Introduction
Non-muscle-invasive bladder cancer (NMIBC) and
muscle-invasive bladder cancer (MIBC) are two subtypes of bladder
transitional cell carcinoma (TCC), which account for ~95% neoplasms
derived from the bladder. The majority of patients with NMIBC
receive transurethral resection, whereas early radical cystectomy
with pelvic node dissection is recommended for patients with MIBC.
Approximately 20% NIMBC cases progress to MIBC and nearly 50% of
patients with MIBC die within 5 years due to distant metastasis
(1,2). There is an urgent need to understand
the molecular mechanisms that lead to TCC metastasis and to
identify potential therapeutic drugs to prolong patient
survival.
Epithelial-mesenchymal transition (EMT) is among the
most well-established theories regarding the mechanism of
metastasis, and targeting EMT has achieved notable breakthroughs in
basic research and clinical trials (3–7). EMT
can be triggered by a number of growth factors and inflammatory
mediators. Transforming growth factor (TGF)-β1 is the most studied
regulator of EMT (5,8). TGF-β1-induced EMT has been extensively
studied in bladder cancer due to high TGF-β1 levels in the blood,
urine and tumor tissues of patients with TCC (9). Following TGF-β1-targeted treatment,
epithelial cells lose apical polarity and obtain spindle-like
mesenchymal morphology with enhanced expression of N-cadherin and
decreased expression of E-cadherin (10). However, the underlying mechanism in
TGF-β1-induced EMT is not fully understood.
Accumulating evidence has suggested that silibinin,
a natural extract from milk thistle, has anti-tumor effects against
various cancer types, including breast, renal, prostate and bladder
cancer (11–17). Silibinin is a potential
chemotherapeutic for bladder cancer due to its outstanding
anti-neoplasm capacities (18).
Beyond its anti-proliferation effect, a study conducted by our
group indicated that silibinin exerts significant anti-metastatic
effects on TCC through dual-blocking of EMT and stemness (19). However, whether TGF-β1-induced EMT
is inhibited by silibinin, and the potential mechanisms involved,
are still unclear.
In the present study, the effects of silibinin on
TGF-β1-induced metastasis and EMT in TCC were investigated in
vitro, focusing on the regulation of prostaglandin-endoperoxide
synthase 2 (COX-2).
Materials and methods
Reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS), penicillin and streptomycin cocktail were
purchased from HyClone; GE Healthcare Life Sciences (Logan, UT,
USA). Primary antibodies anti-E-cadherin (cat. no. 3195),
anti-N-cadherin (cat. no. 14215), anti-Vimentin (cat. no. 5741),
anti-β-catenin (cat. no. 8480), anti-zinc finger E-box binding
homeobox (ZEB)1 (cat. no. 3396), anti-prostaglandin-endoperoxide
synthase 2 (COX-2) (cat. no. 3396), anti-GAPDH (cat. no. 8884) and
secondary antibodies goat anti-rabbit (cat. no. 7074), horse
anti-mouse (cat. no. 7076), TGF-β1 (cat. no. 5154LC) and control
small interfering (si)RNA (cat. no. 6568) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Dilutions for
primary and secondary antibodies were 1:1,000 and 1:3,000
respectively. COX-2 siRNA (cat. no. sc-29279) was from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA) (20). Silibinin, MTT, proteinase and
phosphatase inhibitors cocktail were from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany). Transwell mini-cells were from EMD
Millipore (Billerica, MA, USA) and matrix gel was purchased from BD
Biosciences (Franklin Lakes, NJ, USA). Polyvinylidene difluoride
(PVDF) membrane and enhanced chemiluminescent (ECL) reagents were
from Bio-Rad Laboratories, Inc. (Hercules, CA, USA). DharmaFECT 1
transfection reagent was from GE Healthcare Life Sciences (Little
Chalfont, UK).
Cell culture
T24 cell line was purchased from American Type
Culture Collection (Manassas, VA, USA) and 253J was a gift from Dr
Hsieh JT's laboratory in Southwest University Medical Center
(Dallas, TX, USA). The two cell lines were cultured in DMEM
supplemented with 10% FBS, 100 U/ml penicillin and 100 µg/ml
streptomycin. Cells were incubated in an atmosphere of 95% humidity
at 37°C. Culture medium was replaced every other day, or according
to experimental design requirements.
