Introduction
Cancer upregulated gene 2 (CUG2) has been
identified as a candidate gene whose expression is commonly
increased in various tumor tissues, including ovarian, liver, colon
and lung, playing a crucial role in tumorigenesis (1). CUG2 has been identified as a new
centromere component required for kinetochore function during cell
division (2,3). The oncogenic effect of CUG2 was found
to be similar to that of Ras in a transplant model using NIH3T3
cells (1). Although CUG2
overexpression activates Ras and mitogen-activated protein kinases
(MAPKs), including p38 MAPK, which eventually facilitate oncolytic
retroviral replication (4–6), CUG2 confers resistance to oncolytic
vesicular stomatitis virus infection (7) and induces anticancer drug resistance
through activation of STAT1 (8).
Another study revealed that CUG2 induces epithelial-mesenchymal
transition (EMT) through TGF-β signaling (9). Crosstalk between Sp1 and Smad2/3
mediated by CUG2 or TGF-β plays a crucial role during EMT (9).
Although the transcription factor STAT1 is well
established as an important antiviral agent acting via
IFN-associated intracellular signaling, the role of STAT1 in the
development of cancer is still unclear: A tumor suppressor or
oncogene? On the one hand, STAT1 functions as a tumor suppressor
via the upregulation of caspases (10,11),
cyclin-dependent kinase inhibitor 1A (Cdkn1a; also known as p21)
(12), or the IFN-regulatory factor
1 (IRF1)/p53 pathway (13). On the
other hand, a number of studies have indicated that in certain
cellular contexts, the IFN/STAT1 signaling pathway may facilitate
tumor cell growth (14,15). One study reported that resistance to
ionizing radiation and IFNs are associated with constitutive
overactivity of the IFN/STAT1 pathway in radioresistant tumor cells
(14). Other studies have also
demonstrated that constitutive overexpression of STAT1 is
positively correlated with the protection of tumor cells from
genotoxic stress induced by doxorubicin (16) or cisplatin (17).
Histone deacetylases (HDACs) play important roles in
the maintenance and function of chromatin by regulating the
acetylation state of histones (18). Recent data suggest that HDACs
regulate the acetylation state of many non-histone targets,
including STAT1, MEF2A, and Foxo proteins (19–21).
In particular, overexpression of HDAC4, belonging to the class IIa
family of HDAC, is not only significantly associated with tumor
size in malignant thyroid lesions (22), but also promotes tumor growth by
suppressing p21 expression in colon (23), ovarian (24), and gastric (25) cancer cells. Therefore, HDAC4 has
been suggested to be a useful diagnostic marker for prognosis of
patients with cancer and a potential target for anticancer
therapy.
Since CUG2 has been shown to induce EMT and cancer
stem cell (CSC)-like phenotypes, this study aimed to explore
whether activated STAT1 induced by CUG2 plays a crucial role in
these malignant tumor features besides anticancer drug resistance.
We report that the STAT1-HDAC4 signaling pathway, communicating
with TGF-β signaling, contributes to the increase in cell
migration, invasion, sphere formation, and expression of
stemness-related factors in CUG2-overexpressing cancer cells.
Materials and methods
Cell cultures
Human lung cancer A549 cells (ATCC, Manassas, VA,
USA) stably expressing the vector alone (A549-Vec) or CUG2
(A549-CUG2), and A549-CUG2 cells with stably silenced STAT1
(A549-CUG2-shSTAT1) or the control (A549-CUG2-shVec) were cultured
in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS),
1% penicillin, 1% streptomycin, and G418 (0.5 mg/ml; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) at 37°C with 5% CO2. For
A549-CUG2-shSTAT1 and A549-CUG2-shVec cells, puromycin (1 µg/ml;
Sigma-Aldrich; Merck KGaA) was additionally added to the
medium.
Reagents and antibodies
For immunoblotting, antibodies against STAT1
(#9172), phospho-STAT1 (#9171), Smad2/3 (#5678), and phospho-Smad2
(#3108) were acquired from Cell Signaling Technology (Danvers, MA,
USA). Anti-β-actin (sc-4778), -Klf4 (sc-166229), -HDAC4 (sc-5245),
and -Sp1 (sc-17824) antibodies were obtained from Santa Cruz
Biotechnology (Santa Cruz, CA, USA) and antibodies against
E-cadherin (ab15148), N-cadherin (ab18203), vimentin (ab137321),
Snail (ab180714), Twist (ab175430), Bmi1 (ab126783), Sox2
(ab97959), and Oct4 (ab109183) were obtained from Abcam (Cambridge,
MA, USA). LY2109761 and trichostatin A (TSA) were purchased from
Cayman Chemical (Ann Arbor, MI, USA) and Sigma-Aldrich/Merck KGaA,
respectively. The STAT1-shRNA vector was acquired from Origene
(Rockville, MD, USA).
Cellular fractionation
Cells cultured in 100-mm plates were washed and
harvested with ice-cold PBS, and cell pellets were lysed with 800
µl of TTN buffer [20 mM Tris-HCl (pH 7.4), 0.05% Triton X-100, 150
mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 0.5 mM PMSF, and 1X
protease inhibitor cocktail] on ice for 20 min followed by
centrifugation at 10,000 × g for 15 min. The supernatants
represented the soluble fractions, and the pellets as insoluble
fractions were subsequently solubilized in 800 µl of RIPA buffer
[50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 1%
NP-40, 0.5% deoxycholic acid, 0.1% SDS, 10% glycerol, 0.5 mM PMSF,
and 1X protease inhibitor cocktail] on ice for 30 min and were
centrifuged at 12,000 × g for 15 min. Thereafter, these
supernatants were used as nuclear extracts.
Immunoblotting
Cells were harvested and lysed with lysis buffer
containing 1% NP-40 and protease inhibitors (Sigma-Aldrich; Merck
KGaA) for immunoblotting. Proteins from whole cell lysates were
resolved by 8, 10, or 15% SDS-polyacrylamide gel electrophoresis
(PAGE) and transferred onto nitrocellulose membranes. Primary
antibodies were used at a 1:1,000 or 1:2,000 dilution, and
secondary antibodies conjugated with horseradish peroxidase were
used at a 1:2,000 dilution in 5% nonfat dry milk. After the final
washing, the membranes were evaluated with an enhanced
chemiluminescence reagent using Image Quant LAS 4000 Mini (GE
Healthcare, Tokyo, Japan).
Luciferase reporter assay
A549-CUG2 cells were transfected with TGF-β promoter
vectors (phTG5 and phTG7) (26)
using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). To normalize the transfection efficiency,
a pGK-βgal vector that expresses β-galactosidase under the control
of a phosphoglucokinase promoter was included in the transfection
mixture. At 48 h post-transfection, cells were washed with cold PBS
and lysed in a lysis solution [25 mM Tris (pH 7.8), 2 mM EDTA, 2 mM
DTT, 10% glycerol, and 1% Triton X-100]. Luciferase activity was
measured with a luminometer using a luciferase kit (Promega,
Madison, WI, USA).
Short interfering RNA (siRNA)
transfection
Cells were trypsinized and cultured overnight to
achieve 60–70% confluence before siRNA transfection. Pre-made
STAT1, HDAC4 (Bioneer, Daejeon, Korea), and a negative control
siRNA (Bioneer) were mixed with Lipofectamine 2000. The cells were
incubated with the transfection mixture for 6 h and then rinsed
with medium containing 10% FBS. The cells were incubated for 40 h
before harvesting.
