Introduction
Estrogen regulates the function of many cell types,
mostly by binding its receptors (1). Estrogen receptors (ERs) influence many
physiological processes in mammals (2–5). Two
classes of ERs exist: I) Nuclear ERs, which are ligand-regulated
transcription factors that activate the expression of target genes
in a large variety of responses (6,7), and
II) membrane ERs, which are mostly G-protein-coupled receptors that
mediate nongenomic actions (8,9).
Despite the beneficial actions of endogenous estrogen under
physiological conditions and its preventative effects in certain
cancers, estrogen also exerts deleterious exacerbating effects in
other disease states (10). There
is considerable evidence that circulating estrogens are positively
associated with the risk of breast cancer, accounting for almost
25% of cancer cases in females (11,12).
It is therefore not surprising that there is a wealth of data
demonstrating estrogen to be a key carcinogen for these
malignancies (13).
The direct effect of estrogen on tumor cells and
endothelial cells (ECs) has been proposed as a major factor in
stimulating tumor growth. On the one hand, estrogen regulates the
proliferation, migration and adhesion of tumor cells, while on the
other hand, blood vessel formation is also closely related to the
estrogen level. The vascular growth in the uterus is routinely
observed to be associated with fluctuating estrogen levels under
physiological conditions (14).
Under pathological conditions, estrogen is the key regulatory
factor in tumor angiogenesis (15).
As a result, activation of the ERs on tumor cells and ECs is widely
considered to be the main mechanism underlying how estrogen
enhances tumor growth.
There are clear signs that most immune cells express
ERs, suggesting that these cells, which are mostly derived from
bone marrow, are sensitive to estrogen (16). There is evidence that bone
marrow-derived cells (BMDCs) recruited from the circulating blood
in local angiogenic tissues play a more important role than local
ECs in tumor angiogenesis (17).
Although constituting a minority of the total stromal cell
population in a tumor, BMDCs play a key role in tumor progression
(18). Depletion of BMDCs in tumor
tissues was found to result in delayed tumorigenesis (18). BMDCs are heterogeneous cells derived
from the bone marrow (19), and
certain subtypes of BMDCs [e.g., endothelial progenitor cells
(EPCs)] have been found necessary to mediate estrogen-induced
proangiogenesis in animal models (20,21).
In addition, BMDC-associated ERs were found to be the main target
for estrogen to activate EPCs that were subsequently involved in
tubulogenesis directly or enhance neovascularization by secreting
angiogenic cytokines indirectly (20,21).
In addition, estrogen markedly enhances the genesis of breast
cancers that lack ER expression or express ERs in very few cells
(16,22). This phenomenon implies that the
effect of E2 on the growth of breast cancer is dependent not only
on the local tumor cells but also on the systematic environment of
the tumor cells. However, the exact mechanisms underlying how
estrogen enhances tumor progression are still unclear.
Estradiol (E2) is the predominant estrogen. This
study used E2 and ER-negative breast cancer 4T1 cells to
investigate how estrogen regulates tumor growth. Roles of BMDCs in
estrogen-induced tumor growth were defined. Understanding these
mechanisms is vital for providing a theoretical basis for clinical
E2-induced cancer therapies.
Materials and methods
Cell culture
Mouse mammary tumor cell line 4T1 and human
umbilical vein endothelial cells (HUVECs) were kind gifts from the
Cancer Research Center of Xi'an Jiao Tong University. Cells were
cultured in RPMI-1640 medium supplemented with 10% FBS, penicillin
10,000 IU/ml and streptomycin 10,000 µg/ml. All cell lines were
grown at 37°C and 5% CO2.
Animal model and in vivo tumorigenesis
experiments (23)
All animal procedures were conducted in accordance
with the protocol approved by the Xi'an Jiaotong University Animal
Care and Use Committee and housed under constant temperature,
humidity, and lighting (12-h light per day) and allowed free access
to food and water. Female BALB/c mice (aged, 6–8 weeks) were
subjected to ovariectomy (OVX) or a sham operation (SO). The OVX
mice were randomized into four groups (n=7 in each group): One of
the OVX groups received normal saline as a control (referred to as
‘control’ below), while the remaining three groups were gavaged
with estradiol valerate (E2V, Bayer, Germany) at a doses of 0.27,
0.8, and 2.4 mg/kg/day.
For tumorigenesis experiments, tumors were induced
by implanting 6×105 4T1 cells subcutaneously (16). 4T1-tumor-bearing mice were treated
intragastrically with normal saline or different doses of E2V
continuously. Tumor diameters were measured on days 6, 9 and 12,
and the tumor growth curve in each group was recorded. The maximum
(L) and minimum (W) axes of the tumor were measured,
with the tumor volume calculated as 0.5LW2. After
16 days, mice were euthanized by cervical dislocation after
anesthetization and the tumor tissues were isolated.
Histology and
immunohistochemistry
The tissue sections were stained
immunohistochemically or with hematoxylin and eosin (H&E). The
immunohistochemistry process was performed with CD31 (1:50
dilution; cat. no. ab28364; Abcam, Cambridge, UK) as the primary
antibody in accordance with the instructions provided with the
Histostain-Plus kit (4Abio, Beijing, China).
Cell proliferation assay
17-β E2 (Sigma; Merck KGaA, Darmstadt, Germany) was
solubilized in absolute ethanol. Since BMDCs are mobilized into the
blood circulation and recruited to tumor tissue (24), circulating white blood cells (WBCs)
isolated using red blood cell (RBC) lysis buffer were used as a
substitute for BMDCs in the in vitro experiments. WBCs
(8×105 each group) were treated with RPMI-1640 medium
containing 17-β E2 at several different doses for 72 h at 37°C.
Thereafter, the supernatants were isolated by centrifugation and
used as conditioned media (CM).
ECs or 4T1 cells (3×103 cells/well) were
plated onto a 96-well plate and cultured with normal medium for 24
h, and then replaced with E2-containing medium or CM (200 µl) for
48 h. E2-containing medium or CM was replaced with MTT (5 µg/ml,
Beyotime Institute of Biotechnology, Shanghai, China), and cells
were incubated for another 4 h. The medium was then discarded and
DMSO was added. The 96-well plate was shaken for 10 min to dissolve
the formazan crystals. The absorbance (OD value) was measured at
490 nm using a microplate reader (Bio-Rad 550; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Cultures in normal medium
alone were used as negative controls. The effects of E2 and/or
BMDCs on ECs or 4T1 cell proliferation were expressed as the ‘added
value’ of OD: Added value=(OD value in the experimental
group)-(average OD value in the control group).
The cell proliferation and migration was compared
between E2-free and E2-treated WBC groups using the following four
experimental groups: WBC/0+0 (E2-free WBC CM was mixed with the
same volume of E2-free medium), WBC/1+1 (1-nM E2-treated WBC CM was
mixed with the same volume of 1-nM E2-containing medium), WBC/0+1
(E2-free WBC CM was mixed with the same volume of 1-nM
E2-containing medium), and WBC/1+0 (1-nM E2-treated WBC CM was
mixed with the same volume of E2-free medium). Similarly, the cell
proliferation was compared between E2-free and E2-treated 4T1 cell
groups using another four experimental groups: 4T1/0+0, 4T1/1+1,
4T1/0+1, and 4T1/1+0.
In vitro scratch assay
HUVECs (8×105) were cultured to
confluence on 24-well plates (Corning Costar, Corning, NY, USA).
