Introduction
Pancreatic cancer is widely recognized as one of the
most aggressive malignancies of the digestive system, which is
associated with a poor prognosis. The American Cancer Society
estimates that 55,440 Americans will be diagnosed with pancreatic
cancer in 2018, and that 44,330 will succumb to the disease
(1). It is currently listed as the
fourth leading cause of cancer-associated mortality and is
predicted to become the second by 2030 in the USA (2). In China, 90,100 new cases of
pancreatic cancer and 79,400 cases of pancreatic cancer-associated
mortality were estimated to occur in 2015 (3). Despite considerable advances in
surgical and oncological treatment in recent years, the overall
5-year survival rate for pancreatic cancer is still ~8%. One of the
main factors contributing to the poor prognosis of pancreatic
cancer is late diagnosis, when it has already metastasized to
distant organ sites and is unresectable. In addition, there remains
a lack of an effective therapy for metastatic pancreatic cancer;
therefore, the current status of diagnosis and treatment of
pancreatic cancer is challenging. There is an urgent need to
elucidate the underlying mechanisms of pancreatic cancer, which
regulate its pathogenesis and progression, and provide useful
information for the clinical management of pancreatic cancer.
MicroRNAs (miRNAs/miRs) are non-coding RNA
molecules, 18–22 bases in length, which post-transcriptionally
regulate gene expression through binding to a sequence in the
3′-untranslated region (3′UTR), which has partial or full
complementarity. It has been indicated that miRNAs serve an
important role in regulating cancer-associated processes, such as
proliferation, cell cycle progression, apoptosis, angiogenesis,
invasion and metastasis (4).
According to the distinct cellular function of their targets in
different tumors, miRNAs may function as oncogenes or tumor
suppressors in the development of cancer. miR-148a is a member of
the miR-148/152 family, which is located at chromosome 7p15.2. In
previous studies, miR-148a has been identified as a tumor
suppressor miRNA that is downregulated in various types of cancer,
including breast cancer (5),
gastrointestinal cancers (6,7),
cholangiocarcinoma (8) and
hepatocellular carcinoma (9).
miR-148a is also downregulated in pancreatic cancer tissues and
several pancreatic cell lines (10,11).
In addition, DNA methyltransferase 1 (DNMT1) (12), cholecystokinin B receptor (13), B-cell lymphoma 2 (13), cell division cycle 25B (14) and erb-b2 receptor tyrosine kinase 3
(15) have been identified as
target genes of miR-148a.
DNA methylation is one of the epigenetic mechanisms
affecting cell fate via the regulation of gene expression. Aberrant
hypermethylation of CpG sites is often involved in silencing of
tumor suppressor genes (TSGs). Furthermore, previous studies have
identified CpG island methylation-associated silencing of miRNAs
with tumor suppressor features in human cancer (16,17).
DNMT1 is responsible for the maintenance of the pattern of DNA
methylation. Previous reports have demonstrated that DNMT1 is
overexpressed in pancreatic cancer tissues and cell lines;
furthermore, increased DNMT1 expression is markedly correlated with
poor prognosis (18,19). DNMT1 has also been verified as a
target for miR-148a in gastric (20) and breast cancer (5), and hepatocellular carcinoma (21) and pancreatic cancer (12). Furthermore, hypermethylation of the
miR-148a promoter is associated with DNMT1 overexpression in
pancreatic cancer (12). Therefore,
the interaction between miR-148a and DNMT1 may regulate the
expression of TSGs in pancreatic cancer.
Our previous study demonstrated that miR-148a is
markedly downregulated in human pancreatic ductal adenocarcinoma
(PDAC) cell lines and tissues. In addition, the downregulation of
miR-148a is associated with poor prognosis and
epithelial-mesenchymal transition (EMT). Conversely, restoration of
miR-148a suppresses EMT and invasion of pancreatic cancer cells by
targeting Wnt10b and inhibiting the Wnt/β-catenin signaling pathway
(22). The present study aimed to
reveal the interaction between miR-148a and DNMT1, and its effects
on TSG expression, cell proliferation, migration and invasion of
pancreatic cancer cells. The findings may provide novel insights
into the upstream and downstream regulatory mechanisms of miR-148a
in pancreatic cancer, and may identify a potential target for the
future treatment of pancreatic cancer.
Materials and methods
Tissue samples
A total of 30 paired PDAC and adjacent non-tumorous
tissues (ANT) were obtained from patients with PDAC who underwent
surgery between January 2013 and December 2015 at The First
Affiliated Hospital of Nanchang University (Nanchang, China). None
of the patients received radiotherapy or chemotherapy prior to
surgery. The samples were snap-frozen and stored at −80°C until
analysis. Written informed consent was obtained from all
participants prior to recruitment. The present study was approved
by the Ethics Committee of The First Affiliated Hospital of
Nanchang University.
Cell lines and cell culture
The normal human pancreatic ductal epithelial cell
line HPDE and the pancreatic cancer cell line AsPC-1 were obtained
from the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). AsPC-1 cells were routinely cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.),
penicillin (100 U/ml) and streptomycin (100 µg/ml) in an incubator
containing 95% air and 5% CO2.
5-Aza-2′-deoxycytidine (5-Aza-CdR)
treatment
AsPC-1 cells were seeded in 6-well plates
(5×105 cells/well), and were treated with 5 µM 5-Aza-CdR
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 72 h; 5-Aza-CdR
containing medium was replaced every 24 h. Exponentially growing
cells were used for subsequent assays.
RNA interference
To silence DNMT1 expression, DNMT1-small interfering
RNA (siRNA) (Ambion; Thermo Fisher Scientific, Inc.) was
transfected into AsPC-1 cells. The DNMT1 siRNA sequences were as
follows: Sense, 5′-AGAUGACGGAUGCCUAGAGUU-3′; antisense,
5′-CUCUAGGCAUCCGUCAUCUUU-3′. Negative control (NC) siRNA was also
purchased from Ambion; Thermo Fisher Scientific, Inc. (cat. no.
4390843). The final siRNA concentration was 10 nM. Cells were
plated at 2×105 cells/well in a 6-well plate overnight,
and were then transfected with siRNA at room temperature using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. Cells
were collected for RNA and protein extraction or for further assays
48 h post-transfection.
Synthetic miRNA transfection
AsPC-1 cells were seeded at 2×105 cells/well into a
12-well plate, and were incubated overnight, after which, the cells
were transfected with 100 nM miR-148a mimics (cat. no. 4464066) or
mimics-NC (cat. no. 4464058; Ambion; Thermo Fisher Scientific,
Inc.) for 48 h at room temperature using Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Cells transfected with
Lipofectamine® only were considered a blank control.
Cells were collected for RNA and protein extraction, or for further
assays, 48 h post-transfection.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from AsPC-1 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and was quantified using a NanoDrop spectrophotometer
(NanoDrop; Thermo Fisher Scientific, Inc., Wilmington, DE, USA).
Total RNA (2 µg) was used as a template for synthesizing cDNA using
the M-MLV Reverse Transcriptase kit (Takara Bio, Inc., Otsu,
Japan), according to the manufacturer's protocol. qPCR was
performed using the Applied Biosystems 7500 Fast Real-Time PCR
system with the SYBR-Green PCR kit (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) to detect DNMT1 and
miR-148a expression. The thermocycling conditions were as follows:
94°C for 4 min, followed by 40 cycles at 94°C for 30 sec, 50°C for
30 sec and 72°C for 40 sec, followed by final extension at 72°C for
5 min. β-actin and human U6 RNA served as internal control genes,
respectively. Quantification cycle (Cq) values were used to
quantify the expression levels of each gene, and mRNA or miRNA
levels were calculated according to the 2−∆∆Cq method
(23). The primer sequences are
shown in Table I.
 | Table I.Primer sequences for reverse
transcription-quantitative PCR, BSP and methylation-specific
PCR. |
Table I.