Invasion assay
Invasion assays were performed using 48-well Boyden
chambers (Neuroprobe, Gaithersburg, MD, USA). The lower wells of
the chamber were filled with a standard culture medium. The chamber
was assembled using polycarbonate filters (Neuroprobe) coated with
Matrigel. Cells in a serum-free medium (5×104
cells/well) were seeded in the upper compartment of the chamber.
After incubation for 24 h, cell migration was quantified by
counting the number of migrated cells after staining with
hematoxylin and eosin under a light microscope (Olympus, CKX31-11
PHP, Tokyo, Japan). The data shown are the mean values of three
independent experiments.
Wound healing assay
Cell migration was assessed using a scratch wound
assay. Briefly, cells were cultured in 12-well plates
(5×105 cells/well). When the cells reached 90%
confluence, a single wound was made in the center of the cell
monolayer using a P-200 pipette tip. At 0, 12, and 24 h of
incubation, the wound closure areas were visualized by light
microscopy (Olympus) at a magnification of ×100.
Cell adhesion assay
The wells in 96-well plates were coated with
collagen I solution (100 µg/ml) at 4°C for 12 h and were dried in a
tissue-culture hood. Cells (2×105 cells/well) were
seeded on the well and serum-free RPMI-1640 medium (100 µl) was
added to the well. The cells were incubated at 37°C for 30 min to
allow the cells to adhere to the surface and thereafter the then
any non-adherent cells in the well were washed off with the medium.
After washing, the wells were furnished with medium containing 10%
FBS and the adherent cells were incubated at 37°C for 4 h.
Thereafter, MTT assay was performed for cell counting as previously
described (27).
Sphere formation assay
A549-CUG2 cells were transfected with siRNAs of
STAT1, HDAC4, or the control. At 24 h post-transfection, the cells
were collected and seeded in 24-well ultra-low attachment plates in
a serum-free medium supplemented with 5 µg/ml insulin, 0.4% bovine
serum albumin, 10 ng/ml basic fibroblast growth factor, and 20
ng/ml recombinant human epidermal growth factor for 2, 4 and 6
days. The size and number of spheroids were analyzed under a light
microscope (Olympus). The criterion for sphere formation was set as
spheroids larger than 50 µm in size. The data shown are the mean
values of three independent experiments.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 15
min, permeabilized with cold acetone for 15 min, blocked with 10%
goat serum for 30 min, and incubated with primary antibodies (1:100
dilution) for 30 min at room temperature. After incubation, the
cells were washed extensively with PBS, incubated with Alexa Fluor
418-conjugated goat anti-mouse or donkey anti-rabbit antibody
(1:500 dilution; Molecular Probes, Eugene, OR, USA) in PBS for 30
min at room temperature, and washed 3 times with PBS. For nuclear
staining, cells were incubated with DAPI for 5 min in the dark and
washed 3 times with PBS. The stained cells were mounted using PBS
containing 10% glycerol and photographed using a fluorescence
microscope (Axio Observer D1; Zeiss, Oberkochen, Germany).
Statistical analysis
Data are presented as mean ± standard deviation
(SD). One-way ANOVA or unpaired t-test was used for statistical
analysis (GraphPad Prism 6; GraphPad Software, Inc., La Jolla, CA,
USA). Data were considered statistically significant at the P-value
<0.05.
Results
CUG2-mediated STAT1 activation
increases cell migration and invasion
Since we reported that activation of STAT1 induced
by CUG2 confers resistance to doxorubicin (8), we next explored whether STAT1
contributes to other malignant tumor features. Cell migration was
observed after the center of the monolayer of A549-CUG2 cells
treated with STAT1 siRNA or control siRNA was scratched. STAT1
siRNA inhibited cell migration compared with control siRNA
(Fig. 1A). In addition, when
A549-CUG2 cells treated with STAT1 siRNA were cultured in the upper
chamber coated with Matrigel, the number of invaded cells in the
lower chamber was lower than that treated with control siRNA, after
staining with hematoxylin and eosin (Fig. 1B). Furthermore, we found that STAT1
