Introduction
Osteosarcoma is the most common primary malignant
tumor of the bone found predominantly in children and teenagers and
results in early metastasis and poor prognosis. With the
development of neoadjuvant chemotherapy and surgical improvement,
the five-year survival rate of patients with osteosarcoma is close
to 60% (1). However, adverse drug
reactions and drug resistance of osteosarcomas are becoming more
and more common, and novel drugs are urgently needed to treat
osteosarcoma (2).
Celastrol, a quinone methide triterpenoid, is
isolated from the root of Tripterygium wilfordii and is
primarily used for the treatment of autoimmune diseases and
neurodegenerative diseases (3,4). In
recent years, celastrol has exhibited great potential in inducing
apoptosis of tumor cells and has shown significant progress for
osteosarcoma treatment (5–10). Apoptosis, known as type I-programmed
cell death, mainly involves changes in cell morphology (e.g.,
chromosome condensation or fragmentation), shrinkage or
invagination of the cell membrane and apoptotic bodies (11–15).
Apoptosis is one principal approach to induce tumor death. The
mitochondrial, death receptor and endoplasmic reticulum stress
(ERS) pathways are widely accepted to mediate cell apoptosis.
The endoplasmic reticulum (ER) is a very complex
organelle and functions in protein synthesis, transmembrane
transport, post-translational modification, glycosylation
modification, synthesis of phospholipids and cholesterol and
control of intracellular calcium homeostasis. When ER homeostasis
is disturbed, many unfolded proteins bind to Glucose-regulated
protein 78 (Grp78/Bip) and activate the pathways of PERK,
activating transcription factor 6 (ATF6) and inositol-requiring
enzyme 1 (IRE1) to promote the recovery of ER function and protect
cells. However, extended durations or high strength of ERS will
induce cell apoptosis mainly through three apoptotic pathways,
including CHOP, JNK and caspase-12 (13,16).
The aim of the present study was primarily to
explore the effect of celastrol on the HOS human osteosarcoma cell
line and the related mechanisms (especially ERS, the mitochondrial
pathway and autophagy). We further explored the effect of PERK
signaling on ERS and its relationship with apoptosis and
autophagy.
Materials and methods
Chemicals and cell culture
Celastrol, sodium tauroursodeoxycholate (TUDCA),
thapsigargin (TG) and Hoechst 33258 were purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Celastrol had a
purity higher than 98%. A stock solution (50 mmol/l) was made in
dimethyl sulfoxide (DMSO) (Nanjing KeyGen Biotech, Co., Ltd.,
Nanjing, China) and stored in the dark at −80°C. GSK2656157 was
purchased from Selleck Chemicals (Houston, TX, USA). Dulbecco's
modified Eagle's medium (DMEM), fetal bovine serum (FBS) and
trypsin were all purchased from Gibco™ (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Cell viability and cytotoxicity test kits
[Cell Counting Kit-8 (CCK-8)] and FLUOU-3/AM were obtained from
Dojindo Molecular Technologies (Kumamoto, Japan). The Annexin
V-propidium iodide (PI) double-staining test kit was purchased from
Nanjing KeyGen Biotech. Bip, Calnexin, endoplasmic
oxidoreductin-1-like protein α (Ero1-Lα), protein disulfide
isomerase (PDI), IRE1α, PERK, CHOP, cleaved caspase-3, cytochrome
c, Bax, Bcl-2, LC-3, p62 and β-actin were purchased from
Cell Signaling Technology, Inc., (Danvers, MA, USA). Cleaved
caspase-12 was purchased from Proteintech Group Inc., (Wuhan,
China) and p-PERK was supplied by Santa Cruz Biotechnology (Santa
Cruz, CA, USA). HOS cells were purchased from the American Type
Culture Collection (ATCC; Manassas, VA, USA) and cultured in DMEM
containing 100 µg/ml penicillin, 100 µg/ml streptomycin, and 10%
FBS (complete medium) at 37°C in 5% CO2.
Cytotoxicity assay
The anti-proliferative effect of celastrol on HOS
cells was assessed by after 12 h, they were treated with various
concentrations of celastrol (0, 1.5, 2.5, 3.5 and 4.5 µM) for 24 h.
Subsequently, 10 µl CCK-8 was added to each well, and the plates
were incubated in an incubator for 1 to 2 h at 37°C following the
manufacturer's instructions. A microplate reader was used to detect
the absorbance at 450 nm. Data represented the mean of five
replicates. The cell viability was calculated using the following
equation: Cell viability (%) = Average OD in study group/average OD
in control group ×100%, where OD was the optical density. Each
assay was performed in triplicate. Based on the results of the cell
viability assay, we selected an appropriate celastrol concentration
for subsequent experiments.
