Introduction
Colorectal cancer (CRC) ranks fifth in tumor-related
mortality in China and is also considered one of the most common
digestive tract malignancies throughout the world (1). CRC poses a growing threat to human
health. The lack of effective early diagnosis is of particular
concern, since the majority of patients will miss the best
opportunity for treatment if they are initially diagnosed at a late
stage (2). Thus, it is of great
clinical significance to find new early diagnostic markers as well
as novel therapeutic targets in CRC to improve patient
outcomes.
G protein-coupled receptors (GPCRs) are one of the
largest families of membrane proteins, and typically are comprised
of a large extracellular domain (ECD), a seven-transmembrane
spanning (7TM) domain, and an intracellular domain (ICD) (3). Since more than 30% of drugs currently
target GPCRs, it is important to explore and understand their
biological function in malignant tumors (4). GPR56, the most studied GPCR in
malignant tumors, was first reported to be downregulated in highly
metastatic melanoma cell lines compared with poorly metastatic
lines by Zendman et al in 1999 (5). Since then, research concerning the
role of GPR56 in malignancy has covered many aspects, from its
differential expression to its mechanism of regulation. Many
studies have revealed that GPR56 is expressed as a 3-kb mRNA in
various tumor tissues, with higher levels expressed in esophageal
squamous cell carcinoma (6),
glioblastoma (7), and human
fibrosarcoma (8). In terms of its
mechanism of action, GPR56 has been implicated in proliferation,
migration, angiogenesis, cell adhesion, cell apoptosis, and cell
cycle regulation (9–11). It has also been reported that GPR56
plays an important role in several types of malignances by
interacting with vascular endothelial growth factor (VEGF)
(11), collagen III (12), CD81 (13), and transglutaminase 2 (Tg2)
(14). Furthermore, Jin et
al recently concluded that GPR56 promoted carcinogenesis by
binding progastrin, a pro-angiogenic factor, in mice (15).
Despite the aforementioned studies, the clinical
significance and underlying mechanism of action of GPR56 in
regulating tumorigenesis and metastasis of CRC remains unclear.
Therefore, in this study, we aimed to determine the expression
level and biological function of GPR56 in CRC.
Materials and methods
Tissue samples and cell culture
We collected 110 samples of human CRC and
corresponding non-tumorous colorectal mucosal tissue between June
2010 and June 2013 at the First Affiliated Hospital of Nanjing
Medical University (Nanjing, China). The enrolled patients signed
informed consent forms and none had undergone any neoadjuvant
radiotherapy or chemotherapy. We snap-froze tissue samples in
liquid nitrogen and stored the samples at −80°C until RNA
extraction was performed. The clinicopathological parameters were
defined according to The National Comprehensive Cancer Network
(2015.2). The Research Ethics Committee of the First Affiliated
Hospital of Nanjing Medical University approved this study. We
obtained human CRC cell lines (SW480, HT-29, LOVO, DLD-1 and
HCT116) and the normal human colorectal epithelial cell line
(NCM460) form the American Type Culture Collection (ATTC; Manassas,
VA, USA), and cultured the cells in Dulbecco's modified Eagle's
medium (DMEM) with 10% fetal bovine serum (FBS; both from Wisent
Inc., St-Bruno, Quebec, Canada), 100 U/ml penicillin and 100 µg/ml
streptomycin in a ٥٪ CO2 atmosphere at 37°C. We obtained
the PI3K inhibitor LY294002 from Cell Signaling Technology
(Danvers, MA, USA).
RNA extraction and qRT-PCR
Total RNA was isolated from tissues and cells using
RNAiso Plus (Takara Biotechnology, Inc., Dalian, China), and cDNA
was synthesized using the PrimeScript RT reagent kit (Takara
Biotechnology). qRT-PCR was carried out using StepOnePlus Real-time
PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Foster City, CA, USA) and a SYBR Green PCR kit (Roche Diagnostics,
Indianapolis, IN, USA). Specific oligonucleotide primer sequences
are listed in Table I. The PCR was
performed as follows: 95°C for 30 sec, 40 cycles at 95°C for 5 sec,
60°C for 30 sec; and the dissociation stage at 95°C for 15 sec,
60°C for 1 min and 95°C for 15 sec. The method used for normalizing
the qPCR data was the 2−ΔΔCq method which facilitates
the analysis of relative changes in gene expression in real-time
quantitative PCR experiments (16).
 | Table I.Primer sequences used for qRT-PCR. |
Table I.
Primer sequences used for qRT-PCR.
| Gene | Forward | Reverse |
|---|
| GAPDH |
5′-ACAGTCAGCCGCATCTTCTT-3′ |
5′-GACAAGCTTCCCGTTCTCAG-3′ |
| GPR56 |
5′-AGTTCTGGGCCTTTGGCATTCA-3′ |
5′-AGCACAATGCAAGGCACACAGT-3′ |
Immunohistochemistry
Immunohistochemistry was performed as previously
described (17). The primary
antibody used was GPR56 (diluted 1:200; cat. no. abs133132; Absin
Bioscience, Shanghai, China). The total score was calculated based
on the percentage of positive cells (0, negative; 1, <30%; 2,
31–60%; 3, >60%) multiplied by the staining intensity (0,
negative; 1, weak; 2, moderate; and 3, strong). Scores ≥4 indicated
high GPR56 expression, and scores <4 indicated low GPR56
expression.
siRNA interference and plasmid
transfection
GenePharma Corp. (Shanghai, China) designed and
synthesized the GPR56-targeting small interfering RNAs (siRNAs,
si-GPR56) and the negative control siRNAs (si-NC). The GPR56-siRNA
sequences were as follows: siRNA1, 5′-GCCUGGUGUUUCUGUUCAATT-3′;
siRNA2, 5′-UCACCUCCUUCCAAGGCUUTT-3′; and siRNA3,
5′-CCUGGGCCUUGAUCUUCUUTT-3′. The si-NC sequences were as follows:
5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′
(antisense). The si-GPR56 and si-NC transfection into HCT116 and
DLD-1 cells was conducted using Lipofectamine 3000 (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's protocols. After 48 h, knockdown efficiency was
evaluated. The GPR56 amplification plasmid was obtained from Sangon
Biotech Co., Ltd. (Shanghai, China). The primer sequences were as
follows: Forward, 5′-ACGTAGATATATGACTCCCCAGTCGCTGCT-3′ and reverse,
5′-ACGTGGTACCGTGGCGTTGATCCGGTCCT-3′. The plasmid transfection steps
were roughly similar to siRNA interference: A mixture of Opti-MEM,
Lipofectamine 3000 and overexpression plasmids prepared in
accordance with the instructions were added to a 6-well plate, and
the amplification efficiency was ascertained 48 h later.
Cell proliferation assay
Cell proliferation was assessed using Cell Counting
Kit-8 (Dojindo Molecular Technologies, Tokyo, Japan) assay
according to the manufacturer's protocols. Cells (2×103
cells/well) were plated in 96-well plates and cultured overnight.
Then, 10 µl CCK-٨ reagent was added to each well at ٢٤, ٤٨, ٧٢, and
٩٦ h before a 2-h incubation at 37°C. A microplate reader was used
to detect the absorbance at 450 nm (test wavelength) and 630 nm
(reference wavelength). All experimental procedures were repeated
at least three times.
Mouse tumor xenograft
Twenty female 4-week-old nude mice (BALB/c nude
mice; Vital River, Beijing, China) were purchased from the
Laboratory Animal Centre of Nanjing Medical University (Nanjing,
China). The specific housing conditions were as follows:
Temperature, 21±2°C; humidity, 30–70%; 12-h light/dark cycle; the
ingested food and water were sterile feed and sterilized bottled
water. We selected sixteen mice for our experiments and the average
weight of the mice was ~15 g. All experimental procedures conducted
in vivo were in accordance with the guidelines from the
Animal Ethical and Welfare Committee of Nanjing Medical University.