siRNA-treated cells showed stronger attachment on collagen-coated
wells compared with that of the control siRNA-treated cells, in a
cell attachment assay (Fig. 1C). We
also found that STAT1 silencing reduced expression of biomarkers of
EMT, N-cadherin and vimentin, in A549-CUG2 cells (Fig. 1D). However, STAT1 suppression did
not significantly recover E-cadherin expression in A549-CUG2 cells
(Fig. 1D). Immunofluorescence assay
showed that STAT1 silencing further reduced the intensity of
vimentin staining (Fig. 1E) when
compared with the control siRNA. These results suggest that
activation of STAT1 induced by CUG2 is involved in EMT.
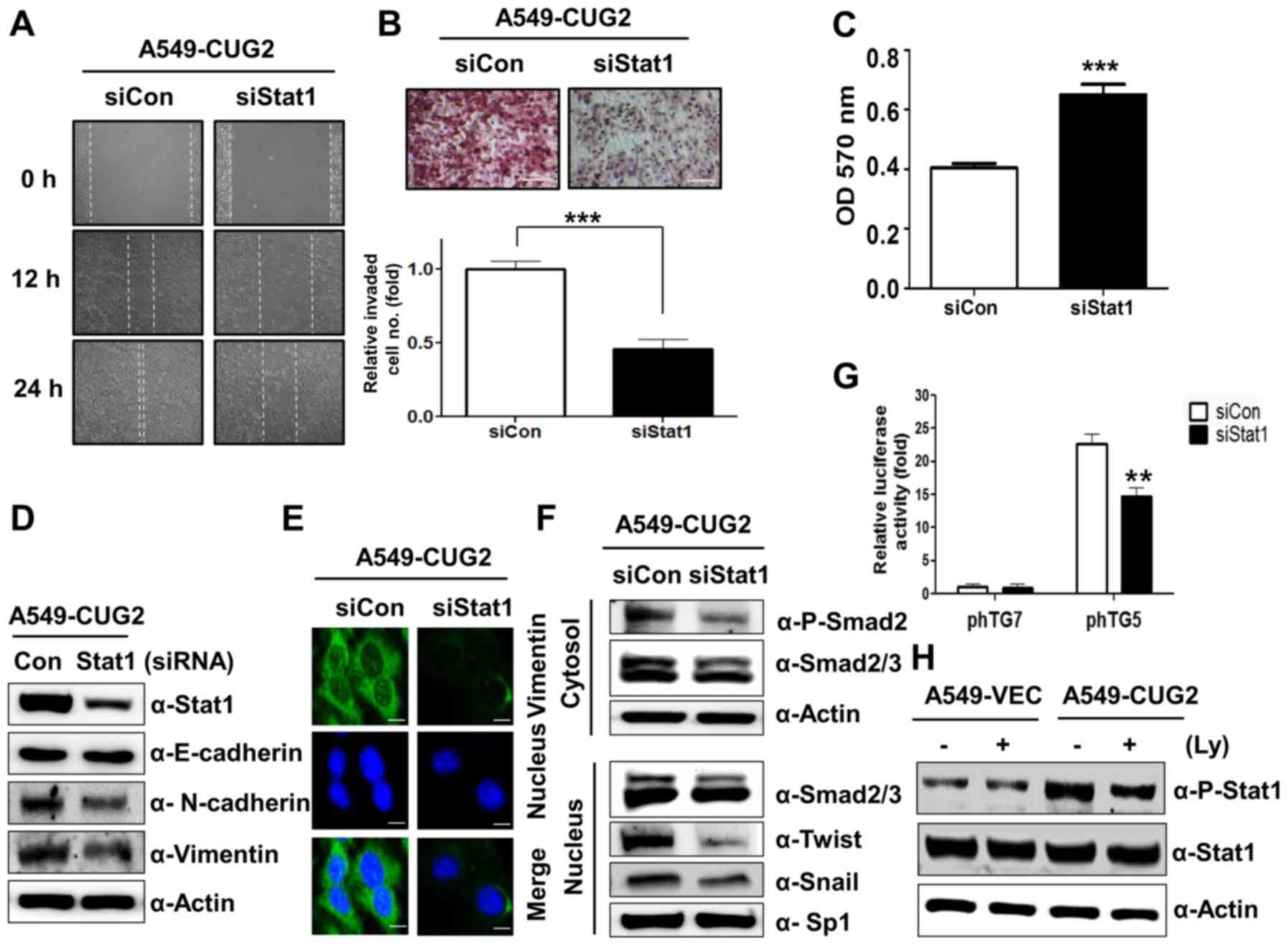 | Figure 1.STAT1 silencing inhibits CUG2-induced
cell migration and invasion. (A) Cell migration was measured by a
wound healing assay at 48 h post-transfection with STAT1 siRNA (500
nM) or control siRNA. (B) An invasion assay was performed using
48-well Boyden chambers coated with Matrigel at 48 h
post-transfection with STAT1 siRNA or control siRNA. Scale bar
indicates 100 µm (***P<0.001). (C) At 48 h post-transfection
with STAT1 siRNA or control siRNA, the A549-CUG2 cells were seeded
in collagen-coated wells. The cells were incubated for 30 min for
attachment and then the attached cells were analyzed by MTT assay
(***P<0.001). (D and F) To examine an effect of STAT1 on levels
of proteins related to EMT and TGF-β signaling, expression of
STAT1, E-cadherin, N-cadherin, and vimentin was detected by
immunoblotting at 48 h post-treatment with STAT1 siRNA or control
siRNA. After transfection and cellular fractionation, expression of
phospho-Smad2, Smad2/3, Snail, and Twist was detected by
immunoblotting. (E) Expression of vimentin was detected by
immunofluorescence using an Alexa Fluor 488-conjugated secondary
antibody (green). DAPI was added for nuclear staining. Scale bar
indicates 10 µm. (G) After A549-CUG2 cells treated with STAT1 siRNA
or control siRNA were transfected with TGF-β promoter vectors (pTG5
and pTG7; 1 µg), luciferase enzyme activities were measured
(**P<0.01). (H) A549-CUG2 cells were treated with LY2109761 (Ly;
10 µM) or DMSO for 24 h, and the cell lysates were prepared for
immunoblotting to detect protein levels of phospho-STAT1 and
STAT1. |
As our recent study showed that TGF-β signaling
plays a critical role in CUG2-induced EMT (9), we aimed to ascertain whether STAT1 is
involved in CUG2-induced TGF-β signaling, leading to EMT. To answer
this question, we transfected A549-CUG2 cells with STAT1 siRNA or
control siRNA and examined expression of TGF-β signaling-related
molecules such as Smad2, Snail, and Twist. As shown in Fig. 1F, we found that STAT1 silencing
reduced phosphorylation of Smad2 in the cytoplasm and expression of
Snail and Twist in the nucleus. These results prompted us to
investigate whether STAT1 could affect TGF-β production. To solve
this question, TGF-β promoter luciferase vector (pTG5) was
introduced (26). STAT1 siRNA
reduced the luciferase activity of TGF-β compared to that with
control siRNA in A549-CUG2 cells, whereas STAT1 siRNA and control
siRNA treatment failed to reduce luciferase activity of TGF-β
promoter lacking Sp1 binding sites (pTG7) (Fig. 1G). The result indicates that both
STAT1 and Sp1 are involved in the synthesis of TGF-β, a critical
mediator of EMT. Conversely, when we suppressed TGF-β signaling
with LY2109761 and examined STAT1 activation, we found that
CUG2-induced phosphorylation of STAT1 was reduced (Fig. 1H), indicating that there is a
crosstalk between the TGF-β and STAT1 signaling pathways.
CUG2-mediated STAT1 activation
increases stemness-related factor expression and sphere
formation
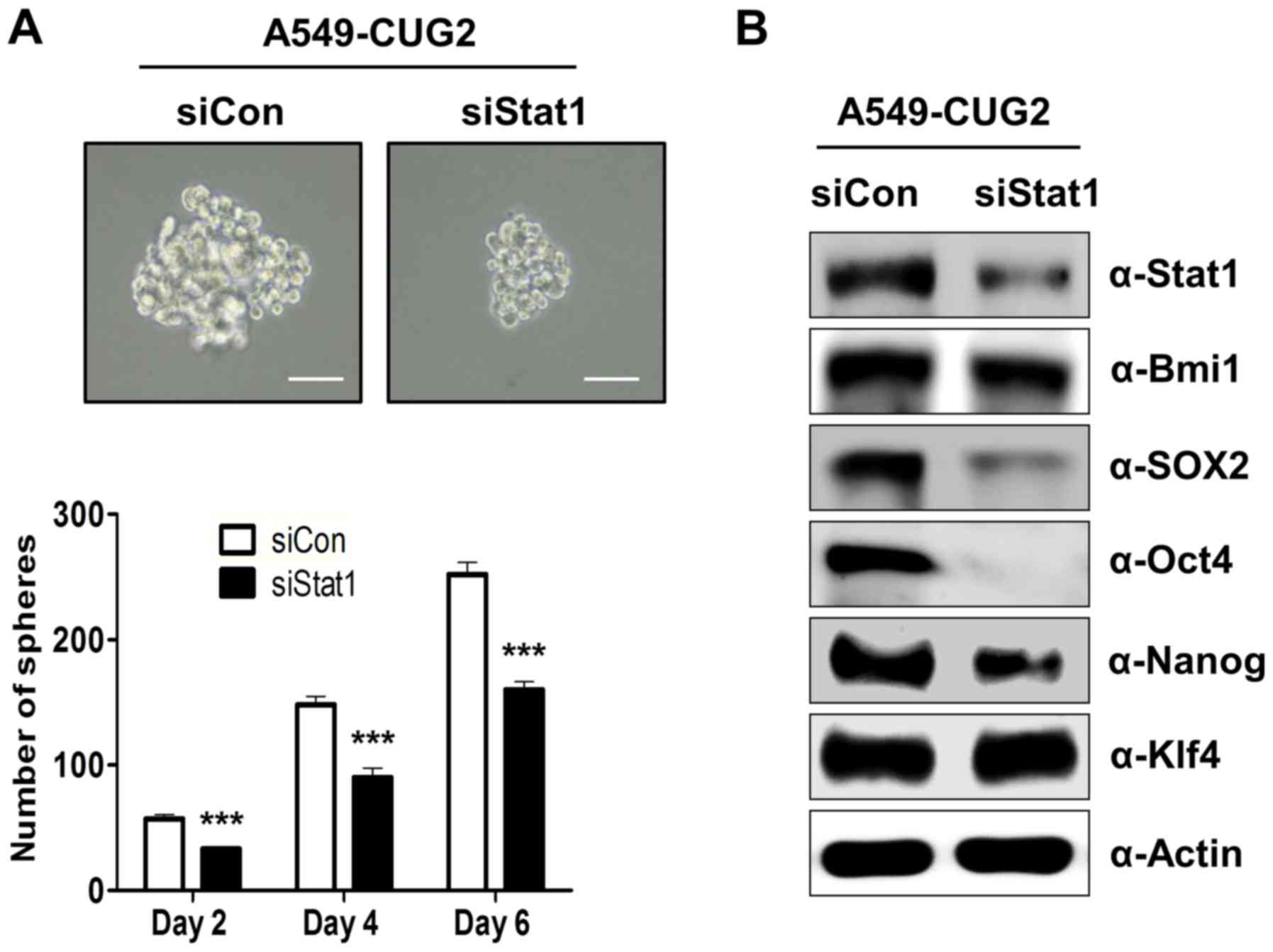
Our recent study showed that CUG2 overexpression
induced CSC-like features, including sphere formation and elevated
expression of stemness-related factors such as Bmi1, Klf4, Oct4,
Sox2 and Nanog (unpublished data). We thus explored whether
activation of STAT1 mediated by CUG2 is involved in the induction
of CSC-like phenotypes. To answer this question, we examined
spheroid forming ability after the suppression of STAT1 expression.