Examination of the cell ultrastructure
by transmission electron microscopy
HOS cells were seeded in 24-well plates at a density
of 5×104 cells/well, and treated with the celastrol
concentration of 3.00 µM for 24 h. Subsequently, the culture medium
was discarded, and the cells were washed three times with
phosphate-buffered saline (PBS) (Nanjing KeyGen Biotech), then
fixed with 1% osmic acid and 2.5% glutaraldehyde, dehydrated using
acetone and gradient ethanol, embedded, solidified, sliced using an
ultramicrotome and stained with 3% uranium acetate-lead citrate.
The cells were observed to evaluate morphological changes and
images were captured with transmission electron microscopy.
Assessment of apoptosis by Hoechst
nuclear staining
HOS cells were seeded in 24-well plates at a density
of 5×104 cells/well, and treated with the celastrol
concentration of 3.00 µM for 24 h. Subsequenlty, the culture medium
was discarded, and the cells were washed three times with PBS.
After the HOS cells were fixed with 4% paraformaldehyde for 5 min,
10 µg/ml Hoechst 33258 (200 µl) was added to the cells at 37°C for
5 min in the dark. The cells were observed to evaluate apoptotic
changes and images were captured with a fluorescence microscope
after being washed three times with PBS.
Apoptosis determined by flow cytometry
(FCM)
HOS cells were seeded in 6-well plates at a density
of 5×106 cells/well. Suspended and adherent cells were
collected after treatment and analyzed with an Annexin V/PI
apoptosis kit using FCM.
Measurement of calcium ions
HOS cells were seeded in 6-well plates at a density
of 5×106 cells/well. Suspended and adherent cells were
collected following treatment and analyzed with a FLUO-3/AM (2 µM)
kit using FCM.
Expression of p-PERK determined by
immunofluorescence staining
HOS cells were seeded in 24-well plates at a density
of 5×104 cells/well, and treated with the celastrol
concentration of 3.00 µM for 24 h. Subsequently, the cells were
washed with PBS, fixed in 4% freshly prepared formaldehyde for 5
min, and permeabilized in PBS with 0.1% Triton X-100 for 5 min at
room temperature. Cells were then incubated for 1 h in blocking
solution (3% bovine serum albumin/0.08% glycine in PBS) and treated
with a rabbit polyclonal antibody against p-PERK (1:1,000; cat. no.
Sc-32577; Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 2 h at
room temperature. The secondary antibody, fluorescein
(FITC)-conjugated AffiniPure goat anti-rabbit (1:200; cat. no.
SA00003-2; ProteinTech Group, Inc., Chicago, IL, USA), was applied
for 1 h at room temperature, and nuclei were counterstained with
DAPI (Beyotime Institute of Biotechnology, Shanghai, China). Slides
were viewed under a fluorescence microscope (Axio Observer A1; Carl
Zeiss AG, Oberkochen, Germany).
Western blot analysis
Western blotting was performed to observe the
protein expression level. Briefly, at various time-points, cells in
the control and celastrol groups were collected and lysed using
radioimmunoprecipitation assay (RIPA) buffer (Keygen Biotech,
Jiangsu, China) containing protease inhibitor cocktail (Selleck
Chemicals, Houston, TX, USA) for 30 min on ice. The following steps
were performed as depicted in our previous study (11).
Statistical analysis
The quantitative data are represented as the mean ±
standard deviation (SD) and comparisons of significant differences
were calculated by one-way analysis of variance (ANOVA) with
Dunnett's test or unpaired Student's t-test. All statistical
analyses were performed using SPSS software (version 22.0; IBM
Corp., Armonk, NY, USA). Statistical significance tests were
two-tailed, and P<0.05 was considered to indicate a
statistically significant difference.
