of SV-HUC-1 cells
transfected with the miR-152 inhibitor was significantly increased,
compared to those transfected with the inhibitor miR-control.
Collectively, our results further confirmed the tumor-suppressive
activity of miR-152 which is epigenetically silenced during human
BC development.
The downregulation of miR-152 mediated by the
methylation of miR-152 promoters was associated with breast cancer
grades and lymph node-metastasis (LN) status (41). Similarly, lower expression of
miR-152 was related to increased tumor sizes and advanced tumor
stages in gastric cancer (7,42). In
addition, DNA hypermethylation on CpG islands was related to the
overexpression of DNMT1 in multistage bladder carcinogenesis
(43). In accordance with these
findings, our results demonstrated that the upregulation of DNMT1
and downregulation of miR-152 were related to the grades and stages
of BC.
Epigenetic dysfunction of genes is an important
feature in human cancer development. Increasing evidence has
indicated that DNA methylation is one of the key regulators in
urinary tumors. DNMT1 plays a vital role in human cell malignant
transformation, indicating that the aberrant expression of DNMT1
may contribute to the development of human cancer (44). Our present study revealed that the
high expression of DNMT1 expression was correlated with low
expression of miR-152 with hypermethylation of miR-152 promoter in
BC cells and tissues. Furthermore, the upregulation of miR-152
expression led to decreased expression of DNMT1. All these findings
indicated a dysregulated DNMT1 expression due to aberrant
expression of miR-152 in BC.
Previous findings revealed that miR-152 induced
aberrant DNA methylation in hepatitis B virus-related
hepatocellular carcinoma (25) and
ovarian cancer by targeting DNA methyltransferase 1 (45). In an arthritic rat model, miR-152
was found to regulate canonical Wnt pathway activation by targeting
DNMT1 3′-UTR (46). Consistent with
these studies, our results indicated that DNMT1 expression was
suppressed by miR-152 via a direct interaction with a highly
conserved region of the DNMT1 3′UTR in BC. Collectively, these
findings demonstrated that miR-152 directly interacted with DNMT1
by targeting its predicted binding site in BC.
Notably, the relationship between miRNA-epigenetics
is enriched by a line of findings indicating that the expression
levels of the epigenetic effectors can be regulated by some miRNAs,
while epigenetic effectors can also modulate the expression of
these miRNAs (47,48). For example, in pancreatic cancer,
the overexpression of miR-148b and miR-152 repressed the expression
of DNMT1, leading to the decreased DNA methylation and
overexpression of tumor-suppressor genes (49). The overexpression of miR-152 in
dairy cow mammary epithelial cells resulted in significantly
lowered expression of DNMT1, which in turn led to a reduction of
global DNA methylation (50).
Conversely, miRNAs can also be modified by DNMTs. The putative CpG
island promoter region of miR-29b/c was revealed to be regulated by
DNA methylation. The increased expression of DNMT3a was related to
silencing of miR-29b/c in gastric carcinogenesis (51). In addition, the overexpression of
DNMT1 led to the hypermethylation of miR-148a and miR-152 genes in
breast cancer. Furthermore, the expression of DNMT1, a direct
target of miR-148a and miR-152, was inversely correlated with the
expression levels of miR-148a/152, indicating a negative feedback
regulatory loop between miR-148a/152 and DNMT1 (41). In a previous study, we identified a
crucial functional crosstalk between miR-152 and DNMT1 via a
double-negative circuit involved in NiS-induced malignant
transformation (23). In the
present study, we confirmed that a high miR-152 level was
associated with low DNMT1 expression in SV-HUC-1 cells, whereas a
low level of miR-152 was related to the high DNMT1 protein
expression in T24 cells and UM-UC-3 cells, indicating a negative
feedback regulation between miR-152 and DNMT1 expression in BC.
In conclusion, we identified a novel regulatory
circuit between miR-152 and DNMT1 and our study revealed that the
tumor-suppressive activity of miR-152 is epigenetically silenced by
directly interacting with the DNMT1 in BC. The discovery of the
relationship between miR-152 and DNMT1 is highly beneficial to
enrich our understanding of BC development, which could be further
explored as effective therapeutic targets and early detection
biomarkers.
Acknowledgements
Not applicable.
Funding
The present study was partly supported by grants
from The National Natural Science Foundation of China (nos.
81472999, 81772699, 81272350 to WJ; no. 81473014 to WZ and no.