We found that STAT1 siRNA restricted the size and number of
spheroids in A549-CUG2 cells compared to the size and number in the
control siRNA cells (Fig. 2A). In
addition, STAT1 silencing significantly reduced Sox2, Oct4, and
Nanog protein levels but failed to decrease Klf4 expression
(Fig. 2B). STAT1 suppression only
marginally diminished Bmi1protein levels (Fig. 2B). These results suggest that STAT1
plays a role in sphere formation and expression of stemness-related
factors such as Sox2, Oct4 and Nanog.
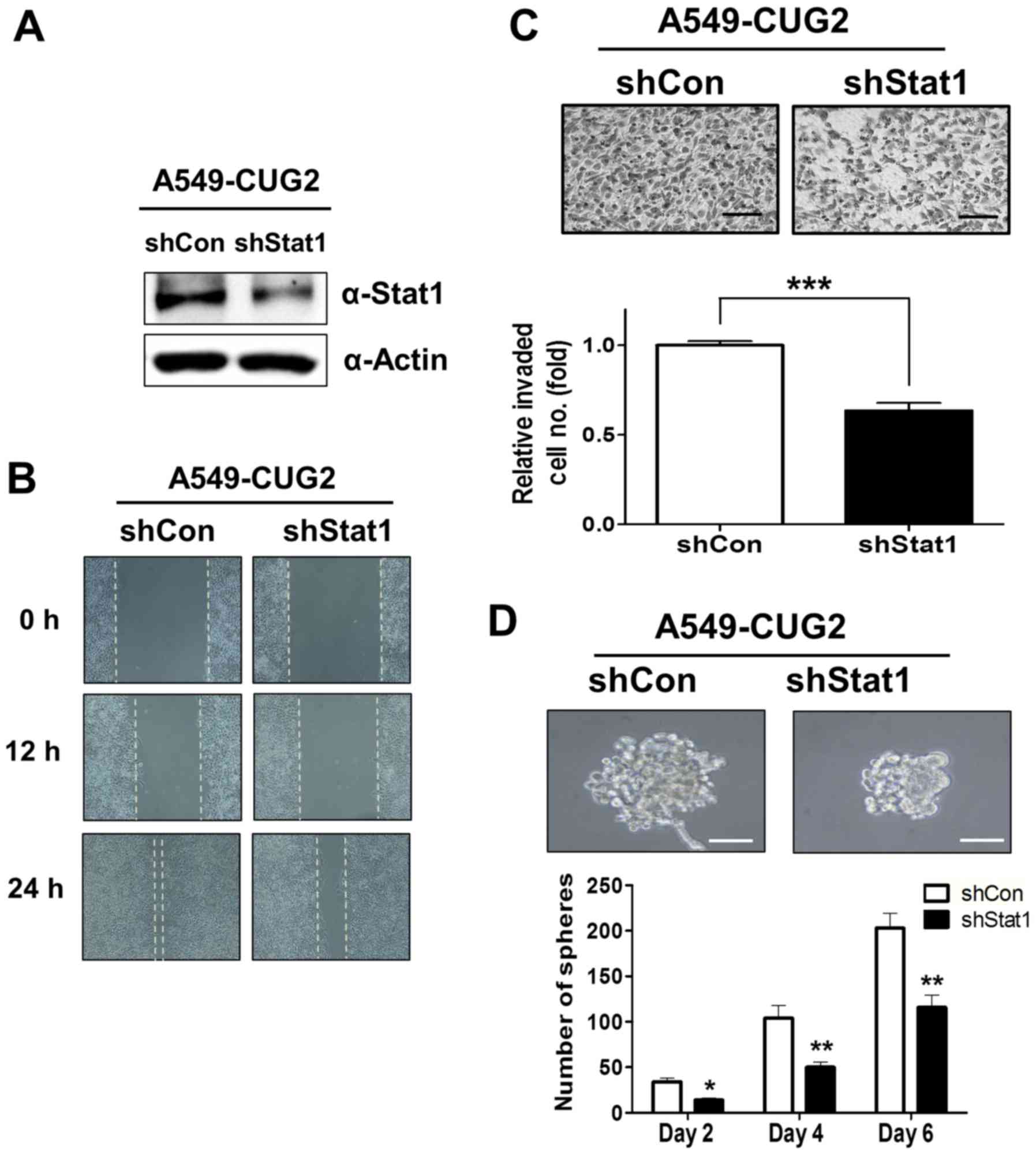
Constitutive suppression of STAT1
inhibits EMT and stemness in A549-CUG2 cells
Finally, to confirm the roles of STAT1 in biological
features such as EMT and stemness, we constructed A549-CUG2 cells
stably silencing STAT1 (A549-CUG2-shSTAT1) which was confirmed by
immnoblotting (Fig. 3A). When cell
migration was examined between A549-CUG2-shSTAT1 cells and the
control (A549-CUG2-shVec) cells with wound-healing assay,
A549-CUG2-shSTAT1 cells showed a slower migration rate than
A549-CUG2-shVec cells (Fig. 3B).
When cell invasion was examined between them, A549-CUG2-shSTAT1
cells significantly exhibited a reduced invasion in lower
plate-wells compared to A549-CUG2-shVec cells (Fig. 3C). In addition, when we compared
sphere forming ability between them, we found that
A549-CUG2-shSTAT1 cells showed smaller size and a fewer number of
spheroid than A549-CUG2-shVec cells (Fig. 3D). These results support that STAT1
is involved in EMT and stemness in A549 cell overexpressing
CUG2.