Primer sequences for reverse
transcription-quantitative PCR, BSP and methylation-specific
PCR.
| Gene | Sequence
(5′-3′) |
|---|
| DNMT1 | F:
CGTGACATTAAGGAGAAGCTG |
|
| R:
CTAGAAGCATTTGCGGTGGAC |
| β-actin | F:
AACCTTCACCTAGCCCCAG |
|
| R:
CTCATCCGATTTGGCTCTTCA |
| miR-148a | F:
TGCGGTCAGTGCACTACAGAAC |
|
| R:
CCAGTGCAGGGTCCGAGGT |
| U6 | F:
CTCGCTTCGGCAGCACA |
|
| R:
AACGCTTCACGAATTTGCGT |
| miR-148a BSP | F:
ATAGGTAGTTTGGTAGAGGTTGGTG |
|
| R:
TATCAAATCAACAAATTCCCTCC |
| miR-148a-M | F:
TAGGGGAGGTTTCGTAAAGC |
|
| R:
CACGAAAACGAATATTCGAAA |
| miR-148a-U | F:
TTTTAGGGGAGGTTTTGTAAAGT |
|
| R:
ACACAAAAACAAATATTCAAAACT |
| RASSF1A-M | F:
GTGTTAACGCGTTGCGTATC |
|
| R:
AACCCCGCGAACTAAAAACGA |
| RASSF1A-U | F:
TTTGGTTGGAGTGTGTTAATGTG |
|
| R:
CAAACCCCACAAACTAAAAACAA |
| p16-M | F:
TTATTAGAGGGTGGGGCGGATCGC |
|
| R:
GACCCCGAACCGCGACCGTAA |
| p16-U | F:
TTATTAGAGGGTGGGGTGGATTGT |
|
| R:
CAACCCCAAACCACAACCATAA |
| ppENK-M | F:
TGTGGGGAGTTATCGAGC |
|
| R:
GCCTTCGCGAAAAAAATCG |
| ppENK-U | F:
TTGTGTGGGGAGTTATTGAGT |
|
| R:
CACCTTCACAAAAAAAATCAATC |
Protein extraction and western blot
analysis
Total proteins were extracted with
radioimmunoprecipitation assay lysis buffer (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and the protein concentration
of each sample was determined using the Bicinchoninic Acid Protein
Assay kit (Thermo Fisher Scientific, Inc.). Equal amounts of
protein (50 µg/lane) were subjected to 10% SDS-PAGE, and were then
transferred to nitrocellulose membranes (EMD Millipore, Billerica,
MA, USA). After blocking with 5% skimmed milk in Tris-buffered
saline-Tween (TBST; 20 mM Tris-HCl, 150 Mm NaCl, 0.1% Tween-20; pH
7.6) for 5 h at room temperature, the membranes were incubated with
primary antibodies overnight at 4°C. After washing with TBST,
membranes were incubated with the appropriate horseradish
peroxidase-conjugated secondary antibody (1:2,000, cat. no.
ab205718; Abcam, Cambridge, MA, USA) for 2 h at room temperature.
Finally, the proteins were visualized using enhanced
chemiluminescence reagents (EMD Millipore). Band intensity was
semi-quantified using Quantity One image analysis software version
4.62 (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The primary
antibodies used were as follows: DNMT1 (1:500, cat. no. ab188453;
Abcam), p16 (1:200, cat. no. ab51243; Abcam), preproenkephalin
(ppENK; 1:100, cat. no. ab98128; Abcam) and Ras association domain
family member 1 (RASSF1A; 1:200, cat. no. ab97749; Abcam).
β-tubulin (1:500, cat. no. ab151318; Abcam) was used as an internal
loading control.
DNA extraction, methylation-specific
PCR (MSP) and bisulfite sequencing PCR (BSP)
Genomic DNA of tissues and cells was extracted using
QIAamp DNA Mini kit (Qiagen GmbH, Hilden, German) and quantified
using the Quantifiler Human DNA Quantification kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Aliquots of genomic
DNA were treated with sodium bisulfite using the EZ DNA
Methylation-Gold™ kit (Zymo Research Corp., Irvine, CA, USA),
according to the manufacturer's protocol. The methylation status of
miR-148a and three TGSs (p16, ppENK and RASSF1A) was analyzed by
MSP. The methylated and unmethylated primers are listed in Table I. Briefly, PCR was conducted in a 50
µl volume containing 10 pmol/µl each primer (1 µl), 10 ng/µl
bisulfte-modifed DNA (5 µl), 10X PCR buffer (5 µl), 10 nM dNTPs (1
µl), 5 U/µl Taq DNA polymerase (0.5 µl; Thermo Fisher Scientific,
Inc.), 25 mM MgCl2 (5 µl) and ddH2O (31.5
µl). Amplification was performed using a thermocycler under the
following conditions: 95°C for 5 min, followed by 35 cycles at 95°C
for 30 sec, 60°C for 60 sec and 72°C for 30 sec, followed by an
extension at 72°C for 7 min. PCR products underwent 3% agarose gel
electrophoresis containing ethidium bromide (Thermo Fisher
Scientific, Inc.) and were then visualized by ultraviolet
illumination. Furthermore, the methylation level of the miR-148a
promoter in AsPC-1 cells treated with 5-Aza-CdR or transfected with
DNMT1-siRNA was analyzed by BSP. The BSP primer was designed by
Methprimer (http://www.urogene.org/methprimer/), according to the
sequence of the miR-148a promoter (2 kb, NC_000007.14:
c25951986-25949987), and the sequence is listed in Table I. The product size was 246 bp with
29 CpGs. PCR amplification was performed according to the protocol
of MSP. Subsequently, the amplified PCR products were purified and
cloned into the pMD19-T vector (Takara Bio, Inc.); five clones of
each cell were sequenced using Sanger sequencing methods (24) by Sangon Biotech Co., Ltd. (Shanghai,
China). The percentage of methylation was calculated
comprehensively and comparatively using CpG viewer, QUMA and
Biq-analyzer (25,26).
Dual luciferase reporter assay
A dual luciferase reporter assay was performed as
previously described (22). The
3′UTR mRNA sequence of the DNMT1 gene containing the miR-148a
binding site was amplified by PCR, in a total volume of 25 µl
containing 100 ng human genomic DNA, and cloned into the pmirGLO
vector (Promega Corporation, Madison, WI, USA) [DNMT1 3′UTR-wild
type (wt)]. The human genomic DNA was extracted from the peripheral
blood of healthy volunteers after obtaining written informed
consent. Mutant DNMT1 3′UTR was generated using the
overlap-extension PCR method (27)
and was cloned into the pmirGLO vector [DNMT1 3′UTR-mutant (mut)].
Subsequently, the AsPC-1 cells (1×105) were seeded in a
48-well plate and co-transfected with DNMT1 3′UTR-wt or DNMT1
3′UTR-mut vector (100 ng) with miR-148a mimics or mimics-NC (100
nM) using Lipofectamine® 2000 at room temperature.
Luciferase activity was detected using the Dual-Luciferase Reporter
Assay system (Promega Corporation) 48 h post-transfection; the
relative luciferase activity was calculated as a ratio of Firefly
luciferase activity versus Renilla luciferase activity. The
experiments were performed independently in triplicate.
Cell proliferation analysis
Cell proliferation was detected by MTT assay
(Sigma-Aldrich; Merck KGaA). Briefly, AsPC-1 cells
(5×103 cells/well in 96-well plates) were transfected
with Lipofectamine® as a blank control, mimics-NC or
miR-148a mimics. After 24, 48, 72, 96 and 120 h, cells were
incubated with MTT (5 mg/ml) for 4 h at 37°C, the supernatants were
discarded and the insoluble formazan was dissolved in dimethyl
sulfoxide. A microplate reader (Bio-Rad Laboratories, Inc.) was
used to determine the optical density (OD) at 490 nm. Inhibition
rate (%) = (OD 490 value of blank - OD 490 value of miR-148a
mimics)/(OD 490 value of blank) × 100%. All experiments were
repeated three times, and the average results were calculated.
