Introduction
Lung cancer is the most commonly diagnosed cancer
and the most common cause of cancer-related death worldwide
(1). Approximately 85% of lung
cancer cases are attributed to smoking, not including those
occurring in nonsmokers exposed to second-hand smoke (2). As a major component in cigarette
smoke, nicotine contributes to tumor progression by activating
angiogenesis, promoting cell proliferation and invasion, and
inhibiting apoptosis, although it does not provoke tumorigenesis
(3,4). Calcium-mediated signal transduction is
suggested to be one of the underlying mechanisms involved in the
tumor-promoting effects induced by nicotine/nicotinic receptors
(5,6). In tumor progression, the diversity of
calcium channels, mostly non-voltage gated calcium channels, on the
plasma membrane is relevant to the differential behaviors in
proliferation and migration of cancer cells (7). Store-operated Ca2+ entry
(SOCE), activated by intracellular Ca2+ store depletion,
is the primary mechanism for Ca2+ influx in non-exitable
cells (8). It has been found that
SOCE remodeling is associated with tumor progression in various
human cancers (9,10).
The Orai channels and Ca2+-permeable
transient receptor potential canonical (TRPC) channels are
Ca2+-permeable channels involved in SOCE upon the
binding of the stromal interaction molecule (STIM) proteins as
Ca2+ sensors (11).
There are three mammalian Orai homologues (Orai1-3) showing
differences in response to the process of Ca2+
depotentiation (12) and six TRPC
isoforms (TRPC1-6) serving as non-selective
Ca2+-permeable cation channels, through which the
Ca2+ current in SOCE is generated by the formations of
Orai1-STIM1 or TRPCs-STIM1 components (13,14).
Hypoxia is a common feature of solid tumor masses
and is essential for the formation of the cellular and physiologic
characteristics of cancer (15).
The association between hypoxia and intracellular
[Ca2+]i regulation has been identified in
hepatoma cells, in which HIF-1α, a key regulator for the adaption
of cancer cells to a low-oxygen microenvironment, enhanced STIM1
transcription and contributed to SOCE and tumorigenesis (16,17).
Furthermore, both inhibition of TRPC6-mediated calcium signaling
and attenuation of HIF-1α signaling elevated the sensitivity of
tumor cells to drugs (18).
In human non-small cell lung cancer cells, it has
been found that nicotine induced HIF-1α overexpression (19). Excessive HIF-1α expression was
associated with the growth of lung cancer A549 cells in
vitro and in vivo (20).
Nevertheless, the association between nicotine-induced HIF-1α
change and SOCE in lung tumor cell growth remains unclear. In this
present study, we evaluated the effects of nicotine on changes in
the expression of store-operated calcium channel (SOCC) components
and SOCE in A549 cells. We demonstrated overexpression of
HIF-1α-mediated SOCC components enhancement of SOCE in the presence
of nicotine.
Materials and methods
Reagents
(−)-Nicotine ditartrate was purchased from
Calbiochem (San Diego, CA, USA; cat. no. 481975). Nifedipine,
cyclopiazonic acid (CPA) and SKF-96365 were obtained from
Sigma-Aldrich Inc. (St. Louis, MO, USA). Fluorescent dye fura-2 AM
was from Invitrogen (Thermo Fisher Scientific, Waltham, MA,
USA).
Cell culture and treatment
The non-small cell lung cancer cell line A549 was
obtained from the American Type Culture Collection (ATCC;
Rockville, MD, USA) and grown in Dulbecco's modified Eagle's medium
(DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin and 100 µg/ml streptomycin at 37°C in a humidified
atmosphere containing 5% CO2. For nicotine treatment,
the cells were seeded into 6-well plates at a density of
1×105 cells per well, grown until 70% confluence and
exchanged with serum-free medium for a 12-h culture to reach growth
arrest. Then, the cells were treated with nicotine in medium
containing 0.1% FBS for the indicated time before being collected
for further analyses.
RNA extraction and quantitative
real-time PCR
Total RNA in cultured cells was extracted with
TRIzol reagent (Invitrogen; Thermo Fisher Scientific) according to
the manufacturer's instructions. Reverse transcription into cDNA
was performed from 1 µg total RNA and quantitative real-time PCR
was carried out by using Scofast™ EvaGreen superMix (Bio-Rad
Laboratories, Hercules, CA, USA) and primers listed in Table I. The relative concentration of each
transcript was calculated using the Pfaffl method (21) and normalized to 18S as an internal
control for each sample.
 | Table I.Primers used for quantitative
RT-PCR. |
Table I.
Primers used for quantitative
RT-PCR.
| TRPC1 | Forward: | 5′
TTGTGGAGGTGGAATTCAGG 3′ |
|---|
|
| Reverse: | 5′ CGTTTGTCA
AGAGGCTCGTC 3′ |
| TRPC2 | Forward: | 5′
TCATGGTCATTGTGCTGCTC 3′ |
|
| Reverse: | 5′
ACTCCACGTCAGCATCATCC 3′ |
| TRPC3 | Forward: | 5′
CAGCCAACACGTTATCAGCA 3′ |
|
| Reverse: | 5′
CCTCAGTTGCTTGGCTCTTG 3′ |
| TRPC4 | Forward: | 5′
CGAAAGGGTTAACCTGCAAA 3′ |
|
| Reverse: | 5′
CAGGGACTGCAGTGTCTCAA 3′ |
| TRPC5 | Forward: | 5′
GTGCTGCTGAACATGCTGAT 3′ |
|
| Reverse: | 5′
GCTTCGTCCTTGCAAACTTC 3′ |
| TRPC6 | Forward: | 5′
CAGACAATGGCGGTCAAGTT 3′ |
|
| Reverse: | 5′
TGGTCCACGCATTATCTTCC 3′ |
| TRPC7 | Forward: | 5′
GTTAAAACCCTGCCAAACGA 3′ |
|
| Reverse: | 5′
TCCCAGATTTCCTTGCATTC 3′ |
| HIF-1α | Forward: | 5′
TGCTTGGTGCTGATTTGTGAACC 3′ |
|
| Reverse: | 5′
CTGTCCTGTGGTGACTTGTCC 3′ |
| Orai1 | Forward: | 5′
ACGTGCACAATCTCAACTCG 3′ |
|
| Reverse: | 5′
AGCACCACCTCAGCTAGGAA 3′ |
| 18S | Forward: | 5′
GCAATTATTCCCCATGAACG 3′ |
|
| Reverse: | 5′
GGCCTCACTAAACCATCCAA 3′ |
RNA interference
For the transient silencing of HIF-1α gene
expression, small interfering RNA (siRNA) targeting to HIF-1α
(siHIF-1α, 5′-CCACCACUGAUGAAUUAAATT-3′) and negative control small
interfering RNA (siNT, 5′-UUCUCCGAACGUGUCACGUTT-3′) were
transfected into A549 cells using HiPerFect Transfection Reagent
(Qiagen, Duesseldorf, Germany) at a final concentration of 5 nM for
24 h. Then the cells were subjected to nicotine treatment for 24 or
48 h.