Confluent cells were scraped with a 200 µl pipette tip and then
incubated with WBC CM (as is shown in Cell proliferation
assay). The scratch was photographed at 0, 6 and 12 h and data
was analysed using Image-Pro Plus (version 6.0).
Sex-mismatched bone-marrow
transplantation (25)
Recipient female mice were lethally irradiated at 8
Gy. Bone marrow was harvested from the femurs and tibias of male
donor mice. Total bone marrow was liberated using 20-gauge needles
to produce a single-cell suspension in RPMI-1640 medium with 10%
FBS. The bone-marrow cell suspensions were centrifuged at 1,000 rpm
for 5 min, and the obtained pellets were resuspended in sterile
RPMI-1640 medium at 3×107 cells/ml. The bone marrow
cells (1×107) were then injected into the lateral tail
vein of a recipient female mouse, after which sulfamethoxazole
solution was administered for 4 weeks. Tumor cell implantation was
performed 8 weeks after bone marrow transplantation (BMT).
Fluorescence in situ
hybridization
Tumor samples obtained from sex-mismatched BMT mice
were embedded in optimal cutting temperature compound and stored at
−80°C. Frozen tissues were sectioned and fixed in paraformaldehyde
for 10 min, and the frozen sections were rinsed with 0.01 M PBS for
10 min. The Y chromosome was detected by Mouse SRY DNA FISH kit
(TBD Science, Tianjin, China) following the manufacturer's
instructions. The sections were finally counterstained with blue
fluorescent 4′-6-diamidino-2-phenylindole (DAPI, Sigma; Merck
KGaA). Images were captured using fluorescence microscopy (Olympus
BX51; Olympus Corp., Tokyo, Japan).
Flow cytometry
Peripheral blood was collected from the mouse
abdominal aorta and then treated with RBC lysis buffer. Leukocytes
(1×106) were incubated with APC-anti-CXCR4 (1:100
dilution; cat. no. 558644; BD Pharmingen; BD Biosciences, Franklin
Lakes, NJ, USA), PE-anti-CD61 (1:80 dilution; cat. no. 104307;
Biolegend, Inc., San Diego, CA, USA), FITC-anti-Sca-1 (1:100
dilution; cat. no. 108115; Biolegend, Inc.), or AF488-anti-CD11b
(1:200 dilution, cat. no. 101205; BioLegend, Inc.) antibodies
following the appropriate protocol. Double staining was also
performed with anti-CXCR4 and anti-CD61 antibodies. Labeled cells
were fixed with paraformaldehyde. Flow cytometry on a FACScan (BD
Biosciences) was used to analyze 10,000 events per sample.
RT-PCR
Total RNA was extracted using RNA fast 2000 kit
(Fastagen, Shanghai, China) following the manufacturer's
instructions. The cDNA was synthesized via Prime Script RT Master
Mix Perfect Real-Time kit (DRR036A; Takara, Tokyo, Japan). The
primer sequences used in this study along with the expected product
sizes are listed in Table I. The
cycling protocol for PCR involved incubating the samples at 94°C
for 2 min followed by 35 cycles of denaturation at 94°C for 30 sec,
annealing at 55°C for 30 sec, and extension at 72°C for 30 sec,
with a final cycle of incubation at 72°C for 2 min. The
amplification products were analyzed by electrophoresis (Beijing
Junyi, Beijing, China) in agarose gels and detected under UV
illumination (Bio-Rad Laboratories) after staining with nucleic
acid dye (DuRed; FanBo Biochemicals, Beijing, China). Images were
analyzed using a quantitative analysis system (Quantity One
Analysis Software, version 4.6.2; Bio-Rad Laboratories).
 | Table I.Primer sequences for RT-PCR. |
Table I.
Primer sequences for RT-PCR.
| Primers | Sequences | Product size
(bp) |
|---|
| Mouse SDF-1
(F) |
5′-AGCCAACGTCAAGCATCTG-3′ | 263 |
| Mouse SDF-1
(R) |
5′-TAATTTCGGGTCAATGCACA-3′ |
|
| Mouse β-actin
(F) |
5′-GAGACCTTCAACACCCCAGC-3′ | 106 |
| Mouse β-actin
(R) | 5′-
ATGTCACGCACGATTTCCC-3′ |
|
| Human β3 (F) |
5′-GCCAGCACCATCTCTTTACC-3′ | 205 |
| Human β3 (R) |
5′-GCACTCTCTCCCTTTGAGGA-3′ |
|
| Human β-actin
(F) |
5′-TGACGTGGACATCCGCAAAG-3′ | 112 |
| Human β-actin
(R) |
5′-CTGGAAGGTGGACAGCGAGG-3′ |
|
Adhesion assay
HUVECs were cultured to confluence on 12-well plates
precoated with gelatin, and then treated with 17-β E2. After 24 h,
the HUVEC monolayers were washed three times with PBS.
Calcein-AM-labeled human WBC (1×105) suspensions were
added to each well and incubated for 60 min at 37°C. The
nonadherent calcein-AM-labeled WBCs were then removed. Adherent
human WBCs were observed using fluorescence microscopy (Leica
Microsystems, Wetzlar, Germany), with the fluorescence intensity
quantified using a fluorescence analyzer (excitation filter 495 nm,
emission filter 515 nm) (PerkinElmer, Inc., Waltham, MA, USA).
Statistical analysis
Data are represented as the mean ± SEM and were
analyzed by a two-tailed Student t-test or one-way analysis of
variance (ANOVA) using GraphPad Prism version 4.03 software
(GraphPad Software, Inc., La Jolla, CA, USA). P-values of 0.05 or
less were regarded as statistically significant.
Results
E2 enhances murine 4T1 breast cancer
growth and angiogenesis in vivo
4T1 cells were implanted subcutaneously into OVX
female mice, and tumor volume were measured every 3 days (Fig. 1A). At 16 days after implantation,
4T1 tumor growth in the OVX mice did not differ significantly from
that in SO mice. As shown in Fig. 1B
and C, treatment with 0.8 and 2.4 mg/kg E2V led to significant
increases in tumor weights, compared with the placebo-treated
control group (P<0.01).