Results
Celastrol decreases HOS cell
viability
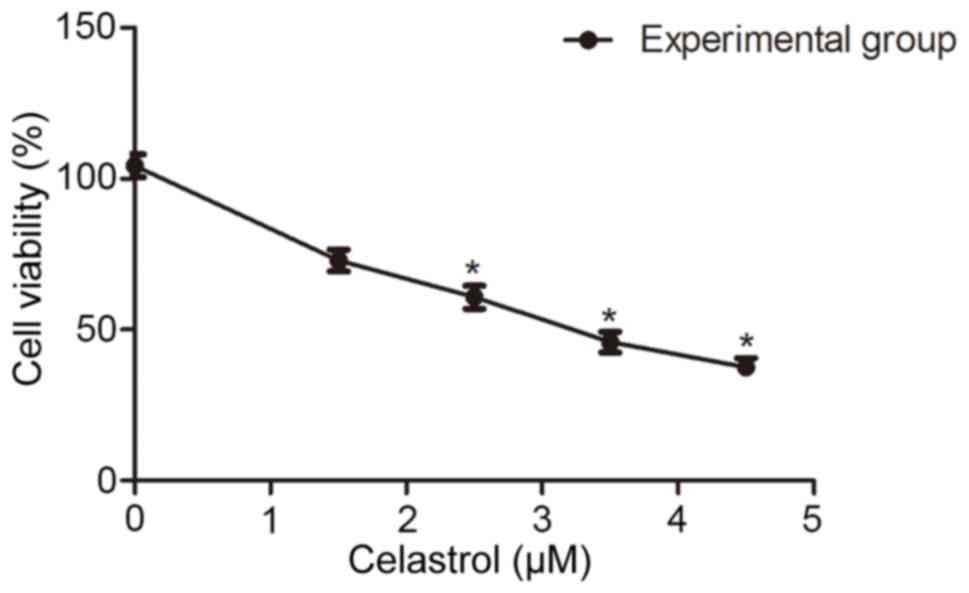
The effect of celastrol on cell viability was
investigated by exposing HOS cells to various concentrations of
celastrol for 24 h (Fig. 1). The
CCK-8 assay demonstrated that celastrol could inhibit the viability
of HOS cells in a dose-dependent manner and revealed that the 50%
inhibitory concentration (IC50) was 3.36 µM for HOS
cells. Therefore, based on previous experiments, we chose a
celastrol concentration of 3.00 µM as the treatment condition for
the following experiments (10).
Celastrol induces apoptosis of HOS
cells
Under inverted phase contrast microscopy, retracted,
round and floating cells were observed, and karyopyknosis,
condensation, karyorrhexis and other typical apoptotic bodies were
observed with Hoechst 33255 nuclear staining following treatment
with celastrol for 24 h (Fig. 2A and
B). However, there were no significant changes in the control
group. FCM revealed that the apoptosis rates of the 6, 12 and 24-h
groups were all higher than that of the control group (P<0.05)
(Fig. 2C and D). These results
indicated that celastrol induced apoptosis of HOS cells.
The ERS pathway and mitochondrial
pathway are involved in apoptosis induced by celastrol in HOS
cells
Under Transmission electron microscopy (TEM), the
HOS cells displayed swelling of the ER following celastrol
treatment for 24 h, whereas the control group exhibited no changes
in the ER (Fig. 3A). FCM
demonstrated that the level of calcium in the celastrol (24 h)
group was significantly higher than that in the control group
(Fig. 3B). In addition, western
blotting revealed that at 6, 12 and 24 h following treatment using
celastrol, the expression levels of ERS-related proteins (Bip,
p-PERK, IRE1α, Calnexin, Ero1-Lα and PDI), ERS-related apoptosis
proteins (CHOP, cleaved caspase-12) and cleaved caspase-3 in HOS
cells were all increased, while PERK was decreased compared to
those of the control group (Fig. 3C and
D, Fig. 4A and B; P<0.05).
These results indicated that ERS was induced by celastrol in HOS
cells, and ERS-induced apoptosis was also involved in HOS cell
apoptosis induced by celastrol. To exclude false-positive results
of the experiment and the influence of DMSO, following treatment
with TG, we observed that the expression levels of Bip, CHOP and
cleaved caspase-12 in HOS cells all increased, while following
treatment with DMSO, all of them decreased, indicating that TG
could induce ERS-induced apoptosis but DMSO could not (Fig. 4C and D; P<0.05).