81600201 to YC), The Key Natural Science Foundation of Guangdong
(no. 2015A030311038 to WJ) and Guangzhou People's Livelihood
Science and Technology Project (no. 201803010052 to WJ), The
Program of Health Department of Guangdong Province (no. C2013025 to
DQ), The Key Science Foundation of Guangzhou (no. 201804020023 to
DQ), The Six Talent Program of Jiangsu (no. 2013WSW-035 to GZ), The
Research Plan of Discipline Distribution Program of Shenzhen
(JCYJ20160328161613864 to JY) and The Technology Research Plan of
Discipline Distribution Program of Shenzhen (JSGG20170414104216477
to JY).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
HZ, DQ and JL conceived and performed the
experiments and analyzed the data. TP, LY, JY, YZ, GZ and YC
performed the experiments. YH, JS, BQ and ML collected the clinical
data and specimens. WJ and WZ conceived the project, analyzed the
data, wrote the manuscript and provided supervision. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
The present study was performed in accordance with
the Declaration of Helsinki. Written informed consent was obtained
from each patient enrolled in this study. The present study was
reviewed and approved by the Ethics Committee of Guangzhou Medical
University in November 2015 (no. 2015–11).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
miR-152
|
microRNA-152
|
|
ATBC
|
adjacent tissues of bladder cancer
|
|
ATCC
|
American Type Culture Collection
|
|
BC
|
bladder cancer
|
|
ChIP
|
chromatin immunoprecipitation
|
|
DAC
|
5-aza-2-deoxycytidine
|
|
DNMT1
|
DNA methyltransferase 1
|
|
DNMTs
|
DNA methyltransferases
|
|
IRB
|
The Institutional Review Board
|
|
m5C
|
5-methylcytosine
|
|
MIBC
|
muscle-invasive bladder cancer
|
|
miRNAs
|
microRNAs
|
|
MLL
|
mixed-lineage leukemia
|
|
MSP
|
methylation-specific PCR
|
|
NMIBC
|
non-muscle-invasive bladder cancer
|
|
NSCLC
|
non-small cell lung cancer
|
|
TCGA
|
The Cancer Genome Atlas
|
|
3′-UTR
|
3′ untranslated region
|
References
|
1
|
Kaufman DS, Shipley WU and Feldman AS:
Bladder cancer. Lancet. 374:239–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cancer Genome Atlas Research N, :
Comprehensive molecular characterization of urothelial bladder
carcinoma. Nature. 507:315–322. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen Q, Chen X, Zhang M, Fan Q, Luo S and
Cao X: miR-137 is frequently downregulated in gastric cancer and is
a negative regulator of Cdc42. Dig Dis Sci. 56:2009–2016. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Croce CM: Causes and consequences of
microRNA dysregulation in cancer. Nat Rev Genet. 10:704–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Y, Song Y, Wang Z, Yue Z, Xu H, Xing
C and Liu Z: Altered expression of MiR-148a and MiR-152 in
gastrointestinal cancers and its clinical significance. J
Gastrointest Surg. 14:1170–1179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang X, Du L, Wang L, Li J, Liu Y, Zheng
G, Qu A, Zhang X, Pan H, Yang Y, et al: Serum microRNA expression
signatures identified from genome-wide microRNA profiling serve as
novel noninvasive biomarkers for diagnosis and recurrence of
bladder cancer. Int J Cancer. 136:854–62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kohler CU, Bryk O, Meier S, Lang K,
Rozynek P, Brüning T and Käfferlein HU: Analyses in human
urothelial cells identify methylation of miR-152, miR-200b and
miR-10a genes as candidate bladder cancer biomarkers. Biochem
Biophys Res Commun. 438:48–53. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davalos V and Esteller M: MicroRNAs and
cancer epigenetics: A macrorevolution. Curr Opin Oncol. 22:35–45.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rouhi A, Mager DL, Humphries RK and
Kuchenbauer F: MiRNAs, epigenetics, and cancer. Mamm Genome.
19:517–525. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Davis CD and Uthus EO: DNA methylation,
cancer susceptibility, and nutrient interactions. Exp Biol Med.
229:988–995. 2004. View Article : Google Scholar
|
|
13
|
Jones PA: DNA methylation errors and
cancer. Cancer Res. 56:2463–2467. 1996.PubMed/NCBI
|
|
14
|
Laird PW and Jaenisch R: DNA methylation
and cancer. Hum Mol Genet. 3:1487–1495. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu L, Wylie RC, Andrews LG and Tollefsbol
TO: Aging, cancer and nutrition: The DNA methylation connection.