Western blotting
Cells were homogenized in RIPA buffer (20 mM Tris,
pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 2 mM
EGTA, 2 mM EDTA, 0.1% SDS) containing protease inhibitor cocktail
(Sigma-Aldrich Inc.). Equal amount of total proteins for each
sample was separated on SDS-PAGE gel, blotted with primary
antibodies against human TRPC1 (1:1,000; rabbit polyclonal; cat.
no. ACC-010; Alomone Labs, Jerusalem, Israel), TRPC6 (1:1,000;
rabbit polyclonal; cat. no. ACC-017; Alomone Labs), Orai1 (1:1,000;
rabbit polyclonal; cat. no. 4281; ProSci, Inc., San Diego, CA,
USA), HIF-1α (1:1,000; mouse monoclone; cat. no. ab113642; Abcam,
Cambridge, UK) or β-actin (1:1,000; mouse monoclone; cat. no.
ab8226; Abcam) and then with the corresponding HRP-conjugated
secondary antibodies (Kirkegaard & Perry Laboratories,
Gaithersburg, MD, USA). Finally, the signals were visualized with
enhanced chemiluminescence reagents (ECL; Bio-Rad
Laboratories).
Proliferation analysis
Cell proliferation was evaluated by using a
colorimetric BrdU cell proliferation assay kit (Roche, South San
Francisco, CA, USA) according to the manufacturer's instructions.
Cell proliferation was quantified by BrdU incorporation and
expressed as a multiple of the value of the control cells.
Scrape-wound migration assay
A scrape-wound migration assay was used to assess
the effects of nicotine on cell mobility. A wound was produced in a
confluent monolayer of A549 cells by scraping the cells with a
pipette tip. Then, the cells were replenished with DMEM containing
0.1% FBS with nicotine (1 µM) and/or SKF-96365 (1 µM) to drive cell
migration. Bright-field images of the wound area were captured at 0
and 24 h post-wounding with a Leica (DMI3000B) microscope (Leica
Microsystems, Frankfurt, Germany) and the total number of pixels in
the empty spaces inside the wound were counted using Adobe
Photoshop CS5. At least three photographs were taken per group at
each time-point. The migration capacity was calculated as the empty
space at 0 h minus the empty space at 24 h and was represented as a
percentage relative to the control.
Measurement of intracellular
[Ca2+]i and SOCE by fura-2 fluorescence
Intracellular [Ca2+]i and SOCE
were measured according to methods described previously (22). A549 cells seeded on coverslips were
incubated with 5 µM fura-2 AM for 1 h in the dark at room
temperature. Then, the coverslips were perfused with physiological
salt solution (PSS, 130 mM NaCl, 5 mM KCl, 1.2 mM MgCl2,
10 mM HEPES and 10 mM glucose) for 10 min to remove the
extracellular fura-2 AM. Basal [Ca2+]i was
determined at 12-sec intervals from the ratio of fura-2
fluorescence emitted at 510 nm after excitation at 340 nm to that
after excitation at 380 nm (F340/F380) measured using a xenon lamp