HDAC4 is involved in the malignant
tumor features of A549-CUG2 cells
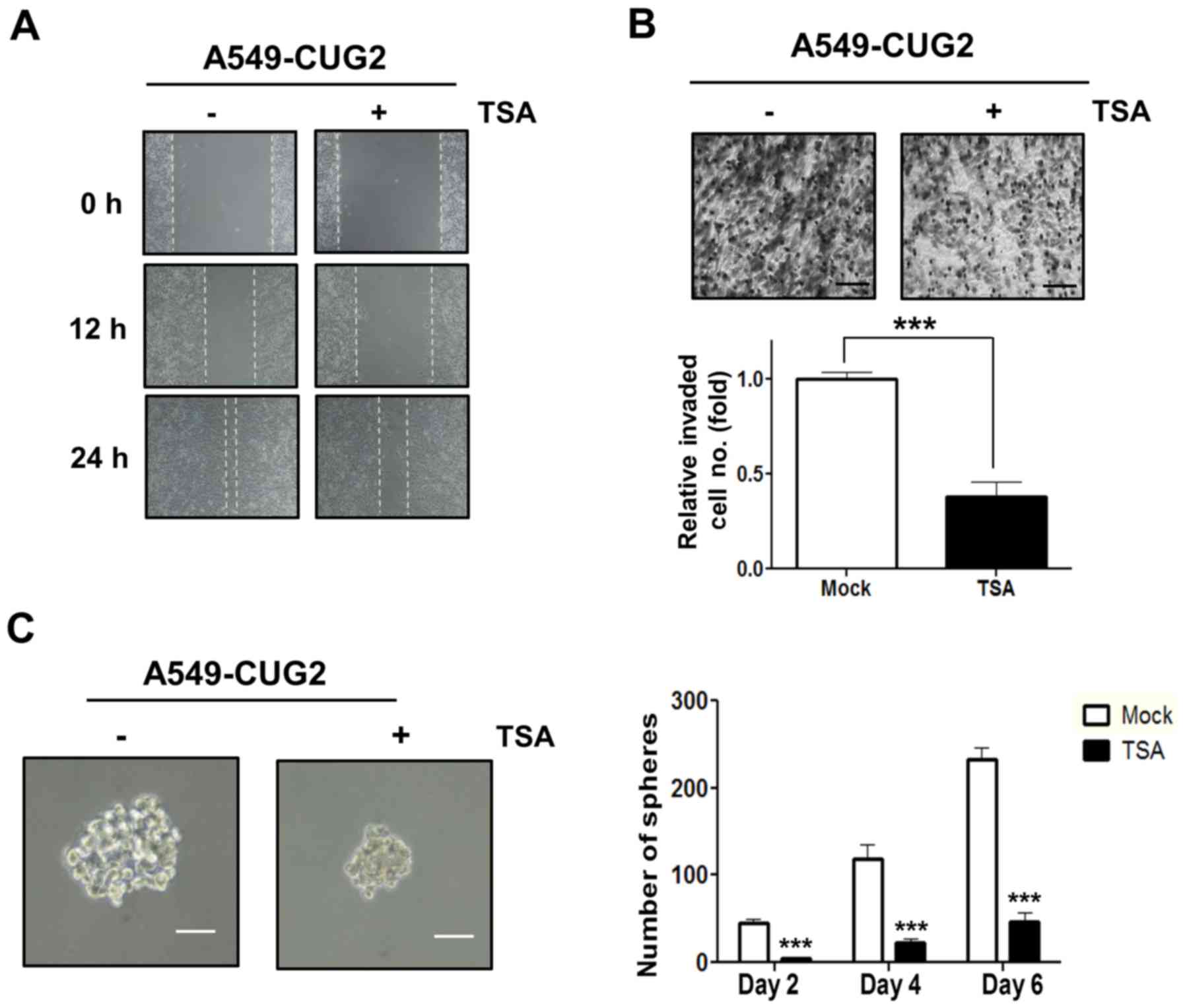
As a recent study showed that activation of STAT1 is
not only regulated by phosphorylation but also acetylation
(28), we introduced TSA, an
inhibitor of HDACs, in A549-CUG2 cells. Treatment with TSA
inhibited cell migration and invasion compared to those with
control DMSO treatment (Fig. 4A and
B). In addition, treatment with TSA diminished the size and
number of spheroids compared to that with DMSO treatment (Fig. 4C). These results indicate that STAT1
acetylation induced by inhibition of HDACs, a less active form of
STAT1, exerts a negative influence on CUG2-mediated cell migration,
invasion, and sphere formation.
Moreover, because STAT1 is a direct substrate of
HDAC4 as a non-histone protein (21), we explored whether HDAC4 plays a
role in CUG2-induced malignant tumor features such as rapid cell
migration, aggressive invasion, and enhanced sphere formation. To
answer this question, we suppressed HDAC4 expression using siRNA in
A549-CUG2 cells and examined the malignant tumor features.
Treatment with HDAC4 siRNA inhibited cell migration and invasion
compared to control RNA treatment (Fig.
5A and B). In addition, we found that HDAC4 siRNA-treated cells
showed stronger attachment on the collagen-coated wells compared
with that of control siRNA-treated cells, in a cell attachment
assay (Fig. 5C). We also found that
HDAC4 suppression inhibited expression of N-cadherin and vimentin
but failed to recover E-cadherin expression as observed with STAT1
silencing (Fig. 5D). When we
examined whether HDAC4 affected rapid cell migration and aggressive
invasion through TGF-β signaling, we found that HDAC4 suppression
decreased the phosphorylation level of Smad2 in the cytoplasm and
expression of Snail and Twist in the nucleus (Fig. 5E). HDAC4 silencing also reduced the
luciferase activity of the TGF-β promoter (Fig. 5F). These results suggest that HDAC4
is involved in EMT through TGF-β signaling in A549-CUG2 cells.
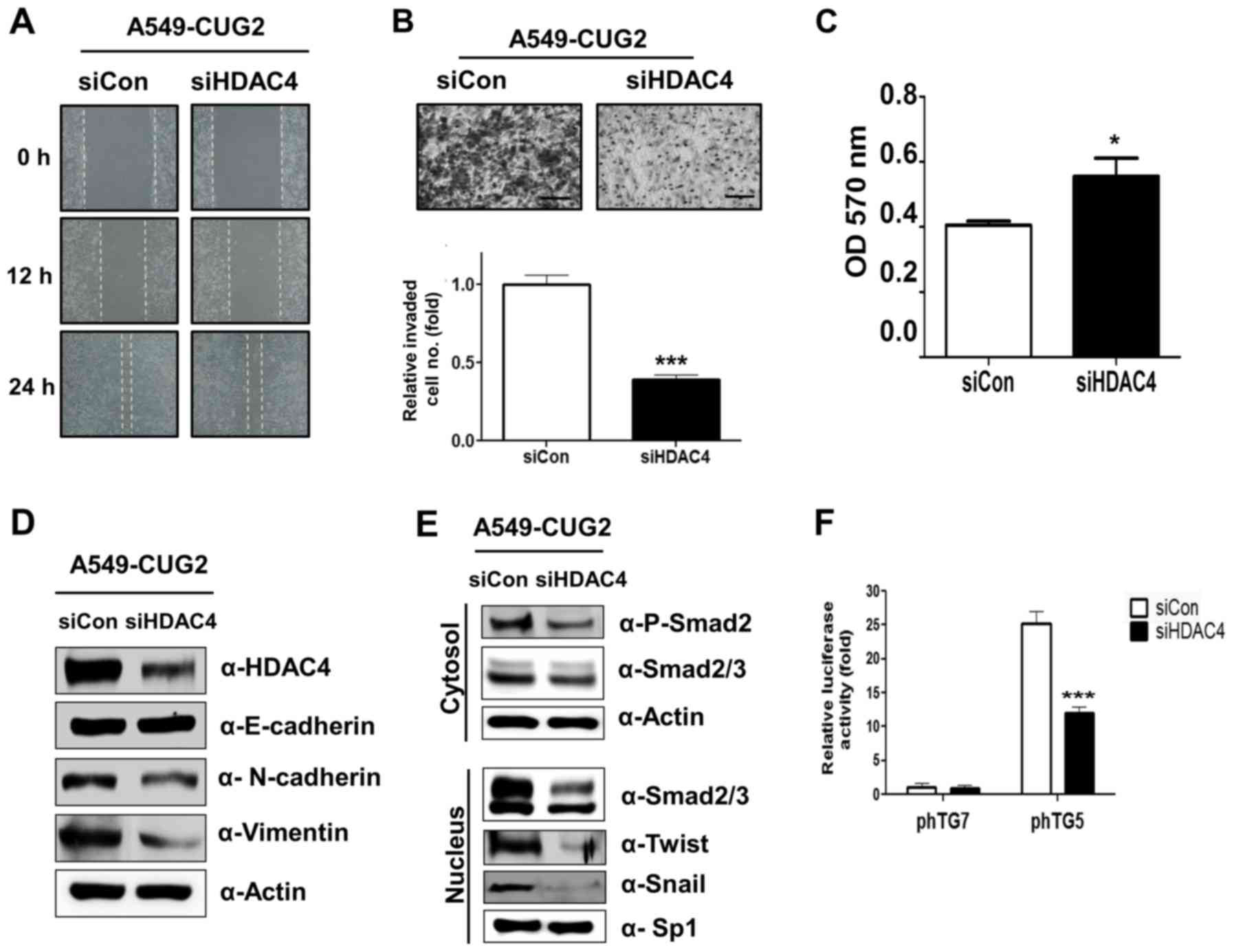 | Figure 5.HDAC4 suppression reduces
CUG2-induced cell migration and invasion. (A) Cell migration was
measured by a wound healing assay at 48 h post-transfection with
HDAC4 siRNA (500 nM) or control siRNA. (B) An invasion assay was
performed after transfection with HDAC4 siRNA or control siRNA
using 48-well Boyden chambers coated with Matrigel. Scale bar
indicates 100 µm (***P<0.001, compared to control siRNA). (C) To