 | Figure 3.ERS is induced by celastrol. (A) For
24 h following treatment, the ER was observed by TEM. (B) The level
of calcium in cells was determined by FCM. P<0.05 vs. the
control group. (C and D) At 6, 12 and 24 h, whole-cell lysates were
prepared to assay Bip, p-PERK, IRE1α, calnexin, Ero1-Lα, PDI and
PERK proteins by western blotting. Data are presented as the mean ±
SD from three independent experiments. *P<0.05 vs. the control
group. ERS, endoplasmic reticulum stress; ER, endoplasmic
reticulum; TEM, transmission electron microscopy; FCM, flow
cytometry; SD, standard deviation. |
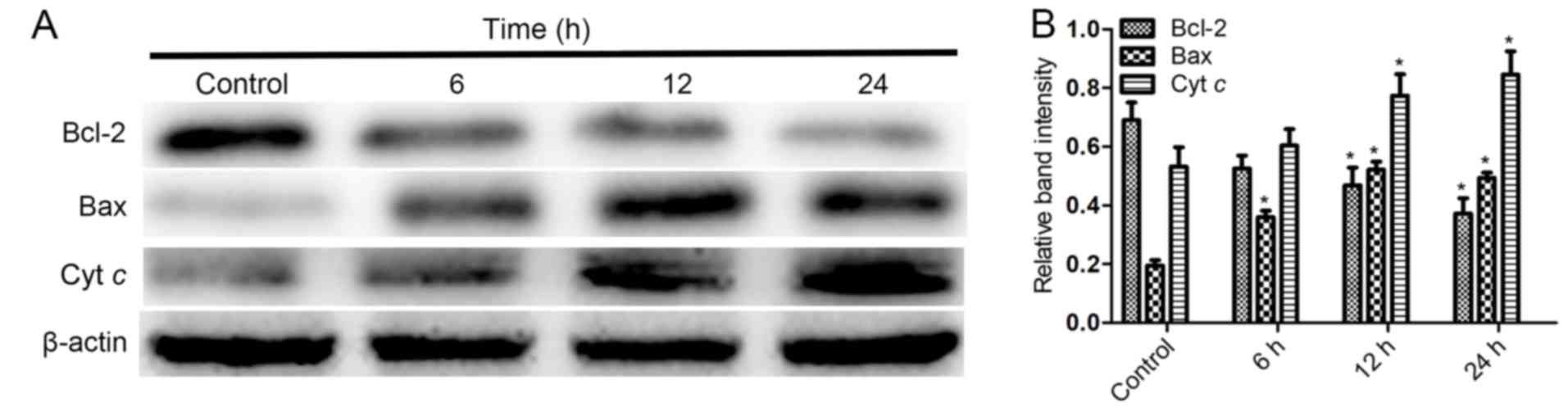
Furthermore, western blotting also revealed that
following treatment with celastrol for 6, 12 and 24 h, Bax and
cytochrome c in the cytoplasm, indexes of mitochondrial
apoptosis, were increased, while Bcl-2 was decreased (Fig. 5A and B; P<0.05). These findings
indicated that the mitochondrial pathway was involved in
celastrol-induced apoptosis in HOS cells.
Apoptosis and autophagy are promoted,
while ERS induced by celastrol is suppressed
TUDCA (500 µM), an inhibitor of ERS, was used to
pretreat HOS cells for 1 h before celastrol treatment (17,18).
FCM demonstrated that pretreatment with TUDCA significantly
increased the apoptosis rate compared to celastrol treatment alone
(Fig. 6A and B; P<0.05).
Furthermore, western blotting revealed that pretreatment with TUDCA
promoted the expression of Lc-3 II and cleaved caspase-3 compared
with celastrol alone treatment, while the expression of Bip,
p-PERK, and p62 was decreased (Fig. 6C
and D; P<0.05). All the results indicated that attenuating
the ERS of HOS cells would promote the autophagy and apoptosis
induced by celastrol.
 | Figure 6.The ERS pathway protects HOS cells
from apoptosis. (A and B) At 24 h, following pretreatment with
TUDCA, the apoptosis rates of HOS cells were determined by FCM.
*P<0.05 vs. the celastrol group. (C and D) At 24 h, following
pretreatment with TUDCA, whole-cell lysates were prepared to assay
Bip, PERK, p-PERK, LC-3, p62, and cleaved caspase-3. Data are
presented as the mean ± SD from three independent experiments.
*P<0.05 vs. the control group. ERS, endoplasmic reticulum
stress; TUDCA, tauroursodeoxycholate; FCM, flow cytometry; SD,
standard deviation. |
Apoptosis is promoted, while the PERK
signaling pathway is suppressed
The unfolded protein response (UPR) protects HOS
cells from apoptosis induced by celastrol. To maintain homeostasis
of HOS cells, a large number of unfolded proteins activate the PERK
signaling pathway by binding to Bip, and PERK, which is an
important transducer, activates downstream molecular partners and
protein secretion related to molecular gene transcription. To
explore whether attenuating the expression of PERK signaling
pathway members would reverse the protective effect of ERS induced
by celastrol and further promote the apoptosis of HOS cells, we
used an inhibitor of PERK (GSK2656157, 5 mM) to pretreat HOS cells
for 1 h before celastrol treatment (19,20).