(Lambda DG4; Sutter Instrument Company, Novato, CA, USA) in 20 to
30 cells.
After basal [Ca2+]i
measurement, the cells were perfused with [Ca2+]-free
PSS containing 0.5 mM EGTA for 5 min to chelate residual
Ca2+ and then with PSS containing 5 µM nifedipine and 10
µM CPA to prevent calcium entry through L-type VOCC and to deplete
SR Ca2+ stores. To assess SOCE, the peak increase in
[Ca2+]i (ratio F340/F380) caused by
restoration of extracellular Ca2+ (2.5 mM
Ca2+ in PSS perfusate containing nifedipine and CPA) was
determined.
Statistical analysis
All experiments were repeated three times. Data were
statistically analyzed using the two-tailed Student's t-test and
are represented as means ± SEM. *P<0.05 and **P<0.01 indicate
a significant and extremely significant difference,
respectively.
Results
Nicotine upregulates the expression of
SOCC components in A549 cells
The calcium channel SOCCs in mammalian cells are
thought to be composed of TRPCs, Orai1 and STIM1 (23). In NSCLC A549 cells, among the seven
TRPC members, TRPC1 and TRPC6 were expressed at relatively high
levels with relatively low levels of TRPC3 and TRPC4. The
expression levels of the other three TRPCs members, TRPC2, TRPC5
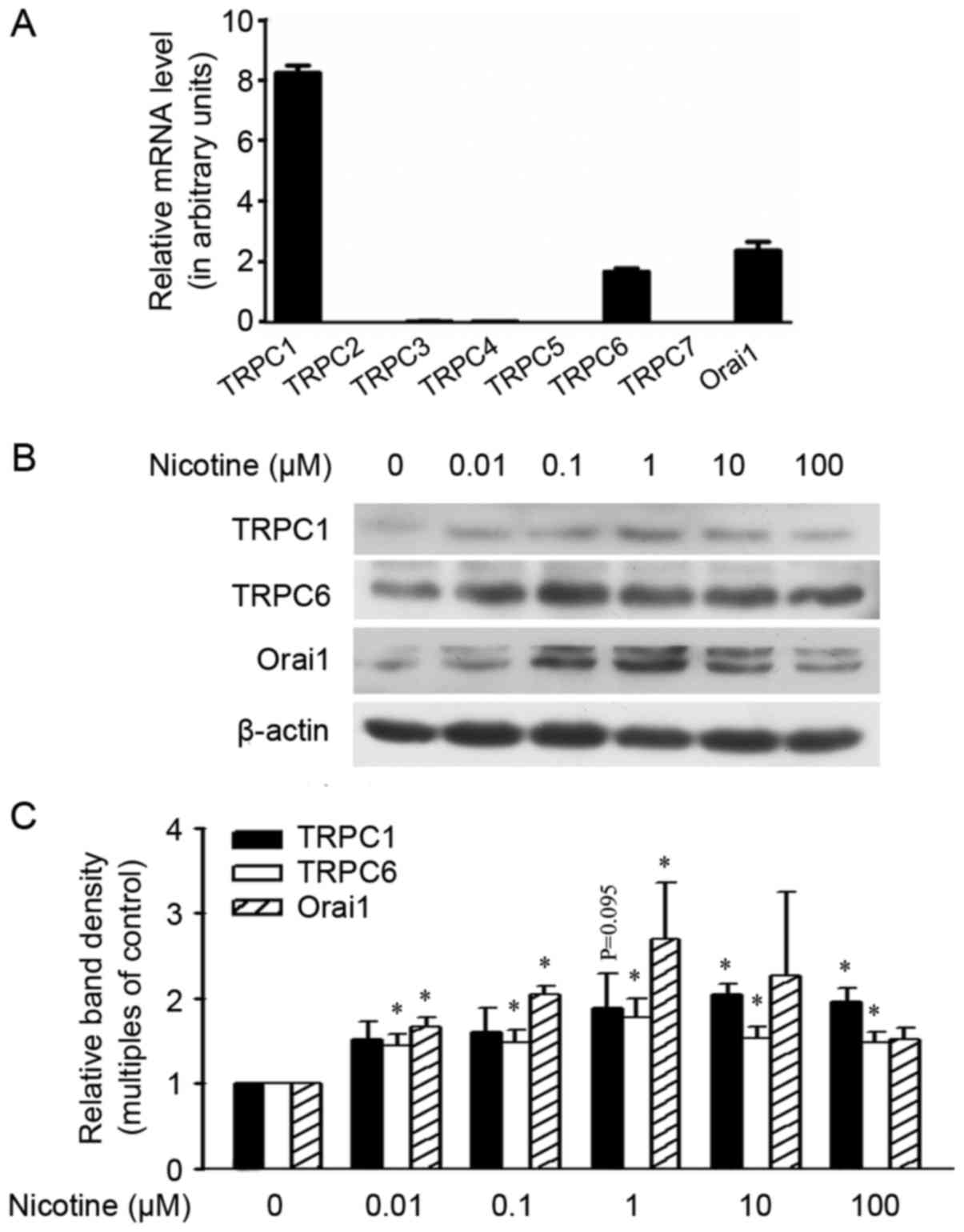
and TRPC7, were not detected in our study (Fig. 1A). The levels of TRPC6 and Orai1 in
A549 cells were upregulated following exposure to nicotine at a
dose as low as 0.01 µM, and were further upregulated by higher
levels of nicotine in a dose-dependent manner (1–100 µM). The
protein TRPC1 was not increased by nicotine at a dose lower than 1
µM, although a trend of upregulation was observed at the
concentration 1 µM. Further increased nicotine dosages of 10 or 100
µM did not induce further upregulation of the proteins TRPC6 and
Orai1 when compared with the dose at 1 µM (Fig. 1B and C), but did induce obvious
cytotoxicity-like cell death. In human smokers, the average peak
plasma nicotine level in smoking is around 10–50 mg/ml (~60–310 nM)
(24). Therefore, we chose to
expose cells to 1 µM nicotine in the present study.
Nicotine increases SOCE and basal
intracellular [Ca2+]i level
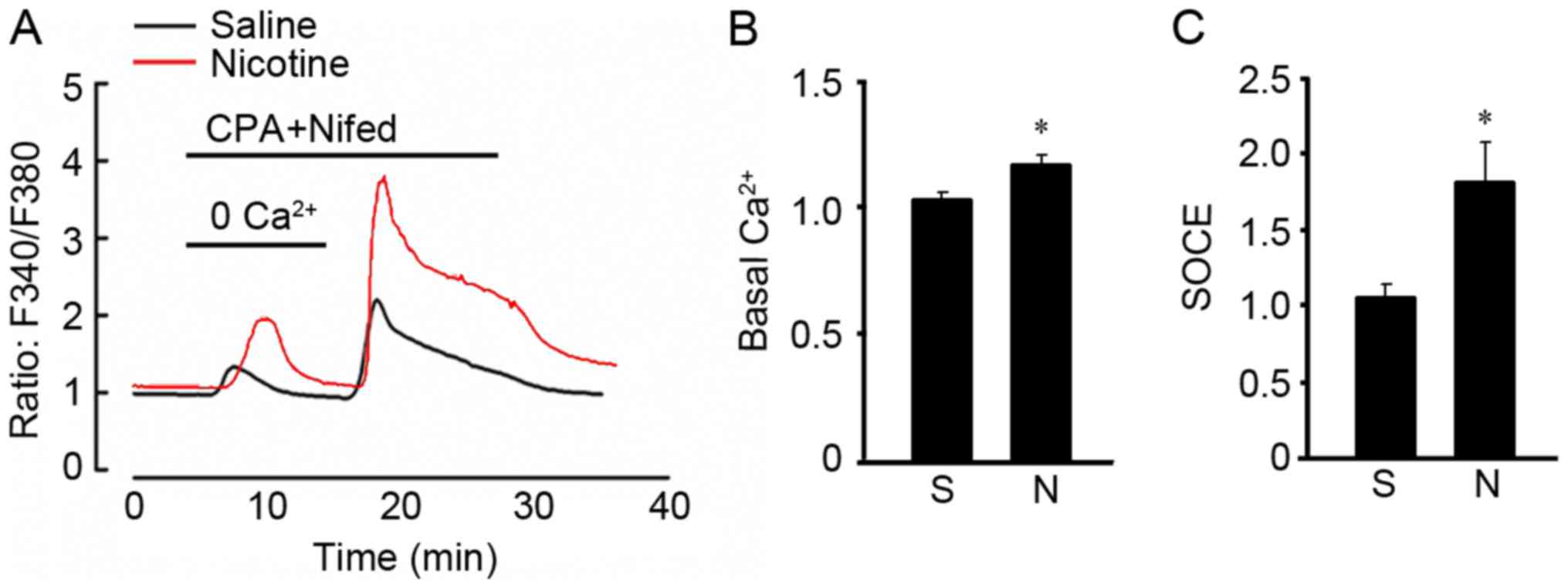
To examine the effects of nicotine on calcium
influx, we measured the SOCE and basal
[Ca2+]i in A549 cells. A 48-h exposure to
nicotine increased basal [Ca2+]i (Fig. 2A and B). After washing the cells
with Ca2+-free PSS containing 10 µM CPA and 5 µM
nifedipine, a low pulse of [Ca2+]i increase
was detected at 10 min which indicated store calcium release and
the peak [Ca2+]i at 20 min reflected SOCE
resulting from the restoration of extracellular Ca2+ at
2.5 mM. Basically, nicotine exposure enhanced basal
[Ca2+]i and SOCE of A549 cells (Fig. 2A and C).