examine an effect of HDAC4 on cell adhesion, A549-CUG2 cells were
seeded in collagen-coated wells at 48 h post-transfection with
HDAC4 siRNA or control siRNA. The cells were incubated for 30 min
for attachment and then the number of the attached cells were
analyzed by MTT assay (*P<0.05). (D) To examine an effect of
HDAC4 on levels of proteins related to EMT, expression of HDAC4,
E-cadherin, N-cadherin and vimentin was detected by immunoblotting
at 48 h post-treatment with HDAC4 or control siRNAs. (E) To examine
a role of HDAC4 in CUG2-induced upregulation of TGF-β signaling,
expression of phospho-Smad2, Smad2/3, Snail, and Twist was detected
by immunoblotting after transfection and cellular fractionation.
(F) To examine a role of HDAC4 in CUG2-induced upregulation of
TGF-β transcriptional activity, A549-CUG2 cells were transfected
with TGF-β promoter vectors (pTG5, pTG7; 1 µg) at 12 h
post-treatment with HDAC4 siRNA or control siRNA. Luciferase enzyme
activities were then measured at 36 h post-transfection
(***P<0.001, compared to control siRNA). |
Furthermore, we also aimed to ascertain whether
HDAC4 plays a role in CSC-like phenotypes, such as sphere formation
and expression of stemness-related factors. HDAC4 silencing
diminished the size and number of spheroids compared to that in the
control (Fig. 6A). HDAC4 siRNA
reduced expression of Sox2 and Nanog compared to that following
control siRNA treatment but failed to decrease the expression of
Bmi1, Oct4 and Klf4 (Fig. 6B).
These results suggest that HDAC4 is involved in the sphere
formation and increases Sox2 and Nanog expression in A549-CUG2
cells.
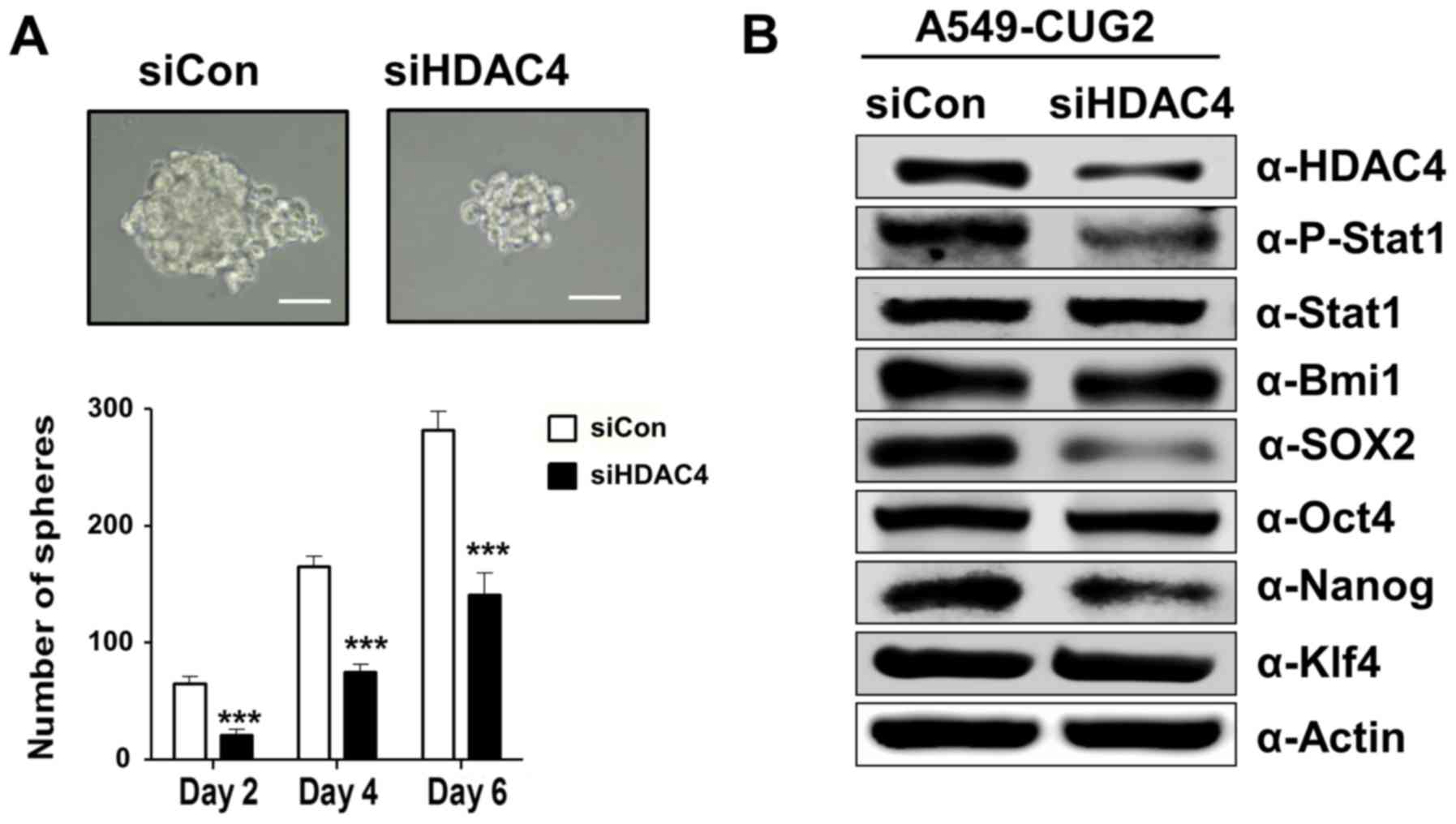 | Figure 6.Suppression of HDAC4 impairs
CUG2-induced sphere formation and expression of Sox2 and Nanog. (A)
After treatment with HDAC4 siRNA or control siRNA, the cells were
seeded in an ultra-low attachment 24-well plate. Spheroid size and
number were evaluated at 2, 4 and 6 days post-seeding. Scale bars
indicate 50 µm. The assay was carried out from three independent
experiments. (***P<0.001, compared to control siRNA). (B)
Expression of HDAC4, phosphorylated STAT1, STAT1, Bmi1, Sox2, Oct4,
Nanog, and Klf4 was detected by immunoblotting at 48 h
post-transfection with HDAC4 siRNA or control siRNA. |
Discussion
Regarding a role of STAT1 in tumorigenesis, some
studies have shown that STAT1 is activated during PDGF signaling
(29). Indeed the studies reported
that both enhanced STAT1 expression and activity occur exclusively
in cells able to undergo EMT (29).
