Introduction
Phosphatase and tensin homolog (PTEN) is a tumor
suppressor and a lipid phosphatase that catalyzes dephosphorylation
of phosphatidylinositol 3,4,5-trisphosphate. PTEN has a high
frequency of mutations in various cancers including prostate cancer
and glioblastoma. PTEN alterations are observed in approximately
half of all malignant tumors and correlate with increased RAC-alpha
serine/threonine-protein (AKT) phosphorylation and initiation of
downstream targets that modulate a wide range of cellular processes
associated with the progression of tumor growth and survival
(1–8).
To investigate the function of PTEN, various
knockout mice have been prepared and the effect of PTEN deficiency
examined (9). The authors also
established a conditional PTEN-deficient mouse model of prostate
cancer driven by the PSA-Cre promoter and demonstrated its
utility in various experimental settings (10–13).
To develop better treatment strategies requires a deeper
understanding the cellular and molecular mechanisms of PTEN
deficiency. However, autochthonous tumors are highly heterogeneous
and are composed of a complex tumor microenvironment that consists
of various cell types including epithelial cancer cells, stromal
fibroblasts, immune endothelial and blood cells, in addition to
other contaminants, all of which contribute to the molecular
characterization of PTEN deficiency. Although small interfering RNA
methodology enables us to induce PTEN-deficiency at the protein
level (14), researchers often take
into consideration the fact that gene expression still remains at a
certain level even under conditions of gene knockdown.
Next-generation genome editing strategies have been
developed and are currently considered some of the best
technological tools to most efficiently characterize gene function
(15). The present study generated
an isogenic PTEN-deficient cell clone from a parental mouse
prostate cancer cell line using the clustered regularly interspaced
short palindromic repeats (CRISPR)/CRISPR-associated protein 9
(CRISPR/Cas9) system and performed comprehensive gene expression
profiling analyses to identify its impact on unique genes
associated with crucial roles in cellular processes, including
cancer development and immunity.
Materials and methods
Cell lines
The mouse prostate cancer cell line, 2924V, which
expresses wild-type PTEN was used in the present study. This cell
line was established from a prostate tumor originating from a 57
week-old PSACre;PtenloxP/loxP conditional
knockout mouse (10). Cells were
cultured at 37°C and 5% CO2 in RPMI-1640 medium
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) supplemented with
10% inactivated fetal bovine serum (HyClone; GE Healthcare Life
Sciences, Logan, UT, USA), 100 IU/ml penicillin, and 100 µg/ml
streptomycin.
PTEN-knockout using the CRISPR/Cas9
system
The CRISPR/Cas9 system was used to disrupt the
expression of the PTEN gene as described previously (16). pSpCas9(BB)-2A-Puro (PX459) was a
gift from Feng Zhang (Broad Institute of MIT, Harvard, MA, USA;
Addgene plasmid #48139; 15). Briefly, a single guide RNA (sgRNA)
sequence was selected using Optimized CRISPR Design (http://crispr.mit.edu/). The sgRNA sequence targeting
PTEN was 5′-GCTAACGATCTCTTTGATGA-3′. The plasmid expressing human
Cas9 and the PTEN sgRNA was prepared by ligating oligonucleotides
into the BbsI site of PX459 (Pten/PX459). To establish a
PTEN knockout clone with a one-nucleotide deletion, 2924V cells
(1×106 cells/dish) were seeded in a 10-cm dish. Cells
were then transfected with 10 µg of PTEN/PX459 using
Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Antibiotic selection (puromycin; 2 µg/ml) was
begun 72 h after transfection and continued for at least 3 days. A
single clone was selected, expanded, and then used for biological
assays. For sequence analysis of the PTEN gene, the following
primer set was used: 5′-CGCTAATCCAGTGTACAGTA-3′ and
5′-CTGCGAGGATTATCCGTCTT-3′.
Morphology
The cells were plated in a 12-well plate. After 24
h, the cell morphology was digitally photographed at ×100
magnification using an inverted microscope system (IX73: Olympus
Corporation, Tokyo, Japan).
MTT assay
Briefly, parent cells, mock cells, and ΔPTEN were
seeded (5×103 cells/well) into a 96-well plate.
Subsequently, 10-µl MTT solution (5 µg/ml; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) was added to each well and cells were
incubated at 37°C in 5% CO2 for 4 h. Next, cell lysis
buffer [10% sodium dodecyl sulfate (SDS) in 0.01 M HCL] was added
to the wells to dissolve the formazan crystals produced by MTT. The
optical density (550 nm) at each time point (day 0, 1, 3, 5, and 7)
is presented as the mean ± standard error of the mean (n=6).
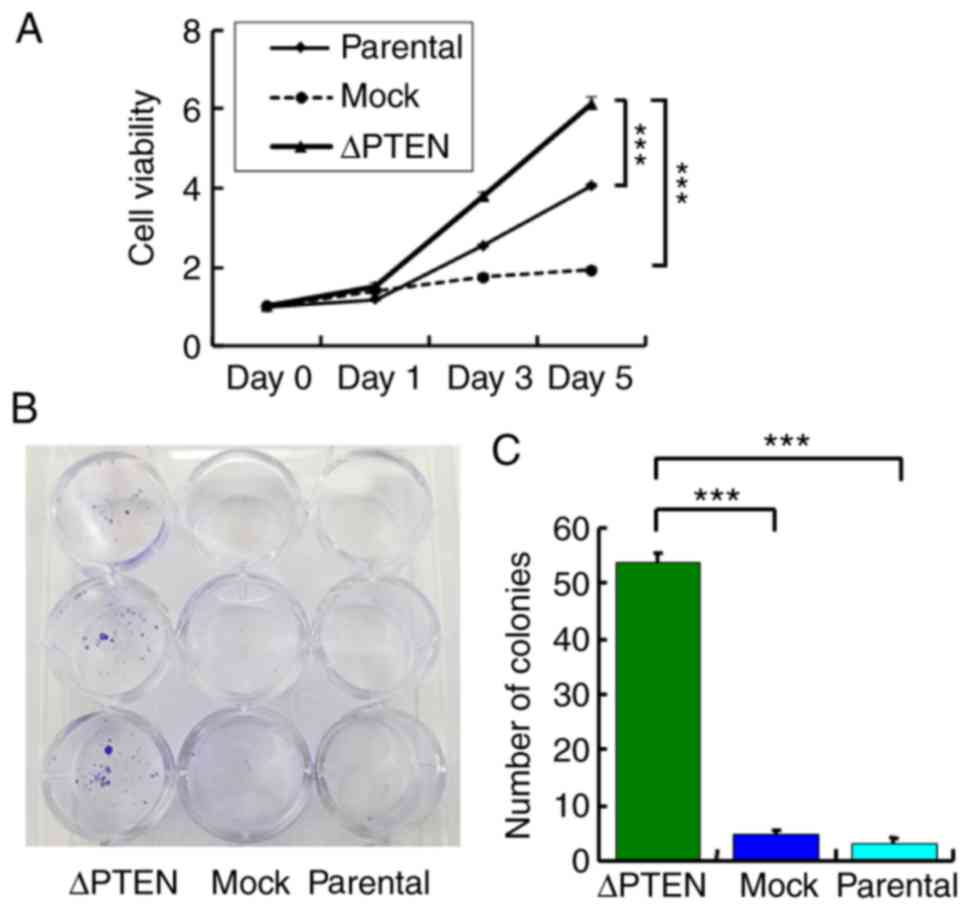
Colony formation assay
Colony formation assays were performed by seeding
500 cells of each line in 12-well plates. After 14 days, cells were
fixed/stained with 3.7% formaldehyde (Wako Pure Chemical
Industries, Ltd., Osaka, Japan) containing 0.2 % (wt/vol) crystal
violet (Sigma-Aldrich; Merck KGaA) at room temperature and number
of colonies were imaged and counted manually. Bar graphs represent
the number of stained colonies. Data are presented as the mean ±
standard error of the mean (n=3).
Western blotting
Cells were lysed in radioimmunoprecipitation assay
buffer (Wako Pure Chemical Industries, Ltd.) supplemented with a
protease inhibitor cocktail (Roche Applied Science, Mannheim,
Germany) for 30 min at 4°C. Insoluble material was removed by
centrifugation at 15,400 × g for 10 min at 4°C. Protein content was
determined using a DC protein assay kit (5000112JA; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) according to the
manufacturer's protocol. Protein lysates were mixed with loading
buffer and boiled for 5 min. A total of 10 µg protein was separated
by electrophoresis on 10% PAGE gels (TEFCO, Tokyo, Japan). Proteins
were then transferred to Immobilon-P membranes (EMD Millipore,
Billerica, MA, USA) and blocked in 5% skim milk in Tris-buffered
saline with 0.01% Tween-20 (TBS-T) for 1 h at room temperature. All
antibodies were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Membranes were incubated with primary
antibodies [PTEN, 1:3,000, #9559; AKT1, 1:1,000, #9272;
phosphorylated (p)-AKT, 1:1,000, #9271; phosphorylated (p)-Rb,
1:3,000, #9307; cyclin D1, 1:3,000, #2922; CDC2, 1:3,000, #9112;
CDK2, 1:3,000, #2546; CDK4, 1:3,000, #2906; CDK6, 1:1,000, #3136;
CDK7, 1:1,000, #2916 and GAPDH 1:5,000, #5174] overnight at 4°C.
After three washes with TBS-T, membranes were incubated with
anti-mouse, #7076/rabbit, #7074, horseradish peroxidase
(HRP)-conjugated secondary antibody (1:5,000) for 1 h at room
temperature followed by a final wash. Proteins were detected by
applying ImmunoStar LD (Wako Pure Chemical Industries, Ltd.) to the
membrane and signals were quantified using ImageQuant LAS-4000 (GE
Healthcare Life Sciences, Little Chalfont, UK) according to the
manufacturer's protocol.