Blockage of SOCE prevents cell
proliferation upon nicotine exposure
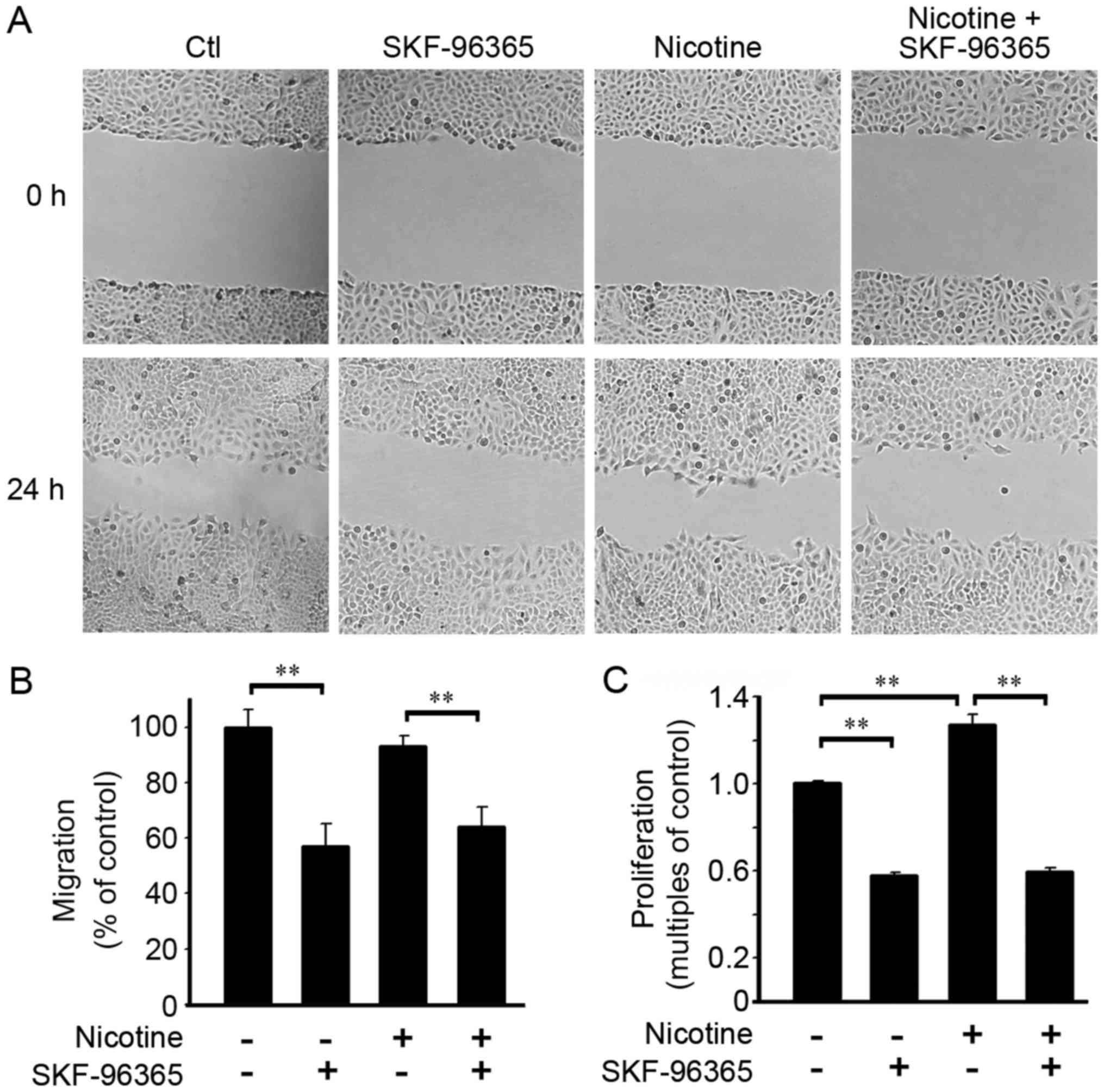
Nicotine has been reported to promote the
proliferation and migration of A549 cells (25). In our study, a scrape-wound
migration assay was conducted to evaluate cell migration capacity.
As shown in Fig. 3A and B, nicotine
(1 µM in 0.1% FBS) did not induce obvious accelerated cell
migration when compared with those cells without nicotine exposure
at 24 h. However, TRPC inhibitor SKF-96365 effectively abrogated
cell migration, no matter whether nicotine was present. The BrdU
incorporation assay showed that nicotine enhanced cell
proliferation at 48 h. The TRPC inhibitor SKF-96365 inhibited both
basal and nicotine triggered cell proliferation (Fig. 3C).