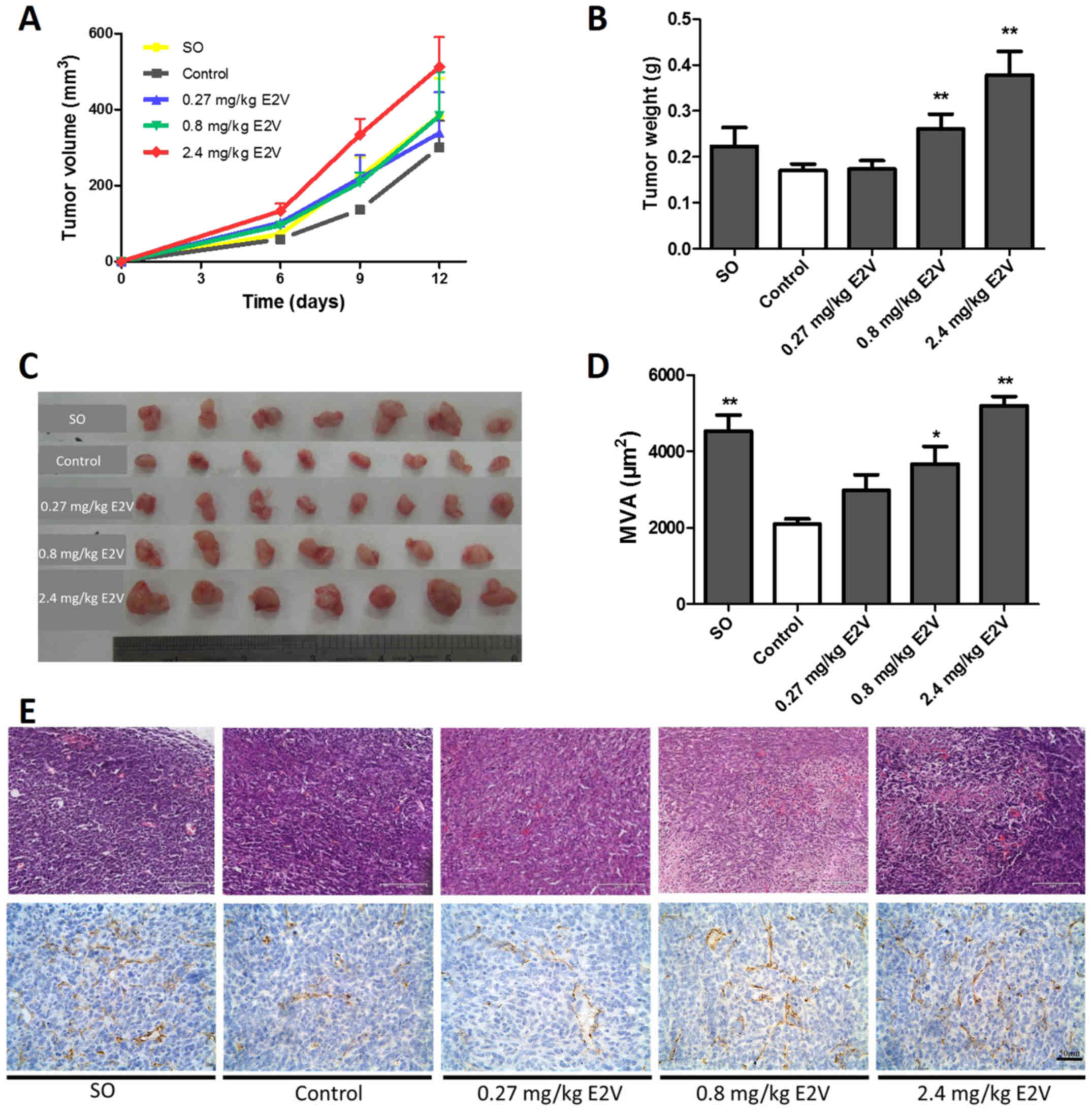 | Figure 1.Increased ER-negative tumor growth
in vivo and vascularization following treatment with E2.
In vivo growth curves (A) and weights (B and C) of 4T1
tumors from SO mice, OVX mice (control), and OVX mice with 0.27,
0.8, and 2.4 mg/kg E2V replacement, n=7 in each group. (D and E)
H&E (scale bars, 145 µm) or anti-CD31-stained tumor tissue
sections (scale bars, 50 µm) and quantification of the
CD31+ MVA. Tumor weights and MVA are indicated as mean
and SEM values. *P<0.05, **P<0.01 vs. control. ER, estrogen
receptor; SO, sham operation; OVX, ovariectomy; H&E,
hematoxylin and eosin; MVA, microvascular area; E2V, estradiol
valerate. |
To observe the effect of E2 on tumor angiogenesis in
ER-negative 4T1 cell-derived tumors, tumor tissues were stained
with H&E staining and immunohistochemistry was conducted
(Fig. 1E). Quantification of tumor
CD31+ vessels revealed that the microvascular area (MVA)
field of view was notably larger in the 0.8 and 2.4 mg/kg E2V
groups than in the OVX group (P<0.05 and P<0.01,
respectively) (Fig. 1D).
E2 and the CM of E2-treated BMDCs
enhances the proliferation of 4T1 cells
E2 affected the proliferation of 4T1 cells in the
concentration range from 0.1 to 100 nM in vitro. The results
showed that 1 or 10 nM E2 significantly increased the proliferation
of 4T1 cells compared with the control (Fig. 2A, P<0.01 or P<0.05,
respectively).
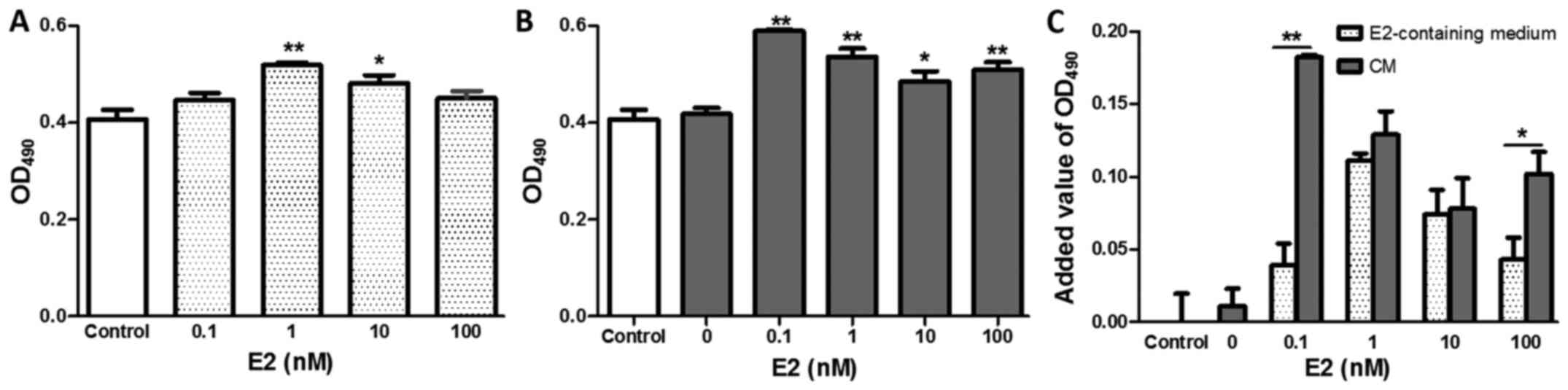 | Figure 2.Effects of E2 and E2-treated BMDCs on
the proliferation of 4T1 cells with E2 at concentrations of 0.1, 1,
10, and 100 nM, respectively. (A) Proliferation of E2-treated 4T1
cells after 48 h of culture measured with the MTT assay. (B)
Proliferation of 4T1 cells after treatment with E2-stimulated BMDC
CM. (C) Comparison of 4T1 cell proliferation between groups treated
with E2 and with CM from BMDCs stimulated with E2. Data are mean
and SEM values. *P<0.05, **P<0.01 vs. the control or E2-free
BMDC CM group. BMDCs, bone marrow-derived cells; E2, 17β-estradiol;
MTT, 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide;
CM, conditioned media. |
BMDCs are mobilized into the blood circulation and
recruited to tumor tissue to enhance tumor angiogenesis and growth.