RNA extraction
For microarray analysis, reverse transcription
(RT)-polymerase chain reaction (PCR) and quantitative (q) PCR,
total RNA was extracted from cell lines with the NucleoSpin RNA kit
(Macherey-Nagel, Düren, Germany) according to the manufacturer's
protocol. RNA extraction for miRNA analysis was performed with the
MiRNeasy Mini Kit (Qiagen GmbH, Hilden, Germany) according to the
manufacturer's protocol.
Microarray analysis
Expression profiling analysis of mRNA was performed
using the Agilent Oligo Microarray Kit (8×60 K; G4852B; Agilent
Technologies, Inc., Santa Clara, CA, USA). Nucleic acid labeling
and hybridization were performed with the One-Color
Microarray-Based Gene Expression Analysis kit (Agilent
Technologies, Inc.) according to the manufacturer's protocol.
Briefly, 100 ng of total RNA was amplified and labeled using the
Low Input Quick Amp Labeling Kit. After labeling, RNA was purified
with RNeasy Mini Kit (Qiagen GmbH). The RNA fragmentation reaction
was performed at 60°C for 30 min, after which the samples were
collected on ice for 1 min, and 2X Hi-RPM Hybridization Buffer was
added to stop the reaction. Samples were further mixed, centrifuged
at 13,000 × g for 1 min at room temperature, placed on ice, and
then loaded onto the array. The arrays were hybridized at 65°C for
17 h. The microarray slides were then washed with Gene Expression
Wash Buffers I and II and scanned with the Agilent Microarray
Scanner-G2505C (Agilent Technologies, Inc.). Feature Extraction
Software Version 11.0.1.1 (Agilent Technologies, Inc.) was used to
extract and analyze the signals and signal intensities were
normalized as previously described (17).
The background signals were normalized and
microarray expression data were rank-ordered according to the
expression levels of the ΔPTEN/mock cells. Differentially expressed
genes between ΔPTEN and mock cells were considered relevant if
there was ≥10-fold change.
Functional enrichment analyses of differentially
expressed genes were carried out using the Panther web-based tool
(18).
To confirm gene expression, RT-qPCR was performed
using the ABI 7900 HT Fast Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.) and calculated using
the 2−ΔΔCq method (19).
Total RNA from the three cell lines were converted to cDNA with the
High Capacity cDNA Reverse Transcription Kit (Applied Biosystems;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The Tet methylcytosine dioxygenase 1 (Tet1), twist family
BHLH transcription factor 2 (Twist2), arginase 1 (Arg1), C-fos
induced growth factor (Figf), Wingless-Type MMTV Integration Site
Family, Member 3 (Wnt3), prostaglandin reductase 1 (Ptgr1),
polypeptide N-acetylgalactosaminyltransferase 14 (Galnt14), and
GAPDH (reference) genes were amplified and detected using
SYBR-Green (Applied Biosystems; Thermo Fisher Scientific, Inc.).
PCR reactions were prepared in a final volume of 20 µl, with 17.6
µl of SYBR-Green PCR Master Mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.), 2.0 µl of DNA, and 0.2 µl each of the 10 pmol/µl
forward and reverse primers. Thermal cycler settings included DNA
polymerase activation at 95°C for 2 min followed by 55 cycles of
denaturation at 95°C for 15 sec, annealing at 60°C for 15 sec, and
extension at 70°C for 20 sec. Each measurement was performed in
triplicate. The quality of the PCR products was monitored by using
the post-PCR melt-curve analysis.
In order to visualize PCR Products, PCR was carried
out changing the number of cycles to 30 cycles. The PCR products
were electrophoresed in 1.5% agarose gel, and stained with GelRed
Nucleic Acid Stain (41003-T; Biotium, Inc., Fremont, CA, USA) and
photographed. Primer sequences are listed in Table I.
 | Table I.Primers used for polymerase chain
reaction. |
Table I.
Primers used for polymerase chain
reaction.
| Gene name | Accession | Relative intensity
of KO/mock | Forward primer | Reverse primer |
|---|
| Tet1 | NM_001253857 | 32 |
ACACAGTGGTGCTAATGCAG |
AGCATGAACGGGAGAATCGG |
| Twist2 | NM_007855 | 13 |
TACAGCAAGAAATCGAGCGAAG |
GCTGAGCTTGTCAGAGGGG |
| Arg1 | NM_007482 | 12 |
CTCCAAGCCAAAGTCCTTAGAG |
GGAGCTGTCATTAGGGACATCA |
| Figf | NM_010216 | 10 |
AACAGATCCGAGCAGCTTCTA |
TTTTGAGCTTCAACCGGCATC |
| Wnt3 | NM_009521 | 10 |
AGCGTAGCAGAAGGTGTGAAG |
CCAGGTGGCCCCTTATGATG |
| Galnt14 | NM_027864 | −19 |
CTCATCAAACTGCTCCCACA |
GCTCTGGATCACCGTACTGC |
| Ptgr1 | NM_025968 | −30 |
CAATCGTTCCTTTTGGGAAG |
CATGAGAGTTGCAGCCAAAA |
miRNA array analysis
miRNA profiling analysis was performed using the
Agilent Mouse miRNA kit (8×60 K; G4872A-070155). miRNA labeling,
hybridization, and washing were carried out according to the
manufacturer's protocol (Agilent Technologies, Inc.). Hybridized
microarrays were scanned with a DNA microarray scanner (Agilent
Microarray Scanner; G2505C) and features were extracted using the
Agilent Feature Extraction image analysis tool (version 11.0.1.1).
Briefly, average values of the spots of each miRNA were
background-subtracted and subjected to further analysis. miRNA
array expression data were rank-ordered according to the expression
levels of the ΔPTEN/mock cells. To confirm miRNA expression level,
RT-qPCR was performed. Total RNA from the three cell lines were
converted to cDNA with the Taqman MicroRNA Reverse Transcription
Kit (Applied Biosystems) according to the manufacturer's protocol.
Realtime PCR was performed using Taqman MicroRNA Assays
(has-miR-210, RT:000512 and snoRNA234; RT:001234 as control;
Applied Biosystems) according to the manufacturer's protocol.
Statistical analysis
At least three replications per experiment and three
independent experiments were performed. The results are expressed
as the mean ± standard error of the mean. Independent-samples
Student's t-test and one-way analysis of variance, followed by the
least significant difference post-hoc test were performed to
analyze data using JMP Ver.13.2.1 (SAS Institute Inc; Tokyo,
Japan). P<0.05 was considered to indicate a statistically
significant difference (*P<0.05; **P<0.01;
***P<0.001).
Results
Establishment of PTEN-knockout cells
using the CRISPR/Cas9 System
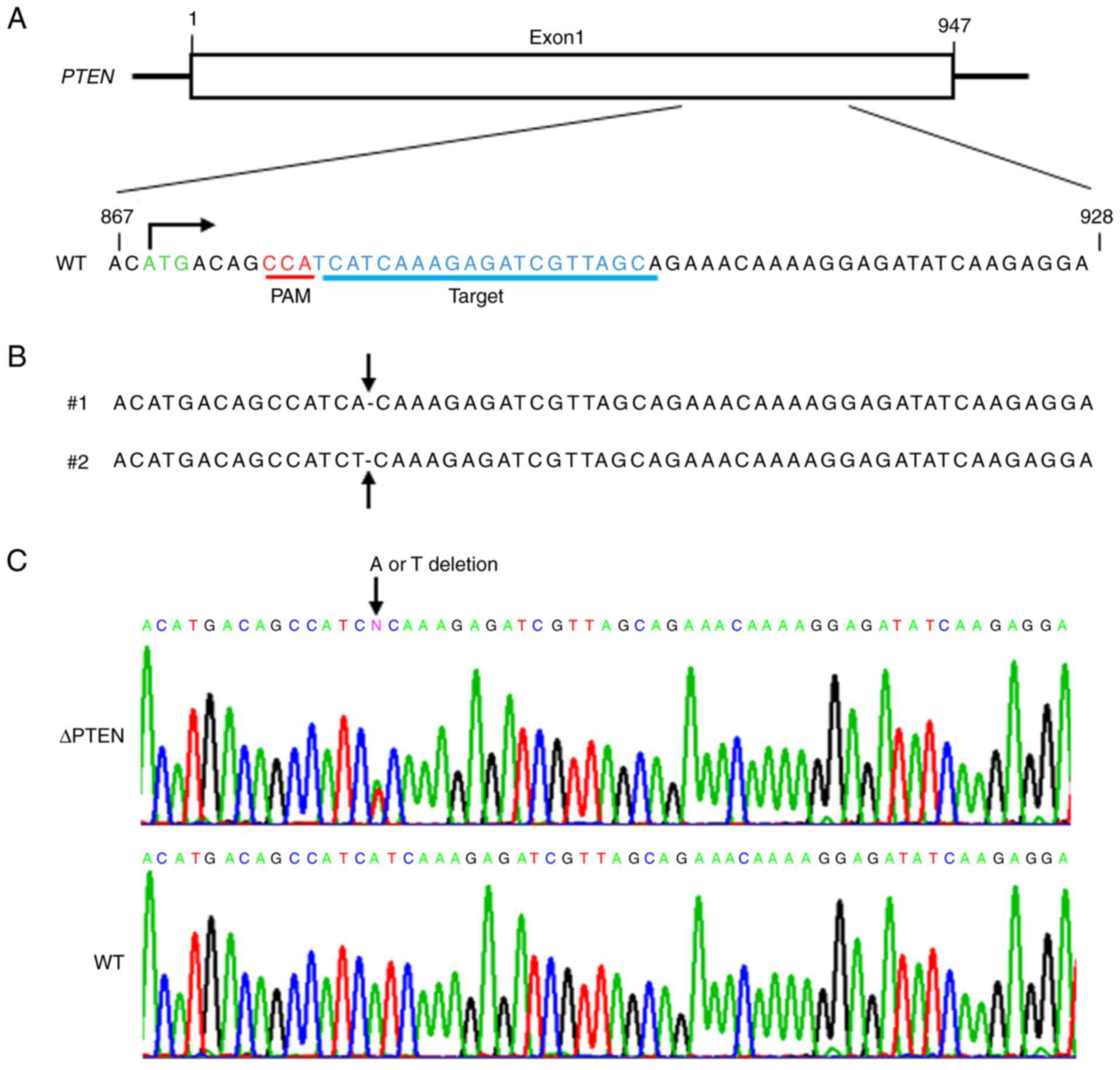
The present study performed genome editing
(targeting sequence presented in Fig.