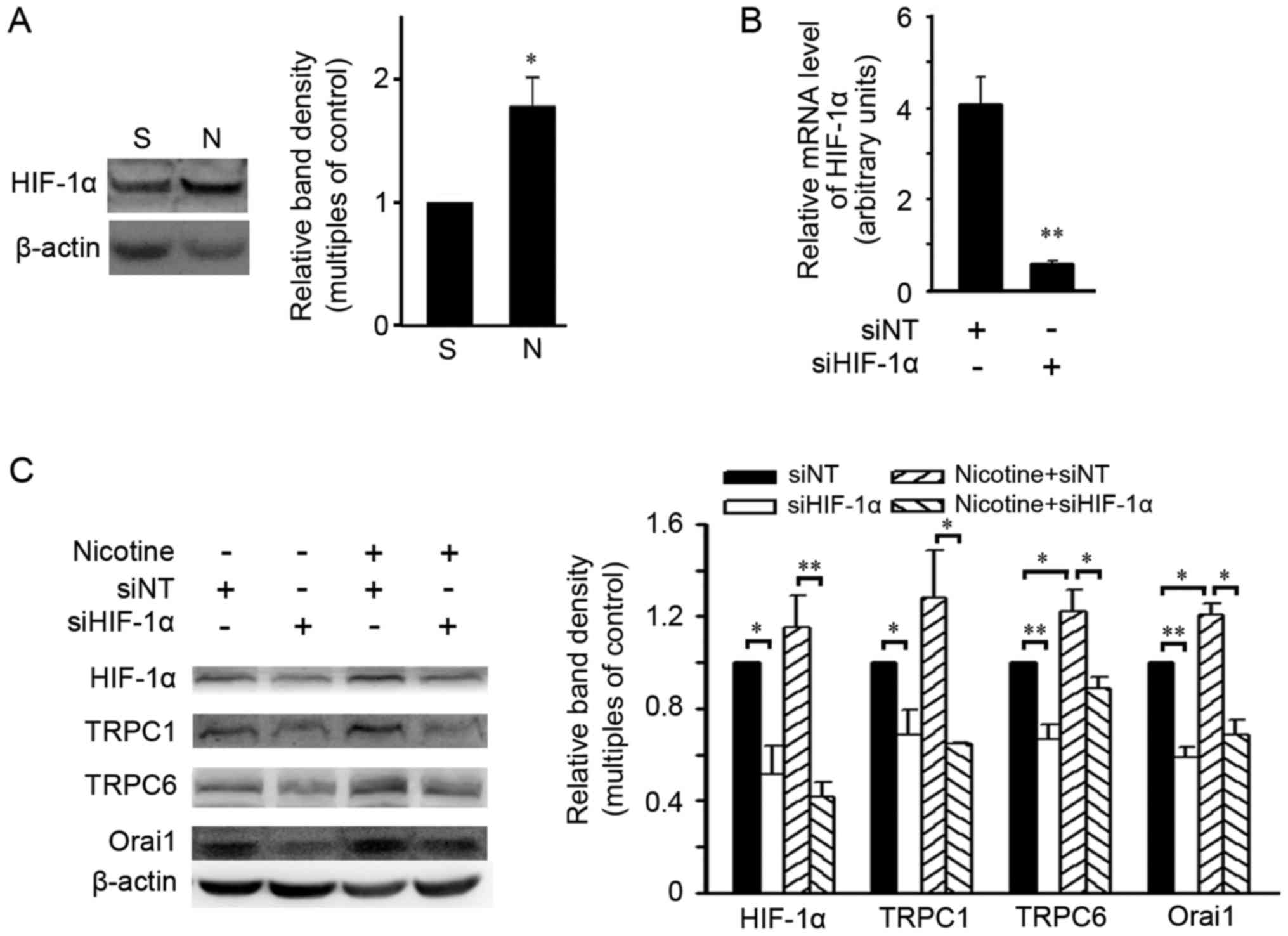
HIF-1α is required for upregulation of
SOCC components induced by nicotine
Since nicotine has been reported to increase HIF-1α
in NSCLC cells and HIF-1α modulated expression of SOCC components
in pulmonary artery smooth muscle cells (26,27),
the effects of HIF-1α on nicotine-triggered expression of SOCC
components were evaluated in A549 cells. As shown in Fig. 4A, nicotine induced an increase in
the HIF-1α level in A549 cells at 48 h. A small interfering RNA
against HIF-1α (siHIF-1α) was used to silence HIF-1α expression
(Fig. 4B). After 48-h nicotine
exposure, siHIF-1α reduced the basal protein levels of HIF-1α and
SOCC components TRPC1, TRPC6 and Orai1, and abolished the
upregulation of these proteins caused by nicotine exposure
(Fig. 4C). Similar upregulatory
effects of HIF-1α on SOCC components were also found in another
NSCLC cell line, NCI-H292, in which the upregulation of SOCC
components upon nicotine exposure were attenuted by siHIF-1α (data
not shown).
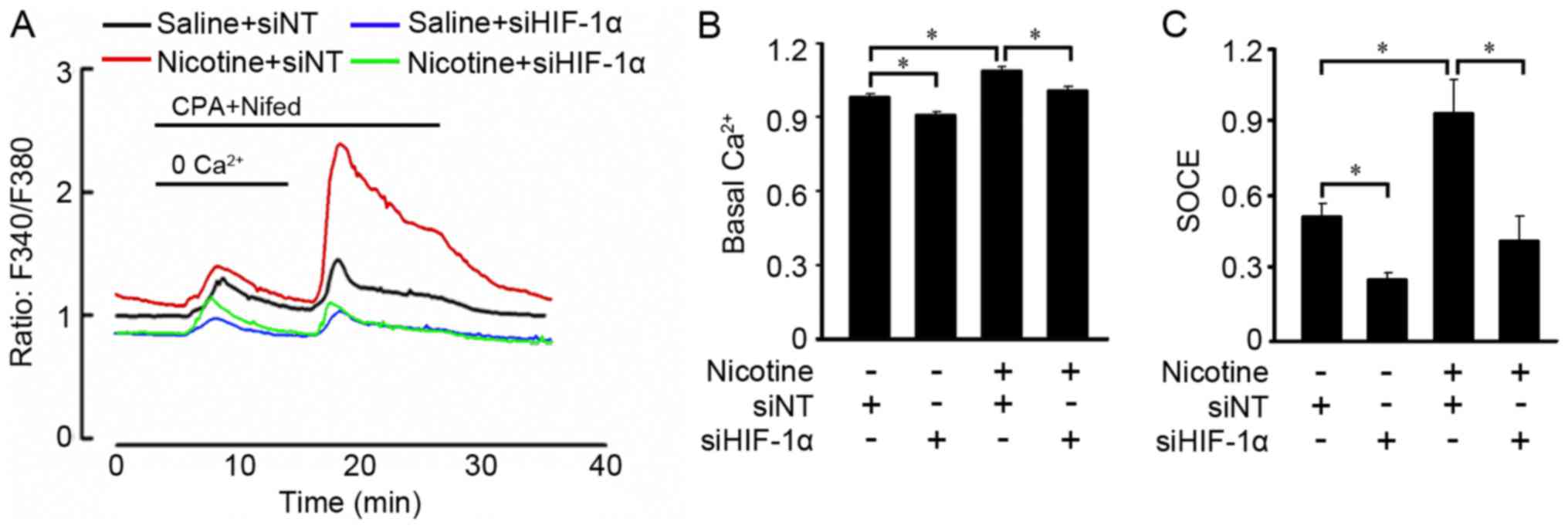
HIF-1α deficiency abolishes the
increases in basal [Ca2+]i and SOCE induced by
nicotine
Since HIF-1α deficiency reduced expression of SOCC
components in A549 cells, we therefore measured the basal
[Ca2+]i and SOCE when HIF-1α expression was
abolished. Downregulation of HIF-1α expression with siHIF-1α
decreased basal [Ca2+]i in the A549 cells and
abolished the increase in basal [Ca2+]i
induced by nicotine (Fig. 5A and
B). Furthermore, HIF-1α deficiency not only reduced SOCE in the
A549 cells, but prevented SOCE increase induced by nicotine
(Fig. 5A and C).