To investigate the role of BMDCs in the E2 enhancement of tumor
growth in vitro, the effects of E2-treated WBC CM on 4T1
cell proliferation were observed. As shown in Fig. 2B, the 4T1 cell proliferation in
E2-stimulated WBC CM groups was significantly increased by 40.9,
28.2, 16.0 and 21.8% for E2 at concentrations of 0.1, 1, 10 and 100
nM, respectively, as compared with the E2-free WBC CM (P<0.01,
P<0.01, P<0.05, P<0.01, respectively). The added value for
OD in the E2-stimulated WBC CM groups for 0.1 or 100 nM E2 was
significantly higher than that at the same concentration of
E2-containing medium groups, as well as that in the E2-free WBC CM
group (Fig. 2C; P<0.01,
P<0.05, respectively). These results suggest that E2 and BMDCs
exert a synergistic effect on enhancing the proliferation of 4T1
cells at a certain concentration.
E2 and the CM of E2-treated BMDCs
facilitate the proliferation and migration of ECs
Stimulation with E2 led to small increases in EC
proliferation in vitro (Fig.
3A). To investigate the role of BMDCs in E2-induced
angiogenesis, the effects of E2-treated WBC CM on EC proliferation
were observed. As shown in Fig. 3B,
although E2-free WBCs CM enhanced EC proliferation by 34.2%
relative to normal medium, E2-treated WBC CM with E2 at
concentrations of 0.1, 1, 10, and 100 nM significantly enhanced EC
proliferation (all P<0.05). Meanwhile, 0.1 to 100 nM E2-treated
WBC CM increased EC proliferation compared with E2-free BMDC CM,
with the enhancement reaching statistical significance in the 10 nM
group (P<0.05). As shown in Fig.
3C, the added values for WBC-mediated E2-induced EC
proliferation were higher than those for the E2-free WBC-mediated
EC group and the E2-stimulated EC group at E2 concentrations
ranging from 0.1 to 10 nM. The differences in EC proliferation
between the E2-stimulated EC groups and E2-treated WBC groups, as
well as the differences between the E2-free WBC group and the
E2-treated WBC groups, suggested a synergistic effect of E2 and
BMDCs on EC proliferation, implying that the E2-induced EC
proliferation was partly mediated by BMDCs at a certain
concentration.
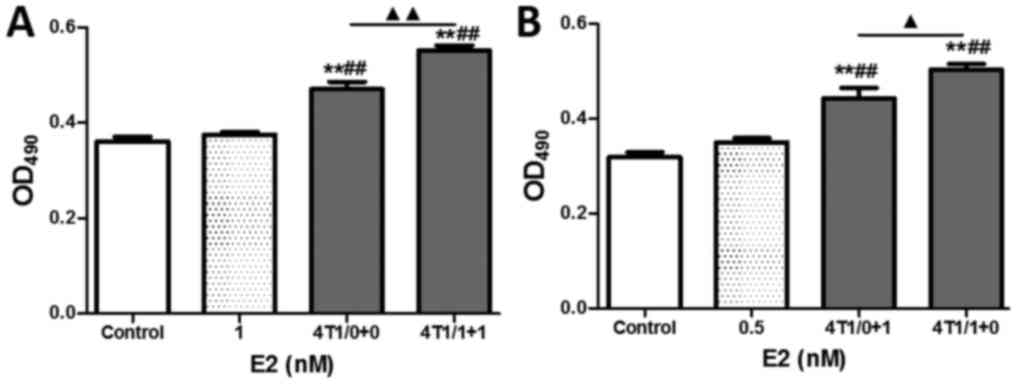
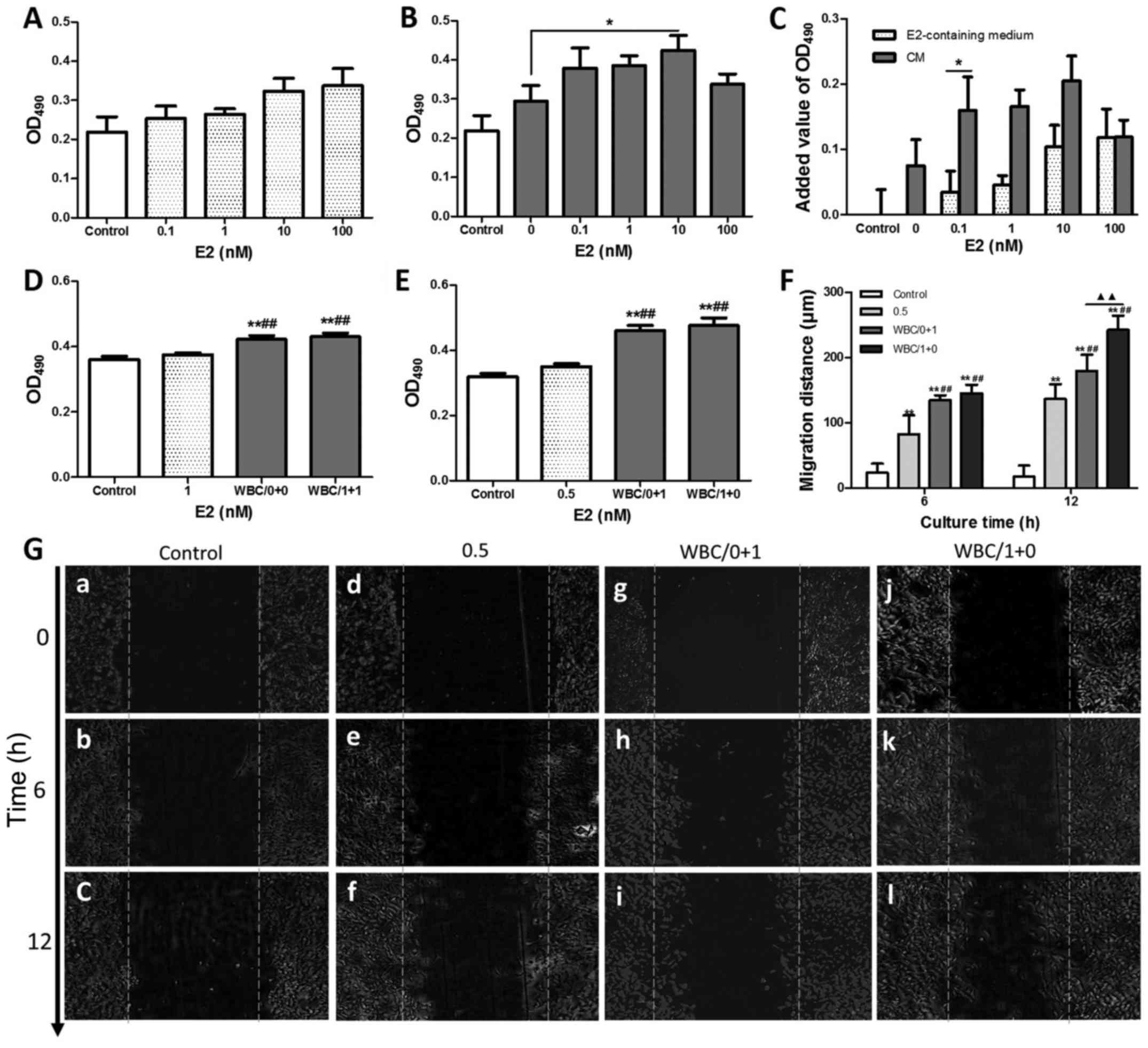 | Figure 3.Effects of E2 and E2-treated BMDCs on
the proliferation and migration of ECs. (A and B) MTT assay of EC
proliferation after treatment with E2 or E2-stimulated BMDC CM for
E2 at concentrations of 0.1, 1, 10 and 100 nM, respectively. (C)
Comparison of EC proliferation between groups treated with E2, and
with CM from BMDCs stimulated with E2. (D) MTT assay of HUVEC
proliferation after 48 h of treatment with E2-containing medium,
E2-free WBC CM or E2-stimulated WBC CM. (E) Comparison of HUVEC
proliferation between groups treated with CM from WBCs stimulated
with or without E2. (F and G) In vitro scratch assay of
HUVEC migration. Data are the mean and SEM values. **P<0.01 vs.
the control; ##P<0.01 vs. the E2-containing medium
groups; ▲▲P<0.01, WBC/0+1 group vs. WBC/1+0 group.