1A) using the CRISPR/Cas9 system and obtained multiple clones
(ΔPTEN). Fig. 1B and C reveal DNA
sequencing of target sites in the resultant clones. These results
demonstrated that targeting PTEN with the CRISPR/Cas9 system
effectively generated mutant-PTEN clones. Next, the present study
assessed the effects of PTEN inactivation in the resulting clones.
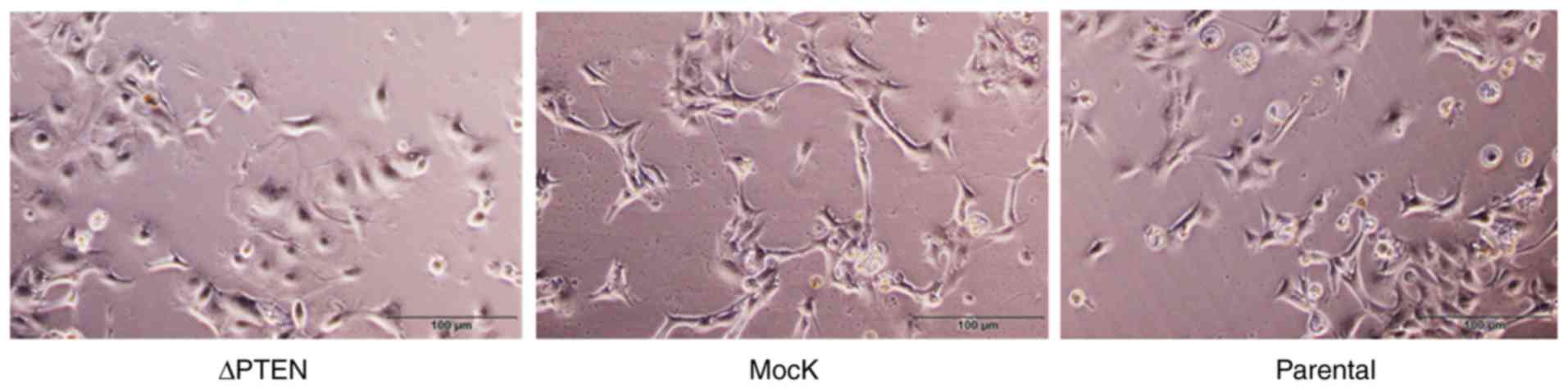
Overall, the general appearance was similar between parental, mock,
and ΔPTEN cells; however, marginal differences were observed in the
morphological appearance of ΔPTEN cells such as heterogeneity of
the cell shape (Fig. 2).
Using western blot analysis, the present study
verified the effective knockout of PTEN in ΔPTEN cells. Fig. 3 shows that the expression of PTEN
was present in the parental strain and mock-transfected cells but
was effectively downregulated in ΔPTEN cells. Accordingly, Akt
phosphorylation levels increased in the absence of PTEN. Activation
of the PI3K/Akt pathway promotes the cellular proliferation of
transformed cells; thus, the expression of molecules involved in
proliferation was then assessed. In ΔPTEN cells, the upregulation
of Akt phosphorylation was associated with the elevated expression
of cyclin D1, cyclin dependent kinase (CDK)4, and decreased
expression of CDK7. In addition, increased phosphorylation of the
retinoblastoma tumor-suppressor protein (RB) was observed, a known
regulator of cellular proliferation, in ΔPTEN cells. As the
expression of molecules involved in the cell cycle was demonstrated
to be enhanced in ΔPTEN cells, the present study then assessed the
effects of PTEN inactivation on cell viability. Fig. 4 shows that the inactivation of the
PTEN function in ΔPTEN cells results in increased cell growth and
enhanced colony formation.
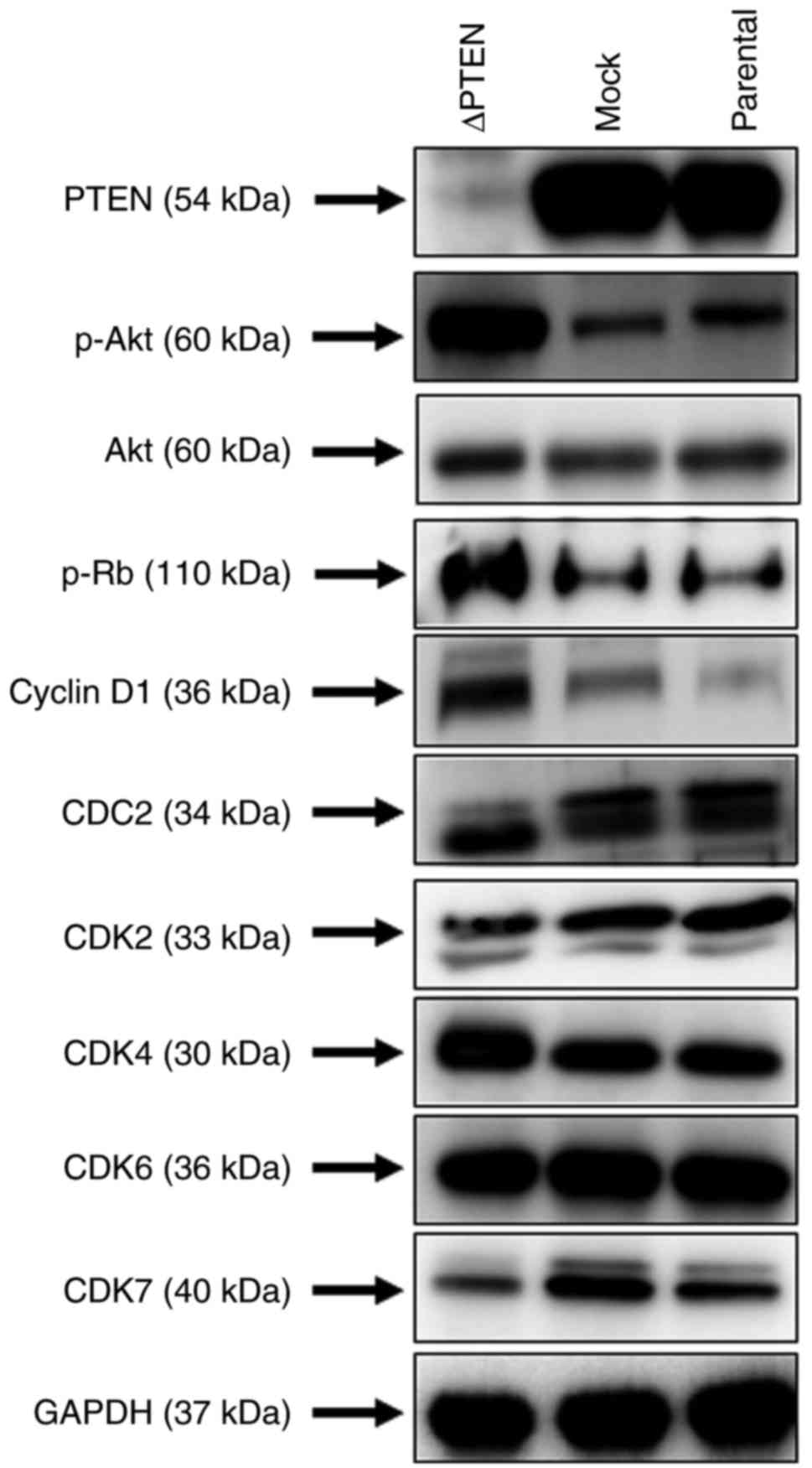 | Figure 3.Western blot analysis of ΔPTEN cells.
Immunoblots showing protein expression of basal PTEN, phospho-Akt,
and the cell cycle-related proteins pRb, Cyclin D1, CDC2, CDK2,
CDK4, CDK6, and CDK7 in ΔPTEN, mock and parental cell lines. GAPDH
was used as a loading control. PTEN, phosphatase and tensin
homolog; ΔPTEN, PTEN-knockout cell line; CDK, cyclin dependent
kinase; CDC2, cyclin-dependent protein kinase 1; p, phosphorylated;
Akt, RAC-alpha serine/threonine-protein kinase; Rb, retinoblastoma
tumor-suppressor protein. |
Microarray and miRNA array analysis of
PTEN-knockout cells
Using gene microarrays, the present study assessed
changes in gene expression following the loss of the PTEN function
and focused on genes that were differentially expressed in ΔPTEN
cells relative to mock-transfected cells and the parental strain
(Fig. 5). Overall, 111 genes were
upregulated by at least 10-fold, including one with a 74-fold
increase in ΔPTEN cells (Table
II). In addition, 23 genes were identified to be substantially
downregulated by ≥10-fold, one with a 139-fold decrease (Table III).