In addition, another study showed that the EGFR-STAT1 axis
participates in the metastasis of pancreatic cancer cells (30). These studies support our finding
that activation of STAT1 under CUG2 overexpression plays a crucial
role in EMT and CSC-like phenotypes. Furthermore, our recent study
showed that TGF-β is also associated with these features induced by
CUG2 (9) (unpublished data). As the
underlying mechanism, we suggest that STAT1 might exert an
influence on TGF-β signaling, which is critical for EMT, as we
identified that STAT1 silencing not only inhibited Smad2
phosphorylation and Snail and Twist expression but also
CUG2-induced TGF-β transcriptional activity. TGF-β signaling
conversely affected STAT1 activation under CUG2 overexpression,
suggesting a crosstalk between the TGF-β and STAT1 signaling
pathways. However, the detailed mechanism will be investigated in
our next study.
Interestingly, we found that STAT1 suppression
inhibited CUG2-induced elevation of stemness-related factors,
specifically Sox2 and Oct4, and Nanog, but not Klf4. Despite the
differential effect of STAT1 on stemness-related factors, we
suggest that activated STAT1 could contribute to CUG2-induced
CSC-like phenotypes. The relationship between STAT1 and stemness
could be supported by the evidence that STAT1 inhibits
transcription of Sonic Hedgehog, which is involved in the
development of breast cancer stem cells (CSCs) when CD24 expression
is suppressed (31).
Another study demonstrated that STAT1 is a direct
substrate of HDAC4 and interacts with HDAC4 (21); thus, we focused on HDAC4, which has
been suggested to be an oncogene owing to the elevated expression
of HDAC4 that contributes to tumor growth by the stabilization of
HIF-1α (19) and reduction of p21
transcription (23). A recent study
reported that HDAC4 positively regulates EMT of esophageal
carcinoma cells by increasing the expression of vimentin and
decreasing the expression of E-cadherin (32). Other reports showed that HDAC4
inhibitors induce apoptosis through ER stress in myeloma cells
(33) and cytotoxicity in
chemoresistant cancer cells (34),
suggesting HDAC4 inhibitors as potential therapeutic drugs against
cancer. These lines of evidence support our finding that
suppression of HDAC4 inhibits CUG2-induced EMT and sphere
formation. Of note, we found that STAT1 knockdown reduces HDAC4
expression whereas HDAC4 silencing inhibits STAT1 phosphorylation
(35). As other studies showed that
STAT1 acetylation inhibited IFNg-induced STAT1 phosphorylation
(36,37), suggesting that STAT1 acetylation
regulates STAT1 signaling, we propose that enhanced STAT1
acetylation due to HDAC4 silencing may reduce STAT1
phosphorylation. Taken together, these reports indicate an
interplay between HDAC4 and STAT1. Here, we report that activation
of STAT1-HDAC4 signaling induced by CUG2 is involved in malignant
tumor features such as EMT and CSC-like phenotypes.
Acknowledgements
Not applicable.
Funding
This study was supported by a 2-year Research Grant
from Pusan National University.
Availability of data and materials
The datasets used the present study are available
from the corresponding author upon reasonable request.
Authors' contributions
SK, OHK, and YHC conceived and designed the study.
SK and CK performed the experiments and SSK was involved in data
compilation and analysis. SK and YHC wrote the paper. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing financial
interest.
References
|
1
|
Lee S, Gang J, Jeon SB, Choo SH, Lee B,
Kim YG, Lee YS, Jung J, Song SY and Koh SS: Molecular cloning and
functional analysis of a novel oncogene, cancer-upregulated gene 2
(CUG2). Biochem Biophys Res Commun. 360:633–639. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hori T, Amano M, Suzuki A, Backer CB,
Welburn JP, Dong Y, McEwen BF, Shang WH, Suzuki E, Okawa K, et al:
CCAN makes multiple contacts with centromeric DNA to provide
distinct pathways to the outer kinetochore. Cell. 135:1039–1052.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim H, Lee M and Lee S, Park B, Koh W, Lee
DJ, Lim DS and Lee S: Cancer-upregulated gene 2 (CUG2), a new
component of centromere complex, is required for kinetochore
function. Mol Cells. 27:697–701. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park EH, Park EH, Cho IR, Srisuttee R, Min
HJ, Oh MJ, Jeong YJ, Jhun BH, Johnston RN, Lee S, et al: CUG2, a
novel oncogene confers reoviral replication through Ras and p38
signaling pathway. Cancer Gene Ther. 17:307–314. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McEntee G, Kyula JN, Mansfield D, Smith H,
Wilkinson M, Gregory C, Roulstone V, Coffey M and Harrington KJ:
Enhanced cytotoxicity of reovirus and radiotherapy in melanoma
cells is mediated through increased viral replication and
mitochondrial apoptotic signalling. Oncotarget. 7:48517–48532.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sborov DW, Nuovo GJ, Stiff A, Mace T,
Lesinski GB, Benson DM Jr, Efebera YA, Rosko AE, Pichiorri F,
Grever MR, et al: A phase I trial of single-agent reolysin in
patients with relapsed multiple myeloma. Clin Cancer Res.
20:5946–5955. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Malilas W, Koh SS, Srisuttee R, Boonying
W, Cho IR, Jeong CS, Johnston RN and Chung YH: Cancer upregulated
gene 2, a novel oncogene, confers resistance to oncolytic vesicular
stomatitis virus through STAT1-OASL2 signaling. Cancer Gene Ther.
20:125–132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malilas W, Koh SS, Kim S, Srisuttee R, Cho
IR, Moon J, Yoo HS, Oh S, Johnston RN and Chung YH: Cancer
upregulated gene 2, a novel oncogene, enhances migration and drug
resistance of colon cancer cells via STAT1 activation. Int J Oncol.
43:1111–1116. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaowinn S, Kim J, Lee J, Shin DH, Kang CD,
Kim DK, Lee S, Kang MK, Koh SS, Kim SJ, et al: Cancer upregulated
gene 2 induces epithelial-mesenchymal transition of human lung
cancer cells via TGF-βeta signaling. Oncotarget. 8:5092–5110. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chin YE, Kitagawa M, Kuida K, Flavell RA
and Fu XY: Activation of the STAT signaling pathway can cause
expression of caspase 1 and apoptosis. Mol Cell Biol. 17:5328–5337.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumar A, Commane M, Flickinger TW, Horvath
CM and Stark GR: Defective TNF-alpha-induced apoptosis in
STAT1-null cells due to low constitutive levels of caspases.