Cell migration and invasion
assays
The migratory and invasive abilities of AsPC-1 cells
were examined using a Transwell chamber (Corning Incorporated,
Corning, NY, USA). Transwell filters coated with Matrigel were used
for the tumor cell invasion assay, whereas Transwell filters
without Matrigel were used for the migration assay. Briefly,
5×105 AsPC-1 cells in serum-free DMEM were plated in the
upper chamber, and DMEM containing 10% FBS was added to the bottom
chamber. After 24 h at 37°C, cells that remained in the upper
chamber were carefully removed using cotton wool. The cells that
had invaded or migrated to the lower surface of the membrane and
the bottom chambers were fixed with methanol for 30 min, and
stained with 0.1% crystal violet for 20 min at room temperature.
Subsequently, the number of stained cells was counted and observed
under an inverted light microscope. Five fields were randomly
selected and the number of cells was expressed as the mean. The
experiment was repeated three times.
Statistical analysis
Statistical analyses were performed using SPSS 22.0
software (IBM Corp., Armonk, NY, USA). Measurement data were
expressed as the means ± standard deviation of three replicate
experiments. Comparative data between the groups were assessed
using a two-tailed Student's t-test, and comparisons among multiple
groups were performed using one-way analysis of variance followed
by the least significant difference post hoc test. Spearman's
correlation test was used for correlation analyses. P<0.05 was
considered to indicate a statistically significant difference.
Results
Overexpression of DNMT1 is responsible
for hypermethylation of the miR-148a gene promoter
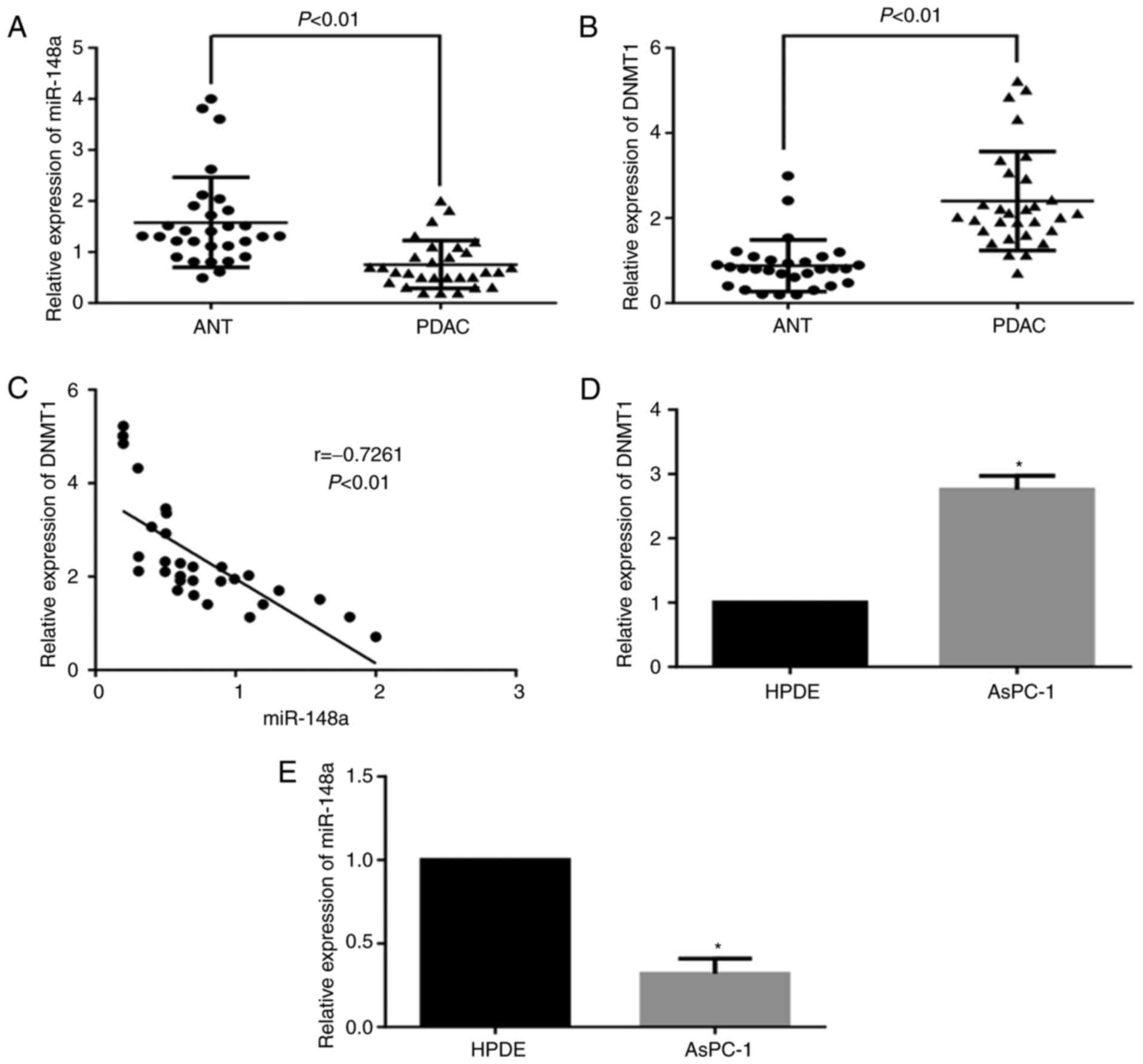
The expression levels of miR-148a were significantly
decreased in PDAC specimens compared with in ANT specimens
(Fig. 1A), whereas the expression
of DNMT1 was significantly increased in PDAC specimens (Fig. 1B). Furthermore, the expression of
miR-148a was negatively correlated with DNMT1 expression
(r=−0.7261, P<0.01; Fig. 1C).
The expression levels of DNMT1 were markedly higher in AsPC-1 cells
than in HPDE cells, whereas the expression levels of miR-148a were
reduced (Fig. 1D and E). To verify
the hypotheses that overexpression of DNMT1 is responsible for the
downregulation of miR-148a in pancreatic cancer, the present study
assessed the methylation status of the miR-148a promoter by MSP in
PDAC tissues and AsPC-1 cells. The results indicated that the
positive rate of miR-148a methylation was significantly higher in
PDAC tissues than in ANT tissues (76.7 vs. 16.7%, P<0.01;
Fig. 2A). In addition, the
methylation status of the miR-148a gene promoter was positive in
AsPC-1 cells, whereas it was negative in HPDE cells (Fig. 2B). After 5-Aza-CdR treatment or
DNMT1-siRNA transfection, the expression of miR-148a was
significantly increased alongside downregulation of DNMT1, and the
miR-148a promoter was unmethylated in AsPC-1 cells (Fig. 2C and D). Furthermore, the methylated
CpG sites of the miR-148a promoter were significantly decreased in
AsPC-1 cells following 5-Aza-CdR treatment or DNMT1-siRNA
transfection (P<0.01 Fig. 2E).
These results suggested that hypermethylation of the miR-148a
promoter suppressed its expression, and overexpression of DNMT1 may
be responsible for hypermethylation of the miR-148a promoter in
pancreatic cancer.