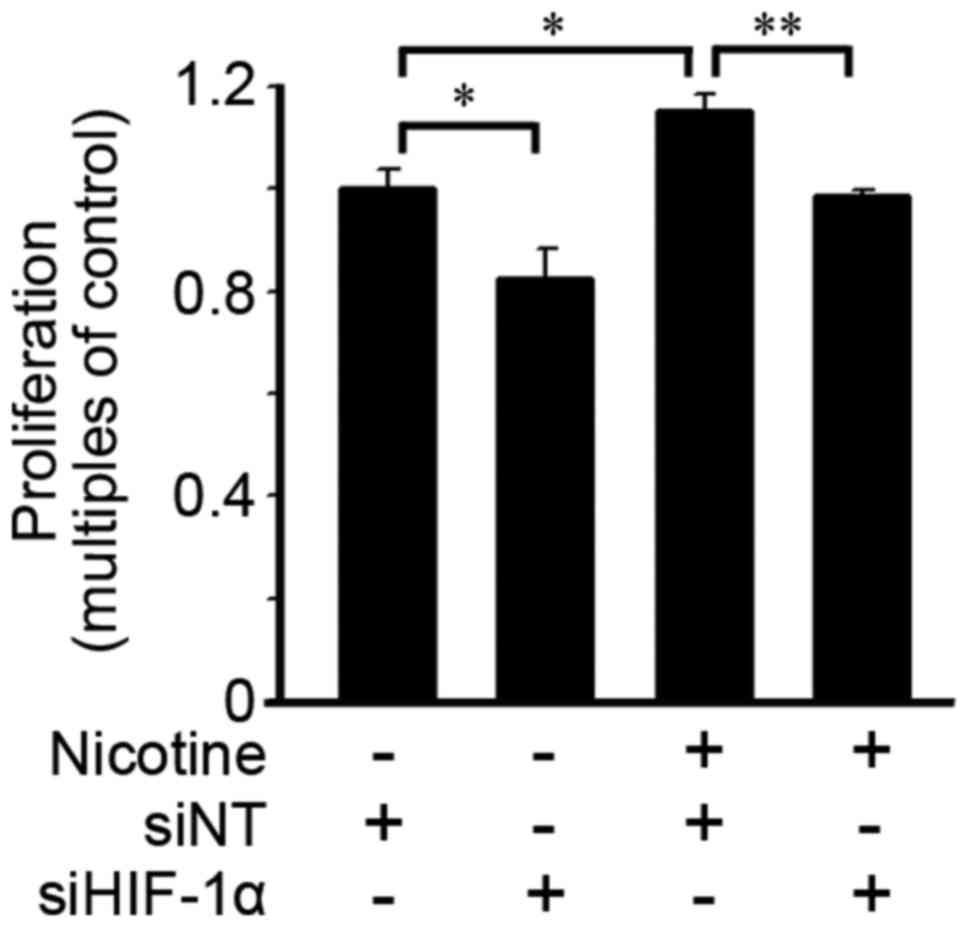
Loss of HIF-1α eliminates
nicotine-induced cell proliferation
The effects of HIF-1α on cell proliferation were
evaluated. As shown in Fig. 6,
HIF-1α deficiency induced by siHIF-1α transfection suppressed cell
proliferation in cells with or without nicotine exposure at 48
h.
Discussion
Lung cancer is a serious life-threatening disease
and cigarette smoking is the primary risk factor. In the present
study, we demonstrated that nicotine, the major component in
cigarette smoke, upregulated the expression of HIF-1α and SOCC
components, and promoted cell proliferation in A549 cells. Enhanced
SOCE and elevated intracellular [Ca2+]i were
associated with the upregulation of HIF-1α. Silencing of HIF-1α or
blocking SOCE abolished the nicotine-induced cell proliferation.
Therefore, HIF-1α mediated the promotive effects of nicotine on
SOCC component expression, SOCE and cell proliferation in lung
cancer cells.
Intracellular [Ca2+]i
regulated by SOCE is essential for modulating cell migration and
proliferation in normal and cancer cells. Suppression of SOCE by
abolishing SOCC component expression was found to prevent the
proliferation and invasion of lung cancer cells (28–30).
In the present study, we determined the expression of SOCC
components in A549 cells. The expression of TRPC isoforms in A549
cells is consistent with the expressional profile of TRPCs in NSCLC
tissue, in which the mRNA levels of TRPC1, 3, 4 and 6 are
detectable and the levels of TRPC2, 5 and 7 are below the detection
limit (31).
Nicotine participates in the progression of lung
cancer through the non-neuronal cholinergic system mediated by the
nicotine acetylcholine receptor (32,33).
Normal bronchial epithelial cells express α3-, α4-, α5- and
α7-nAChR subunits which modulate Ca2+ metabolism
(34). The receptors nAChRs are
functionally conserved in mediating nicotine responses on TRPCs in
neurons from worms to mammals (35). However, to date it is not known
which nAChR subunit mediates the functions of nicotine in
regulating expression of SOCC components in epithelial cells. The
candidates could be α5- and α7-nAChR, as it has been suggested that
the dysregulations of α5- and α7-nAChR in lung cancer tissues are
associated with different influences of nicotine on the
tumorigenesis of different lung cancer types (36).
The present study demonstrated that HIF-1α mediated
the upregulatory effects of nicotine on TRPCs and Orai1. It has
been confirmed that nicotine upregulates HIF-1α expression through
binding α5-nAChR and activating the downstream Erk1/2 or PI3K/Akt
signaling in NSCLC (26,37). Actually, the genes encoding TRPCs
could be target genes of HIF-1α in modulating the growth of various
types of cells, since, except for nicotine, HIF-1α is believed to
be a common mediator of various factors in enhancing the expression
of TRPCs, SOCC and intracellular [Ca2+] in pulmonary
arterial smooth muscle cells and cardiomyocytes suffering hypoxia,
which result in cell proliferation and migration or tissue
hypertrophy (27,38,39).
Although the association between Orai1 dysexpression
and lung cancer progression remains to be evaluated, Orai1
elevation is related to enhanced tumor cell proliferation and
invasion (40). Our results not
only demonstrated the involvement of Orai1 in nicotine-triggered
lung cancer cell proliferation, but showed that Orai1 may be
a target gene of HIF-1α. Furthermore, like in other cell types, it
is reasonable to suppose that the functions of Orai1 on SOCE in
lung tumor cells might not be limited as a channel forming protein,
but as a regulator for SOCC complex formation, including regulating
the recruitment of TRPC1 (41) or
the formation of ternary complex of TRPC-Orai1-STIM1 (42).