Cells [HCT116 and DLD-1, 2×106 cells in 100 µl
phosphate-buffered saline (PBS)] were injected into the groins of
the nude mice. The mice with tumor implantation were randomly
divided into two groups (NC and siRNA) after 4 weeks. Once every 48
h, an intratumoral injection of si-GPR56 or si-NC mixed with the
reagent (in vivo-jetPEI®; Polyplus, New York, NY,
USA) was performed according to the manufacturer's instructions.
After 4 weeks, the nude mice were sacrificed by cervical
dislocation and xenograft tumors were dissected. The volume of the
implanted tumors was calculated by using the formula:
Volume=(width2 x length)/2. The maximum tumor volumes
were as follows: si-NC (HCT116): 1,456 mm3; si-NC
(DLD-1): 1,115 mm3; si-RNA2 (HCT116): 755
mm3; si-RNA2 (DLD-1): 643 mm3.
Flow cytometric analysis
We detected apoptosis using an Annexin V-FITC
Apoptosis Detection kit (BD Biosciences, Franklin Lakes, NJ, USA)
according to the manufacturer's protocols after a 48-h siRNA
transfection. The cells were kept on ice and then analyzed by a
flow cytometer (Becton-Dickinson, San Jose, CA, USA). The
experiment was repeated three times. The HCT116 and DLD-1 cells
transfected with si-NC or si-RNA2 were stored for 48 h for cell
cycle analysis. Cells were stained with propidium iodide
(Sigma-Aldrich; Merck, Darmstadt, Germany) and RNase A (Takara
Biotechnology, Dalian, China) for 30 min at room temperature after
being fixed with 70% ethanol at −20°C overnight. Assessment of the
cell cycle distribution was conducted using flow cytometry
(FACSCalibur; BD Biosciences).
Transwell assay
Cell migration and invasion were performed in a
24-well Transwell plate with polycarbonate sterile chambers (8 µm
filters; BD Biosciences) with or without Matrigel coating. Cells
were seeded in the upper chamber at a density of ٢x١٠4
cells with 100 µl serum-free DMEM, whereas complete culture medium
containing ١٠٪ fetal bovine serum was added to the lower chamber.
After incubation for ٢٤ h at 37°C, we counted the migrated and
invaded cells on the membrane using a light microscope and recorded
the results as the means ± standard deviation (SD). The experiment
was performed in triplicate.
Western blot analysis
Western blotting was performed as previously
described (18). The primary
antibodies included were as follows: GPR56 (cat. no. sc-390192),
E-cadherin (cat. no. sc-71009), vimentin (cat. no. sc-80975), PI3K
(cat. no. sc-293172), AKT (cat. no. sc-135829) and GAPDH (cat. no.
sc-47724). These antibodies were purchased from Santa Cruz
Biotechnology, Inc., (Dallas, TX, USA) and the dilution ratio was
1:1,000. Secondary antibodies were HRP goat anti-mouse IgG (H+L)
(1:5,000; cat. no. 115-035-003; Jackson ImmunoResearch; Shanghai
Rebiosci Biotech Co., Ltd., Shanghai, China); p-PI3K (cat. no.
4228) and p-AKT (1:1,000; cat. no. 130308; both from Cell Signaling
Technology). Secondary antibodies were goat anti-rabbit IgG (H+L)
(1:5,000; cat. no. 111-035-003; Jackson ImmunoResearch; Shanghai
Rebiosci Biotech Co., Ltd.
Statistical analysis
Statistical computations were performed using the
Statistical Program for Social Sciences (SPSS) 20.0 (IBM Corp.,
Armonk, NY, USA) and Graph Pad Prism 5.0 software (GraphPad
Software, Inc., La Jolla, CA, USA). The Chi-square tests were
performed to investigate the associations between GPR56 expression
and clinicopathological factors. Differences between the two groups
were assessed by Student's t-test, whereas differences between
multiple groups were evaluated by one-way ANOVA followed by
Student-Newman-Keuls (SNK) post hoc test. Survival data were
analyzed using Kaplan-Meier survival curves and log-rank test. Data
were expressed as the mean ± SD. A P-value <0.05 was considered
to indicate a statistically significant difference.
Results
GPR56 is overexpressed in CRC and is
associated with clinicopathological factors
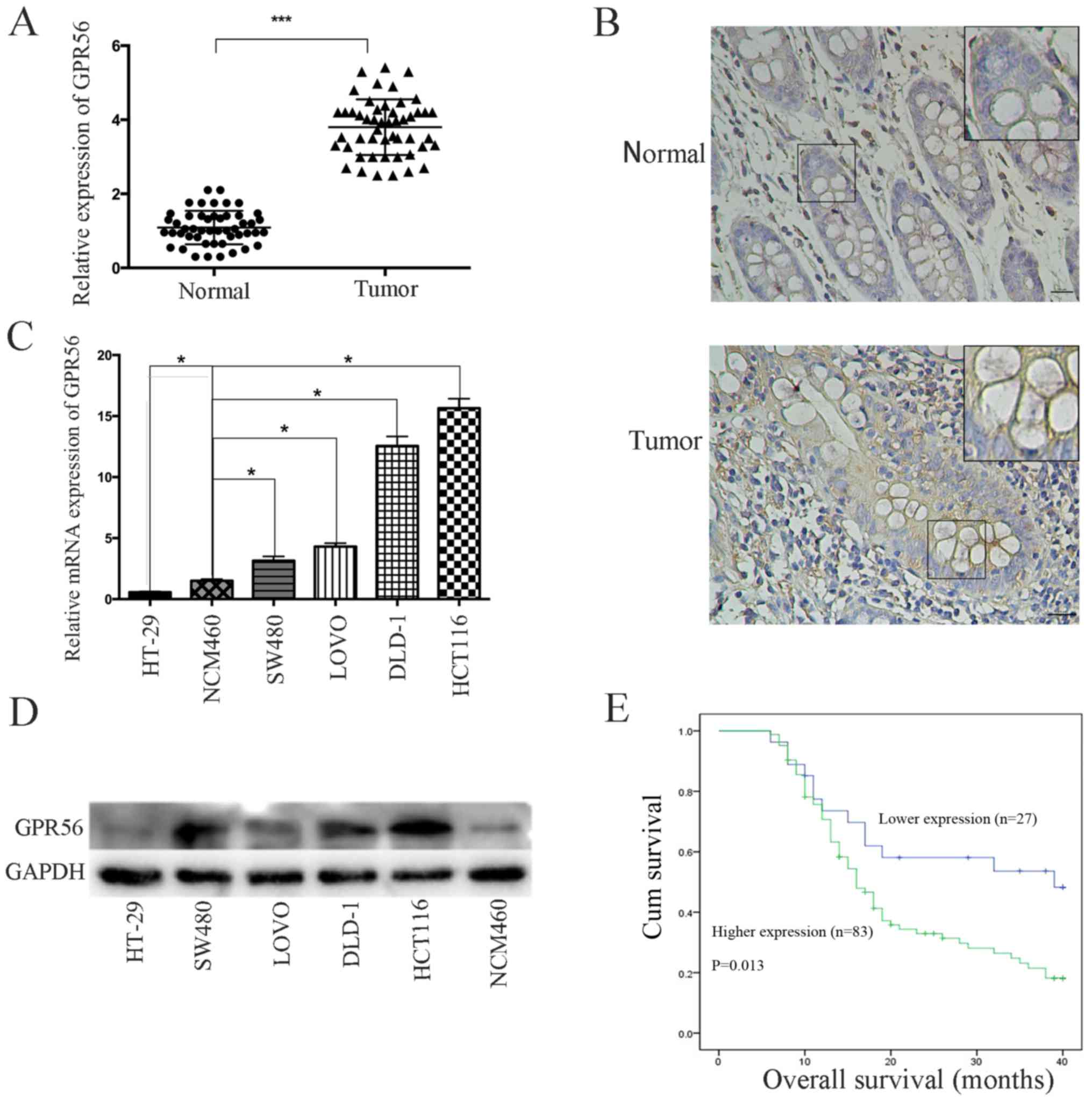
First, we assessed the expression of GPR56 by
qRT-PCR in 110 pairs of CRC and matched normal adjacent tissues to
examine the role of GPR56 in CRC progression. The expression of
GPR56 was significantly higher in tumor tissues than adjacent
non-tumorous tissues (P<0.001; Fig.