E2, 17β-estradiol; BMDCs, bone marrow-derived cells; ECs,
endothelial cells; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide; CM, conditioned media; HUVECs, human
umbilical vein endothelial cells; WBC, white blood cell; OD,
optical density; WBC/0+0 (E2-free WBC CM was mixed with the same
volume of E2-free medium); WBC/1+1 (1-nM E2-treated WBC CM was
mixed with the same volume of 1-nM E2-containing medium); WBC/0+1
(E2-free WBC CM was mixed with the same volume of 1-nM
E2-containing medium) and WBC/1+0 (1-nM E2-treated WBC CM was mixed
with the same volume of E2-free medium). |
To investigate the synergistic action between E2 and
BMDCs in enhancing HUVEC functions further, a more refined analysis
was designed to observe HUVEC proliferation and migration with or
without 1 nM E2-treated WBC CM. As shown in Fig. 3D, although both E2-free WBC CM and
E2-treated WBC CM significantly enhanced the proliferation of
HUVECs in comparison to control or E2-treated ECs (all P<0.01),
no distinct increases were observed between the E2-free and
E2-treated groups regardless of whether culturing was performed
with either normal medium or CM for 48 h.
Moreover, in order to eliminate the disturbance of
HUVEC proliferation caused by the E2 concentrations in the medium,
the final E2 concentration was adjusted to 0.5 nM with CM and/or
normal medium. A small difference was found between the 0.5 nM E2
normal-media group and the control, indicating that E2 induced a
slight increase in HUVEC proliferation. The marked enhancement of
proliferation of HUVECs in groups of WBC/0+1 and WBC/1+0 compared
with the E2 group indicated that BMDC CM significantly enhanced the
effect of HUVECs on proliferation (all P<0.01). However, no
significant difference in HUVEC proliferation was found between the
WBC/0+1 and WBC/1+0 groups, implying that the enhancement of HUVEC
proliferation was associated with BMDC CM rather than with E2
(Fig. 3E).
In contrast, the migration of HUVECs increased
markedly after 6 and 12 h of culture in the 0.5 nM E2 group,
WBC/0+1 group, and WBC/1+0 group, compared with the controls
(Fig. 3F and G, all P<0.01).
Moreover, the medium containing WBC CM significantly enhanced HUVEC
migration by 63.4 and 31.6% compared with 0.5 nM E2 after 6 and 12
h of culture, respectively (all P<0.01), while the medium
containing E2-treated WBC CM (0.5 nM E2 in medium) markedly
enhanced HUVEC migration compared with BMDC CM medium after 12 h of
culture (P<0.01). These results indicate that BMDCs play an
important role in mediating the enhancement of EC migration by
E2.
E2-treated 4T1 cells enhance the
proliferation of HUVECs
As shown in Fig. 4A,
although no obvious difference in HUVEC proliferation was found
between the E2-treatment and control groups, both E2-free 4T1 CM
and E2-treated 4T1 CM significantly enhanced HUVEC proliferation
compared with the control and E2-treatment groups (all P<0.01).
To eliminate disturbances from differences in the E2 concentration
in the medium, the final E2 concentration was adjusted to 0.5 nM
with 4T1 CM and/or normal medium. The results showed that a
significant increase of 13.5% was observed between the 4T1/0+1
group and the 4T1/1+0 group (Fig.
4B, P<0.05), implying that E2 treatment markedly enhanced
the HUVEC proliferation induced by 4T1 cells. These findings
suggest that the E2 enhancement of angiogenesis was partly mediated
by 4T1 tumor cells.
E2 induces the mobilization of
CXCR4+, β3+, Sca-1+ and
CXCR4+β3+ BMDCs
The numbers of proangiogenic BMDCs in circulating
blood, such as CXCR4+, β3+ (also
known as CD61), CXCR4+β3+,
CD11b+, and Sca-1+ BMDCs, were assessed with
flow cytometry. The results showed that CXCR4+ BMDCs and
β3+ BMDCs decreased sharply in mouse
circulating blood after OVX (Fig. 5A
and B). The CXCR4+ BMDC counts were significantly
rescued by 94.8 and 86.7% in mice after administering 0.27 and 0.8
mg/kg E2V, respectively, compared with control mice (P<0.01).
The number of circulating β3+ BMDCs was
markedly increased in the 2.4 mg/kg E2V group compared with the
control group (P<0.01). The numbers of Sca-1+ BMDCs
were notably increased by 35.3 and 22.2% in the 0.8 and 2.4 mg/kg
E2V groups, respectively, compared with the control group (Fig. 5C, P<0.01 and P<0.05). As shown
in Fig. 5D, a slight increase in
CD11b+ BMDC counts in the 0.27 mg/kg group and mild
decreases in CD11b+ BMDC counts in the 0.8 and 2.4 mg/kg
groups were observed in circulating blood following E2V treatment,
but none of these results reached statistical significance. The
counts of circulating CXCR4+/CD61+ BMDCs in
the 0.8 and 2.4 mg/kg E2V groups were significantly increased,
compared with the control group (Fig.
5E and F, P<0.05).
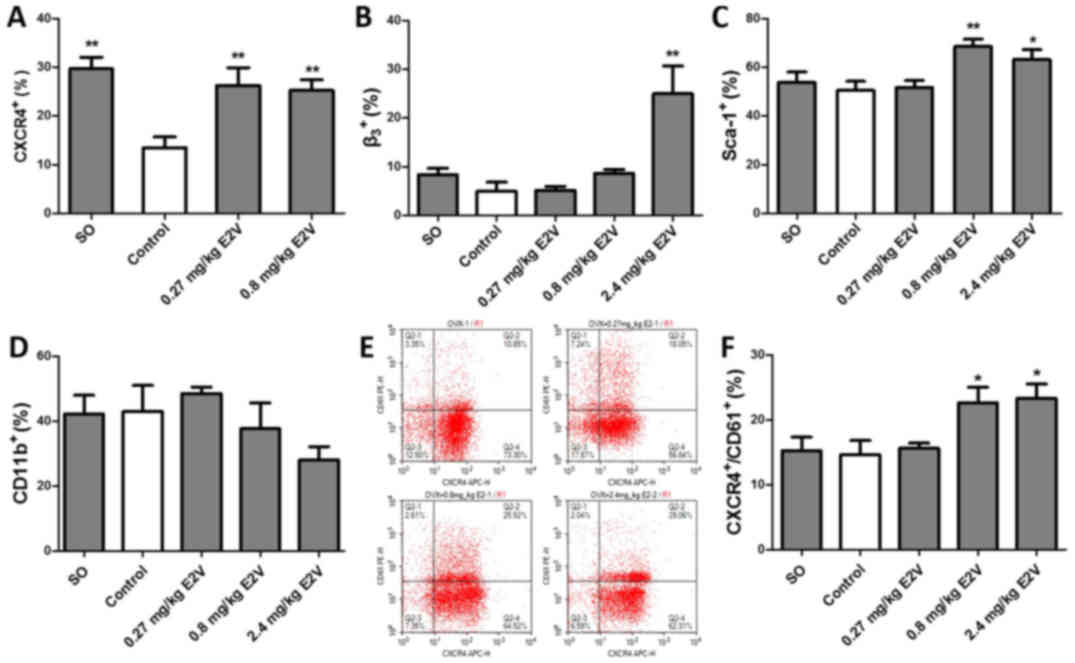 | Figure 5.Effects of E2 on mobilization of
CXCR4+, β3+, Sca-1+ and
CXCR4+CD61 (β3)+ BMDCs in circulating blood.