Based on our results, it is comprehensible to
suggest that HIF-1α upregulation upon nicotine stimulation
eventually promotes lung tumor cell proliferation by increasing
expression of SOCC components and intracellular [Ca2+].
Actually, nicotine-activated signaling mediated by PKC, NF-κB, Srk,
PI3K/Akt, Raf-1, ERK1/2 and p90RSK are related with increases in
cell proliferation, migration, invasion or inhibition of cell
apoptosis (43–46). However, we did not observe an
obvious change in migration within 24 to 48 h upon nicotine
exposure, although inhibition of SOCE abolished cell migration
(Fig. 3B and data not shown).
Probably this was due to the lower concentration of nicotine used
in this study when compared with other studies, which may need a
longer time to trigger a change in migration capacity (25,47).
In summary, the present study demonstrated that
nicotine upregulated the expression of SOCC components TRPC6 and
Orai1 by increasing HIF-1α expression in NSCLC cells, which
eventually led to enhanced SOCE, elevated intracellular
[Ca2+]i and promotion of cell proliferation.
These findings suggest that HIF-1α-SOCE signaling plays a pivotal
role in pro-tumor functions of nicotine in NSCLC.
Acknowledgements
This research was supported by the National Natural
Science Foundation of China (81071917, 81170052, 81070043,
81173112, 81220108001), the Guangdong Natural Science Foundation
team grant (1035101200300000), the Guangdong Province Universities
and Colleges Pearl River Scholar Funded Scheme (2014), the Key
Project of the Department of Education of Guangdong Province
(cxzd1142), the Guangzhou Department of Education Team Grant for
Innovation (13C08), the Changjiang Scholars and Innovative Research
Team in University grant (IRT0961), and Guangzhou Department of
Education Yangcheng Scholarships (10A058S, 12A001S).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Warren GW and Cummings KM: Tobacco and
lung cancer: Risks, trends, and outcomes in patients with cancer.
Am Soc Clin Oncol Educ Book. 359–364. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grozio A, Catassi A, Cavalieri Z, Paleari
L, Cesario A and Russo P: Nicotine, lung and cancer. Anticancer
Agents Med Chem. 7:461–466. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cardinale A, Nastrucci C, Cesario A and
Russo P: Nicotine: Specific role in angiogenesis, proliferation and
apoptosis. Crit Rev Toxicol. 42:68–89. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Improgo MR, Tapper AR and Gardner PD:
Nicotinic acetylcholine receptor-mediated mechanisms in lung
cancer. Biochem Pharmacol. 82:1015–1021. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carlisle DL, Liu X, Hopkins TM, Swick MC,
Dhir R and Siegfried JM: Nicotine activates cell-signaling pathways
through muscle-type and neuronal nicotinic acetylcholine receptors
in non-small cell lung cancer cells. Pulm Pharmacol Ther.
20:629–641. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Déliot N and Constantin B: Plasma membrane
calcium channels in cancer: Alterations and consequences for cell
proliferation and migration. Biochim Biophys Acta. 1848:2512–2522.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Parekh AB and Putney JW Jr: Store-operated
calcium channels. Physiol Rev. 85:757–810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen YF, Chen YT, Chiu WT and Shen MR:
Remodeling of calcium signaling in tumor progression. J Biomed Sci.
20:232013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Villalobos C, Sobradillo D,
Hernández-Morales M and Nuñez L: Remodeling of calcium entry
pathways in cancer. Adv Exp Med Biol. 898:449–466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Smyth JT, Hwang SY, Tomita T, DeHaven WI,
Mercer JC and Putney JW: Activation and regulation of
store-operated calcium entry. J Cell Mol Med. 14:2337–2349. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
DeHaven WI, Smyth JT, Boyles RR and Putney
JW Jr: Calcium inhibition and calcium potentiation of Orai1, Orai2,
and Orai3 calcium release-activated calcium channels. J Biol Chem.
282:17548–17556. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ambudkar IS, de Souza LB and Ong HL:
TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell
Calcium. 63:33–39. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yazbeck P, Tauseef M, Kruse K, Amin MR,
Sheikh R, Feske S, Komarova Y and Mehta D: STIM1 phosphorylation at
Y361 recruits Orai1 to STIM1 puncta and induces Ca2+
entry. Sci Rep. 7:427582017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ruan K, Song G and Ouyang G: Role of
hypoxia in the hallmarks of human cancer. J Cell Biochem.
107:1053–1062. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Y, Guo B, Xie Q, Ye D, Zhang D, Zhu Y,
Chen H and Zhu B: STIM1 mediates hypoxia-driven
hepatocarcinogenesis via interaction with HIF-1. Cell Rep.
12:388–395. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Semenza GL, Agani F, Feldser D, Iyer N,
Kotch L, Laughner E and Yu A: Hypoxia, HIF-1, and the
pathophysiology of common human diseases. Adv Exp Med Biol.
475:123–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wen L, Liang C, Chen E, Chen W, Liang F,
Zhi X, Wei T, Xue F, Li G, Yang Q, et al: Regulation of multi-drug
resistance in hepatocellular carcinoma cells is TRPC6/calcium
dependent. Sci Rep. 6:232692016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo L, Li L, Wang W, Pan Z, Zhou Q and Wu
Z: Mitochondrial reactive oxygen species mediates nicotine-induced
hypoxia-inducible factor-1α expression in human non-small cell lung
cancer cells. Biochim Biophys Acta. 1822:852–861. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li W, Chen YQ, Shen YB, Shu HM, Wang XJ,
Zhao CL and Chen CJ: HIF-1α knockdown by miRNA decreases survivin
expression and inhibits A549 cell growth in vitro and in vivo. Int
J Mol Med. 32:271–280. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang J, Shimoda LA and Sylvester JT:
Capacitative calcium entry and TRPC channel proteins are expressed
in rat distal pulmonary arterial smooth muscle. Am J Physiol Lung
Cell Mol Physiol. 286:L848–L858. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smyth JT, Dehaven WI, Jones BF, Mercer JC,
Trebak M, Vazquez G and Putney JW Jr: Emerging perspectives in
store-operated Ca2+ entry: Roles of Orai, Stim and TRP.
Biochim Biophys Acta. 1763:1147–1160. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Calderon LE, Liu S, Arnold N, Breakall B,
Rollins J and Ndinguri M: Bromoenol lactone attenuates
nicotine-induced breast cancer cell proliferation and migration.
PLoS One. 10:e01432772015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nair S, Bora-Singhal N, Perumal D and
Chellappan S: Nicotine-mediated invasion and migration of non-small
cell lung carcinoma cells by modulating STMN3 and
GSPT1 genes in an ID1-dependent manner. Mol Cancer.