1A). These findings were confirmed by immunohistochemical
analysis (Fig. 1B; Table II). Next, we determined the
association of GPR56 expression level with the clinicopathological
features of patients with CRC (Table
III). We divided the 110 pairs of CRC tissue samples into two
experimental groups (high, n=83; low, n=27) based on the mean GPR56
levels. The expression level of GPR56 was significantly associated
with the following clinicopathological factors: TNM stage
(P=0.017), lymph node metastasis (P=0.009), depth of invasion
(P=0.023), and distant metastasis (P=0.019). Yet, we found no
association between GPR56 levels and other factors, including age
(P=0.336), sex (P=0.322), tumor diameter (P=0.224),
carcinoembryonic antigen (P=0.224), and primary tumor site
(P=0.312). We also examined GPR56 expression levels in CRC cell
lines, namely, LOVO, DLD-1, SW480, HT-29, HCT116, and in normal
colon epithelial cells (NCM460) by qRT-PCR and western blotting.
GPR56 mRNA and protein expression levels were significantly higher
in all CRC cell lines apart from HT-29 when compared with NCM460
cells (Fig. 1C and D).
| Table II.Analysis of GPR56 expression in
colorectal carcinoma and adjacent normal tissues by IHC. |
Table II.
Analysis of GPR56 expression in
colorectal carcinoma and adjacent normal tissues by IHC.
| Tissue type | Total scores | No. of samples | χ2 | P-value |
|---|
| Carcinoma | 0-3 | 4 | 8.286 | 0.004<0.05 |
|
| 4-6 | 16 |
|
|
| Adjacent
normal | 0-3 | 13 |
|
|
|
| 4-6 | 7 |
|
|
| Table III.Association of GPR56 expression with
clinicopathological factors in colorectal cancer. |
Table III.
Association of GPR56 expression with
clinicopathological factors in colorectal cancer.
|
|
| GPR56
expression |
|
|---|
|
|
|
|
|
|---|
|
Characteristics | No. of
patients | Low | High | P-valuea |
|---|
| Age (years) |
|
<65 | 49 | 10 | 39 | 0.336 |
|
≥65 | 61 | 17 | 44 |
|
| Sex |
|
Male | 48 | 14 | 34 | 0.322 |
|
Female | 62 | 13 | 49 |
|
| Tumor diameter
(cm) |
|
<5 | 60 | 12 | 35 | 0.224 |
| ≥5 | 50 | 15 | 48 |
|
| TNM stage |
|
I/II | 64 | 21 | 43 | 0.017a |
|
III/IV | 46 | 6 | 40 |
|
| Lymph node
metastasis |
|
Positive | 44 | 5 | 39 | 0.009a |
|
Negative | 66 | 22 | 44 |
|
| Depth of
invasion |
|
T1+T2 | 41 | 15 | 26 | 0.023a |
|
T3+T4 | 69 | 12 | 57 |
|
| Distant
metastasis |
|
Positive | 21 | 1 | 20 | 0.019a |
|
Negative | 89 | 26 | 63 |
|
| CEA |
|
<4.7 | 46 | 14 | 32 | 0.224 |
|
≥4.7 | 64 | 13 | 51 |
|
| Primary tumor
site |
|
Colon | 50 | 10 | 40 | 0.312 |
|
Rectum | 60 | 17 | 43 |
|
High expression of GPR56 predicts poor
prognosis in patients with CRC
To further investigate the relationship between
GPR56 expression and prognosis of CRC patients, Kaplan-Meier
analysis was used to evaluate the correlation between GPR56
expression and overall survival (OS). We found that the OS for
patients with low GPR56 levels was significantly better than those