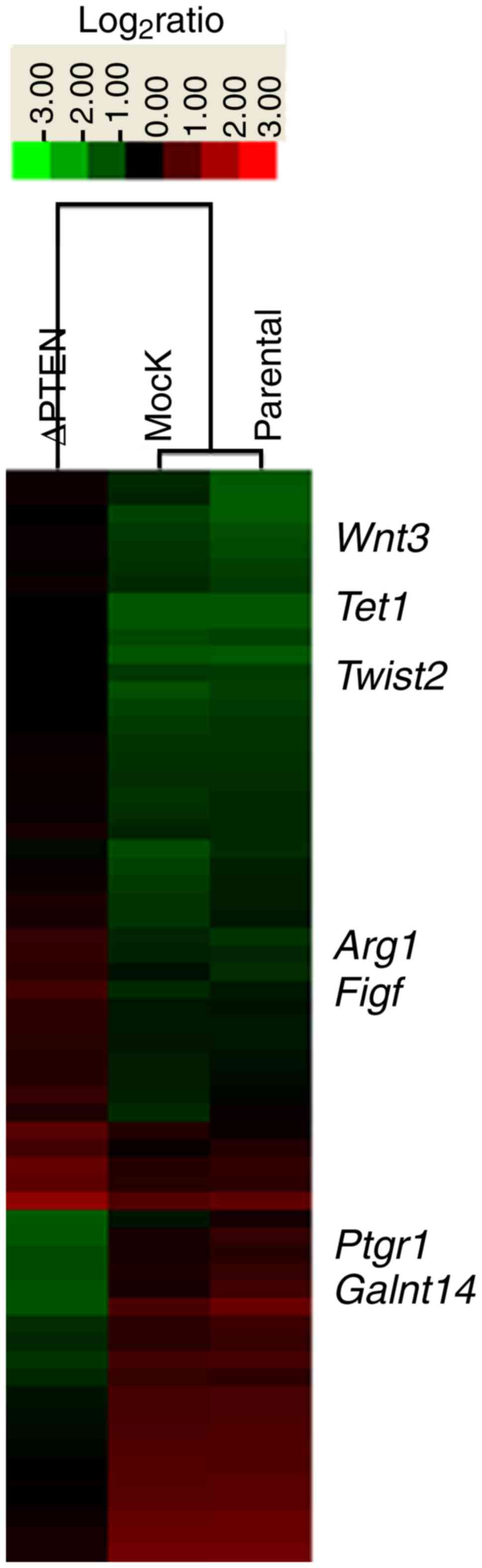 | Figure 5.Microarray analysis identification of
novel genes associated with PTEN expression. Heat map representing
gene expression changes (at least 10-fold) in ΔPTEN, mock, and
parental cells. PTEN, phosphatase and tensin homolog; ΔPTEN,
PTEN-knockout cell line; Tet1, Tet methylcytosine dioxygenase 1;
Figf, C-fos-induced growth factor; Twist2, twist family BHLH
transcription factor 2; Arg1, Arginase 1; Ptgr1, prostaglandin
reductase 1; Galnt14, polypeptide N-acetylgalactosaminyltransferase
14. |
| Table II.Genes that are upregulated 10-fold or
more in phosphatase and tensin homolog knockout cells. |
Table II.
Genes that are upregulated 10-fold or
more in phosphatase and tensin homolog knockout cells.
| Gene symbol | Accession | UniGene | Function | Relative intensity
of KO/mock |
|---|
| Sprr2d | NM_011470 | Mm.87820 | Small proline-rich
protein 2D | 74 |
| Serpinb6b | NM_011454 | Mm.36526 | Serine (or
cysteine) peptidase inhibitor, clade B, member 6b | 36 |
| Lifr | NM_013584 | Mm.149720 | Leukemia inhibitory
factor receptor | 36 |
| Tet1 | NM_001253857 | Mm.491965 | Tet methylcytosine
dioxygenase 1 | 32 |
| Sprr2e | NM_011471 | Mm.261596 | Small proline-rich
protein 2E | 29 |
| Dppa2 | NM_028615 | Mm.27857 | Developmental
pluripotency associated 2 | 29 |
| BB612626 | BB612626 | Mm.463155 | Not indicated | 26 |
| Gap43 | NM_008083 | Mm.1222 | Growth associated
protein 43 | 25 |
| Sprr1b | NM_009265 | Mm.140151 | Small proline-rich
protein 1B | 23 |
| Tspan10 | NM_145363 | Mm.209875 | Tetraspanin 10 | 21 |
| Tex15 | NM_031374 | Mm.280624 | Testis expressed
gene 15 | 20 |
| Prl7a2 | NM_011168 | Mm.153999 | Prolactin family 7,
subfamily a, member 2 | 19 |
| Gm9519 | XR_391191 | Mm.389803 | Not indicated | 19 |
| Nlrc5 | NM_001033207 | Mm.334720 | NLR family, CARD
domain containing 5 | 19 |
| Gm26579 | XR_380816 | Mm.356554 | Not indicated | 19 |
| Aqp9 | NM_001271843 | Mm.335570 | Aquaporin 9 | 18 |
| Sp110 | NM_175397 | Mm.335802 | Sp110 nuclear body
protein | 18 |
| Gm29824 | XR_870586 | Mm.135986 | ncRNA | 17 |
| Sdr16c6 | BC125450 | Mm.298546 | Short chain
dehydrogenase/reductase family 16C, member 6 | 17 |
| Prl7a1 | NM_008930 | Mm.196424 | Prolactin family 7,
subfamily a, member 1 | 17 |
| Cfhr2 | NM_001025575 | Mm.439660 | Complement factor
H-related 2 | 17 |
| Fa2h | NM_178086 | Mm.41083 | Fatty acid
2-hydroxylase | 17 |
| Zfp352 | NM_153102 | Mm.214642 | Zinc finger protein
352 | 16 |
| Cyp7a1 | NM_007824 | Mm.57029 | Cytochrome P450,
family 7, subfamily a, polypeptide 1 | 16 |
| Tvp23a | NM_001013778 | Mm.325732 | Trans-golgi network
vesicle protein 23A | 15 |
| Mmel1 | NM_013783 | Mm.116944 | Membrane
metallo-endopeptidase-like 1 | 15 |
| Ivl | NM_008412 | Mm.207365 | Involucrin | 15 |
| Sprr2h | NM_011474 | Mm.10693 | Small proline-rich
protein 2H | 15 |
| AV154423 | AV154423 | Mm.486971 | Not indicated | 15 |
| Phf11d | NM_199015 | Mm.479285 | PHD finger protein
11D | 15 |
| Col8a1 | NM_007739 | Mm.130388 | Collagen, type
VIII, alpha 1 | 14 |
| Evi2a | NM_001033711 | Mm.439665 | Ecotropic viral
integration site 2a | 14 |
| Samd15 | NM_001290288 | Mm.302304 | Sterile alpha motif
domain containing 15 | 14 |
| Gm13119 | NM_001034101 | Mm.389596 | Not indicated | 14 |
| Trp63 | NM_011641 | Mm.20894 | Transformation
related protein 63 | 14 |
| AA623943 | XM_006534607 | not indicated | Expressed sequence
AA623943 | 14 |
| Defb3 | NM_013756 | Mm.103651 | Defensin beta
3 | 14 |
| Gm30692 | XR_863174 | not indicated | ncRNA | 14 |
| Gm32204 | XM_006497575 | not indicated | Not indicated | 14 |
| Serpinb2 | NM_011111 | Mm.271870 | Serine (or
cysteine) peptidase inhibitor, clade B, member 2 | 14 |
| Rsad2 | NM_021384 | Mm.24045 | Radical S-adenosyl
methionine domain containing 2 | 14 |
| Gm31115 | XR_390648 | not indicated | Not indicated | 14 |
| Slco4a1 | NM_148933 | Mm.133687 | Solute carrier
organic anion transporter family, member 4a1 | 14 |
| Sod3 | NM_011435 | Mm.2407 | Superoxide
dismutase 3, extracellular | 13 |
| Ccdc153 | NM_001081369 | Mm.347681 | Coiled-coil domain
containing 153 | 13 |
| Lce1d | NM_027137 | Mm.176243 | Late cornified
envelope 1D | 13 |
| Twist2 | NM_007855 | Mm.9474 | Twist basic
helix-loop-helix transcription factor 2 | 13 |
| Plcb1 | NM_019677 | Mm.330607 | Phospholipase C,
beta 1 | 13 |
| Olfr376 | NM_001172686 | Mm.236410 | Olfactory receptor
376 | 13 |
| Gm19619 | NR_040428 | Mm.125059 | Not indicated | 13 |
| Dnm3 | NM_172646 | Mm.441620 | Dynamin 3 | 13 |
| Chst5 | NM_019950 | Mm.432506 | Carbohydrate
(N-acetylglucosamine 6-O) sulfotransferase 5 | 13 |
| BF119155 | BF119155 | Mm.432156 | Not indicated | 12 |
| Arg1 | NM_007482 | Mm.154144 | Arginase,
liver | 12 |
| Cdh7 | AK034096 | Mm.487119 | Cadherin 7, type
2 | 12 |
| Ugt1a6b | NM_201410 | Mm.300095 | UDP
glucuronosyltransferase 1 family, polypeptide A6B | 12 |
| Chrna5 | NM_176844 | Mm.103778 | Cholinergic
receptor, nicotinic, alpha polypeptide 5 | 12 |
| Phf11a | NM_172603 | Mm.254918 | PHD finger protein
11A | 12 |
| Sh3tc2 | NM_172628 | Mm.262320 | SH3 domain and
tetratricopeptide repeats 2 | 12 |
| Armc2 | NM_001034858 | Mm.211320 | Armadillo repeat
containing 2 | 12 |
| Fgf21 | NM_020013 | Mm.143736 | Fibroblast growth
factor 21 | 12 |
| Ccdc109b | NM_025779 | Mm.31056 | Coiled-coil domain
containing 109B | 12 |
| Klk7 | NM_011872 | Mm.251227 | Kallikrein
related-peptidase 7 (chymotryptic, stratum corneum) | 12 |
| Sh3gl3 | NM_017400 | Mm.432002 | SH3-domain
GRB2-like 3 | 11 |
| Gm17202 | XR_389494 | not indicated | Not indicated | 11 |