Mech Ageing Dev. 124:989–998. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maruyama R, Toyooka S, Toyooka KO, Harada
K, Virmani AK, Zöchbauer-Müller S, Farinas AJ, Vakar-Lopez F, Minna
JD, Sagalowsky A, et al: Aberrant promoter methylation profile of
bladder cancer and its relationship to clinicopathological
features. Cancer Res. 61:8659–8663. 2001.PubMed/NCBI
|
|
17
|
Fabbri M and Calin GA: Epigenetics and
miRNAs in human cancer. Adv Genet. 70:87–99. 2010.PubMed/NCBI
|
|
18
|
Kunej T, Godnic I, Ferdin J, Horvat S,
Dovc P and Calin GA: Epigenetic regulation of microRNAs in cancer:
An integrated review of literature. Mutat Res. 717:77–84. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Melo SA and Esteller M: Dysregulation of
microRNAs in cancer: Playing with fire. FEBS Lett. 585:2087–2099.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Balaguer F, Link A, Lozano JJ, Cuatrecasas
M, Nagasaka T, Boland CR and Goel A: Epigenetic silencing of
miR-137 is an early event in colorectal carcinogenesis. Cancer Res.
70:6609–6618. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fabbri M, Garzon R, Cimmino A, Liu Z,
Zanesi N, Callegari E, Liu S, Alder H, Costinean S,
Fernandez-Cymering C, et al: MicroRNA-29 family reverts aberrant
methylation in lung cancer by targeting DNA methyltransferases 3A
and 3B. Proc Natl Acad Sci USA. 104:15805–15810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Varambally S, Cao Q, Mani RS, Shankar S,
Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al:
Genomic loss of microRNA-101 leads to overexpression of histone
methyltransferase EZH2 in cancer. Science. 322:1695–1699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ji W, Yang L, Yuan J, Yang L, Zhang M, Qi
D, Duan X, Xuan A, Zhang W, Lu J, et al: MicroRNA-152 targets DNA
methyltransferase 1 in NiS-transformed cells via a feedback
mechanism. Carcinogenesis. 34:446–453. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qi D, Li J, Que B, Su J, Li M, Zhang C,
Yang M, Zhou G and Ji W: Long non-coding RNA DBCCR1-003 regulate
the expression of DBCCR1 via DNMT1 in bladder cancer. Cancer Cell
Int. 16:812016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang J, Wang Y, Guo Y and Sun S:
Downregulated microRNA-152 induces aberrant DNA methylation in
hepatitis B virus-related hepatocellular carcinoma by targeting DNA
methyltransferase 1. Hepatology. 52:60–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCq method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wilson AS, Power BE and Molloy PL: DNA
hypomethylation and human diseases. Biochim Biophys Acta.
1775:138–162. 2007.PubMed/NCBI
|
|
28
|
Lewis BP, Shih IH, Jones-Rhoades MW,
Bartel DP and Burge CB: Prediction of mammalian microRNA targets.
Cell. 115:787–798. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Egger G, Liang G, Aparicio A and Jones PA:
Epigenetics in human disease and prospects for epigenetic therapy.
Nature. 429:457–463. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sutherland JE and Costa M: Epigenetics and
the environment. Ann NY Acad Sci. 983:151–160. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tsuchiya N and Nakagama H: MicroRNA, SND1,
and alterations in translational regulation in colon
carcinogenesis. Mutat Res. 693:94–100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Low SY, Ho YK, Too HP, Yap CT and Ng WH:
MicroRNA as potential modulators in chemoresistant high-grade
gliomas. J Clin Neurosci. 21:395–400. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nugent M: MicroRNA function and
dysregulation in bone tumors: The evidence to date. Cancer Manag
Res. 6:15–25. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gregory RI and Shiekhattar R: MicroRNA
biogenesis and cancer. Cancer Res. 65:3509–3512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stumpel DJ, Schotte D, Lange-Turenhout EA,
Schneider P, Seslija L, de Menezes RX, Marquez VE, Pieters R, den
Boer ML and Stam RW: Hypermethylation of specific microRNA genes in
MLL-rearranged infant acute lymphoblastic leukemia: Major matters
at a micro scale. Leukemia. 25:429–439. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Miao CG, Qin D, Du CL, Ye H, Shi WJ, Xiong
YY, Zhang XL, Yu H, Dou JF, Ma ST, et al: DNMT1 activates the
canonical Wnt signaling in rheumatoid arthritis model rats via a
crucial functional crosstalk between miR-152 and the DNMT1, MeCP2.