with high GPR56 levels (P=0.013; Fig.
1E). These data indicated that GPR56 may be helpful for
evaluating the prognosis of CRC patients.
GPR56 knockdown decreases CRC cell
proliferation
We selected the HCT116 and DLD-1 cell lines, which
had higher GPR56 expression levels compared to the other CRC cell
lines (LOVO, HT-29, SW480), to examine whether GPR56 levels and
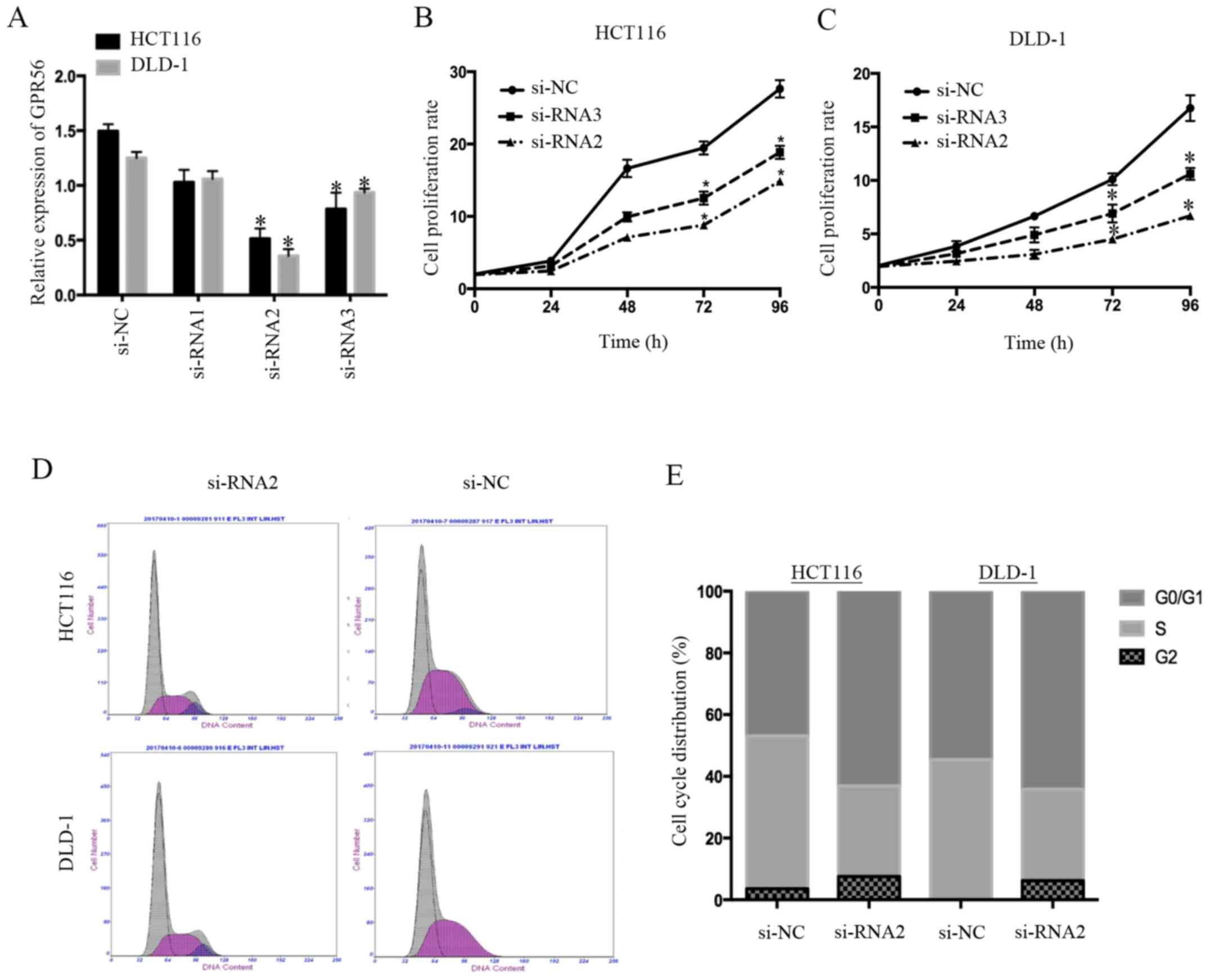
cancer cell proliferation were associated. We decreased GPR56
expression levels in the HCT116 and DLD-1 cells using specific
siRNA targeting GPR56 (si-RNA1, si-RNA2 and si-RNA3). Cells
transfected with si-RNA2 and si-RNA3 had a significantly decreased
GPR56 mRNA expression level compared with the negative control
(si-NC) group in both cell lines (P<0.05; Fig. 2A). Next, we performed Cell Counting
Kit-8 (CCK-8) assays to identify the function of GPR56 in the
proliferation of CRC cells. We found that cells in which GPR56 was
knocked down exhibited decreased proliferation compared to the
controls (P<0.05; Fig. 2B and
C). Since the effect of si-RNA2 was more marked than that of
si-RNA3, we only used si-RNA2 in the subsequent experiments. The
effects of GPR56 on the cell cycle were further examined via flow
cytometry. There were increased cells in the G0/G1 phase and
decreased cells in the S phase in the GPR56-knockdown group
compared to the controls (Fig. 2D and
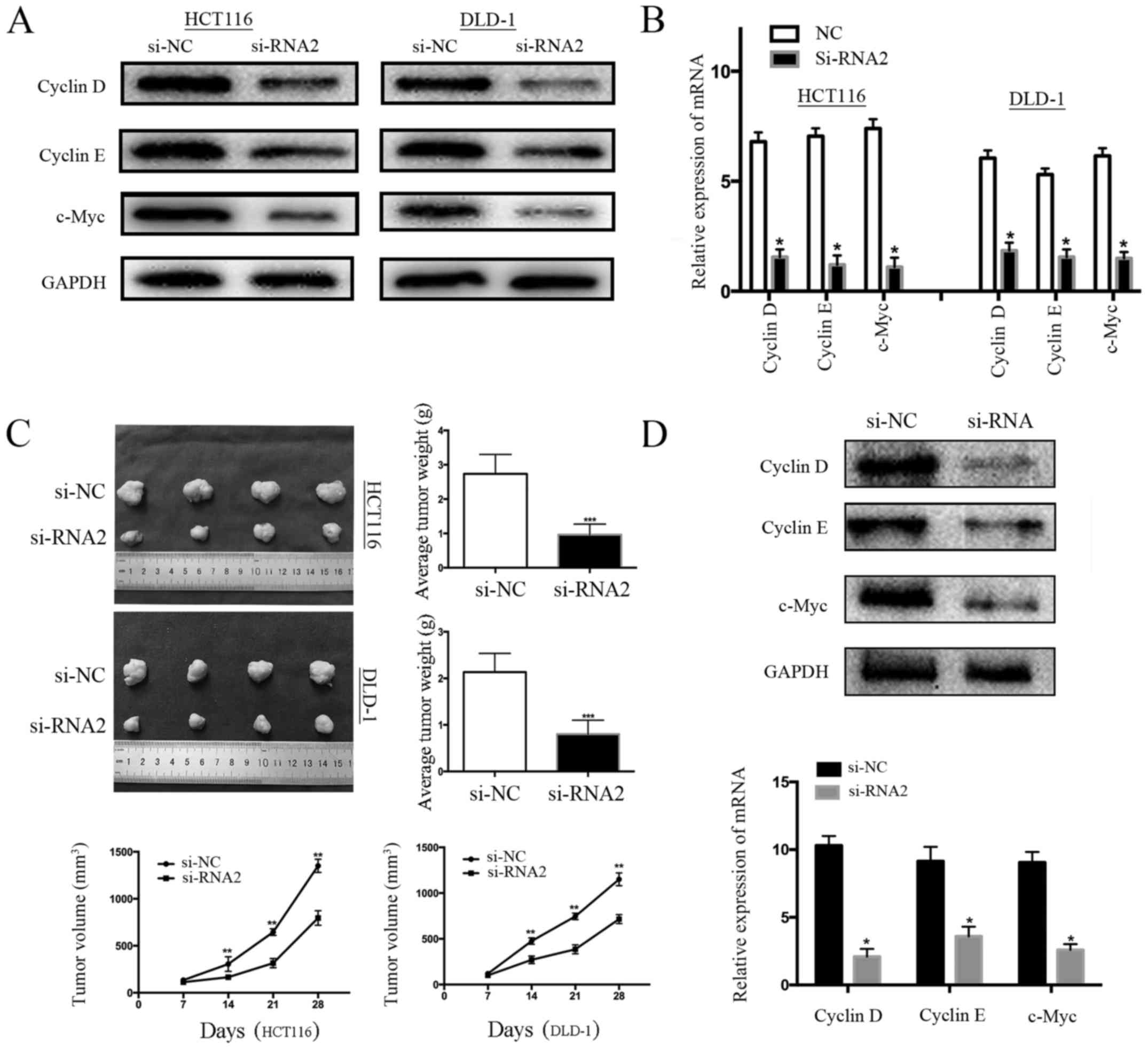
E). Subsequently, western blot analysis and qRT-PCR were
performed to investigate protein and mRNA expression levels of the
target proteins associated with the cell cycle distribution (i.e.,
cyclin D, cyclin E, and c-Myc). We found that protein and mRNA
levels of cyclin D, cyclin E, and c-Myc in the control cells (si-NC
cells) were higher than in the GPR56-knockdown cells (Fig. 3A and B). To investigate whether
GPR56 knockdown could impair tumor formation in vivo, HCT116
and DLD-1 cells were injected into the groins of nude mice. After 4
weeks, intratumoral injection with si-GPR56 or si-NC was performed
once every 48 h. After 4 weeks, compared with the si-NC group, the
tumor weight of the si-RNA2 group exhibited a significant decrease
and similar to the results of the in vitro cell
proliferation experiment, the tumor growth rate was significantly
lower in the interfering group (si-RNA2) than in the negative
control group (si-NC) (Fig. 3C). In
addition, the expression of cell-cycle proteins in tumor tissues
formed by HCT116 in nude mice was detected by western blotting and
qRT-PCR. The results revealed that the protein and mRNA levels of
cyclin D, cyclin E, and c-Myc in the control mice (si-NC) were
higher than in the GPR56-knockdown mice, which was similar with the
results of experiment in human colorectal cancer cell lines
(Fig. 3D). Collectively, these
results indicated that GPR56 played an oncogenic role by
stimulating CRC proliferation.