Wound healing assay
Cells were seeded in 6-well plates. At ~80%
confluency, cells were treated with TGF-β1 (5 ng/ml), silibinin (50
µM) or both for 24 h. Then, wounds were scratched using a 200-µl
pipette tip and cell monolayers were rinsed with pre-warmed PBS
(37°C) 3 times. The cells were then aspirated and cultured in 2 ml
fresh serum-free medium containing the appropriate treatments.
Images were captured using an inverted microscope at ×40
magnification.
Transwell migration and invasion
assay
Cells were pre-treated with TGF-β1 (5 ng/ml),
silibinin (50 µM) or both for 24 h, then cells were digested and
centrifuged at room temperature at 200 × g for 5 min. The cell
number was counted using a hemocytometer. For the Transwell
invasion assay, Matrigel was diluted in serum-free medium (1:5) and
pipetted onto the inner membrane of the Transwell mini-cells. Cells
were re-suspended in serum-free medium. Cells (2×104 or
8×104) were seeded to the upper chamber for the
migration and invasion assays, respectively, in a final volume of
500 µl. The lower chamber was filled with 800 µl complete medium
with 10% FBS and the medium in the upper chambers contained TGF-β1
(5 ng/ml), silibinin (50 µM) or both. After 24 h of incubation,
cells were fixed in 4% paraformaldehyde and stained with 0.1%
crystal violet at room temperature for 15 min. Migrated or invaded
cells in the membrane were observed using a light microscope
(Olympus, Tokyo, Japan) at ×100 magnification. For each mini-cell,
five images were randomly captured and cell number was counted
using ImageJ software, version 6.0 (National Institutes of Health,
Bethesda, MD, USA).
MTT assay
Cells (3,000/well) were seeded into a 96-well plate
and incubated in the cell culture incubator overnight. The medium
was replaced with fresh complete medium with/without silibinin (50
µM) and/or TGF-β1 (5 ng/ml). After 48 h, MTT was added to each well
(final concentration 0.5 mg/ml) 2–4 h prior to harvesting. The
medium was removed and 150 µl dimethyl sulfoxide was added to each
well. Samples were vortexed gently to dissolve the precipitates.
The optical density value was read using a BioTek plate reader at a
490 nm wavelength (BioTek Instruments, Inc., Winooski, VT,
USA).
Cell counting assay
Cells (100,000/well) were seeded to the wells of a
6-well plate and incubated overnight. The medium was replaced with
fresh complete medium with/without silibinin (50 µM) and/or TGF-β1
(5 ng/ml) and after 48 h, cells were scraped and resuspended in 5
ml PBS. Cells were then counted using the Beckman Coulter Z2 cell
and particle counter (Beckman Coulter, Inc., Brea, CA, USA).
mRNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Protocols used for mRNA isolation, cDNA reversing
and RT-qPCR were as previously described (13). Briefly, mRNA was isolated by RNA
Fast 200 isolation kit (Fastagen Biotech, Shanghai, China) and then
reverse transcribed to cDNA using Takara PrimeScript™ RT Master Mix
(Perfect Real-Time) (cat. no. RR036A; Takara Bio, Inc., Otsu,
Japan), the thermocycling conditions were 37°C for 15 min
(reverse-transcription), 85°C for 5 sec (for heat inactivation of
reverse transcriptase) and 4°C (end of reverse transcriptase). A
reaction solution which consisted of primers, cDNA and SYBR
advantage qPCR premix (cat. no. 639676; Takara Bio, Inc.) was made
and loaded to Bio-Rad CFX96 real-time PCR machine (Bio-Rad
Laboratories, Inc.). The protocol utilized was initial denaturation
(95°C for 30 sec, 1 cycle), PCR (95°C for 5 sec, 55°C for 30 sec
and 72°C for 30 sec, 40 cycles). Experiments were conducted in
triplicate. Primer sequences are listed in Table I. The 2−ΔΔCq method was
used to analyze relative gene expression (21).