Following treatment with GSK2656157, the proliferation of HOS cells
was further inhibited compared with that of the celastrol group;
karyopyknosis, condensation, karyorrhexis and other typical
apoptotic bodies were more evident; and the apoptosis rates further
increased, while immunofluorescence revealed that the expression of
p-PERK was significantly attenuated (Fig. 7A-D; P<0.05). Following
pretreatment with GSK2656157, the expression of PERK was inhibited,
while cleaved caspase-3 was increased. All these results
demonstrated that inhibiting the expression of PERK strongly
promoted apoptosis induced by celastrol. In addition, we found that
pretreatment with GSK2656157 attenuated autophagy and suppressed
the expression of PERK (Fig. 7E-F;
P<0.05).
 | Figure 7.The PERK signaling pathway among ERS
pathways has a protective effect on HOS cells. HOS cells were
pretreated with GSK2656157 for 1 h in the presence or absence of
celastrol. Some cells received no pretreatment or only GSK2656157.
After the corresponding treatment, (A) the expression of PERK was
determined by fluorescence microscopy (magnification, ×200). (B)
The apoptotic morphological changes of HOS cells were observed
under fluorescence microscopy (magnification, ×200). (C) The
apoptotic rates were determined by FCM. *P<0.05 vs. the
celastrol group. (D) Cell viability was determined using the CCK-8
assay. (E and F) At 24 h following treatment, whole-cell lysates
were prepared to assay PERK, p-PERK, Bip, LC-3, p62, and cleaved
caspase-3 by western blotting. Data are presented as the mean ± SD
from three independent experiments. *P<0.05 vs. the control
group. ERS, endoplasmic reticulum stress; FCM, flow cytometry;
CCK-8, Cell Counting Kit-8; SD, standard deviation. |
Discussion
Despite the advances in medical technology for
patients with osteosarcoma, long-term survival remains stagnant,
and novel anti-osteosarcoma drugs are urgently needed. In addition,
numerous studies have revealed that many components extracted from
traditional Chinese medicine have antitumor effects (3,8,10).
Celastrol, extracted from the root of T. wilfordii, has been
revealed to inhibit the proliferation of tumors, and the
mitochondrial apoptotic pathway and death receptor apoptosis
pathway were involved in the celastrol-induced apoptosis (19,20).
Research has reported the effect of celastrol on osteosarcoma,
which has demonstrated great antitumor potential. Yu et al
(21) found that celastrol
negatively regulated cell invasion and migration ability of human
osteosarcoma via the downregulation of the PI3K/Akt/NF-κB signaling
pathway in vitro. However, there is still a lack of studies
on the anticancer effect and the mechanism of celastrol on
autophagy and apoptosis of osteosarcoma. Our results revealed that
celastrol could inhibit cell viability, induce morphological
changes of apoptosis and increase the apoptosis rate of HOS cells
compared with control cells.
The functions of the ER, including protein
synthesis, transmembrane transport, and modification following
translation, play a key role in cell survival. The ER also serves
as the largest intracellular calcium library and regulates
intracellular calcium homeostasis. The concentration of calcium
ions in the cytoplasm increases when the ER is damaged. Following
treatment with celastrol, evident swelling and dysfunction of the
ER appeared, with a significant increase in the cytoplasmic free
calcium ion level, which indicated that the ER of HOS cells was
damaged (22–25). The ER is also the receptor of
cellular stress. When the ER is stimulated by damage factors,
misfolded or folded proteins accumulate so that ERS is induced and
the UPR is further promoted. When the UPR is induced, the heat
shock protein Bip is normally located in the ER, the ERS
transduction proteins IRE-1, ATF6 and PERK convert from a binding
state into a dissociative state, and PERK becomes phosphorylated
while the level of PERK decreases to activate downstream chaperone
proteins and gene transcription related to protein secretion, which
will improve the processing capacity of the ER for unfolded or
misfolded proteins (26,27). Thus, Bip is used as a marker protein
for ERS. ERS will activate calnexin, Ero1-Lα, PDI and other
chaperone proteins to promote the proper folding of unfolded or
misfolded proteins, which alleviates the breakdown of cells. The
results of this experiment revealed that the expression of Bip,
calnexin, Ero1-Lα, PDI, IRE1α and p-PERK increased, while PERK
decreased, indicating that ERS was induced by celastrol in HOS
cells.