GPR56 knockdown promotes apoptosis of
CRC cells in vitro
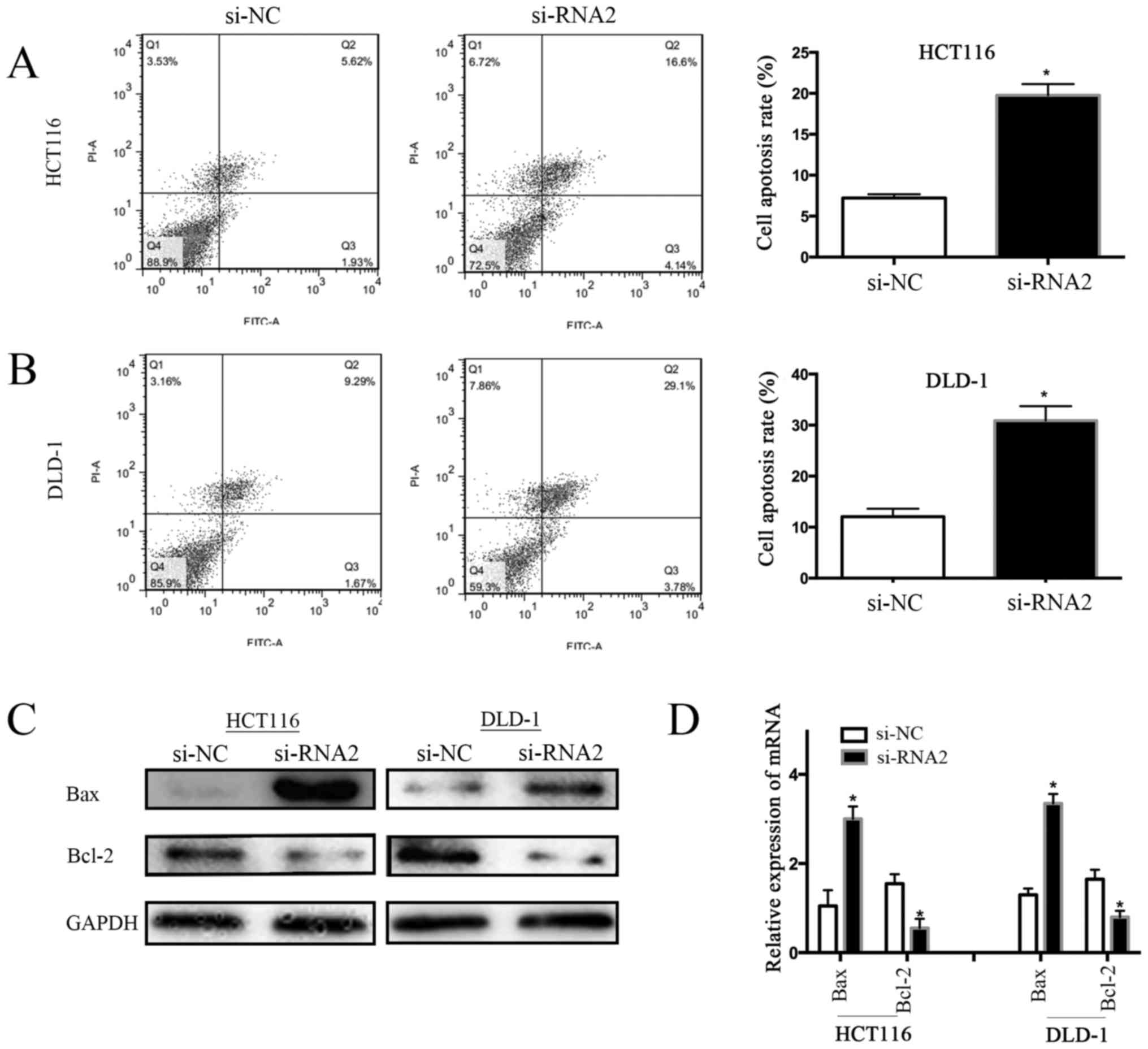
Next, we investigated the association between GPR56
and CRC cell apoptosis by flow cytometry. We found that the cell
apoptosis rate was lower in GPR56-knockdown cells than in control
cells (si-NC) (i.e., 7.55% vs. 20.74% for the HCT116 cell line; and
10.96%, vs. 32.88% for the DLD-1 cell line). Compared to
GPR56-knockdown cells, the rate of apoptosis increased by 13.19% in
HCT116 cells and by 21.92% in DLD-1 cells (Fig. 4A and B; P<0.05). Bcl-2 is a known
apoptosis inhibitor, while Bax is a pro-apoptotic member of the
Bcl-2 family. Therefore, we assessed the expression level of Bcl-2
and Bax proteins in CRC cells (HCT116 and DLD-1) with and without
GPR56 knockdown. Western blotting revealed that GPR56 knockdown
decreased Bcl-2 levels and increased Bax levels in the HCT116 and
DLD-1 cell lines compared to the controls (si-NC) (Fig. 4C). In addition, qRT-PCR analysis of
the mRNA expression of Bcl-2 and Bax revealed similar results
(P<0.05; Fig. 4D). These results
indicated that knockdown of GPR56 activated the apoptosis of CRC
cells, possibly by regulating the Bcl-2/Bax ratio.
GPR56 knockdown inhibits the migration
and invasion of CRC cells in vitro
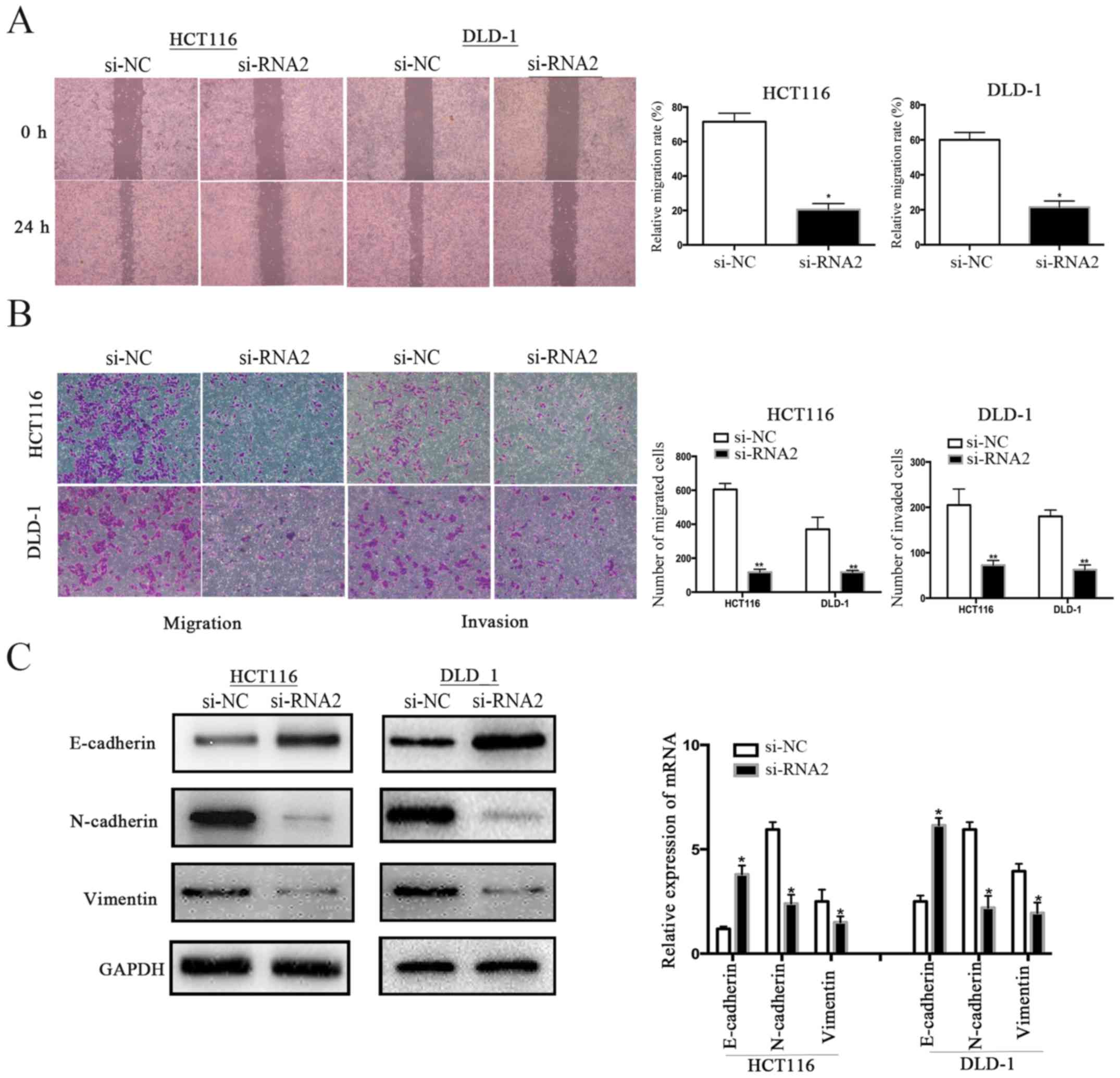
We performed wound healing and Transwell assays
using HCT116 cells and DLD-1 cells to determine the function of
GPR56 in CRC cell motility. The wound healing assay revealed a
relatively weak migration ability of HCT116 and DLD-1 cells in
which GPR56 was knocked down compared to the control group (si-NC)
(Fig. 5A). Furthermore, Transwell
migration and invasion assays revealed that knockdown of GPR56
significantly inhibited the migration and invasion abilities of
both HCT116 and DLD-1 cells (Fig.