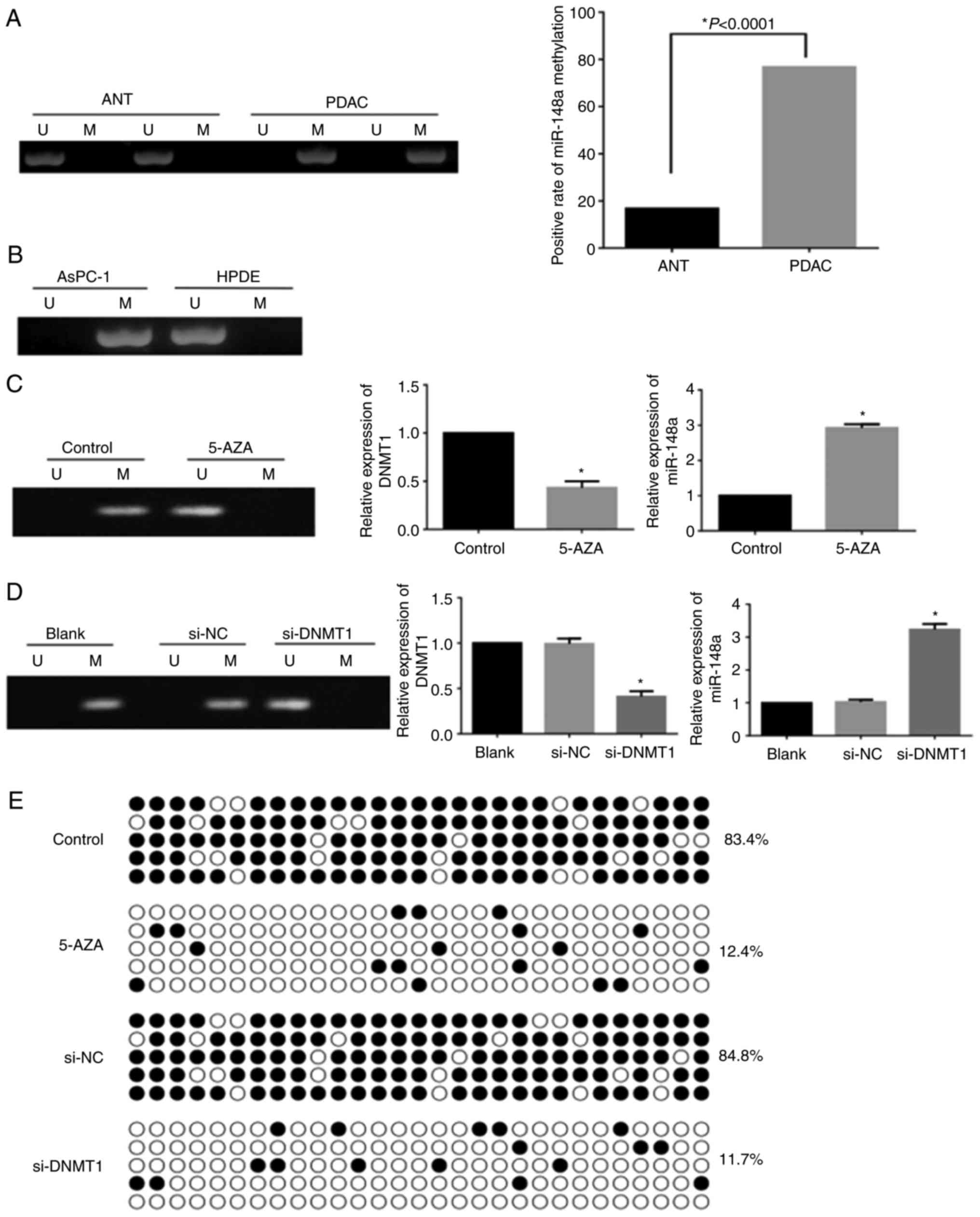 | Figure 2.Methylation status of the miR-148a
promoter. (A) Methylation status of the miR-148a promoter in ANT
and PDAC specimens. (B) Methylation status of the miR-148a promoter
in HPDE and AsPC-1 cells. (C) Effects of 5-Aza-CdR on methylation
status of the miR-148a promoter, and DNMT1 and miR-148a expression
(*P<0.01). (D) Effects of si-DNMT1 on methylation status of the
miR-148a promoter, and DNMT1 and miR-148a expression (*P<0.01).
(E) Bisulfite sequencing polymerase chain reaction analysis of CpG
sites in the miR-148a promoter in AsPC-1 cells with or without
5-Aza-CdR treatment and si-DNMT1 transfection. Open and filled
circles represent U and M CpG sites, respectively. Each horizontal
row represents a single clone. There are 29 CpG sites. 5-AZA,
5-Aza-2′-deoxycytidine; ANT, adjacent non-tumorous; DNMT1, DNA
methyltransferase 1; M, methylated; miR-148a, microRNA-148a; NC,
negative control; PDAC, pancreatic ductal adenocarcinoma; si, small
interfering RNA; U, unmethylated. |
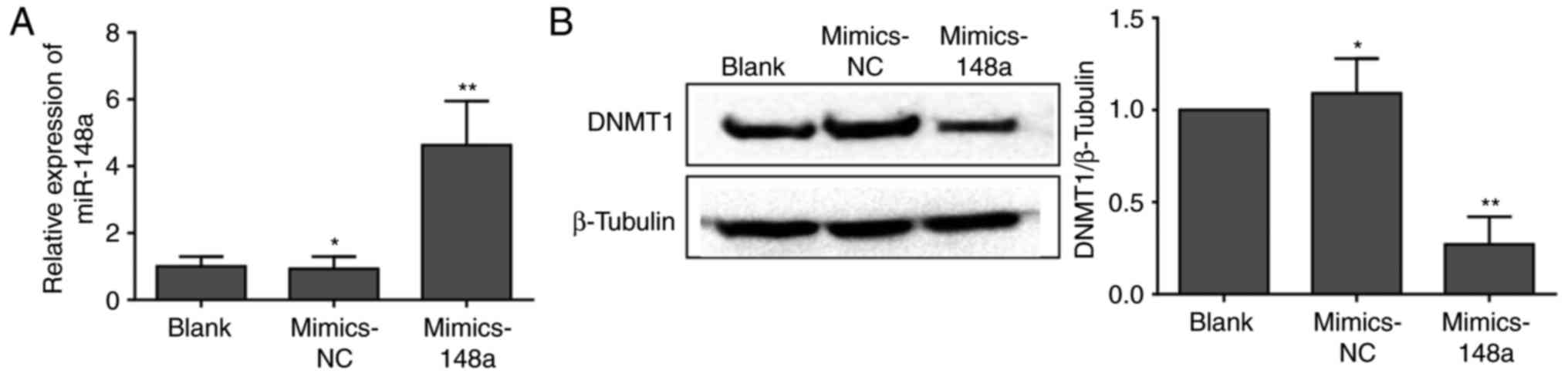
Restoration of miR-148a expression
suppresses DNMT1 expression in vitro
To investigate the effects of miR-148a on the
regulation of DNMT1 expression, AsPC-1 cells in the control groups
were transfected with Lipofectamine® (blank) or
mimics-NC, whereas cells in the experimental group were transfected
with miR-148a mimics. As shown in Fig.
3A, miR-148a expression was significantly higher following
miR-148a mimics transfection. To further investigate a
miRNA-dependent mechanism of DNMT1 regulation, the protein
expression levels of DNMT1 were detected by western blot analysis
following transfection with miR-148a mimics. As expected, DNMT1
expression was significantly decreased in AsPC-1 cells transfected
with miR-148a mimics (Fig. 3B).