| Iigp1 | NM_021792 | Mm.261140 | Interferon
inducible GTPase 1 | 11 |
| AF067061 | NM_199060 | Mm.247428 | cDNA sequence
AF067061 | 11 |
| Plce1 | NM_019588 | Mm.34031 | Phospholipase C,
epsilon 1 | 11 |
| Tprg | NM_175165 | Mm.126851 | Transformation
related protein 63 regulated | 11 |
| Spint3 | NM_001177401 | Mm.234248 | Serine peptidase
inhibitor, Kunitz type, 3 | 11 |
| LOC102634459 | XR_387206 | not indicated | Not indicated | 11 |
| Gm4340 | NM_001177535 | Mm.339215 | Not indicated | 11 |
| Arhgap9 | NM_001285785 | Mm.227198 | Rho GTPase
activating protein 9 | 11 |
| Aldh1a3 | NM_053080 | Mm.140988 | Aldehyde
dehydrogenase family 1, subfamily A3 | 11 |
| Adh7 | NM_009626 | Mm.8473 | Alcohol
dehydrogenase 7 (class IV), mu or sigma polypeptide | 11 |
| Prl3d2 | NM_172155 | Mm.458451 | Prolactin family 3,
subfamily d, member 1 | 11 |
| Lama1 | AK051116 | Mm.303386 | Laminin, alpha
1 | 11 |
| Gm7978 | NM_001270457 | Mm.380154 | Not indicated | 11 |
| BB114814 | XM_006508432 | Mm.304207 | Expressed sequence
BB114814 | 11 |
| D5Ertd577e | NM_177187 | Mm.348793 | DNA segment, Chr 5,
ERATO Doi 577, expressed | 11 |
| Lrrn4 | NM_177303 | Mm.386903 | Leucine rich repeat
neuronal 4 | 11 |
| Trav12-1 | BC038136 | Mm.333502 | T cell receptor
alpha variable 12-1 | 11 |
| CV783874 | CV783874 | Mm.441595 | Not indicated | 11 |
| Igfbp5 | NM_010518 | Mm.405761 | Insulin-like growth
factor binding protein 5 | 11 |
| Gm3259 | NM_001270456 | Mm.402477 | Not indicated | 10 |
| Gm4665 | AK132957 | Mm.437523 | Not indicated | 10 |
| Sox11 | NM_009234 | Mm.41702 | SRY (sex
determining region Y)-box 11 | 10 |
| Tcstv1 | NM_018756 | Mm.302175 | 2-Cell-stage,
variable group, member 1 | 10 |
| Sprr3 | NM_011478 | Mm.57092 | Small proline-rich
protein 3 | 10 |
| Podn | NM_001285956 | Mm.74710 | Podocan | 10 |
| Ifit3b | NM_001005858 | Mm.271850 | Interferon-induced
protein with tetratricopeptide repeats 3B | 10 |
| Gabra1 | NM_010250 | Mm.439668 | Gamma-aminobutyric
acid (GABA) 1 A receptor, subunit alpha | 10 |
| Ifi44 | NM_133871 | Mm.30756 | Interferon-induced
protein 44 | 10 |
| a | NM_015770 | Mm.315593 | Nonagouti | 10 |
| Rd3l | XM_006515764 | Mm.134213 | Retinal
degeneration 3-like | 10 |
| Figf | NM_010216 | Mm.297978 | Vascular
endothelial growth factor D | 10 |
| B430212C06Rik | NR_033214 | Mm.491997 | Not indicated | 10 |
| AK034098 | AK034098 | Mm.446266 | Not indicated | 10 |
| Bank1 | NM_001033350 | Mm.30832 | B cell scaffold
protein with ankyrin repeats 1 | 10 |
| Sel1l3 | NM_172710 | Mm.235020 | Sel-1 suppressor of
lin-12-like 3 | 10 |
| Sgsm1 | NM_172718 | Mm.200203 | Small G protein
signaling modulator 1 | 10 |
| Krt13 | NM_010662 | Mm.4646 | Keratin 13 | 10 |
| Tpo | NM_009417 | Mm.4991 | Thyroid
peroxidase | 10 |
| Tnfrsf18 | NM_009400 | Mm.491989 | Tumor necrosis
factor receptor superfamily, member 18 |
|
| Klra2 | NM_008462 | Mm.4783 | Killer cell
lectin-like receptor, subfamily A, member 2 |
|
| Pcdh7 | NM_018764 | Mm.332387 | Protocadherin
7 | 10 |
| Cbr2 | NM_007621 | Mm.21454 | Carbonyl reductase
2 | 10 |
| Wnt3 | NM_009521 | Mm.159091 | Wingless-type MMTV
integration site family, member 3 |
|
| AK042637 | AK042637 | not indicated | Not indicated | 10 |
| CO811058 | CO811058 | Mm.421039 | Not indicated | 10 |
| St18 | NM_173868 | Mm.234612 | Suppression of
tumorigenicity 18 | 10 |
| Table III.Genes that are downregulated 10-fold
or more in phosphatase and tensin homolog knockout cells. |
Table III.
Genes that are downregulated 10-fold
or more in phosphatase and tensin homolog knockout cells.
| Gene symbol | Accession | UniGene | Function | Relative intensity
of mock/KO |
|---|
| Wtip | NM_207212 | Mm.422738 | WT1-interacting
protein | 139 |
| Gm32139 | XR_001782443 | not indicated | not indicated | 38 |
| Gm38426 | NR_103491 | Mm.437155 | not indicated | 37 |
| Ptgr1 | NM_025968 | Mm.34497 | prostaglandin
reductase | 30 |
| Pla2g16 | NM_139269 | Mm.274810 | phospholipase A2,
group XVI | 29 |
| Galnt14 | NM_027864 | Mm.271953 |
UDP-N-acetyl-alpha-D-galactosamine:polypeptide
N-acetylgalactosaminyltransferase 14 | 19 |
| Dhrs4 | NM_030686 | Mm.27427 |
dehydrogenase/reductase (SDR family)
member 4 | 19 |
| Fam124a | NM_001243857 | Mm.291864 | family with
sequence similarity 124, member A | 18 |
| Robo1 | NM_019413 | Mm.310772 | roundabout guidance
receptor 1 | 16 |
| Evc2 | NM_145920 | Mm.25506 | Ellis van Creveld
syndrome 2 | 15 |
| Spats2l | NM_144882 | Mm.159989 | spermatogenesis
associated, serine-rich 2-like | 14 |
| Ttc12 | NM_172770 | Mm.177413 | tetratricopeptide
repeat domain 12 | 14 |
| Gxylt2 | NM_198612 | Mm.272037 | glucoside
xylosyltransferase 2 | 14 |
| Tmem173 | NM_028261 | Mm.45995 | transmembrane
protein 173 | 13 |
| Gm2030 | NM_001100445 | Mm.411645 | not indicated | 13 |
| Gm16404 | NM_001220497 | Mm.380174 | not indicated | 13 |
| Gm1993 | NM_001102677 | Mm.484626 | not indicated | 12 |
| Gm10487 | NM_001100609 | Mm.483167 | not indicated | 12 |
| Gm5168 | NM_001025607 | Mm.370361 | not indicated | 12 |
| Slx | NM_001136476 | Mm.489202 | Sycp3 like
X-linked | 12 |
| Scml2 | NM_001290651 | Mm.159173 | sex comb on
midleg-like 2 | 11 |
| Gm14625 | NM_001220498 | Mm.477978 | not indicated | 11 |
| Ppic | NM_008908 | Mm.4587 | peptidylprolyl
isomerase C | 10 |
The mRNA expression levels of Tet1, Twist2, Arg1,
Figf, Wnt3, Ptgr1, and Galnt14 were measured by RT-qPCR
to confirm the results of the microarray analysis (Fig. 6A). The expression patterns of the
RT-PCR analysis were parallel to those in the microarray, thereby
verifying the results (Fig.
6B).