5B). Thus, we demonstrated that GPR56 played an important role
in the migration and invasion of CRC.
GPR56 induces epithelial-mesenchymal
transition phenotypes
Epithelial-mesenchymal transition (EMT) is a vital
process for primary tumor cells to gain migration ability. We
assessed whether GPR56 had any influence on EMT in CRC cells to
examine the function of GPR56 in CRC cell proliferation and
migration. We determined the protein and mRNA expression levels of
E-cadherin, N-cadherin, and vimentin in GPR56-knockdown cell lines
compared to the controls (si-NC). We determined that GPR56
knockdown reversed the EMT-related protein levels in both HCT116
and DLD-1 compared to the controls. Specifically, epithelial marker
E-cadherin was highly expressed whereas the expression of the
mesenchymal markers N-cadherin and vimentin was significantly
decreased (Fig. 5C).
GPR56 promotes CRC cell migration by
inducing EMT via regulation of the PI3K/AKT signaling pathway
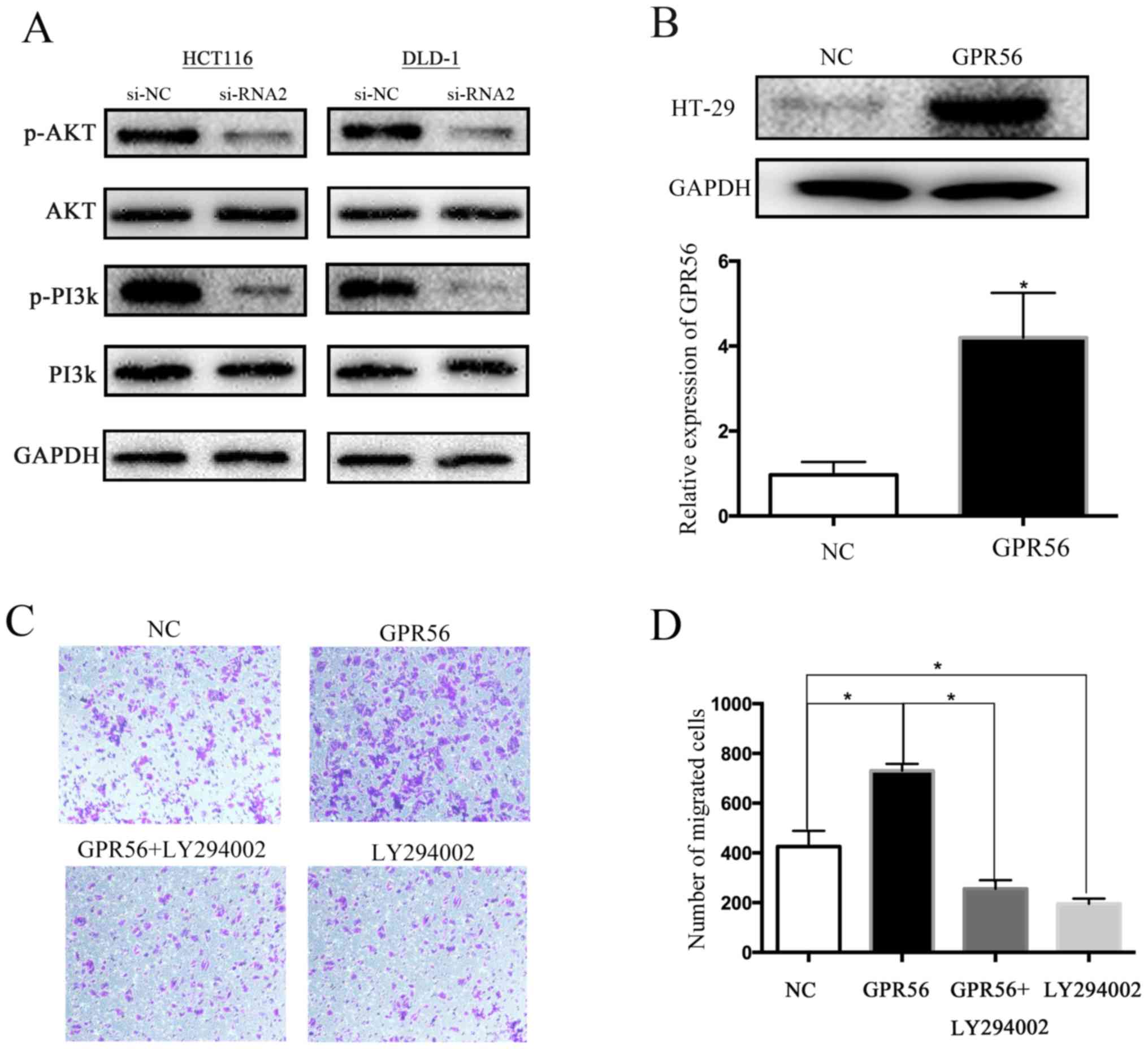
First, we examined the association between GPR56
expression and PI3K/AKT signaling. Western blotting revealed that
GPR56 knockdown markedly decreased phosphorylated PI3K and AKT
levels but not that of non-phosphorylated PI3K and AKT (Fig. 6A). Considering there are a series of
oncogenes reported to promote EMT via activation of the PI3K/AKT
signaling pathway in various malignancies, we attempted to uncover
the relationship between the PI3K/AKT signaling pathway and EMT in
CRC. We chose the HT-29 cell line, which had a low expression of
GPR56, for these experiments and then overexpressed the
GPR56-target gene using a special amplification plasmid (Fig. 6B). As predicted, overexpression of
GPR56 significantly increased cell migration in a Transwell assay;
however, the addition of the PI3K/AKT-specific inhibitor LY294002
decreased the cell migration capacity (Fig. 6C and D).