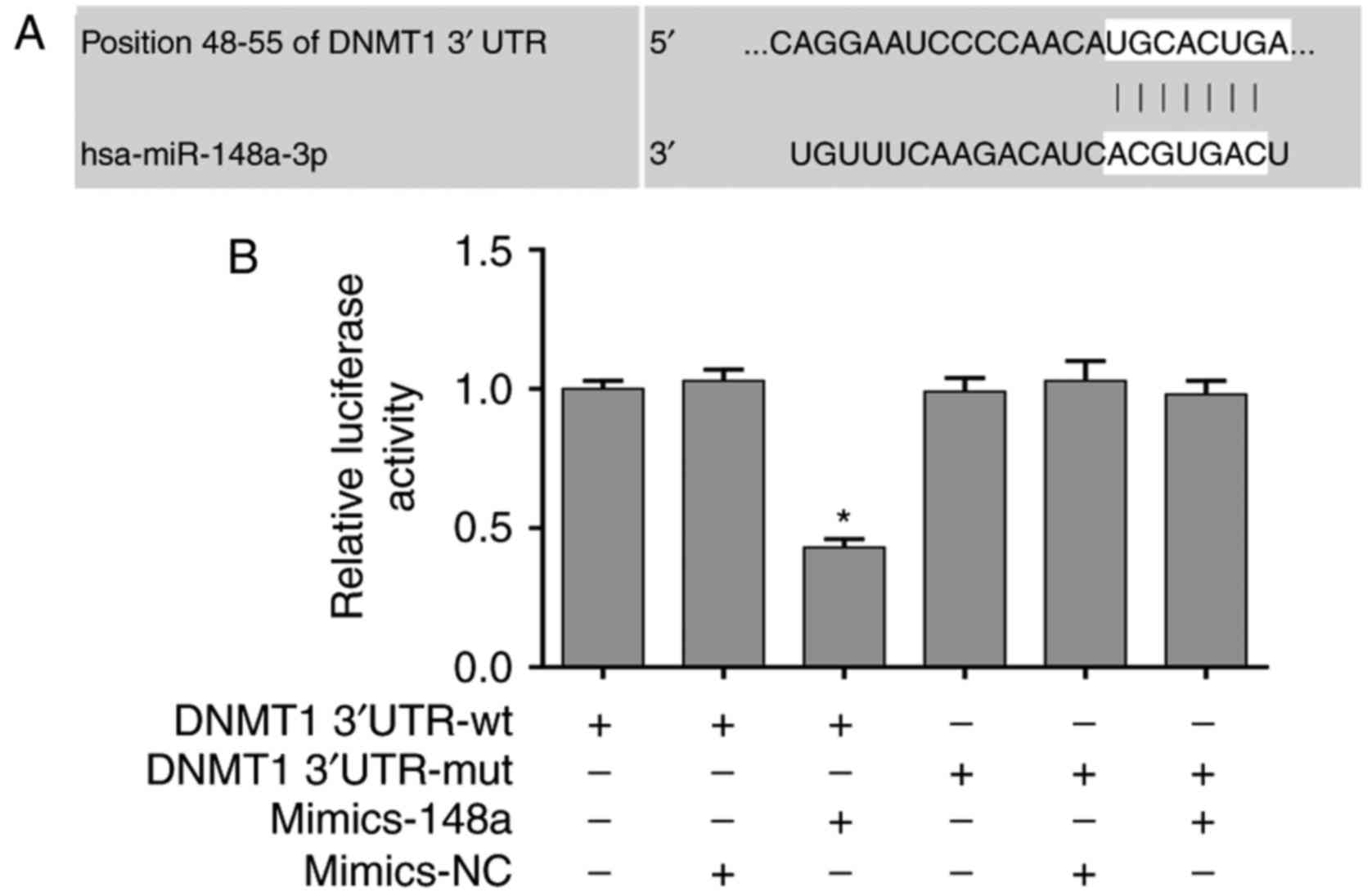
miR-148a directly targets DNMT1
3′UTR
The present study revealed that DNMT1 expression was
significantly inhibited by miR-148a in AsPC-1 cells. In addition,
DNMT1 was identified as a potential target of miR-148a using the
bioinformatics prediction tools TargetScan 7.1 (http://www.targetscan.org) and miRBase (http://www.mirbase.org/) (Fig. 4A). Therefore, a dual luciferase
reporter assay was performed to validate direct binding of miR-148a
to DNMT1 mRNA 3′UTR. The results indicated that miR-148a mimics
significantly decreased the luciferase activity of the DNMT1
3′UTR-wt vector, but did not affect the activity of the DNMT1
3′UTR-mut vector (Fig. 4B), thus
suggesting that the 3′UTR of DNMT1 was a functional target site for
miR-148a-induced silencing of DNMT1.
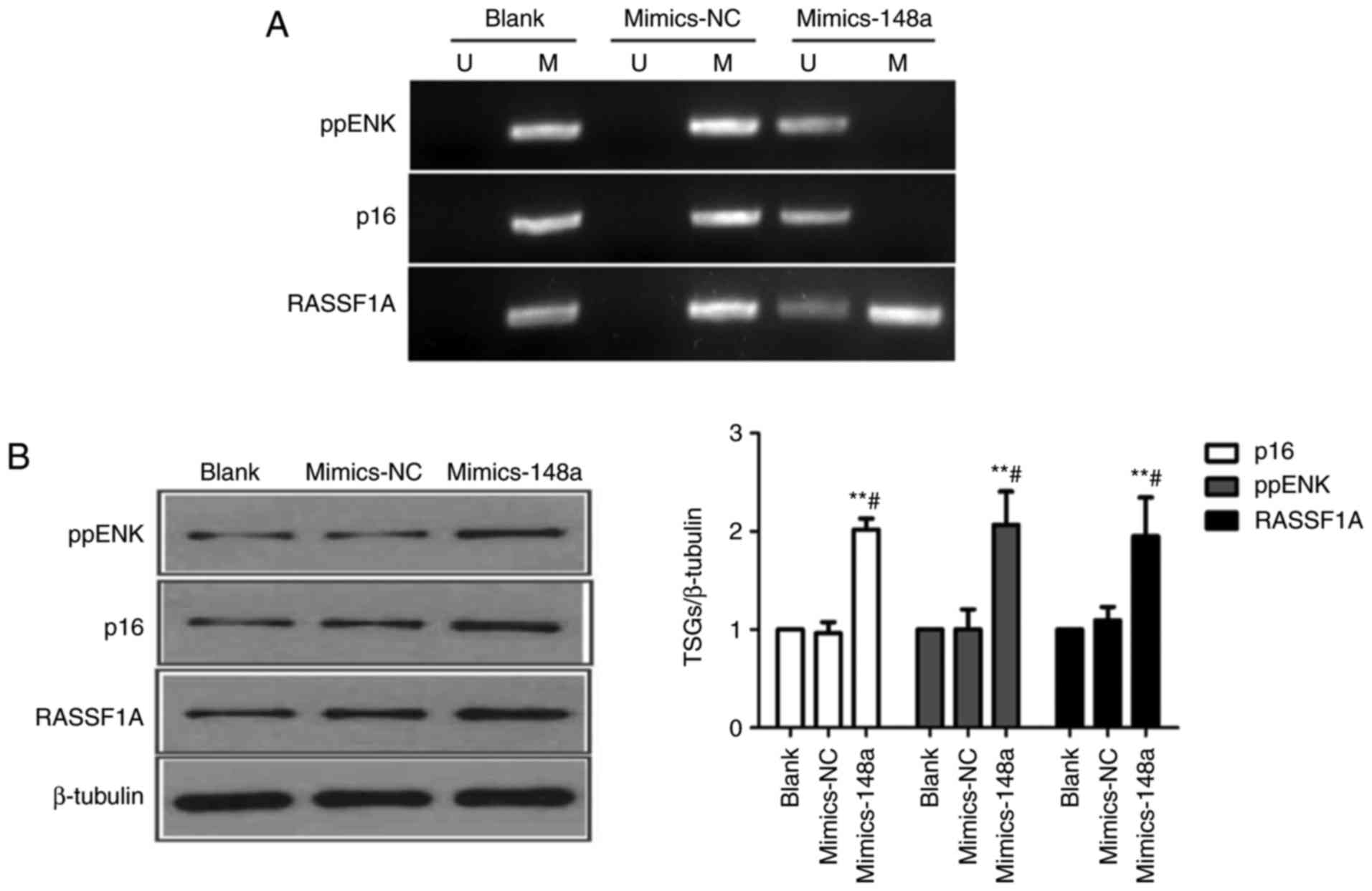
Restoration of miR-148a decreases the methylation
levels of p16, ppENK and RASSF1A promoter regions, and reactivates
their expression in AsPC-1 cells. DNMT1 is constitutively expressed
in proliferating cells, and functions as a maintenance enzyme to
ensure that methylation patterns are copied to daughter cells
during DNA replication (28). To
further investigate whether miR-148a could reactivate TSGs by
targeting DNMT1, AsPC-1 cells were transfected with
Lipofectamine® (blank), mimics-NC or miR-148a mimics.