13:1732014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Q, Tang X, Zhang ZF, Velikina R, Shi
S and Le AD: Nicotine induces hypoxia-inducible factor-1alpha
expression in human lung cancer cells via nicotinic acetylcholine
receptor-mediated signaling pathways. Clin Cancer Res.
13:4686–4694. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Lu W, Yang K, Wang Y, Zhang J, Jia
J, Yun X, Tian L, Chen Y, Jiang Q, et al: Peroxisome
proliferator-activated receptor γ inhibits pulmonary hypertension
targeting store-operated calcium entry. J Mol Med. 93:327–342.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sweeney M, Yu Y, Platoshyn O, Zhang S,
McDaniel SS and Yuan JX: Inhibition of endogenous TRP1 decreases
capacitative Ca2+ entry and attenuates pulmonary artery
smooth muscle cell proliferation. Am J Physiol Lung Cell Mol
Physiol. 283:L144–L155. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Choi DL, Jang SJ, Cho S, Choi HE, Rim HK,
Lee KT and Lee JY: Inhibition of cellular proliferation and
induction of apoptosis in human lung adenocarcinoma A549 cells by
T-type calcium channel antagonist. Bioorg Med Chem Lett.
24:1565–1570. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang LL, Liu BC, Lu XY, Yan Y, Zhai YJ,
Bao Q, Doetsch PW, Deng X, Thai TL, Alli AA, et al: Inhibition of
TRPC6 reduces non-small cell lung cancer cell proliferation and
invasion. Oncotarget. 8:5123–5134. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang Q, He J, Lu W, Yin W, Yang H, Xu X
and Wang D: Expression of transient receptor potential canonical
channel proteins in human non-small cell lung cancer. Zhongguo Fei
Ai Za Zhi. 13:612–616. 2010.(In Chinese). PubMed/NCBI
|
|
32
|
Shiraishi K, Kohno T, Kunitoh H, Watanabe
S, Goto K, Nishiwaki Y, Shimada Y, Hirose H, Saito I, Kuchiba A, et
al: Contribution of nicotine acetylcholine receptor polymorphisms
to lung cancer risk in a smoking-independent manner in the
Japanese. Carcinogenesis. 30:65–70. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Millar NS and Gotti C: Diversity of
vertebrate nicotinic acetylcholine receptors. Neuropharmacology.
56:237–246. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zia S, Ndoye A, Nguyen VT and Grando SA:
Nicotine enhances expression of the alpha 3, alpha 4, alpha 5, and
alpha 7 nicotinic receptors modulating calcium metabolism and
regulating adhesion and motility of respiratory epithelial cells.
Res Commun Mol Pathol Pharmacol. 97:243–262. 1997.PubMed/NCBI
|
|
35
|
Feng Z, Li W, Ward A, Piggott BJ, Larkspur
ER, Sternberg PW and Xu XZ: A C. elegans model of
nicotine-dependent behavior: Regulation by TRP-family channels.
Cell. 127:621–633. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bordas A, Cedillo JL, Arnalich F,
Esteban-Rodriguez I, Guerra-Pastrián L, de Castro J, Martín-Sánchez
C, Atienza G, Fernández-Capitan C, Rios JJ and Montiel C:
Expression patterns for nicotinic acetylcholine receptor subunit
genes in smoking-related lung cancers. Oncotarget. 8:67878–67890.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ma X, Jia Y, Zu S, Li R, Jia Y, Zhao Y,
Xiao D, Dang N and Wang Y: α5 Nicotinic acetylcholine receptor
mediates nicotine-induced HIF-1α and VEGF expression in non-small
cell lung cancer. Toxicol Appl Pharmacol. 278:172–179. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang J, Fu X, Yang K, Jiang Q, Chen Y, Jia
J, Duan X, Wang EW, He J, Ran P, et al: Hypoxia inducible
factor-1-dependent up-regulation of BMP4 mediates hypoxia-induced
increase of TRPC expression in PASMCs. Cardiovasc Res. 107:108–118.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chu W, Wan L, Zhao D, Qu X, Cai F, Huo R,
Wang N, Zhu J, Zhang C, Zheng F, et al: Mild hypoxia-induced
cardiomyocyte hypertrophy via up-regulation of HIF-1α-mediated TRPC
signalling. J Cell Mol Med. 16:2022–2034. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xia J, Wang H, Huang H, Sun L, Dong S,
Huang N, Shi M, Bin J, Liao Y and Liao W: Elevated Orai1 and STIM1
expressions upregulate MACC1 expression to promote tumor cell
proliferation, metabolism, migration, and invasion in human gastric
cancer. Cancer Lett. 381:31–40. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cheng KT, Liu X, Ong HL, Swaim W and
Ambudkar IS: Local Ca2+ entry via Orai1 regulates plasma
membrane recruitment of TRPC1 and controls cytosolic
Ca2+ signals required for specific cell functions. PLoS
Biol. 9:e10010252011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Berna-Erro A, Redondo PC and Rosado JA:
Store-operated Ca2+ entry. Adv Exp Med Biol.
740:349–382. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Catassi A, Servent D, Paleari L, Cesario A
and Russo P: Multiple roles of nicotine on cell proliferation and
inhibition of apoptosis: Implications on lung carcinogenesis. Mutat
Res. 659:221–231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Paleari L, Catassi A, Ciarlo M, Cavalieri
Z, Bruzzo C, Servent D, Cesario A, Chessa L, Cilli M, Piccardi F,
et al: Role of alpha7-nicotinic acetylcholine receptor in human
non-small cell lung cancer proliferation. Cell Prolif. 41:936–959.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dasgupta P, Rizwani W, Pillai S, Kinkade
R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, et
al: Nicotine induces cell proliferation, invasion and
epithelial-mesenchymal transition in a variety of human cancer cell
lines. Int J Cancer. 124:36–45. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Egleton RD, Brown KC and Dasgupta P:
Nicotinic acetylcholine receptors in cancer: Multiple roles in
proliferation and inhibition of apoptosis. Trends Pharmacol Sci.
29:151–158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shi J, Liu F, Zhang W, Liu X, Lin B and
Tang X: Epigallocatechin-3-gallate inhibits nicotine-induced
migration and invasion by the suppression of angiogenesis and
epithelial-mesenchymal transition in non-small cell lung cancer
cells. Oncol Rep. 33:2972–2980. 2015. View Article : Google Scholar : PubMed/NCBI
|