(A) Percentage of circulating CXCR4+ cells in 4T1
tumor-bearing SO mice, control mice, and E2V-treated OVX mice
(0.27, 0.8, and 2.4 mg/kg). (B) Percentage of circulating
β3+ cells. (C) Percentage of circulating
Sca-1+ cells. (D) Percentage of circulating
CD11b+ cells. (E and F) Percentage of circulating
CXCR4+CD61+ cells. Data are mean and SEM
values. *P<0.05, **P<0.01 vs. the control. E2, 17β-estradiol;
BMDCs, bone marrow-derived cells; SO, sham operation; OVX,
ovariectomy; E2V, estradiol valerate. |
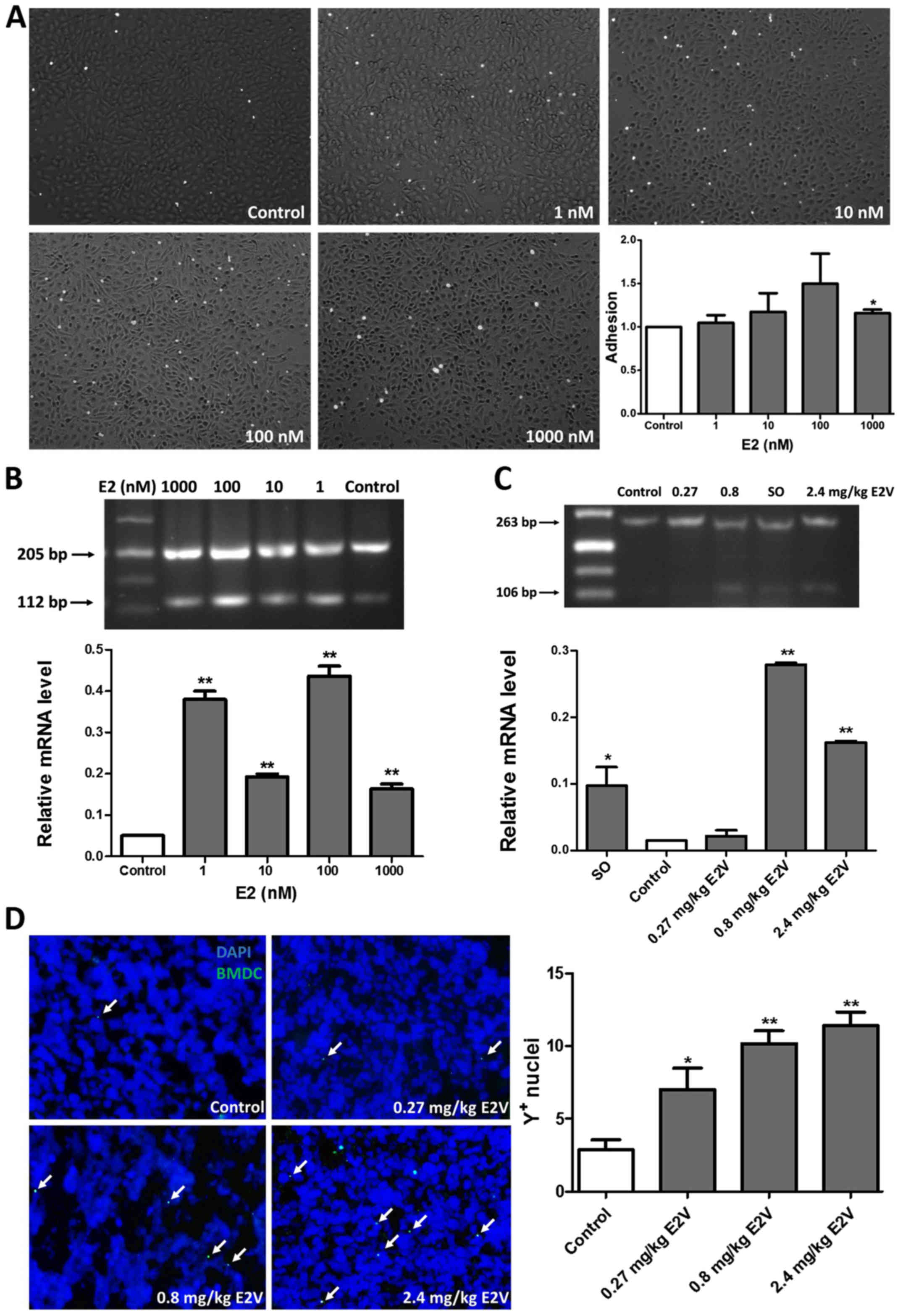
E2 enhances BMDC adhesion to ECs in
vitro
The adhesion assay results showed that more BMDCs
attached to an E2-pretreated EC monolayer than to an untreated EC
monolayer, with the enhancement reaching statistical significance
at 1,000 nM (Fig. 6A, P<0.05).
Furthermore, transcriptional expression of integrin β3
mRNA was higher than that in the control following treatment of
HUVECs with 1, 10, 100, and 1,000 nM E2, respectively, in
vitro (Fig. 6B, P<0.01),
indicating that the enhanced adhesion of BMDCs to ECs was
attributable to the stimulation of expression of integrin
β3 on ECs by E2.
E2 augments SDF-1 mRNA expression in
tumor tissues and enhances the recruitment and retention of BMDCs
in 4T1 tumor tissues
As shown in Fig. 6C,
the expression level of SDF-1 mRNA in tumor tissues was
significantly higher in the 0.8 and 2.4 mg/kg E2V groups than in
the control group (P<0.01). To confirm that the recruitment of
BMDCs in tumor tissues was related to E2, the retention of BMDCs in
tumor tissues was analyzed in sex-mismatched BMT mouse models. The
densities of Y-chromosome-positive cells in tumor tissues from the
0.27, 0.8, and 2.4 mg/kg E2V groups were increased by 141.4, 251.7
and 293.1%, respectively, compared with the control group
(P<0.05, Fig. 6D).
Discussion
Evidence has been subsequently obtained of a central
role of estrogen in the progression of breast cancers irrespective
of whether they are positive or negative for ERs (16). These results imply that estrogen
might provoke a systemic host response to induce tumor progression
not only via tumor cells themselves in vivo.
There are multiple lines of evidence that estrogen
directly modulates angiogenesis via ECs (14). The present experiments demonstrated
that E2 enhanced murine ER-negative 4T1 tumor growth and
angiogenesis in a dose-dependent manner. Nevertheless, BMDCs play a
pivotal role in stimulating progression in tumors (26). ERs were found to be expressed in
multiple BMDCs (16), and thus
BMDCs might be involved in the development of E2-induced breast
tumors.