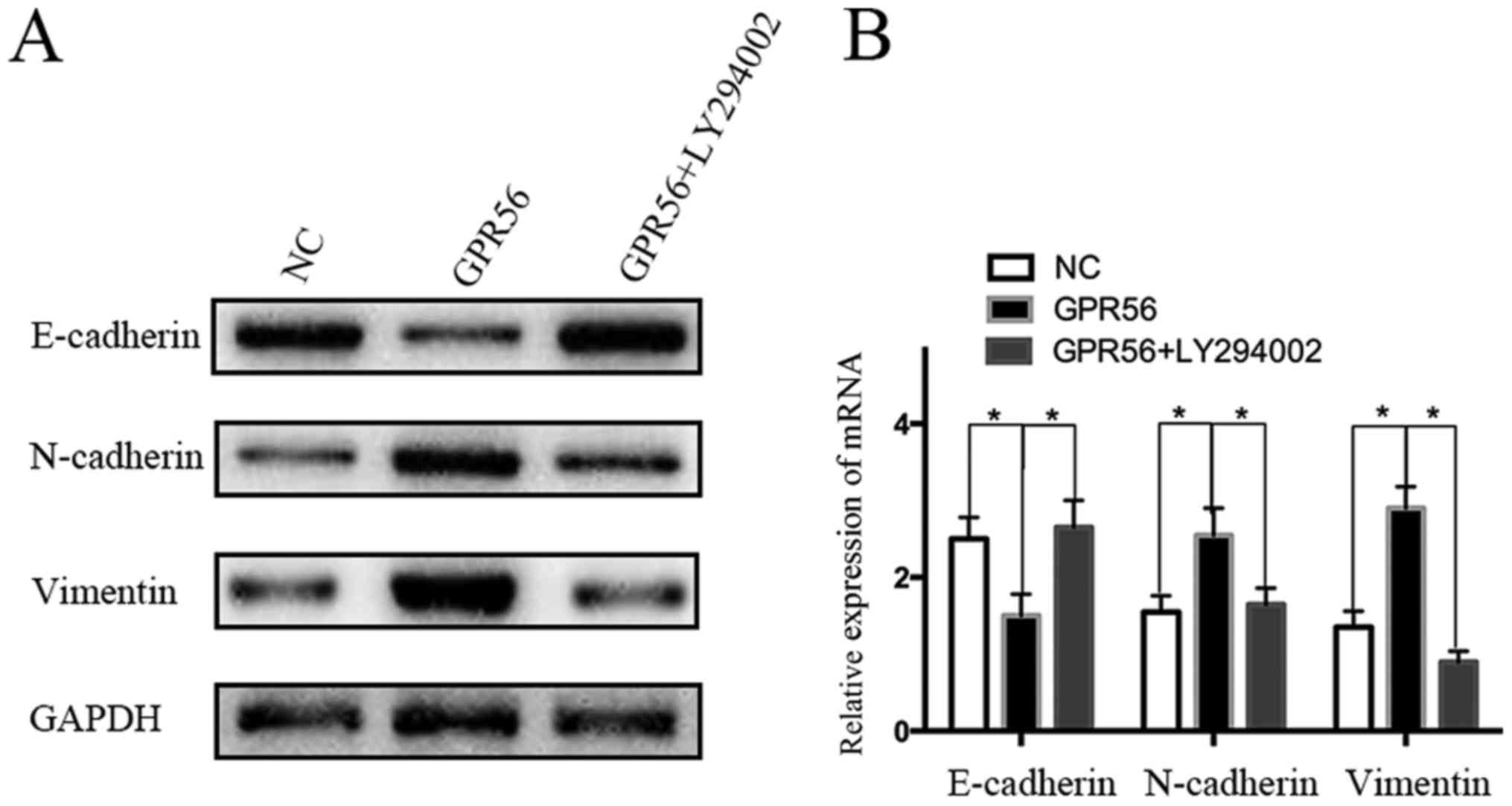
In addition, western blotting and qRT-PCR analysis
revealed that GPR56 overexpression upregulated vimentin and
N-cadherin expression and downregulated E-cadherin expression.
Conversely, when cells overexpressing GPR56 were treated with
LY294002, the typical change in EMT was reversed (Fig. 7A and B). To summarize, these results
preliminarily demonstrated that GPR56 promoted EMT via PI3K/AKT
signaling, which led to the increased migration ability of CRC
cells.
Discussion
GPCRs have recently become a hot topic in tumor
research (19–21). They are reported to be vital
regulators of various cellular functions such as cell adhesion,
migration, polarity, and guidance. Although the expression of GPR56
has been demonstrated to be increased, deceased, or silenced in
either tumor cells or the stromal cells of the tumor
microenvironment (22), its precise
role and related mechanism in CRC remains unclear. Our study
revealed for the first time the GPR56-related regulatory mechanisms
that may be involved in the development and metastasis of CRC.
Considering GPR56 was previously found to be
upregulated in a series of cancer types [such as esophageal
squamous cell carcinoma, human fibrosarcoma, and human epithelial
ovarian cancer (23)], we
investigated whether high expression of GPR56 had a similar effect
in CRC progression. Our findings indicated that GPR56 was
overexpressed at both the mRNA and protein levels in CRC and its
high expression was significantly associated with the malignant
progression of the primary tumor. We also found that patients with
high expression of GPR56 had a relatively poor prognosis compared
to those with low expression. These results demonstrated that GPR56
has the potential to be a clinical marker for both diagnosis and
prognosis evaluation in CRC.
We also investigated how GPR56 functions as an
oncogene in CRC cells. We found that GPR56 played a vital role in
changing the proliferation, migration and invasion abilities of CRC
cells. GPR56 knockdown also caused CRC cells to arrest at the G0/G1
phase and promoted apoptosis in both HCT116 and DLD-1 cell lines.
Collectively, our results indicated that high expression of GPR56
was positively associated with the metastatic potential of primary
colorectal tumors.
Since EMT is a vital mechanism in the metastasis of
primary cancers (24), we
hypothesized that GPR56 may play a role in this process. EMT is a
process whereby epithelial cells are transformed into motile
mesenchymal cells. It typically occurs during morphogenesis in
embryonic development, and is silent in the adult body; however,
EMT can be recovered under a variety of pathological conditions,
including wound healing, fibrosis, and cancer metastasis (25). Thus, we assessed the expression of
EMT-induced markers after knockdown of GPR56 in two CRC cell lines.
Depletion of GPR56 evidently weakened N-cadherin and vimentin
expression, but increased the expression of E-cadherin. This
indicated that GPR56 promoted metastasis of CRC by regulating the
expression of genes related to EMT.
The PI3K/AKT signaling pathway is a vital pathway
related to cancer progression and invasion (26). Crosstalk between PI3K/AKT signaling
and EMT in cancers has been reported (27–30).
Thus, we investigated whether PI3K/AKT-mediated EMT was involved in
GPR56-mediated migration and invasion in CRC. We determined that
overexpression of GPR56 promoted the migration of CRC cells and
this promotion could be inhibited by LY294002, a specific inhibitor
of PI3K/AKT signaling. Conversely, depletion of GPR56 caused a
significant decrease in phosphorylated PI3K and AKT. In addition,
when cells that expressed high levels of GPR56 were treated with
LY294002, EMT was reversed. These observations indicated that high
expression of GPR56 affected CRC migration by stimulating EMT via a
PI3K/AKT-mediated mechanism.
In conclusion, in the present study, high GPR56
expression was detected in CRC tissues and cell lines at both the
protein and mRNA levels. Since GPR56 has an oncogenic role, it has
the potential to be a useful biomarker of CRC. Furthermore, GPR56
promoted CRC cell proliferation, migration, and invasion, and was
critical in CRC metastasis by stimulating EMT via activation of
PI3K/AKT signaling. However, this investigation still has some
flaws. We found that GPR56 was highly expressed in most colorectal
cancer cell lines (LOVO, DLD-1, SW480, HCT116) but expressed at a
relatively low level in HT-29 cells when compared to a normal cell
line (NCM460). The molecular mechanism behind this phenomenon
remains unclear. We conjectured that it may be related to
differences between cell lines and multigene interactions in
cancers. The precise underlying mechanism of the interaction
between GPR56 and the progression of CRC requires further
investigation.
Acknowledgements
We thank Hailong Zhao for his assistance with the
statistical analysis. We also thank our colleagues at the Public
Laboratory of General Surgery, the First Affiliated Hospital of
Nanjing Medical University for their technical assistance.
Funding
The present study was supported in part by the
Jiangsu Key Medical Discipline (General Surgery) (no.