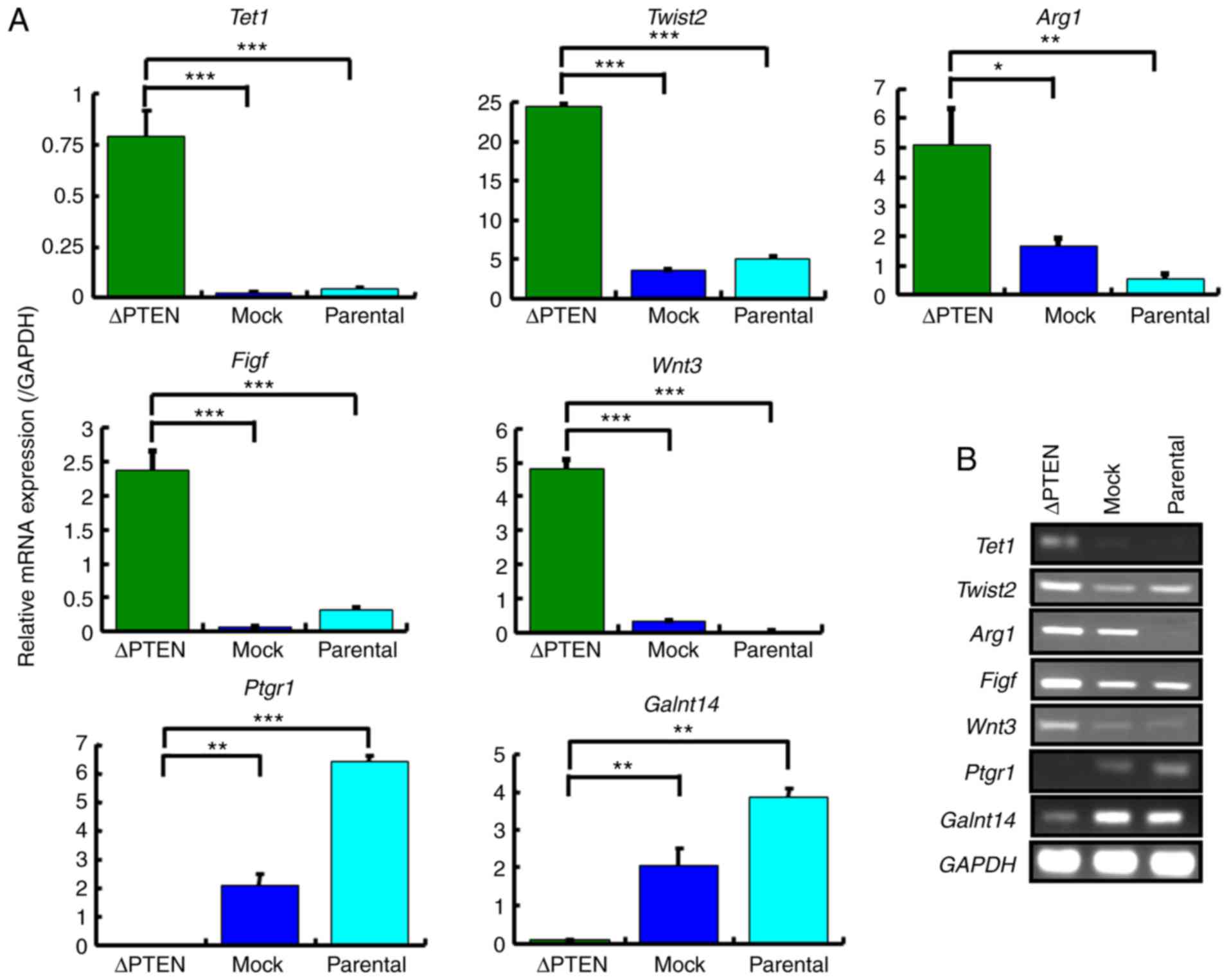 | Figure 6.Confirmation of mRNA expression
levels in selected genes with a ≥10-fold change in the microarray
analysis. (A) Expression levels of Tet1, Twist2, Arg1, Figf,
Wnt3, Ptgr1, and Galnt14 in ΔPTEN, mock, and parental
cell lines were analyzed using the qRT-PCR method. The relative
gene expression levels are shown after normalization to GAPDH mRNA
expression. (B) RT-PCR analysis of the mRNA expression of Tet1,
Twist2, Arg1, Figf, Wnt3, Ptgr1 and Galnt14 in ΔPTEN, mock, and
parental cell lines. *P<0.05; **P<0.01; ***P<0.001. PTEN,
phosphatase and tensin homolog; ΔPTEN, PTEN-knockout cell line;
Tet1, Tet methylcytosine dioxygenase 1; Figf, C-fos-induced growth
factor; Twist2 twist family BHLH transcription factor 2; Arg1,
Arginase 1; Ptgr1, prostaglandin reductase 1; Galnt14, polypeptide
N-acetylgalactosaminyltransferase 14; qRT-PCR, quantitative reverse
transcription- polymerase chain reaction. |
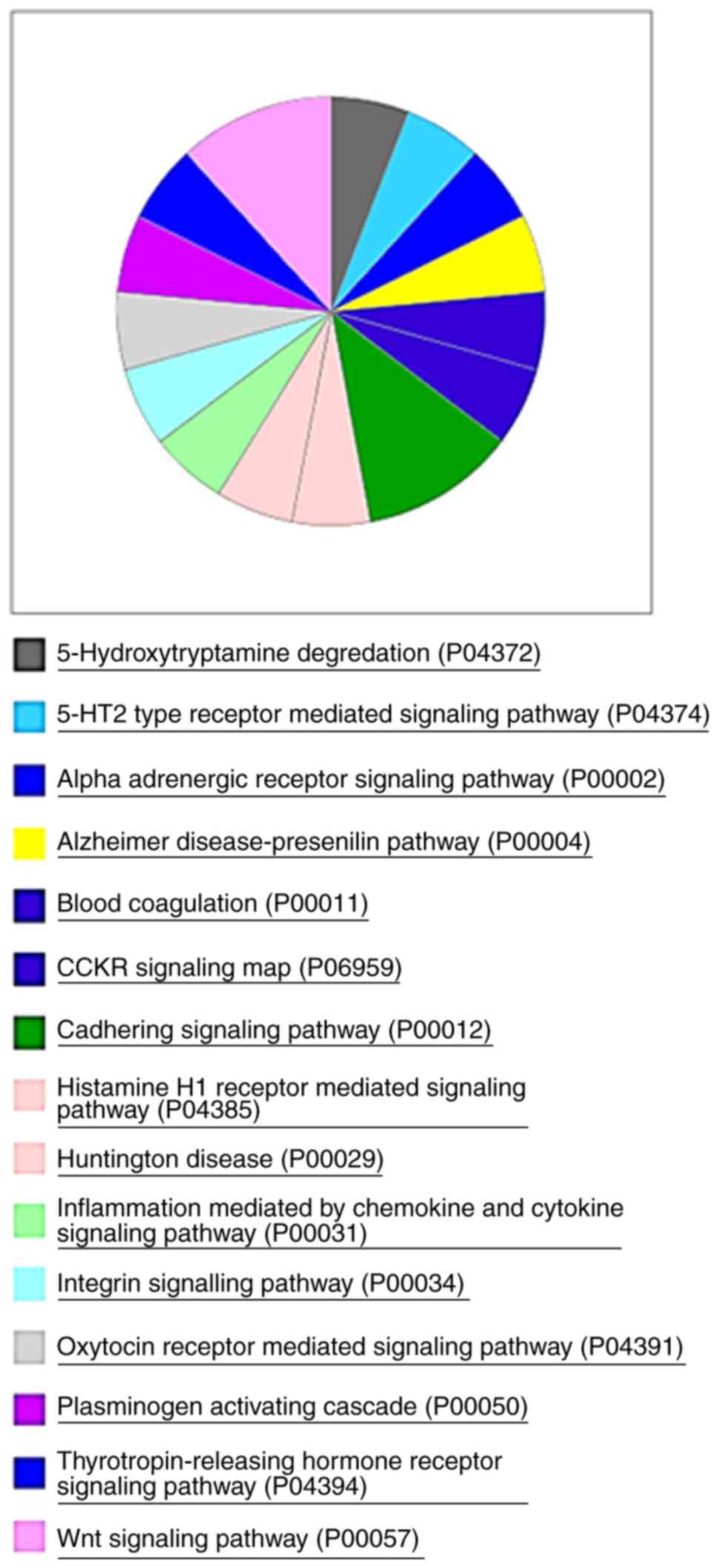
Next, the present study performed gene enrichment
profiling to gain better insight into the genes differentially
expressed after PTEN-knockout in the model. The analysis using the
Panther annotation database revealed that 64 of the 111 upregulated
genes corresponded to 15 pathways (Fig.
7).
Furthermore, the expression of non-coding RNAs, in
particular miRNAs, were assessed to further delve into the impact
of PTEN inactivation and altered gene expression. Notably, the
miRNA expression analysis in this study revealed that only the
mmu-miR-210-3p expression increased (≥10-fold) due to
PTEN-knockout. Corroborating our initial observation, the RT-qPCR
analysis presented an evident increase of mmu-miR-210-3p in ΔPTEN
cells compared with the parental strain and mock-transfected cells
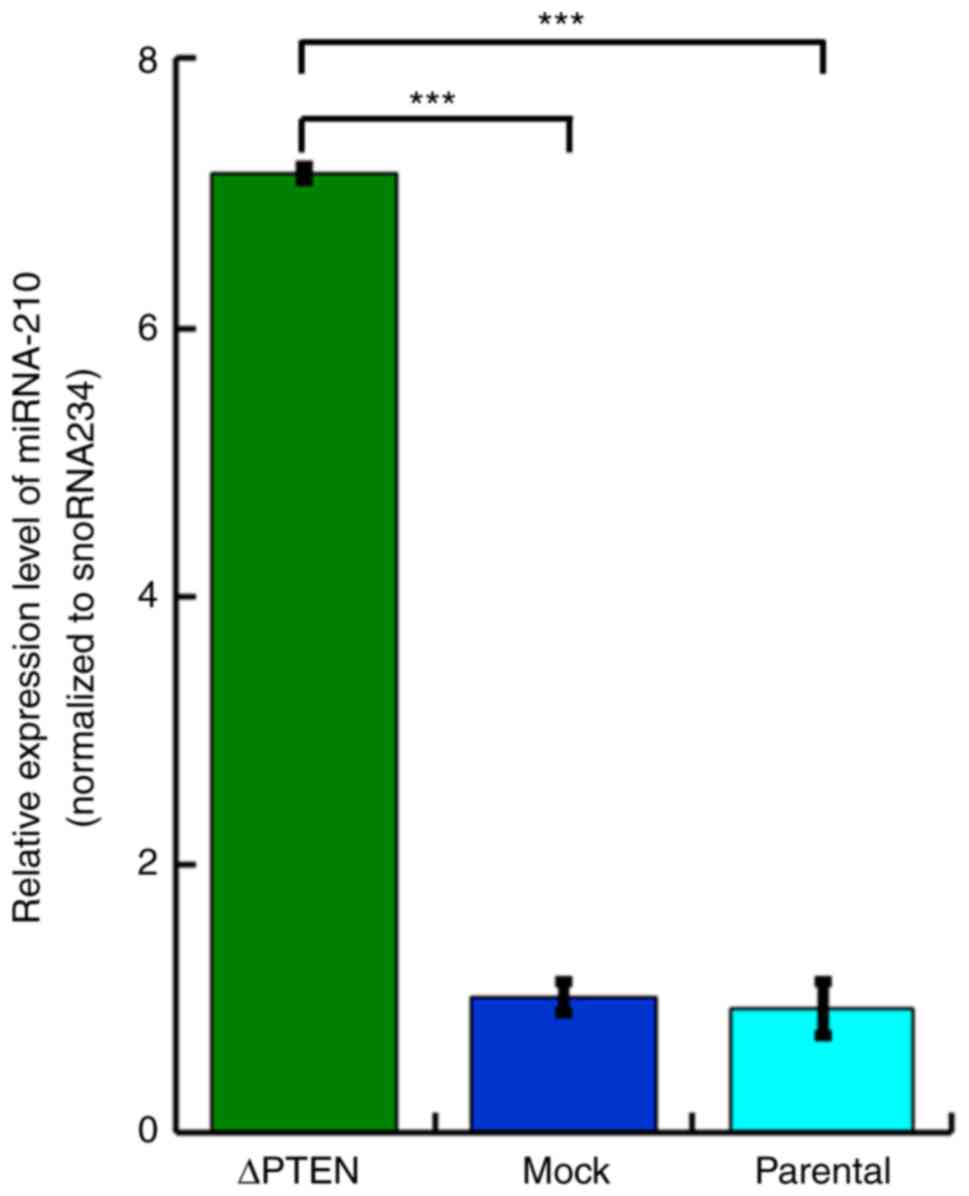
(Fig. 8).