Subsequently, the methylation levels of three pancreatic
cancer-associated TSGs, p16, ppENK and RASSF1A, were detected by
MSP. As shown in Fig. 5, the
methylation status of p16, ppENK and RASSF1A was positive. In
addition, the basal expression levels of these three proteins were
detected by western blot analysis in AsPC-1 cells. The results
indicated that restoration of miR-148a altered the methylation
status of p16, ppENK and RASSF1A promoter regions (p16 and ppENK,
from methylated to unmethylated; RASSF1A, from methylated to
partially methylated), and significantly increased the protein
expression levels of p16, ppENK and RASSF1A in AsPC-1 cells
(Fig. 5A and B).
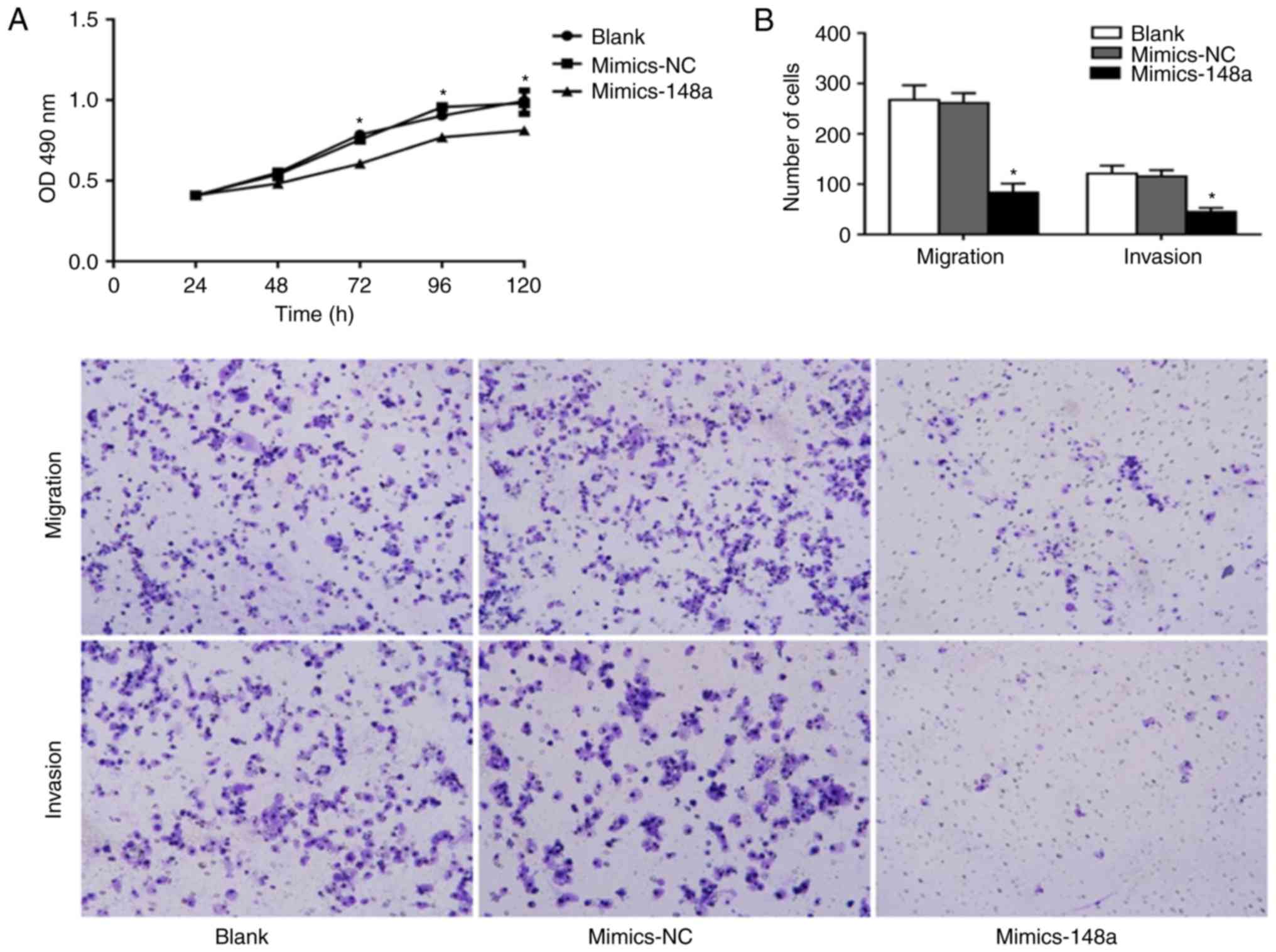
miR-148a inhibits cell proliferation,
migration and invasion of AsPC-1 cells
To determine the effects of miR-148a on cell
proliferation, migration and invasion, AsPC-1 cells were
transfected with Lipofectamine® (blank), mimics-NC or
miR-148a mimics. Subsequently, the MTT assay was used to evaluate
cellular proliferation, and Transwell assays were used to measure
the migratory and invasive ability of AsPC-1 cells. Compared with
the two control groups, the miR-148a mimics group exhibited
ascending cell proliferation at 48, 72, 96 and 120 h
post-transfection (Fig. 6A). In
addition, the migratory and invasive abilities of AsPC-1 cells were
significantly reduced following miR-148a mimics transfection
(Fig. 6B). These findings indicated
that miR-148a overexpression may inhibit cell proliferation,
migration and invasion of AsPC-1 cells.
Discussion
The molecular pathogenesis of pancreatic cancer is
the result of a complex multi-stage and multi-step process, which
is associated with numerous genes. The inactivation of TSGs and
aberrant expression of miRNAs are two important molecular
mechanisms that contribute to the progression of pancreatic cancer
(29,30); however, their intrinsic association
remains poorly understood. DNA methylation is an epigenetic
mechanism that occurs following the addition of a methyl group to
DNA, thereby often modifying the function of the gene and
regulating gene expression. DNMT1 serves a role in maintaining
methylation patterns during the DNA methylation process. The
overexpression of DNMT1 has been reported to be responsible for
hypermethylation and silencing of some cancer-associated miRNAs and
TSGs (31–34). Furthermore, DNMT1 has been
identified as the target of some miRNAs. Therefore, it was
hypothesized that DNMT1 might be a functional link between miRNAs
and TSGs. The present study revealed that DNMT1 was aberrantly
upregulated in PDAC, and was responsible for hypermethylation of
the miR-148a promoter. Furthermore, restoration of miR-148a
reactivated the TSGs p16, ppENK and RASSF1A by targeting DNMT1, and
inhibited cell proliferation, migration and invasion of AsPC-1
pancreatic cancer cells.
miR-148a is a member of the miR-148/152 family,
which has been investigated in numerous types of cancer. In
addition, its aberrant expression has a significant role in
tumorigenesis and metastasis. miR-148a has been identified as a
tumor suppressor miRNA in several types of cancer (5–9);
however, the antitumor effects of miR-148a remain controversial in
pancreatic cancer. At least four studies have revealed that
re-expression of miR-148a inhibits cell proliferation, apoptosis,
migration and invasion in pancreatic cancer (12–15).
However, Delpu et al (35)
demonstrated that miR-148a does not impact PDAC proliferation in
vitro and in vivo. Our previous study revealed that
upregulation of miR-148a suppresses cell migration and invasion of
BxPC-3 pancreatic cancer cells (22). The present study revealed that
re-expression of miR-148a in AsPC-1 pancreatic cancer cells, which
was mediated by miR-148a mimics transfection, could inhibit tumor
cell growth, migration and invasion. Therefore, the tumor
suppressor role of miR-148a in pancreatic cancer has been
validated.
Numerous TSGs are aberrantly methylated and silenced
in pancreatic cancer, including p16, ppENK and RASSF1A (36). p16 is a cyclin-dependent kinase
inhibitor that slows down the cell cycle by inhibiting progression
from G1 phase to S phase (37). ppENK is a neuropeptide transmitter
gene that encodes Met-enkephalin, which is a topically active
inhibitor of cancer that interacts with the opioid growth factor
receptor (38). The protein encoded
by the RASSF1A gene has a key role in mediating numerous cellular
processes, including apoptosis, cell migration, cell survival and
microtubule stabilization (39).