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Sequences
(5′-3′) |
|---|
| GAPDH |
|
| F |
CGACCACTTTGTCAAGCTCA |
| R |
AGGGGAGATTCAGTGTGGTG |
| E-cadherin |
|
| F |
CGAGAGCTACACGTTCACGG |
| R |
GGGTGTCGAGGGAAAAATAGG |
| N-cadherin |
|
| F |
ACAGTGGCCACCTACAAAGG |
| R |
CCGAGATGGGGTTGATAATG |
| β-catenin |
|
| F |
ATGGCTACTCAAGCTGAC |
| R |
CAGCACTTTCAGCACTCTGC |
| ZEB1 |
|
| F |
GCACCTGAAGAGGACCAGAG |
| R |
TGCATCTGGTGTTCCATTTT |
| Vimentin |
|
| F |
GAGAACTTTGCCGTTGAAGC |
| R |
GCTTCCTGTAGGTGGCAATC |
| COX-2 |
|
| F |
ATCACAGGCTTCCATTGACC |
| R |
CAGGATAGAGCTCCACAGCA |
Western-blotting
Cells were harvested using 1X
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Jiangsu, China) containing proteinase and
phosphatase inhibitor cocktail and boiled with SDS loading buffer.
Protein concentration was quantified using a BCA quantification kit
(Pierce; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Total
protein from each sample (30 µg) was separated by 10% SDS-PAGE.
Proteins were then transferred to a PVDF membrane. After blocking
for 1 h in 5% skim milk at room temperature, membranes were
incubated with primary antibodies for 2 h at room temperature.
Membranes were washed 3 times with Tris-buffered saline Tween-20
(TBST) and then incubated with secondary antibodies for 1 h at room
temperature. Following 3 washes in TBST, membranes were immersed in
ECL mix for 5 min. Protein bands were detected and quantified using
a Bio-Rad ChemiDoc system, version 4.0 (Bio-Rad Laboratories,
Inc.).
siRNA transfection
siRNAs were transfected into cells using DharmaFECT
1 transfection reagent according to the manufacturer's protocol.
Briefly, culture medium in the 6-well plate was replaced with 2 ml
serum-free medium 1 h before transfection. A total of 10 µl siRNAs
and 4 µl transfection reagents were diluted in 200 µl separate
serum-free medium and then mixed together and incubated at room
temperature for 10 min. The transfection complex mixture was then