ERS is a protective mechanism for cells, but when
prolonged excessive stress or dysfunction occurs, apoptosis may be
induced (28–30). Apoptosis mediated through ERS is
mainly induced by the PERK/EIF2α signaling pathway activation of
the ERS-specific transcription factors CHOP/GADD153. Apoptosis is
mediated by the specific transcription factors CHOP/GADD153 of ERS
through the PERK/EIF2α signaling pathway or by the specific
caspase-12 of ERS to activate apoptosis initiation factor caspase-9
and apoptosis executive factor caspase-3. Thus, elevated expression
levels of CHOP and caspase-12 are markers of ERS-mediated apoptosis
in ERS cells (31). The results of
western blotting revealed that following treatment with celastrol,
the expression of CHOP, cleaved caspase-12 and cleaved caspase-3
markedly increased, which indicated that ERS was involved in the
apoptosis induced by celastrol. Furthermore, TG and DMSO were used
as a positive control test to exclude false-positive results and
the impact of DMSO. The mitochondrial pathway is an important
mechanism in apoptosis for tumors. In addition, Bcl-2, Bax and
cytochrome c are regarded as the indexes of the
mitochondrial pathway. The results of western blotting revealed
that following treatment for 6, 12 and 24 h, the expression of Bax
and cytochrome c was increased and Bcl-2 was decreased,
which indicated that the mitochondrial pathway was involved in the
apoptosis of HOS cells induced by celastrol.
Moderate ERS facilitated intracellular environmental
homeostasis and maintain cell survival, while long-lasting ERS
injured cells. We clarified the specific role of ERS in HOS cells,
and following pretreatment with TUDCA, FCM and western blotting
revealed that apoptosis and autophagy induced by celastrol
increased, while ERS was attenuated. In our previous research, we
have found that external factors could induce cytoprotective
autophagy against apoptosis or enhance pro-apoptotic and
pro-autophagic effects on human osteosarcoma cells (11,32).
In the present study, the pro-apoptotic and pro-autophagic effects
of celastrol on human osteosarcoma cells have already been
confirmed by Li et al (10).
Therefore, the results demonstrated that ERS inhibited autophagy
and apoptosis and played a protective role in HOS cells. To
suppress the protective function of ERS, further promote apoptosis
and enhance the chemotherapeutic effect of celastrol, we sought to
reverse the target of HOS-cell resistance to celastrol. Studies
have found that PERK is the key factor of the PERK/EIF2α signaling
pathway (33,34). Salaroglio et al (13) reported that PERK molecules mediated
colon cancer cell resistance to ERS and chemotherapeutic agents.
Feng et al (35) found that
the activation of PERK signaling molecules attenuated the ischemic
injury of neuronal cells after melatonin preconditioning (35). Following pretreatment with
GSK2656157, the results of CCK-8, fluorescence staining with
Hoechst 33258, FCM and western blotting demonstrated that following
inhibition of p-PERK expression, apoptosis induced by celastrol was
further increased, which reversed the tolerance of HOS cells to
celastrol. All the results indicated that the PERK/EIF2α signaling
pathway of ERS played a protective role in the apoptosis of HOS
cells induced by celastrol. Inhibition of the expression of p-PERK
attenuated the effect of ERS and further promoted apoptosis of HOS
cells. Pretreatment with TUDCA inhibited the occurrence of ERS and
the phosphorylation of PERK and promoted autophagy, while
pretreatment with GSK2656157 inhibited the phosphorylation of PERK
but attenuated autophagy. ERS, the PERK/EIF2α signaling pathway and
autophagy are usually triggered simultaneously due to the same
stimulus, but their complicated relationships are controversial.
Thus, the details and relationships among ERS, the PERK/EIF2α
signaling pathway and autophagy are still unclear and warrant
further studies in more than one cell line and in vivo
experiments.
In conclusion, the ERS pathway, the mitochondrial
pathway and autophagy were involved in the apoptosis of HOS cells
induced by celastrol. The ERS pathway not only protected HOS cells
from apoptosis but also mediated apoptosis. Further research found
that the expression of p-PERK increased, while ERS was induced,
which contributed to the resistance to celastrol. These results
will enrich our understanding of the mechanisms mediating
celastrol-induced tumor cell death and the relationship among ERS,
autophagy and apoptosis. Further studies are required to determine
how to regulate the relationship of ERS, apoptosis and autophagy to
enhance the anti-osteosarcoma effect of celastrol.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (81572634).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
YC carried out the experimental work and the data
collection and interpretation. YO, YT and HL participated in the
design and coordination of experimental work and acquisition of
data. HYi, SZ and HYu participated in the study design, data
collection, analysis of data and preparation of the manuscript. ZZ
and BH carried out the study design, the analysis and
interpretation of data and drafted the manuscript. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ando K, Heymann MF, Stresing V, Mori K,
Rédini F and Heymann D: Current therapeutic strategies and novel
approaches in osteosarcoma. Cancers (Basel). 5:591–616. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arai K, Sakamoto R, Kubota D and Kondo T:
Proteomic approach toward molecular backgrounds of drug resistance
of osteosarcoma cells in spheroid culture system. Proteomics.