The RASSF1A gene has also been revealed to arrest the cell cycle by
inhibiting accumulation of cyclin D1 (40). Attri et al (41) reported that p16 expression is
decreased in 80% (20/25) of pancreatic cancer cases, 65% (13/20) of
which is caused by high methylation of the promoter region. Dammann
et al (42) revealed that
RASSF1A hypermethylation is detected in 64% of PDAC tissues and
87.5% of eight pancreatic cancer cell lines; in addition, p16
hypermethylation is detected in 43% of PDAC tissues and 63% of
eight pancreatic cancer cell lines. Fukushima et al
(43) demonstrated that 93.3%
(14/15) of invasive PDAC cases exhibit ppENK methylation, whereas
no methylation is detected in noncancerous pancreatic epithelium.
In the present study, restoration of miR-148a altered the
methylation status of p16, ppENK and RASSF1A promoter regions, and
reactivated their protein expression in AsPC-1 cells. These results
could explain, at least in part, the tumor suppressor effects of
miR-148a. To further investigate the potential mechanism underlying
the regulatory effects of miR-148a on the expression of these TSGs,
the present study focused on DNMT1 due to its important role in the
process of DNA methylation. Our previous study demonstrated that
DNMT1-siRNA could inhibit cell growth and promote apoptosis of
BxPC-3 pancreatic cancer cells, and its mechanisms were associated
with demethylation of the TSGs p16, ppENK and RASSF1A (44). The present findings clearly
suggested that DNMT1 was a direct target gene of miR-148a. This
conclusion was based on numerous experimental data. Firstly, the
expression of DNMT1 in PDAC tissues was negatively correlated with
miR-148a. Secondly, miR-148a upregulation significantly
downregulated DNMT1 expression in AsPC-1 cells. Finally, the
luciferase reporter assay using the DNMT1 3′UTR-wt reporter
indicated that miR-148a mimics significantly reduced luciferase
activity.
The mechanism underlying aberrant downregulation of
miR-148a in pancreatic cancer is another issue of concern. miR-148a
has been reported to be silenced by promoter hypermethylation in
gastrointestinal, hepatocellular and nasopharyngeal carcinoma
(20,45–47).
Hanoun et al (48) revealed
that hypermethylation of the promoter region encoding miR-148a is
responsible for its repression, not only in PDAC samples, but also
in pancreatic intraepithelial neoplasia. Furthermore, the interplay
between miR-148a and DNMT1 has been detected in PDAC, and the
increased expression of miR-148a arrests UTR methylation of p27,
giving rise to increased levels of p27, which is a cyclin-dependent
kinase inhibitor that possesses tumor suppressor activity (12). The present results demonstrated that
DNMT1 was overexpressed in PDAC tissues and AsPC-1 cells, which was
accompanied by hypermethylation of the miR-148a promoter. In AsPC-1
cells treated with 5-Aza-CdR or DNMT1-siRNA, DNMT1 was
downregulated, miR-148a expression was significantly increased, and
the miR-148a promoter was demethylated. Furthermore, the results of
BSP indicated that the methylated CpG sites of miR-148a were
significantly decreased in 5-Aza-CdR-treated or
DNMT1-siRNA-transfected AsPC-1 cells. Therefore, it may be
concluded that overexpression of DNMT1 leads to the
hypermethylation and silencing of miR-148a in pancreatic
cancer.
In conclusion, the present study demonstrated that
DNMT1 was aberrantly upregulated in pancreatic cancer, and its
overexpression was responsible for hypermethylation of the miR-148a
promoter. Furthermore, restoration of miR-148a reactivated the TSGs
p16, ppENK and RASSF1A by targeting DNMT1. These results indicated
that an interaction exists between miR-148a and DNMT1 in pancreatic
cancer. Notably, overexpression of miR-148a significantly inhibited
cell proliferation, migration and invasion in pancreatic cancer
cells. These results suggested the existence of a miR-148a-DNMT1
regulatory circuit and indicated that miR-148a may be a potential
therapeutic target in pancreatic cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81660401), the
Natural Science Foundation of Jiangxi Province (grant no.
20161BAB205242) and the Scientific Research Foundation of the
Education Office Jiangxi Province (grant no. GJJ14018).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
WX and YL designed the study. LH and GS wrote the
manuscript, collected clinical information and performed
statistical analyses. LH, GS, LP, ZW and WX performed the
experiments. HX assisted with the dual-luciferase reporter assays.
YT conducted the pathological diagnosis of the pancreatic tissues
and assisted with the Transwell assays. All authors read and
approved the manuscript, and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Written informed consent was provided by all
participants and volunteers prior to recruitment. The present study
was approved by the Ethics Committee of the First Affiliated
Hospital of Nanchang University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the United States. Cancer Res. 74:2913–2921.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan SH and Wang LH: Regulation of cancer
metastasis by microRNAs. J Biomed Sci. 22:92015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu Q, Jiang Y, Yin Y, Li Q, He J, Jing Y,
Qi YT, Xu Q, Li W, Lu B, et al: A regulatory circuit of
miR-148a/152 and DNMT1 in modulating cell transformation and tumor
angiogenesis through IGF-IR and IRS1. J Mol Cell Biol. 5:3–13.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang H, Li Y, Huang Q, Ren X, Hu H, Sheng
H and Lai M: MiR-148a promotes apoptosis by targeting Bcl-2 in
colorectal cancer. Cell Death Differ. 18:1702–1710. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sakamoto N, Naito Y, Oue N, Sentani K,
Uraoka N, Oo Zarni H, Yanagihara K, Aoyagi K, Sasaki H and Yasui W:
MicroRNA-148a is downregulated in gastric cancer, targets MMP7, and
indicates tumor invasiveness and poor prognosis. Cancer Sci.
105:236–243. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Braconi C, Huang N and Patel T:
MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor
suppressor gene expression by interleukin-6 in human malignant
cholangiocytes. Hepatology. 51:881–890. 2010.PubMed/NCBI
|
|
9
|
Heo MJ, Kim YM, Koo JH, Yang YM, An J, Lee
SK, Lee SJ, Kim KM, Park JW and Kim SG: microRNA-148a dysregulation
discriminates poor prognosis of hepatocellular carcinoma in
association with USP4 overexpression. Oncotarget. 5:2792–2806.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bloomston M, Frankel WL, Petrocca F,
Volinia S, Alder H, Hagan JP, Liu CG, Bhatt D, Taccioli C and Croce
CM: MicroRNA expression patterns to differentiate pancreatic
adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA.
297:1901–1908. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Szafranska AE, Davison TS, John J, Cannon
T, Sipos B, Maghnouj A, Labourier E and Haln SA: MicroRNA
expression alterations are linked to tumorigenesis and
non-neoplastic processes in pancreatic ductal adenocarcinoma.
Oncogene. 26:4442–4452. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhan Q, Fang Y, Deng X, Chen H, Jin J, Lu
X, Peng C, Li H and Shen B: The interplay between miR-148a and
DNMT1 might be exploited for pancreatic cancer therapy. Cancer
Invest. 33:267–275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang R, Li M, Zang W, Chen X, Wang Y, Li
P, Du Y, Zhao G and Li L: MiR-148a regulates the growth and
apoptosis in pancreatic cancer by targeting CCKBR and Bcl-2. Tumour
Biol. 35:837–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liffers ST, Munding JB, Vogt M, Kuhlmann
JD, Verdoodt B, Nambiar S, Maghnouj A, Mimohammadsadegh A, Hahn SA
and Tannapfel A: MicroRNA-148a is down-regulated in human
pancreatic ductal adenocarcinomas and regulates cell survival by
targeting CDC25B. Lab Invest. 91:1472–1479. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feng H, Wang Y, Su J, Liang H, Zhang CY,
Chen X and Yao W: MicroRNA-148a suppresses the proliferation and
migration of pancreatic cancer cells by down-regulating ErbB3.