added to the culture medium and mixed well by shaking several times
to make the final concentration of 50 nM. The medium was replaced
by complete medium after 24 h of incubation. mRNA was isolated 48 h
after transfection, while protein was harvested at 72 h after
transfection.
Statistical analysis
All experiments were performed at least 3 times.
SPSS software, version 19.0 (IBM SPSS, Armonk, NY, USA) was used to
perform the statistical analysis. Analysis of variance followed by
post-hoc Student-Newman-Keuls test was utilized for statistical
analysis between or among groups. When the comparison involved only
2 groups, a Student's t-test was used. P<0.05 was considered to
indicate a statistically significant difference.
Results
Silibinin attenuates TGF-β1-induced
migration and invasion in TCC
TGF-β1 has been reported to promote metastasis in
various cancer models in vitro and in vivo, including
bladder cancer. Additionally, silibinin has been demonstrated to be
a potent metastasis inhibitor. Using TCC cell lines T24 and 253J,
with high metastatic and invasive potential, as the model system
in vitro, the present study aimed to explore the potential
effects of silibinin on TGF-β1-induced migration and invasion in
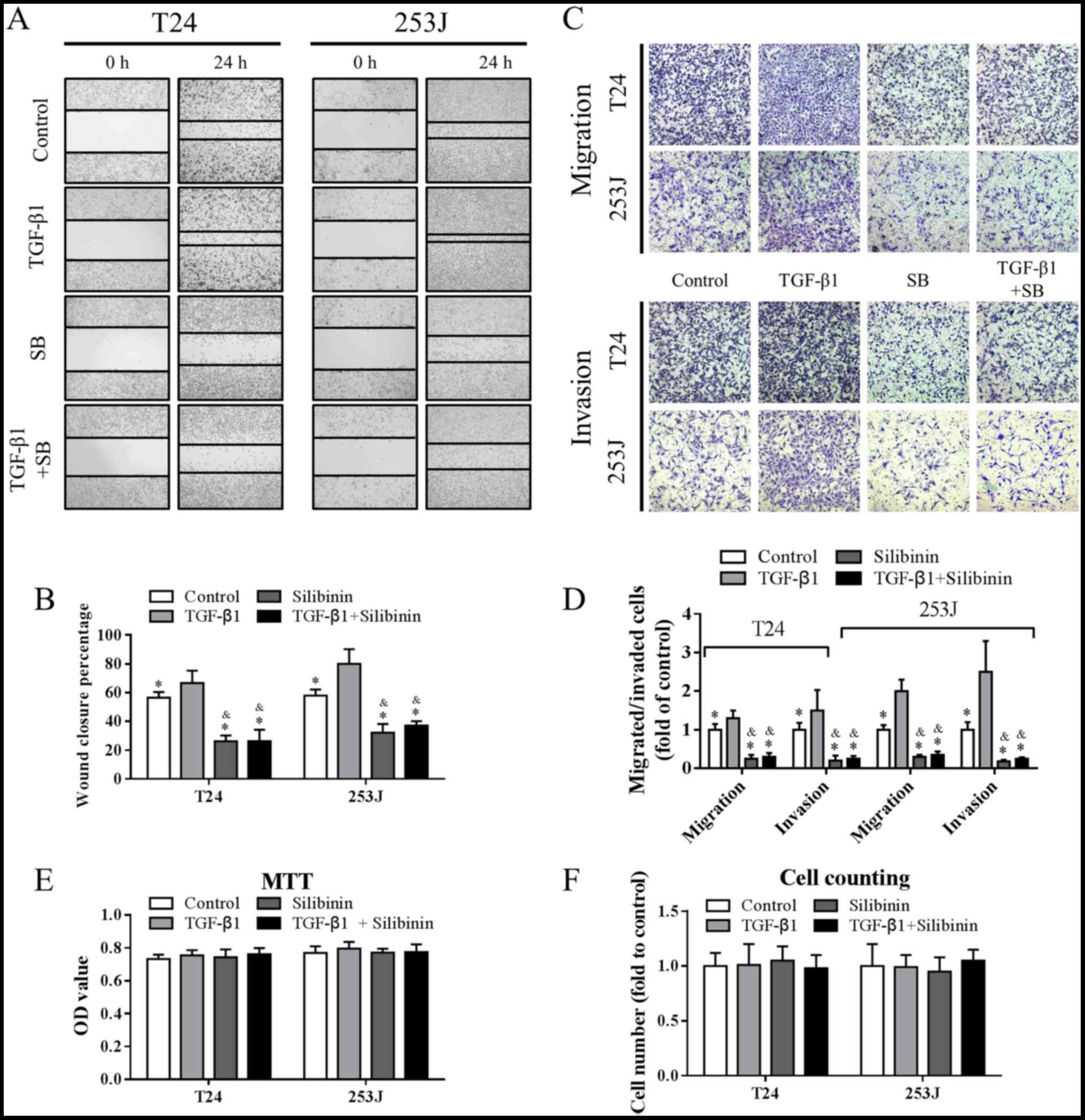
TCC. As determined by the results of a wound healing assay,
presented in Fig. 1A and B, 5 ng/ml
TGF-β1 promoted cell migration, while 50 µM silibinin inhibited
migration, which was consistent with the literature (22). This effect was further confirmed by
Transwell migration and invasion assays (Fig. 1C and D). Additionally, TGF-β1 at 5
ng/ml and/or silibinin at 50 µM treatment for 48 h had no impact on
cell growth (Fig. 1E and F). These
data suggested that TGF-β1 induced migration and invasion, and that
these were attenuated by silibinin.
Silibinin attenuates TGF-β1-induced
migration and invasion via EMT inhibition
EMT induction is one of the main mechanisms that
contributes to increased metastatic potential promoted by TGF-β1.