13:2351–2360. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hu M, Luo Q, Alitongbieke G, Chong S, Xu
C, Xie L, Chen X, Zhang D, Zhou Y, Wang Z, et al: Celastrol-induced
Nur 77 interaction with TRAF2 alleviates inflammation by promoting
mitochondrial ubiquitination and autophagy. Mol Cell. 66(141–153):
e62017.
|
|
4
|
Zhang R, Zhang N, Zhang H, Liu C, Dong X,
Wang X, Zhu Y, Xu C, Liu L, Yang S, et al: Celastrol prevents
cadmium-induced neuronal cell death by blocking reactive oxygen
species-mediated mammalian target of rapamycin pathway. Br J
Pharmacol. 174:82–100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li PP, He W, Yuan PF, Song SS, Lu JT and
Wei W: Celastrol induces mitochondria-mediated apoptosis in
hepatocellular carcinoma Bel-7402 cells. Am J Chin Med. 43:137–148.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ni H, Zhao W, Kong X, Li H and Ouyang J:
NF-kappa B modulation is involved in celastrol induced human
multiple myeloma cell apoptosis. PLoS One. 9:e958462014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sethi G, Ahn KS, Pandey MK and Aggarwal
BB: Celastrol, a novel triterpene, potentiates TNF-induced
apoptosis and suppresses invasion of tumor cells by inhibiting
NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB
activation. Blood. 109:2727–2735. 2007.PubMed/NCBI
|
|
8
|
Li Z, Zhang J, Tang J and Wang R:
Celastrol increases osteosarcoma cell lysis by γδ T cells through
up-regulation of death receptors. Oncotarget. 7:84388–84397.
2016.PubMed/NCBI
|
|
9
|
Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY,
Kwon YJ and Choi KS: Release of Ca2+ from the
endoplasmic reticulum and its subsequent influx into mitochondria
trigger celastrol-induced paraptosis in cancer cells. Oncotarget.
5:6816–6831. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li HY, Zhang J, Sun LL, Li BH, Gao HL, Xie
T, Zhang N and Ye ZM: Celastrol induces apoptosis and autophagy via
the ROS/JNK signaling pathway in human osteosarcoma cells: An in
vitro and in vivo study. Cell Death Dis. 6:e16042015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang Q, Ou YS, Tao Y, Yin H and Tu PH:
Apoptosis and autophagy induced by pyropheophorbide-α methyl
ester-mediated photodynamic therapy in human osteosarcoma MG-63
cells. Apoptosis. 21:749–760. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chiang CK, Wang CC, Lu TF, Huang KH, Sheu
ML, Liu SH and Hung KY: Involvement of Endoplasmic Reticulum
Stress, Autophagy, and Apoptosis in Advanced Glycation End
Products-Induced Glomerular Mesangial Cell Injury. Sci Rep.
6:341672016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Salaroglio IC, Panada E, Moiso E,
Buondonno I, Provero P, Rubinstein M, Kopecka J and Riganti C: PERK
induces resistance to cell death elicited by endoplasmic reticulum
stress and chemotherapy. Mol Cancer. 16:912017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng X, Xu F, Liang H, Cao H, Cai M, Xu W
and Weng L: SIRT1/HSF1/HSP pathway is essential for
exenatide-alleviated, lipid-induced hepatic endoplasmic reticulum
stress. Hepatology. 66:809–824. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kimura K, Mamane A, Sasaki T, Sato K,
Takagi J, Niwayama R, Hufnagel L, Shimamoto Y, Joanny JF, Uchida S,
et al: Endoplasmic-reticulum-mediated microtubule alignment governs
cytoplasmic streaming. Nat Cell Biol. 19:399–406. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qi L, Tsai B and Arvan P: New insights
into the physiological role of endoplasmic reticulum-associated
degradation. Trends Cell Biol. 27:430–440. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rani S, Sreenivasaiah PK, Kim JO, Lee MY,
Kang WS, Kim YS, Ahn Y, Park WJ, Cho C and Kim DH:
Tauroursodeoxycholic acid (TUDCA) attenuates pressure
overload-induced cardiac remodeling by reducing endoplasmic
reticulum stress. PLoS One. 12:e01760712017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Uppala JK, Gani AR and Ramaiah KVA:
Chemical chaperone, TUDCA unlike PBA, mitigates protein aggregation
efficiently and resists ER and non-ER stress induced HepG2 cell
death. Sci Rep. 7:38312017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang X, Yang J, Chen M, Li L, Huan F, Li
A, Liu Y, Xia Y, Duan JA and Ma S: Metabolomics profiles delineate
uridine deficiency contributes to mitochondria-mediated apoptosis
induced by celastrol in human acute promyelocytic leukemia cells.