Pancreas. 45:1263–1271. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lopez-Serra P and Esteller M: DNA
methylation-associated silencing of tumor-suppressor microRNAs in
cancer. Oncogene. 31:1609–1622. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xie K, Liu J, Chen J, Dong J, Ma H, Liu Y
and Hu Z: Methylation-associated silencing of microRNA-34b in
hepatocellular carcinoma cancer. Gene. 543:101–107. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peng DF, Kanai Y, Sawada M, Ushijima S,
Hiraoka N, Kosuge T and Hirohashi S: Increased DNA
methyltransferase 1 (DNMT1) protein expression in precancerous
conditions and ductal carcinomas of the pancreas. Cancer Sci.
96:403–408. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li A, Omura N, Hong SM and Goggins M:
Pancreatic cancer DNMT1 expression and sensitivity to DNMT1
inhibitors. Cancer Biol Ther. 9:321–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu A, Xia J, Zuo J, Jin S, Zhou H, Yao L,
Huang H and Han Z: MicroRNA-148a is silenced by hypermethylation
and interacts with DNA methyltransferase 1 in gastric cancer. Med
Oncol. 29:2701–2709. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Long XR, He Y, Huang C and Li J:
MicroRNA-148a is silenced by hypermethylation and interacts with
DNA methyltransferase 1 in hepatocellular carcinogenesis. Int J
Oncol. 44:1915–1922. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peng L, Liu Z, Xiao J, Tu Y, Wan Z, Xiong
H, Li Y and Xiao W: MicroRNA-148a suppresses epithelial-mesenchymal
transition and invasion of pancreatic cancer cells by targeting
Wnt10b and inhibiting the Wnt/β-catenin signaling pathway. Oncol
Rep. 38:301–308. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sanger F, Nicklen S and Coulson AR: DNA
sequencing with chain-terminating inhibitors. Proc Natl Acad Sci
USA. 74:5463–5467. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kumaki Y, Oda M and Okano M: QUMA:
Quantification tool for methylation analysis. Necleic Acids Res.
36:W170–W175. 2008. View Article : Google Scholar
|
|
26
|
Bock C, Reither S, Mikeska T, Paulsen M,
Walter J and Lengauer T: BiQ Analyzer: Visualization and quality
control for DNA methylation data from bisulfite sequencing.
Bioinformatics. 21:4067–4068. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee J, Shin MK, Ryu DK, Kim S and Ryu WS:
Insertion and deletion mutagenesis by overlap extension PCR.
Methods Mol Biol. 634:137–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bestor TH: The DNA methyltransferases of
mammals. Hum Mol Genet. 9:2395–2402. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cowan RW and Maitra A: Genetic progression
of pancreatic cancer. Cancer J. 20:80–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yonemori K, Kurahara H, Maemura K and
Natsugoe S: MicroRNA in pancreatic cancer. J Hum Genet. 62:33–40.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang L, Luo P, Song Q and Fei X:
DNMT1/miR-200a/GOLM1 signaling pathway regulates lung
adenocarcinoma cells proliferation. Biomed Pharmacother.
99:839–847. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ning X, Shi Z, Liu X, Zhang A, Han L,
Jiang K, Kang C and Zhang Q: DNMT1 and EZH2 mediated methylation
silences the microRNA-200b/a/429 gene and promotes tumor
progression. Cancer Lett. 59:198–205. 2015. View Article : Google Scholar
|
|
33
|
Peng DF, Kanai Y, Sawada M, Ushijima S,
Hiraoka N, Kitazawa S and Hirohashi S: DNA methylation of multiple
tumor-related genes in association with overexpression of DNA
methyltransferase 1 (DNMT1) during multistage carcinogenesis of the
pancreas. Carcinogenesis. 27:1160–1168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu B, Song J, Luan J, Sun X, Bai J, Wang
H, Li A, Zhang L, Feng X and Du Z: Promoter methylation status of
tumor suppressor genes and inhibition of expression of DNA
methyltransferase 1 in non-small cell lung cancer. Exp Biol Med.
241:1531–1539. 2016. View Article : Google Scholar
|
|
35
|
Delpu Y, Lulka H, Sicard F, Saint-Laurent
N, Lopez F, Hanoun N, Buscail L, Cordelier P and Torrisani J: The
rescue of miR-148a expression in pancreatic cancer: An
inappropriate therapeutic tool. PLoS One. 8:e555132013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pan FP, Zhou HK, Bu HQ, Chen ZQ, Zhang H,
Xu LP, Tang J, Yu QJ, Chu YQ, Pan J, et al: Emodin enhances the
demethylation by 5-AZa-CdR of pancreatic cancer tumor-suppressor
genes P16, RASSF1A and ppENK. Oncol Rep. 35:1941–1949. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Witkiewicz AK, Knudsen KE, Dicker AP and
Knudsen ES: The meaning of p16ink4a expression in tumors:
Functional significance, clinical associations and future
developments. Cell Cycle. 10:2497–2503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang L, Yang H, Li J, Hao J and Qian J:
ppENK gene methylation status in the development of pancreatic
carcinoma. Gastroenterol Res Pract. 2013:1309272013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vos MD, Martinez A, Elam C, Dallol A,
Taylor BJ, Latif F and Clark GJ: A role for the RASSF1A tumor
suppressor in the regulation of tubulin polymerization and genomic
stability. Cancer Res. 64:4244–4250. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shivakumar L, Minna J, Sakamaki T, Pestell
R and White MA: The RASSF1A tumor suppressor blocks cell cycle
progression and inhibits cyclin D1 accumulation. Mol Cell Biol.
22:4309–4318. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Attri J, Srinivasan R, Majumdar S, Radotra
BD and Wig J: Alterations of tumor suppressor gene p16INK4a in
pancreatic ductal carcinoma. BMC Gastroenterol. 5:222005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dammann R, Schagdarsurengin U, Liu L, Otto
N, Gimm O, Dralle H, Boehm BO, Pfeifer GP and Hoang-Vu C: Frequent
RASSF1A promoter hypermethylation and K-ras mutations in pancreatic
carcinoma. Oncogene. 22:3806–3812. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fukushima N, Sato N, Ueki T, Rosty C,
Walter KM, Wilentz RE, Yeo CJ, Hruban RH and Goggins M: Aberrant
methylation of preproenkephalin and p16 genes in pancreatic
intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am
J Pathol. 160:1573–1581. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xiao WD, Li Y, Li XM, Cai J, Zeng LS and
Hu W: RNA interference-mediated silencing of the DNMT1 gene
inhibits cell proliferation in human pancreatic carcinoma cell line
BxPC-3. Shijie Huaren Xiaohua Zazhi. 19:3397–3401. 2011.(In
Chinese).
|
|
45
|
Sun J, Song Y, Wang Z, Wang G, Gao P, Chen
X, Gao Z and Xu H: Clinical significance of promoter region
hypermethylation of microRNA-148a in gastrointestinal cancers. Onco
Targets Ther. 7:853–863. 2014.PubMed/NCBI
|
|
46
|
Long XR, He Y, Huang C and Li J:
MicroRNA-148a is silenced by hypermethylation and interacts with
DNA methyltransferase in hepatocellular carcinogenesis. Int J
Oncol. 44:1915–1922. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li HP, Huang HY, Lai YR, Huang JX, Chang
KP, Hsueh C and Chang YS: Silencing of miRNA-148a by
hypermethylation activates the integrin-mediated signaling pathway
in nasopharyngeal carcinoma. Oncotarget. 5:7610–7624. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hanoun N, Delpu Y, Suriawinata AA, Bournet
B, Bureau C, Selves J, Tsongalis GJ, Dufresne M, Buscail L,
Cordelier P and Torrisani J: The silencing of microRNA 148a
production by DNA hypermethylation is an early event in pancreatic
carcinogenesis. Clin Chem. 56:1107–1118. 2010. View Article : Google Scholar : PubMed/NCBI
|