Discussion
The present study investigated the changes in
carcinogenesis-associated genes caused by PTEN deficiency in a
mouse prostate cancer model. A PTEN-deficient mouse prostate cancer
isogenic cell line was established using the CRISPR/Cas9 system.
The phosphorylation of Akt and the expression of cyclin D1 were
elevated in PTEN-deficient cells. Although the expression of CDK7
wa srevealed to be decreased, the reason remains unknown. The
expression levels of cyclinD1 and CDK4 that increased as
demonstrated by western blotting, were not recognized as much
different from that of parental cell or mock cell in microarray
analysis. The reason for this increase in the expression currently
remains unclear. Furthermore, microarray and miRNA arrays were used
to assess the changes in gene expression. These results suggested
that the PTEN expression is essential for normal gene
expression.
The present study observed alterations in the
expression levels of various genes due to the loss of the PTEN
gene, which may be attributed to an increase in the expression of
TET1. Demethylation is increased in several genes due to the
enhanced expression of TET1, a dioxygenase that catalyzes the
conversion of the modified genomic base 5-methylcytosine (5 mc)
into 5-hydroxymethylcytosine (5hmc). Reportedly, Nanog is an
example of a gene induced by Tet1 expression. Not only does Nanog
have a role in the maintenance of mouse ES cells (20), but also in the maintenance of cancer
stem cells, suppression of apoptosis, promotion of cancer
progression and metastasis, and angiogenesis (21). These findings suggest that PTEN
deficiency drives TET1 and TET1-gene regulation during
carcinogenesis; however, the underlying reasons for an increase in
the TET1 expression in PTEN deficiency remain unclear.
In this model, we established the upregulation of
Twist2, Figf, Wnt3, and Arg1 in response to
Pten-knockout; these genes play vital roles in the
development and progression of cancer and promote the immune
suppression in the surrounding tumor microenvironment.
Twist2, a member of the basic helix-loop-helix
(BHLH) transcription factors, is overexpressed in several cancer
types. Previously, studies have reported the correlation of Twist
expression with head and neck squamous cell carcinomas, cervical
carcinomas, and ovarian cancer (22–24).
Yang et al reported that hypoxia inducible factor-1 (HIF-1)
promotes EMT through direct regulation of Twist expression
(25). Twist is a master regulator
of gastrulation and mesoderm specification and is implicated to be
essential in the mediation of cancer metastasis (25). In this study, the expression of
Twist was enhanced by the absence of functional PTEN, suggesting
transcriptional regulation by mechanisms mediated by proteins other
than HIF-1.
Figf, also termed vascular endothelial growth factor
(VEGF)-D is a member of the platelet-derived growth factor
(PDGF)/VEGF family and is active in angiogenesis and endothelial
cell growth. Reportedly, the normal VEGF-D expression is detected
in the lung, heart, skeletal muscle, skin, adrenal gland, and
gastrointestinal tract (26–28).
In addition, VEGF-D is upregulated in glioblastoma (29), melanoma (30), colorectal carcinoma (31), breast carcinoma (32), and cervical intraepithelial
neoplasia (33). Tian et al
reported that PTEN suppresses VEGF expression in HCC through both
phosphatase-dependent and -independent mechanisms (34). The present study also implicated
that PTEN regulates VEGF expression through both phosphatase- and
HIF-1a- -independent mechanisms. Kaushal et al (35) reported that VEGF-D expression is
observed in all prostate cancer tissues and that it is increased in
samples at advanced stages compared with those at the early stage;
however, they did not report the correlation between VEGF-D
expression and PTEN. In 20% of prostate cancers, PTEN is deleted
(36). PTEN-deficient prostate
cancers express VEGF-D at an early stage; therefore, it is
imperative to compare the expression between tumors with and
without PTEN.
The Wingless-type MMTV integration site family genes
comprise structurally related genes that encode a secreted
signaling protein implicated in oncogenesis and several
developmental processes, including the regulation of cell fate and
patterning during embryogenesis (37,38).
In normal tissues, Wnt3 RNA is primarily detected in the
testis, skin, and brain. Reportedly, Wnt3 is associated with cancer
progression, invasion, and malignant conversion in cancer of the
gastric (39), lungs (40,41),
and hepatocellular carcinoma (42,43).
In ΔPTEN cells, the cyclin D1 expression was upregulated. In
addition, PTEN loss has been reported to augment cyclin D1
expression, although the underlying mechanism remains only
partially understood. However, Zang et al reported that the
Wnt/b-catenin pathway stimulates increased cyclin D1 levels in
lymph node metastasis in papillary thyroid cancer (44). These results suggest an association
between the PTEN-loss-induced Wnt3 expression and the subsequent
upregulation of cyclin D1.
In mammals, two arginase isoforms (Arg1 and Arg2)
have been identified. Arg1 is known as the liver type and is
expressed in hepatocytes (45). In
mice, Arg1 can also be expressed in myeloid cells under the
stimulation of T helper 2 cytokines interleukin (IL)-4 and IL-13
(46), transforming growth factor-b
(47), macrophage-stimulating
protein (48), or GM-CSF (49). Although immune responses are
controlled by amino acid metabolism, Rodriguez et al
reported that arginase produced by tumor-infiltrating macrophages
repressed the expression of the T-cell receptor CD3z, resulting in
the suppression of antigen-specific T-cell responses (46). In this study, it was observed that
Arg1 expression increased following Pten deficiency; however, to
the best of the author's knowledge, there are no reports
demonstrating that Arg1 is highly expressed in any cancer other
than liver cancer. Recent cancer treatment strategies have targeted
arginine metabolism (50,51), thereby making Arg1 an attractive
therapeutic approach for PTEN-deficient tumors.
The present study also analyzed differential
expression of miRNA and observed an increased expression of
mmu-miR-210-3p in the PTEN-deficient cell line. Reportedly, the
expression of miR-210 is induced by HIF-1, which is known to be
upregulated in PTEN-deficient cells (52). The expression levels of the target
genes of miR-210 were not observed to be altered in the present
study. Studies have suggested that miR-210 is involved in multiple
processes, including angiogenesis, metastasis, oncogenesis,
decreased cell division, and tumor suppression (53,54).
However, the exact function remains unclear and requires further
investigation.
In conclusion, the present study established a mouse
prostate cancer cell model of PTEN deficiency and provided evidence
of altered expression of genes associated with the deregulation of
signaling processes implicated in human cancers. Gene expression
profiling suggested enhanced regulation cancer hallmarks, including
cell proliferation, angiogenesis, metastasis, and
immunosuppression. However, further studies are warranted to
dissect and elucidate molecular mechanisms involved in promoting
PTEN-deficient cancer progression.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
AT, KY, SK, AO, HU, MADV, YK, SS, RU, TN and YH
equally took part in the conception and design of the study,
acquisition and interpretation of data, drafting the article and
final approval of the version to be published.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Toker A and Cantley LC: Signalling through
the lipid products of phosphoinositide-3-OH kinase. Nature.
12:673–676. 1997. View
Article : Google Scholar
|
|
2
|
Li J, Yen C, Liaw D, Podsypanina K, Bose
S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al:
PTEN, a putative protein tyrosine phosphatase gene mutated
in human brain, breast, and prostate cancer. Science. 28:1943–1947.
1997. View Article : Google Scholar
|
|
3
|
Steck PA, Pershouse MA, Jasser SA, Yung
WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T,
et al: Identification of a candidate tumour suppressor gene,
MMAC1, at chromosome 10q23.3 that is mutated in multiple
advanced cancers. Nat. Genet. 15:356–362. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tamura M, Gu J, Matsumoto K, Aota S,
Parsons R and Yamada KM: Inhibition of cell migration, spreading,
and focal adhesions by tumor suppressor PTEN. Science.
280:1614–1617. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yamada KM and Araki M: Tumor suppressor
PTEN: Modulator of cell signaling, growth, migration, and
apoptosis. J Cell Sci. 114:2375–2382. 2001.PubMed/NCBI
|
|
6
|
Waite KA and Eng C: Protean PTEN: Form and
function. Am J Hum Genet. 70:829–844. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McCall P, Witton CJ, Grimsley S, Nielsen
KV and Edwards J: Is PTEN loss associated with clinical outcome
measures in human prostate cancer? Br J Cancer. 99:1296–1301. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davies MA: Regulation, role, and targeting
of Akt in cancer. J Clin Oncol. 29:4715–4717. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carnero A and Paramio JM: The
PTEN/PI3K/AKT pathway in vivo, cancer mouse models. Front Oncol.
4:2522014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Velasco MA, Tanaka M, Yamamoto Y,
Hatanaka Y, Koike H, Nishio K, Yoshikawa K and Uemura H: Androgen
deprivation induces phenotypic plasticity and promotes resistance
to molecular targeted therapy in a PTEN-deficient mouse
model of prostate cancer. Carcinogenesis. 35:2142–2153. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamamoto Y, De Velasco MA, Kura Y, Nozawa
M, Hatanaka Y, Oki T, Ozeki T, Shimizu N, Minami T, Yoshimura K, et
al: Evaluation of in vivo responses of sorafenib therapy in a
preclinical mouse model of PTEN-deficient of prostate
cancer. J Transl Med. 8:1502015. View Article : Google Scholar
|
|
12
|
Koike H, Nozawa M, De Velasco MA, Kura Y,
Ando N, Fukushima E, Yamamoto Y, Hatanaka Y, Yoshikawa K, Nishio K
and Uemura H: Conditional PTEN-deficient mice as a prostate cancer
chemoprevention model. Asian Pac J Cancer Prev. 16:1827–1831. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
De Velasco MA, Kura Y, Yoshikawa K, Nishio
K, Davies BR and Uemura H: Efficacy of targeted AKT inhibition in
genetically engineered mouse models of PTEN-deficient
prostate cancer. Oncotarget. 29:15959–15976. 2016.
|
|
14
|
Jia L, Ji S, Maillet JC and Zhang X: PTEN
suppression promotes neurite development exclusively in
differentiating PC12 cells via PI3-kinase and MAP kinase signaling.