Int Immunopharmacol. 28:344–353. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu C, Li J, Ding Q, Cheng G, Zhou H, Tao
L, Cai H, Li P, Cao Q, Ju X, et al: miR-152 controls migration and
invasive potential by targeting TGFα in prostate cancer cell lines.
Prostate. 73:1082–1089. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cheng Z, Ma R, Tan W and Zhang L: MiR-152
suppresses the proliferation and invasion of NSCLC cells by
inhibiting FGF2. Exp Mol Med. 46:e1122014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ma J, Yao Y, Wang P, Liu Y, Zhao L, Li Z,
Li Z and Xue Y: MiR-152 functions as a tumor suppressor in
glioblastoma stem cells by targeting Kruppel-like factor 4. Cancer
Lett. 355:85–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen S, Sanjana NE, Zheng K, Shalem O, Lee
K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R, et al:
Genome-wide CRISPR screen in a mouse model of tumor growth and
metastasis. Cell. 160:1246–1260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xu Q, Jiang Y, Yin Y, Li Q, He J, Jing Y,
Qi YT, Xu Q, Li W, Lu B, et al: A regulatory circuit of
miR-148a/152 and DNMT1 in modulating cell transformation and tumor
angiogenesis through IGF-IR and IRS1. J Mol Cell Biol. 5:3–13.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zheng B, Liang L, Wang C, Huang S, Cao X,
Zha R, Liu L, Jia D, Tian Q, Wu J, et al: MicroRNA-148a suppresses
tumor cell invasion and metastasis by downregulating ROCK1 in
gastric cancer. Clin Cancer Res. 17:7574–7583. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nakagawa T, Kanai Y, Ushijima S, Kitamura
T, Kakizoe T and Hirohashi S: DNA hypermethylation on multiple CpG
islands associated with increased DNA methyltransferase DNMT1
protein expression during multistage urothelial carcinogenesis. J
Urol. 173:1767–1771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Szyf M and Detich N: Regulation of the DNA
methylation machinery and its role in cellular transformation. Prog
Nucleic Acid Res Mol Biol. 69:47–79. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xiang Y, Ma N, Wang D, Zhang Y, Zhou J, Wu
G, Zhao R, Huang H, Wang X, Qiao Y, et al: MiR-152 and miR-185
co-contribute to ovarian cancer cells cisplatin sensitivity by
targeting DNMT1 directly: A novel epigenetic therapy independent of
decitabine. Oncogene. 33:378–386. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Miao CG, Yang YY, He X, Huang C, Huang Y,
Qin D, Du CL and Li J: MicroRNA-152 modulates the canonical Wnt
pathway activation by targeting DNA methyltransferase 1 in
arthritic rat model. Biochimie. 106:149–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wiklund ED, Kjems J and Clark SJ:
Epigenetic architecture and miRNA: Reciprocal regulators.
Epigenomics. 2:823–840. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sato F, Tsuchiya S, Meltzer SJ and Shimizu
K: MicroRNAs and epigenetics. FEBS J. 278:1598–1609. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Azizi M, Teimoori-Toolabi L, Arzanani MK,
Azadmanesh K, Fard-Esfahani P and Zeinali S: MicroRNA-148b and
microRNA-152 reactivate tumor suppressor genes through suppression
of DNA methyltransferase-1 gene in pancreatic cancer cell lines.
Cancer Biol Ther. 15:419–427. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang J, Bian Y, Wang Z, Li D, Wang C, Li Q
and Gao X: MicroRNA-152 regulates DNA methyltransferase 1 and is
involved in the development and lactation of mammary glands in
dairy cows. PLoS One. 9:e1013582014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cui H, Wang L, Gong P, Zhao C, Zhang S,
Zhang K, Zhou R, Zhao Z and Fan H: Deregulation between miR-29b/c
and DNMT3A is associated with epigenetic silencing of the CDH1
gene, affecting cell migration and invasion in gastric cancer. PLoS
One. 10:e01239262015. View Article : Google Scholar : PubMed/NCBI
|