Oncotarget. 7:46557–46572. 2016.PubMed/NCBI
|
|
20
|
Kapoor S: Tumor growth attenuating effect
of celastrol in systemic malignancies. Int J Cancer. 139:14312016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu X, Wang Q, Zhou X, Fu C, Cheng M, Guo
R, Liu H, Zhang B and Dai M: Celastrol negatively regulates cell
invasion and migration ability of human osteosarcoma via
downregulation of the PI3K/Akt/NF-κB signaling pathway in vitro.
Oncol Lett. 12:3423–3428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Santofimia-Castano P, Izquierdo-Alvarez A,
Plaza-Davila M, Martinez-Ruiz A, Fernandez-Bermejo M,
Mateos-Rodriguez JM, Salido GM and Gonzalez A: Ebselen impairs
cellular oxidative state and induces endoplasmic reticulum stress
and activation of crucial mitogen-activated protein kinases in
pancreatic tumour AR42J cells. J Cell Biochem. 119:1122–1133. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takata T, Ihara H, Hatano N, Tsuchiya Y,
Akaike T and Watanabe Y: Reactive sulfur species inactivate
Ca2+/calmodulin-dependent protein kinase IV via
S-polysulfidation of its active-site cysteine residue. Biochem J.
474:2547–2562. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bononi A, Giorgi C, Patergnani S, Larson
D, Verbruggen K, Tanji M, Pellegrini L, Signorato V, Olivetto F,
Pastorino S, et al: BAP1 regulates IP3R3-mediated Ca2+
flux to mitochondria suppressing cell transformation. Nature.
546:549–553. 2017.PubMed/NCBI
|
|
25
|
Scorrano L, Oakes SA, Opferman JT, Cheng
EH, Sorcinelli MD, Pozzan T and Korsmeyer SJ: BAX and BAK
regulation of endoplasmic reticulum Ca2+: A control
point for apoptosis. Science. 300:135–139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hetz C and Saxena S: ER stress and the
unfolded protein response in neurodegeneration. Nat Rev Neurol.
13:477–491. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Badiola N, Penas C, Miñano-Molina A,
Barneda-Zahonero B, Fadó R, Sánchez-Opazo G, Comella JX, Sabriá J,
Zhu C, Blomgren K, et al: Induction of ER stress in response to
oxygen-glucose deprivation of cortical cultures involves the
activation of the PERK and IRE-1 pathways and of caspase-12. Cell
Death Dis. 2:e1492011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cerezo M, Lehraiki A, Millet A, Rouaud F,
Plaisant M, Jaune E, Botton T, Ronco C, Abbe P, Amdouni H, et al:
Compounds triggering ER stress exert anti-melanoma effects and
overcome BRAF inhibitor resistance. Cancer Cell. 29:805–819. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
López I, Tournillon AS, Martins Prado R,
Karakostis K, Malbert-Colas L, Nylander K and Fåhraeus R:
p53-mediated suppression of BiP triggers BIK-induced apoptosis
during prolonged endoplasmic reticulum stress. Cell Death Differ.
24:1717–1729. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guha P, Kaptan E, Gade P, Kalvakolanu DV
and Ahmed H: Tunicamycin induced endoplasmic reticulum stress
promotes apoptosis of prostate cancer cells by activating mTORC1.
Oncotarget. 8:68191–68207. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Arozena Acevedo A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tu P, Huang Q, Ou Y, Du X, Li K, Tao Y and
Yin H: Aloe-emodin-mediated photodynamic therapy induces autophagy
and apoptosis in human osteosarcoma cell line MG 63 through the
ROS/JNK signaling pathway. Oncol Rep. 35:3209–3215. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gupta A, Hossain MM, Miller N, Kerin M,
Callagy G and Gupta S: NCOA3 coactivator is a transcriptional
target of XBP1 and regulates PERK-eIF2α-ATF4 signalling in breast
cancer. Oncogene. 35:5860–5871. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chiu TL and Su CC: Tanshinone IIA
increases protein expression levels of PERK, ATF6, IRE1α, CHOP,
caspase 3 and caspase 12 in pancreatic cancer BxPC 3 cell derived
xenograft tumors. Mol Med Rep. 15:3259–3263. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng D, Wang B, Wang L, Abraham N, Tao K,
Huang L, Shi W, Dong Y and Qu Y: Pre-ischemia melatonin treatment
alleviated acute neuronal injury after ischemic stroke by
inhibiting endoplasmic reticulum stress-dependent autophagy via
PERK and IRE1 signalings. J Pineal Res. 62:e123952017. View Article : Google Scholar
|