J Cell Biochem. 111:1390–1400. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khan FA, Pandupuspitasari NS, Chun-Jie H,
Ao Z, Jamal M, Zohaib A, Khan FA, Hakim MR and Shujun Z:
CRISPR/Cas9 therapeutics: A cure for cancer and other genetic
diseases. Oncotarget. 7:52541–52552. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 8:2281–2308. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizuno S, Hanamura I, Ota A, Karnan S,
Narita T, Ri M, Mizutani M, Goto M, Gotou M, Tsunekawa N, et al:
Overexpression of salivary-type amylase reduces the sensitivity to
bortezomib in multiple myeloma cells. Int J Hematol. 102:569–578.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mi H, Huang X, Muruganujan A, Tang H,
Mills C, Kang D and Thomas PD: PANTHER version 11: Expanded
annotation data from Gene Ontology and Reactome pathways, and data
analysis tool enhancements. Nucleic Acids Res. 45:D183–D189. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ito S, D'Alessio AC, Taranova OV, Hong K,
Sowers LC and Zhang Y: Role of Tet proteins in 5mC to 5hmC
conversion, ES cell self renewal and inner cell mass specification.
Nature. 26:1129–1133. 2010. View Article : Google Scholar
|
|
21
|
Gawlik-Rzemieniewska N and Bednarek I: The
role of NANOG transcriptional factor in the development of
malignant phenotype of cancer cells. Cancer Biol Ther. 17:1–10.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gasparotto D, Polesel J, Marzotto A,
Colladel R, Piccinin S, Modena P, Grizzo A, Sulfaro S, Serraino D,
Barzan L, et al: Overexpression of TWIST2 correlates with poor
prognosis in head and neck squamous cell carcinomas. Oncotarget.
2:1165–1175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Y, Wang W, Wang W, Yang R, Wang T, Su
T, Weng D, Tao T, Li W, Ma D, et al: Correlation of TWIST2
up-regulation and epithelial-mesenchymal transition during
tumorigenesis and progression of cervical carcinoma. Gynecol Oncol.
124:112–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mao Y, Xu J, Song G and Yin H: Twist2
promotes ovarian cancer cell survival through activation of Akt.
Oncol Lett. 6:169–174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang
SY, Liu CJ, Teng SC and Wu KJ: Direct regulation of TWIST by
HIF-1alpha promotes metastasis. Nat Cell Biol. 10:295–305. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yamada Y, Nezu J, Shimane M and Hirata Y:
Molecular cloning of a novel vascular endothelial growth factor,
VEGF-D. Genomics. 42:483–488. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Achen MG, Jeltsch M, Kukk E, Mäkinen T,
Vitali A, Wilks AF, Alitalo K and Stacker SA: Vascular endothelial
growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF
receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci
USA. 95:548–553. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Trompezinski S, Berthier-Vergnes O, Denis
A, Schmitt D and Viac J: Comparative expression of vascular
endothelial growth factor family members, VEGF-B, -C and -D, by
normal human keratinocytes and fibroblasts. Exp Dermatol.
13:198–105. 2004. View Article : Google Scholar
|
|
29
|
Debinski W, Slagle-Webb B, Achen MG,
Stacker SA, Tulchinsky E, Gillespie GY and Gibo DM: VEGF-D is an
X-linked/AP-1 regulated putative onco-angiogen in human
glioblastoma multiforme. Mol Med. 7:598–608. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Achen MG, Williams RA, Minekus MP,
Thornton GE, Stenvers K, Rogers PA, Lederman F, Roufail S and
Stacker SA: Localization of vascular endothelial growth factor-D in
malignant melanoma suggests a role in tumour angiogenesis. J
Pathol. 193:147–154. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
White JD, Hewett PW, Kosuge D, McCulloch
T, Enholm BC, Carmichael J and Murray JC: Vascular endothelial
growth factor-D expression is an independent prognostic marker for
survival in colorectal carcinoma. Cancer Res. 62:1669–1675.
2002.PubMed/NCBI
|
|
32
|
Nakamura Y, Yasuoka H, Tsujimoto M, Yang
Q, Imabun S, Nakahara M, Nakao K, Nakamura M, Mori I and Kakudo K:
Prognostic significance of vascular endothelial growth factor D in
breast carcinoma with longterm follow-up. Clin Cancer Res.
9:716–721. 2003.PubMed/NCBI
|
|
33
|
Van Trappen PO, Steele D, Lowe DG, Baithun
S, Beasley N, Thiele W, Weich H, Krishnan J, Shepherd JH, Pepper
MS, et al: Expression of vascular endothelial growth factor
(VEGF)-C and VEGF-D, and their receptor VEGFR-3, during different
stages of cervical carcinogenesis. J Pathol. 201:544–554. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tian T, Nan KJ, Wang SH, Liang X, Lu CX,
Guo H, Wang W and Ruan ZP: PTEN regulates angiogenesis and VEGF
expression through phosphatase-dependent and -independent
mechanisms in HepG2 cells. Carcinogenesis. 31:1211–1219. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kaushal V, Mukunyadzi P, Dennis RA, Siegel
ER, Johnson DE and Kohli M: Stage-specific characterization of the
vascular endothelial growth factor axis in prostate cancer:
Expression of lymphangiogenic markers is associated with
advanced-stage disease. Clin Cancer Res. 11:584–593.
2005.PubMed/NCBI
|
|
36
|
Netto GJ: Molecular updates in prostate
cancer. Surg Pathol. 8:561–580. 2015. View Article : Google Scholar
|
|
37
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
He S, Lu Y, Liu X, Huang X, Keller ET,
Qian CN and Zhang J: Wnt3a: Functions and implications in cancer.
Chin J Cancer. 34:554–562. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang HS, Nie X, Wu RB, Yuan HW, Ma YH, Liu
XL, Zhang JY, Deng XL, Na Q, Jin HY, et al: Downregulation of human
Wnt3 in gastric cancer suppresses cell proliferation and induces
apoptosis. Onco Targets Ther. 27:3849–3860. 2016. View Article : Google Scholar
|
|
40
|
Nakashima N, Liu D, Huang CL, Ueno M,
Zhang X and Yokomise H: Wnt3 gene expression promotes tumor
progression in non-small cell lung cancer. Lung Cancer. 76:228–234.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xing Z, Wang HY, Su WY, Liu YF, Wang XX,
Zhan P, Lv TF and Song Y: Wnt3 knockdown sensitizes human non-small
cell type lung cancer (NSCLC) cells to cisplatin via regulating the
cell proliferation and apoptosis. Eur Rev Med Pharmacol Sci.
22:1323–1332. 2018.PubMed/NCBI
|
|
42
|
Kim M, Lee HC, Tsedensodnom O, Hartley R,
Lim YS, Yu E, Merle P and Wands JR: Functional interaction between
Wnt3 and Frizzled-7 leads to activation of the Wnt/beta-catenin
signaling pathway in hepatocellular carcinoma cells. J Hepatol.
48:780–791. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nambotin SB, Tomimaru Y, Merle P, Wands JR
and Kim M: Functional consequences of WNT3/Frizzled7-mediated
signaling in non-transformed hepatic cells. Oncogenesis.
22:e312012. View Article : Google Scholar
|
|
44
|
Zhang J, Gill AJ, Issacs JD, Atmore B,
Johns A, Delbridge LW, Lai R and McMullen TP: The Wnt/β-catenin
pathway drives increased cyclin D1 levels in lymph node metastasis
in papillary thyroid cancer. Hum Pathol. 43:1044–1050. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bronte V and Zanovello P: Regulation of
immune responses by L-arginine metabolism. Nat Rev Immunol.
5:641–654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rodriguez PC, Quiceno DG, Zabaleta J,
Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J,
Sotomayor EM, et al: Arginase I production in the tumor
microenvironment by mature myeloid cells inhibits T-cell receptor
expression and antigen-specific T-cell responses. Cancer Res.
64:5839–5849. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Boutard V, Havouis R, Fouqueray B,
Philippe C, Moulinoux JP and Baud L: Transforming growth factor-β
stimulates arginase activity in macrophages. Implications for the
regulation of macrophage cytotoxicity. J Immunol. 155:2077–2084.
1995.PubMed/NCBI
|
|
48
|
Morrison AC and Correll PH: Activation of
the stem cell derived tyrosine kinase/RON receptor tyrosine kinase
by macrophage-stimulating protein results in the induction of
arginase activity in murine peritoneal macrophages. J Immunol.
168:853–860. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jost MM, Ninci E, Meder B, Kempf C, Van
Royen N, Hua J, Berger B, Hoefer I, Modolell M and Buschmann I:
Divergent effects of GM-CSF and TGFbeta1 on bone marrow-derived
macrophage arginase-1 activity, MCP-1 expression, and matrix
metalloproteinase-12: A potential role during arteriogenesis. FASEB
J. 17:2281–2283. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Qiu F, Huang J and Sui M: Targeting
arginine metabolism pathway to treat arginine-dependent cancers.
Cancer Lett. 364:1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fultang L, Vardon A, De Santo C and Mussai
F: Molecular basis and current strategies of therapeutic arginine
depletion for cancer. Int J Cancer. 139:501–509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Huang X, Ding L, Bennewith KL, Tong RT,
Welford SM, Ang KK, Story M, Le QT and Giaccia AJ: Hypoxia
inducible mir-210 regulates normoxic gene expression involved in
tumor initiation. Mol Cell. 35:856–867. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dang K and Myers KA: The role of
hypoxia-induced miR-210 in cancer progression. Int J Mol Sci.
16:6353–6372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ying Q, Liang L, Guo W, Zha R, Tian Q,
Huang S, Yao J, Ding J, Bao M, Ge C, et al: Hypoxia-inducible
microRNA-210 augments the metastatic potential of tumor cells by
targeting vacuole membrane protein 1 in hepatocellular carcinoma.
Hepatology. 54:2064–2075. 2011. View Article : Google Scholar : PubMed/NCBI
|