These observations prompted the present study to
observe interactions among BMDCs, ECs, and 4T1 tumor cells, as well
as the effects of E2 on this process. Meanwhile, considering that
tissue-resident BMDCs are recruited from circulating blood, WBCs
were employed to observe the role of BMDCs in tumor progression and
angiogenesis in the experiments. We found that E2 significantly
enhanced ER-negative 4T1 tumor cell proliferation and that CM from
E2-treated BMDCs-but not BMDC CM-enhanced the E2-induced
enhancement of 4T1 tumor cell proliferation at certain
concentrations. As shown in Fig. 2A and
B, the greatest values of OD were observed at 1 nM
E2-containing medium group and at 0.1 nM E2-treated WBC CM group
respectively, which were not in sync. Meanwhile, the data in both
(E2 and CM) mediums did not show linear dose-response dependence
relationships. As a result, the increases of OD value between the
E2 and E2-containing condition medium groups showed significance at
the concentrations of 0.1 and 100 nM, but not at 1 and 10 nM.
Moreover, although E2 enhanced the EC proliferation only slightly,
BMDCs enhanced E2-induced EC proliferation significantly at certain
concentrations. And E2-treated WBC CM promoted the proliferation of
ECs in a nonlinear dose-effect relationship manner. The results
suggest that there may be other factors involved in this process,
and angiogenesis-related cytokines should be assessed using
immunoassay methods such as ELISA, western blotting, IHC and
protein array. Additionally, even though BMDC CM increased the
enhancement of HUVEC proliferation, no obvious difference was found
between the BMDC CM and E2-treated BMDC CM groups. In contrast,
both E2 and BMDC CM greatly increased HUVEC migration in comparison
with the control, and E2-treated BMDC CM markedly increased the
enhancement of HUVEC migration as compared with either the E2 or
BMDC CM group. These results reveal that, on the one hand, CM of
BMDCs markedly affected the proliferation and migration of ECs, but
not the proliferation of 4T1 cells, while on the other hand, CM of
BMDCs stimulated with E2 distinctly enhanced the proliferation of
4T1 cells and the function of ECs. Meanwhile, E2 significantly
enhanced 4T1 cell proliferation and EC migration.
Moreover, the present study also found that E2
greatly enhanced the BMDC-induced facilitation of HUVEC migration,
but not proliferation, with E2 also significantly enhancing
4T1-stimulated EC proliferation. Therefore, E2 enhances tumor
growth and angiogenesis by stimulating 4T1 cell proliferation and
HUVEC migration directly, and also by stimulating BMDCs to enhance
this process indirectly. These results imply that BMDCs may play a
key role in mediating E2-induced tumorigenesis and
angiogenesis.
The number of BMDCs in 4T1 tumor tissues was
observed using a BMT mouse model. These findings confirmed the
assumption that E2 increases the recruitment of BMDCs in tumor
tissues, which, along with the observation that BMDCs mediate the
E2-induced enhancement of tumor cells and EC function in
vitro, at least partly elucidated the mechanism via which E2
enhances ER-negative tumor progression and angiogenesis through
intensifying the recruitment of BMDCs in local tissues.
BMDCs are mobilized from bone marrow and released
into circulating blood, and then are recruited into tumor tissues
by passing through the blood vessel endothelium (26). Adhesion molecules such as integrin
β3 (CD61) on the cell surface were found to play an
important role in this process (27). Integrin β3 has the
potential to control tumor growth by regulating BMDC adhesion to
the vascular endothelium (24).
Cytokines and chemokines released by tumor tissues attract BMDCs
into tumor tissues (28), of which
SDF-1 and its receptor CXCR4 on BMDCs are key partners for BMDC
recruitment and trafficking (29).
Our previous research demonstrated that almost all of the BMDCs in
tumor tissues exhibit positivity for CXCR4. Furthermore, it was
reported that CXCR4+, β3+ and
CD11b+ myeloid cell homing into tumor tissues
accelerates tumor angiogenesis and growth (24,30,31).
Thus, β3+, CXCR4+ and
CD11b+ BMDCs in circulating blood were analyzed in the
present study, with the mRNA transcriptions of β3
expressed on ECs and SDF-1 in tumor tissues being determined.
However, the SO group exhibited a higher level of CXCR4+
but it did not have altered subpopulations of
β3+,
CXCR4+β3+, CD11b+, and
Sca-1+ BMDCs, which is probably owing to fluctuation of
estrogen levels.
Increases in proangiogenic β3+
and/or CXCR4+ BMDCs in blood, the enhancement of
β3 transcription in ECs and SDF-1 mRNA transcriptions in
tumor tissues, and the enhancement of adhesion of BMDCs to ECs were
found in the present study, which yielded a schematic explanation
of how E2 augments BMDCs in tumor tissues. These findings also
confirmed previous reports of E2 enhancing ER-negative tumor growth
and angiogenesis by influencing the mobilization and recruitment of
a proangiogenic population of bone marrow-derived myeloid cells
(16).
Previous studies have shown that tumor cells are
capable of secreting cytokines so as to stimulate vascular
development in response to E2 treatment (32). Our results also indicated that the
proliferation of ECs was significantly upregulated by ER-negative
4T1 tumor cells in vitro, and that this process was markedly
enhanced by E2. E2 usually binds to classical nuclear ERs that
regulate the expression of genes related to the cell cycle and
proliferation (33). However, E2
was also found to combine with GPER, as distinguished from the
classic ERs, and trigger HIF1α-dependent VEGF expression by
activating the GPER, EGFR, ERK, and c-fos signaling
pathways; the increased VEGF then leads to E2-supported
angiogenesis (32). Hence, the
above-described experiments revealed enhancement-based interactions
involving BMDCs, tumor cells and vascular ECs, and an enhancement
role of E2 on this process.
Collectively, the results described herein show that
E2 enhances the mobilization of proangiogenic CXCR4+,
β3+ and
CXCR4+β3+ BMDCs from bone marrow
into circulating blood. E2 also enhances the transcriptions of
integrin β3 mRNA on ECs and SDF-1 in tumor tissues,
thereby facilitating the adhesion of BMDCs to ECs, increasing the
recruitment of BMDCs, and resulting in an increased retention of
BMDCs in tumor tissues. BMDCs significantly affect the
proliferation and migration of ECs, and 4T1 cells markedly enhance
the proliferation of ECs, which may improve angiogenesis. These
findings also demonstrate that E2 can significantly enhance 4T1
cell proliferation and EC migration directly. Moreover, E2
significantly potentiates the BMDC-induced increase in EC migration
and obviously intensifies 4T1-stimulated EC proliferation
indirectly. These data represent preliminary evidence of the role
of E2 in protumoral processes and may yield novel strategies for
use in the development of new drugs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (no. 81071765 and 81372379),
the Natural Science Foundation of Shaanxi Province (no.
2016JM8037), and the Research Foundation of The First Affiliated
Hospital of Xi'an Jiaotong University (no. 2017MS-09).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
WF and YZ designed the study. YZ, XueL, JC and XZ
performed the experiments. QZ, XF and PZ analyzed the data. WM,
XuaL, FT and KC compiled the figures. YZ and WF wrote the paper.