Science. 278:1630–1632. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chin YE, Kitagawa M, Su WC, You ZH,
Iwamoto Y and Fu XY: Cell growth arrest and induction of
cyclin-dependent kinase inhibitor p21WAF1/CIP1 mediated
by STAT1. Science. 272:719–722. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Townsend PA, Scarabelli TM, Davidson SM,
Knight RA, Latchman DS and Stephanou A: STAT-1 interacts with p53
to enhance DNA damage-induced apoptosis. J Biol Chem.
279:5811–5820. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Khodarev NN, Beckett M, Labay E, Darga T,
Roizman B and Weichselbaum RR: STAT1 is overexpressed in tumors
selected for radioresistance and confers protection from radiation
in transduced sensitive cells. Proc Natl Acad Sci USA.
101:1714–1719. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weichselbaum RR, Ishwaran H, Yoon T,
Nuyten DS, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike
B, et al: An interferon-related gene signature for DNA damage
resistance is a predictive marker for chemotherapy and radiation
for breast cancer. Proc Natl Acad Sci USA. 105:18490–18495. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fryknas M, Dhar S, Oberg F, Rickardson L,
Rydaker M, Goransson H, Gustafsson M, Pettersson U, Nygren P,
Larsson R and Isaksson A: STAT1 signaling is associated with
acquired crossresistance to doxorubicin and radiation in myeloma
cell lines. Int J Cancer. 120:189–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roberts D, Schick J, Conway S, Biade S,
Laub PB, Stevenson JP, Hamilton TC, O'Dwyer PJ and Johnson SW:
Identification of genes associated with platinum drug sensitivity
and resistance in human ovarian cancer cells. Br J Cancer.
92:1149–1158. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang XJ and Seto E: HATs and HDACs: From
structure, function and regulation to novel strategies for therapy
and prevention. Oncogene. 26:5310–5318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Geng H, Harvey CT, Pittsenbarger J, Liu Q,
Beer TM, Xue C and Qian DZ: HDAC4 protein regulates HIF1α protein
lysine acetylation and cancer cell response to hypoxia. J Biol
Chem. 286:38095–38102. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mihaylova MM, Vasquez DS, Ravnskjaer K,
Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M and
Shaw RJ: Class IIa histone deacetylases are hormone-activated
regulators of FOXO and mammalian glucose homeostasis. Cell.
145:607–621. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stronach EA, Alfraidi A, Rama N, Datler C,
Studd JB, Agarwal R, Guney TG, Gourley C, Hennessy BT, Mills GB, et
al: HDAC4-regulated STAT1 activation mediates platinum resistance
in ovarian cancer. Cancer Res. 71:4412–4422. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Giaginis C, Alexandrou P, Delladetsima I,
Giannopoulou I, Patsouris E and Theocharis S: Clinical significance
of histone deacetylase (HDAC)-1, HDAC-2, HDAC-4, and HDAC-6
expression in human malignant and benign thyroid lesions. Tumour
Biol. 35:61–71. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wilson AJ, Byun DS, Nasser S, Murray LB,
Ayyanar K, Arango D, Figueroa M, Melnick A, Kao GD, Augenlicht LH
and Mariadason JM: HDAC4 promotes growth of colon cancer cells via
repression of p21. Mol Biol Cell. 19:4062–4075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shen YF, Wei AM, Kou Q, Zhu QY and Zhang
L: Histone deacetylase 4 increases progressive epithelial ovarian
cancer cells via repression of p21 on fibrillar collagen matrices.
Oncol Rep. 35:948–954. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kang ZH, Wang CY, Zhang WL, Zhang JT, Yuan
CH, Zhao PW, Lin YY, Hong S, Li CY and Wang L: Histone deacetylase
HDAC4 promotes gastric cancer SGC-7901 cells progression via p21
repression. PLoS One. 9:e988942014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim SJ, Glick A, Sporn MB and Roberts AB:
Characterization of the promoter region of the human transforming
growth factor-beta 1 gene. J Biol Chem. 264:402–408.
1989.PubMed/NCBI
|
|
27
|
Zhang Y, Wang W, Wang Y, Huang X, Zhang Z,
Chen B, Xie W, Li S, Shen S and Peng B: NEK2 promotes
hepatocellular carcinoma migration and invasion through modulation
of the epithelial-mesenchymal transition. Oncol Rep. 39:1023–1033.
2018.PubMed/NCBI
|
|
28
|
Kramer OH and Heinzel T:
Phosphorylation-acetylation switch in the regulation of STAT1
signaling. Mol Cell Endocrinol. 315:40–48. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jechlinger M, Sommer A, Moriggl R, Seither
P, Kraut N, Capodiecci P, Donovan M, Cordon-Cardo C, Beug H and
Grunert S: Autocrine PDGFR signaling promotes mammary cancer
metastasis. J Clin Invest. 116:1561–1570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seshacharyulu P, Ponnusamy MP, Rachagani
S, Lakshmanan I, Haridas D, Yan Y, Ganti AK and Batra SK: Targeting
EGF-receptor(s)-STAT1 axis attenuates tumor growth and metastasis
through downregulation of MUC4 mucin in human pancreatic cancer.
Oncotarget. 6:5164–5181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Suyama K, Onishi H, Imaizumi A, Shinkai K,
Umebayashi M, Kubo M, Mizuuchi Y, Oda Y, Tanaka M, Nakamura M, et
al: CD24 suppresses malignant phenotype by downregulation of SHH
transcription through STAT1 inhibition in breast cancer cells.
Cancer Lett. 374:44–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zeng LS, Yang XZ, Wen YF, Mail SJ, Wang
MH, Zhang MY, Zheng XF and Wang HY: Overexpressed HDAC4 is
associated with poor survival and promotes tumor progression in
esophageal carcinoma. Aging. 8:1236–1249. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kikuchi S, Suzuki R, Ohguchi H, Yoshida Y,
Lu D, Cottini F, Jakubikova J, Bianchi G, Harada T, Gorgun G, et
al: Class IIa HDAC inhibition enhances ER stress-mediated cell
death in multiple myeloma. Leukemia. 29:1918–1927. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marek L, Hamacher A, Hansen FK, Kuna K,
Gohlke H, Kassack MU and Kurz T: Histone deacetylase (HDAC)
inhibitors with a novel connecting unit linker region reveal a
selectivity profile for HDAC4 and HDAC5 with improved activity
against chemoresistant cancer cells. J Med Chem. 56:427–436. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kaowinn S, Jun SW, Kim CS, Shin DM, Hwang
YH, Kim K, Shin B, Kaewpiboon C, Jeong HH, Koh SS, et al: Increased
EGFR expression induced by a novel oncogene, CUG2, confers
resistance to doxorubicin through Stat1-HDAC4 signaling. Cell
Oncol. 40:549–561. 2017. View Article : Google Scholar
|
|
36
|
Kramer OH, Knauer SK, Greiner G, Jandt E,
Reichardt S, Gührs KH, Stauber RH, Böhmer FD and Heinzel T: A
phosphorylation-acetylation switch regulates STAT1 signaling. Genes
Dev. 23:223–235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ginter T, Bier C, Knauer SK, Sughra K,
Hildebrand D, Munz T, Liebe T, Heller R, Henke A, Stauber RH, et
al: Histone deacetylase inhibitors block IFNγ-induced STAT1
phosphorylation. Cell Signal. 24:1453–1460. 2012. View Article : Google Scholar : PubMed/NCBI
|