ZDXKA2016005).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
BJ and YF conceived and designed the study. BJ and
YS performed the experiments. BJ wrote the paper. BJ and YF
reviewed and edited the manuscript. DJ, WQ, ZZ, QW, YZ and CZ were
also involved in the conception of the study and gave their advice
in the process of the research. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The Research Ethics Committee of the First
Affiliated Hospital of Nanjing Medical University approved this
study. The enrolled patients signed informed consent forms. All
experimental procedures conducted in vivo were in accordance
with the guidelines from the Animal Ethical and Welfare Committee
of Nanjing Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yao H and Zhang Z: Standardized diagnosis
and treatment of colorectal liver metastasis. Zhonghua Wei Chang
Wai Ke Za Zhi. 20:753–757. 2017.(In Chinese). PubMed/NCBI
|
|
3
|
Aust G, Zhu D, Van Meir EG and Xu L:
Adhesion GPCRs in tumorigenesis. Handb Exp Pharmacol. 234:369–396.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Drews J: Drug discovery: A historical
perspective. Science. 287:1960–1964. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zendman AJ, Cornelissen IM, Weidle UH,
Ruiter DJ and van Muijen GN: TM7XN1, a novel human EGF-TM7-like
cDNA, detected with mRNA differential display using human melanoma
cell lines with different metastatic potential. FEBS Lett.
446:292–298. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kausar T, Sharma R, Hasan MR, Tripathi SC,
Saraya A, Chattopadhyay TK, Gupta SD and Ralhan R: Clinical
significance of GPR56, transglutaminase 2, and NF-κB in esophageal
squamous cell carcinoma. Cancer Invest. 29:42–48. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shashidhar S, Lorente G, Nagavarapu U,
Nelson A, Kuo J, Cummins J, Nikolich K, Urfer R and Foehr ED: GPR56
is a GPCR that is overexpressed in gliomas and functions in tumor
cell adhesion. Oncogene. 24:1673–1682. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mori A, Arii S, Furutani M, Hanaki K,
Takeda Y, Moriga T, Kondo Y, Rivas Gorrin MJ and Imamura M:
Vascular endothelial growth factor-induced tumor angiogenesis and
tumorigenicity in relation to metastasis in a HT1080 human
fibrosarcoma cell model. Int J Cancer. 80:738–743. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saito Y, Kaneda K, Suekane A, Ichihara E,
Nakahata S, Yamakawa N, Nagai K, Mizuno N, Kogawa K, Miura I, et
al: Maintenance of the hematopoietic stem cell pool in bone marrow
niches by EVI1-regulated GPR56. Leukemia. 27:1637–1649. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chiang NY, Peng YM, Juang HH, Chen TC, Pan
HL, Chang GW and Lin HH: GPR56/ADGRG1 activation promotes melanoma
cell migration via NTF Dissociation and CTF-mediated
galpha12/13/RhoA signaling. J Invest Dermatol. 137:727–736. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang L, Chen G, Mohanty S, Scott G, Fazal
F, Rahman A, Begum S, Hynes RO and Xu L: GPR56 Regulates VEGF
production and angiogenesis during melanoma progression. Cancer
Res. 71:5558–5568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Luo R, Jin Z, Deng Y, Strokes N and Piao
X: Disease-associated mutations prevent GPR56-collagen III
interaction. PLoS One. 7:e298182012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Little KD, Hemler ME and Stipp CS: Dynamic
regulation of a GPCR-tetraspanin-G protein complex on intact cells:
Central role of CD81 in facilitating GPR56-Galpha q/11 association.
Mol Biol Cell. 15:2375–2387. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang L, Friedland S, Corson N and Xu L:
GPR56 inhibits melanoma growth by internalizing and degrading its
ligand TG2. Cancer Res. 74:1022–1031. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jin G, Sakitani K, Wang H, Jin Y,
Dubeykovskiy A, Worthley DL, Tailor Y and Wang TC: The G-protein
coupled receptor 56, expressed in colonic stem and cancer cells,
binds progastrin to promote proliferation and carcinogenesis.
Oncotarget. 8:40606–40619. 2017.PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang S, Zhang C, Zhang Z, Qian W and Sun
Y, Ji B, Zhang Y, Zhu C, Ji D, Wang Q and Sun Y: Transcriptome
analysis in primary colorectal cancer tissues from patients with
and without liver metastases using next-generation sequencing.
Cancer Med. 6:1976–1987. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun Y, Ji B, Feng Y, Ji D, Zhu C, Wang S,
Zhang C, Zhang D and Sun Y: TRIM59 facilitates the proliferation of
colorectal cancer and promotes metastasis via the PI3K/AKT pathway.
Oncol Rep. 38:43–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kishore A and Hall RA: Disease-associated
extracellular loop mutations in the adhesion G protein-coupled
receptor G1 (ADGRG1; GPR56) differentially regulate downstream
signaling. J Biol Chem. 292:9711–9720. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tseng WY, Wu Jan YJ, Yang TY, Chiang NY,
Tsai WP, Gordon S, Chang GW, Kuo CF, Luo SF and Lin HH: High levels
of soluble GPR56/ADGRG1 are associated with positive rheumatoid
factor and elevated tumor necrosis factor in patients with
rheumatoid arthritis. J Microbiol Immunol Infect pii: S1684-1182.
30094. 2017.
|
|
21
|
Mehta P and Piao X: Adhesion G-protein
coupled receptors and extracellular matrix proteins: Roles in
myelination and glial cell development. Dev Dyn. 246:275–284. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ke N, Sundaram R, Liu G, Chionis J, Fan W,
Rogers C, Awad T, Grifman M, Yu D, Wong-Staal F and Li QX: Orphan G
protein-coupled receptor GPR56 plays a role in cell transformation
and tumorigenesis involving the cell adhesion pathway. Mol Cancer
Ther. 6:1840–1850. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu Z, Huang Z, Yang W, Li Z, Xing S, Li
H, Hu B and Li P: Expression of orphan GPR56 correlates with tumor
progression in human epithelial ovarian cancer. Neoplasma.
64:32–39. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang L, Wang X and Lai M: Modulation of
epithelial-to-mesenchymal cancerous transition by natural products.
Fitoterapia. 106:247–255. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu PL, Liu WL, Chang JM, Chen YH, Liu YP,
Kuo HF, Hsieh CC, Ding YS, Chen WW and Chong IW: MicroRNA-200c
inhibits epithelial-mesenchymal transition, invasion, and migration
of lung cancer by targeting HMGB1. PLoS One. 12:e01808442017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang SC, Chai DS, Chen CB, Wang ZY and
Wang L: HPIP promotes thyroid cancer cell growth, migration and EMT
through activating PI3K/AKT signaling pathway. Biomed Pharmacother.
75:33–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wei S, Wang L, Zhang L, Li B, Li Z, Zhang
Q, Wang J, Chen L, Sun G, Li Q, et al: ZNF143 enhances metastasis
of gastric cancer by promoting the process of EMT through PI3K/AKT
signaling pathway. Tumour Biol. 37:12813–12821. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song Y, Li ZX, Liu X, Wang R, Li LW and
Zhang Q: The Wnt/β-catenin and PI3K/Akt signaling pathways promote
EMT in gastric cancer by epigenetic regulation via H3 lysine 27
acetylation. Tumour Biol. 39:10104283177126172017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fu H, He Y, Qi L, Chen L, Luo Y, Chen L,
Li Y, Zhang N and Guo H: cPLA2α activates PI3K/AKT and inhibits
Smad2/3 during epithelial-mesenchymal transition of hepatocellular
carcinoma cells. Cancer Lett. 403:260–270. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu Q, Liu X, Liu Z, Wang Y, Tu J, Li L,
Bao H, Yang L and Tu K: MicroRNA-1296 inhibits metastasis and
epithelial-mesenchymal transition of hepatocellular carcinoma by
targeting SRPK1-mediated PI3K/AKT pathway. Mol Cancer. 16:1032017.
View Article : Google Scholar : PubMed/NCBI
|