All authors approved the final version and agreed to be accountable
for all aspects of the work in ensuring that questions related to
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All animal procedures were done in accordance with
the protocol approved by the Xi'an Jiaotong University Animal Care
and Use Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BMDCs
|
bone marrow-derived cells
|
|
BMT
|
bone marrow transplantation
|
|
CM
|
conditioned media
|
|
E2
|
17β-estradiol
|
|
E2V
|
estradiol valerate
|
|
ECs
|
endothelial cells
|
|
ERs
|
estrogen receptors
|
|
H&E
|
hematoxylin and eosin
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
MVA
|
microvascular area
|
|
OD
|
optical density
|
|
OVX
|
ovariectomy
|
|
SDF-1
|
stromal cell-derived factor-1
|
|
SO
|
sham operation
|
|
WBCs
|
white blood cells
|
References
|
1
|
O'Lone R, Frith MC, Karlsson EK and Hansen
U: Genomic targets of nuclear estrogen receptors. Mol Endocrinol.
18:1859–1875. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deroo BJ and Korach KS: Estrogen receptors
and human disease. J Clin Invest. 116:561–570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mauvais-Jarvis F, Clegg DJ and Hevener AL:
The role of estrogens in control of energy balance and glucose
homeostasis. Endocr Rev. 34:309–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cui J, Shen Y and Li R: Estrogen synthesis
and signaling pathways during aging: From periphery to brain.
Trends Mol Med. 19:197–209. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nelson ER, Wardell SE and McDonnell DP:
The molecular mechanisms underlying the pharmacological actions of
estrogens, SERMs and oxysterols: Implications for the treatment and
prevention of osteoporosis. Bone. 53:42–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ascenzi P, Bocedi A and Marino M:
Structure-function relationship of estrogen receptor alpha and
beta: Impact on human health. Mol Aspects Med. 27:299–402. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hamilton KJ, Arao Y and Korach KS:
Estrogen hormone physiology: Reproductive findings from estrogen
receptor mutant mice. Reprod Biol. 14:3–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Björnström L and Sjöberg M: Mechanisms of
estrogen receptor signaling: Convergence of genomic and nongenomic
actions on target genes. Mol Endocrinol. 19:833–842. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Santollo J and Daniels D: Multiple
estrogen receptor subtypes influence ingestive behavior in female
rodents. Physiol Behav. 152:431–437. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yager JD and Davidson NE: Estrogen
carcinogenesis in breast cancer. N Engl J Med. 354:270–282. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McGuire S: World cancer report 2014.
Geneva, Switzerland: World health organization, international
agency for research on cancer, WHO Press, 2015. Adv Nutr.
7:418–419. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Endogenous Hormones and Breast Cancer
Collaborative Group, . Key TJ, Appleby PN, Reeves GK, Travis RC,
Alberg AJ, Barricarte A, Berrino F, Krogh V, Sieri S, et al: Sex
hormones and risk of breast cancer in premenopausal women: A
collaborative reanalysis of individual participant data from seven
prospective studies. Lancet Oncol. 14:1009–1019. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dunn BK, Wickerham DL and Ford LG:
Prevention of hormone-related cancers: Breast cancer. J Clin Oncol.
23:357–367. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Losordo DW and Isner JM: Estrogen and
angiogenesis: A review. Arterioscler Thromb Vasc Biol. 21:6–12.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arnal JF, Fontaine C, Billon-Galés A,
Favre J, Laurell H, Lenfant F and Gourdy P: Estrogen receptors and
endothelium. Arterioscler Thromb Vasc Biol. 30:1506–1512. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Iyer V, Klebba I, McCready J, Arendt LM,
Betancur-Boissel M, Wu MF, Zhang X, Lewis MT and Kuperwasser C:
Estrogen promotes ER-negative tumor growth and angiogenesis through
mobilization of bone marrow-derived monocytes. Cancer Res.
72:2705–2713. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Feng W, Madajka M, Kerr BA, Mahabeleshwar
GH, Whiteheart SW and Byzova TV: A novel role for platelet
secretion in angiogenesis: Mediating bone marrow-derived cell
mobilization and homing. Blood. 117:3893–3902. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murdoch C, Muthana M, Coffelt SB and Lewis
CE: The role of myeloid cells in the promotion of tumour
angiogenesis. Nat Rev Cancer. 8:618–631. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rizvi AZ, Swain JR, Davies PS, Bailey AS,
Decker AD, Willenbring H, Grompe M, Fleming WH and Wong MH: Bone
marrow-derived cells fuse with normal and transformed intestinal
stem cells. Proc Natl Acad Sci USA. 103:6321–6325. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ruifrok WP, de Boer RA, Iwakura A, Silver
M, Kusano K, Tio RA and Losordo DW: Estradiol-induced, endothelial
progenitor cell-mediated neovascularization in male mice with
hind-limb ischemia. Vasc Med. 14:29–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hamada H, Kim MK, Iwakura A, Ii M, Thorne
T, Qin G, Asai J, Tsutsumi Y, Sekiguchi H, Silver M, et al:
Estrogen receptors alpha and beta mediate contribution of bone
marrow-derived endothelial progenitor cells to functional recovery
after myocardial infarction. Circulation. 114:2261–2270. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Péqueux C, Raymond-Letron I, Blacher S,
Boudou F, Adlanmerini M, Fouque MJ, Rochaix P, Noël A, Foidart JM,
Krust A, et al: Stromal estrogen receptor-alpha promotes tumor
growth by normalizing an increased angiogenesis. Cancer Res.
72:3010–3019. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ström JO, Theodorsson A, Ingberg E,
Isaksson IM and Theodorsson E: Ovariectomy and 17β-estradiol
replacement in rats and mice: A visual demonstration. J Vis Exp.
64:e40132012.
|
|
24
|
Feng W, McCabe NP, Mahabeleshwar GH,
Somanath PR, Phillips DR and Byzova TV: The angiogenic response is
dictated by beta3 integrin on bone marrow-derived cells. J Cell
Biol. 183:1145–1157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Houghton J, Stoicov C, Nomura S, Rogers
AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR and Wang TC:
Gastric cancer originating from bone marrow-derived cells. Science.
306:1568–1571. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shaked Y and Voest EE: Bone marrow derived
cells in tumor angiogenesis and growth: are they the good, the bad
or the evil? Biochim Biophys Acta. 1796:1–4. 2009.PubMed/NCBI
|
|
27
|
Mahabeleshwar GH, Feng W, Phillips DR and
Byzova TV: Integrin signaling is critical for pathological
angiogenesis. J Exp Med. 203:2495–2507. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Melero-Martin JM and Dudley AC: Concise
review: Vascular stem cells and tumor angiogenesis. Stem Cells.
29:163–168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sainz J and Sata M: CXCR4, a key modulator
of vascular progenitor cells. Arterioscler Thromb Vasc Biol.
27:263–265. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Teicher BA and Fricker SP: CXCL12
(SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 16:2927–2931.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahn GO, Tseng D, Liao CH, Dorie MJ,
Czechowicz A and Brown JM: Inhibition of Mac-1 (CD11b/CD18)
enhances tumor response to radiation by reducing myeloid cell
recruitment. Proc Natl Acad Sci USA. 107:8363–8368. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
De Francesco EM, Pellegrino M, Santolla
MF, Lappano R, Ricchio E, Abonante S and Maggiolini M: GPER
mediates activation of HIF1α/VEGF signaling by estrogens. Cancer
Res. 74:4053–4064. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heldring N, Pike A, Andersson S, Matthews
J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M and
Gustafsson JA: Estrogen receptors: How do they signal and what are
their targets. Physiol Rev. 87:905–931. 2007. View Article : Google Scholar : PubMed/NCBI
|