As silibinin attenuated TGF-β1-induced migration and invasion, the
present study subsequently investigated the potential mechanisms
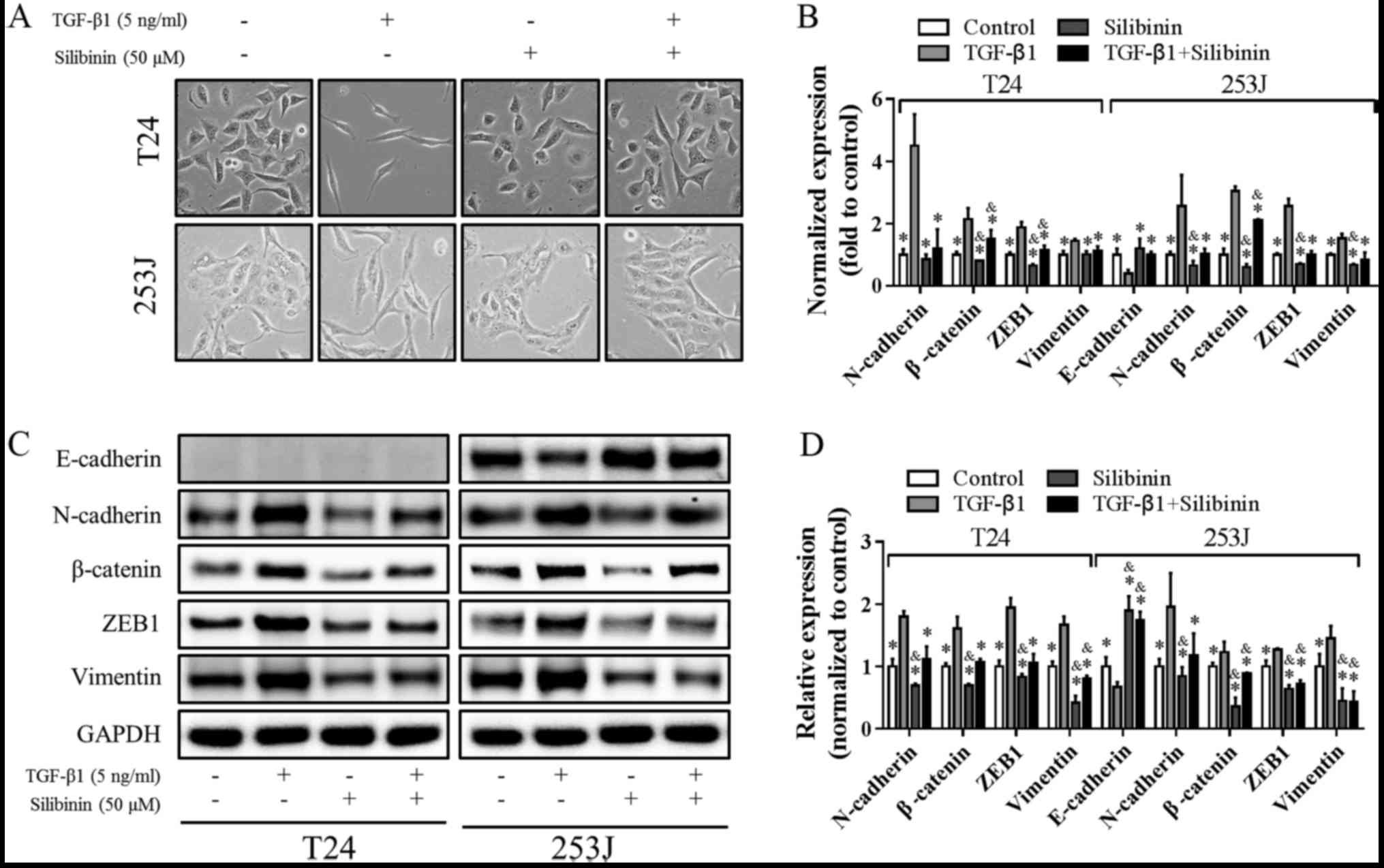
involved, focusing on EMT signaling. As presented in Fig. 2A, following TGF-β1 treatment, cells
exhibited a spindle-like shape with mesenchymal morphology and were
more isolated from each other. Interestingly, this change in
phenotype was reversed by co-treatment with silibinin and TGF-β1.
Silibinin treatment alone did not markedly affect cell morphology.
To further confirm the change in phenotype, levels of EMT markers
were determined using RT-qPCR. TGF-β1 increased the expression of
the mesenchymal markers, N-cadherin, Vimentin, β-catenin and ZEB1,
and decreased the expression of the epithelial marker, E-cadherin.
When silibinin treatment was combined with TGF-β1, the changes in
EMT markers induced by TGF-β1 were attenuated (Fig. 2B). The changes in expression of
these EMT-associated genes were further confirmed at the protein
level by western blotting analysis, and similar effects were
observed (Fig. 2C and D). Notably,
E-cadherin was undetectable in the T24 cell line at the mRNA and
protein level, which is in accordance with the literature (23). Collectively, these results indicated
that EMT inhibition may be the underlying mechanism involved in the
inhibitory effects of silibinin on migration and invasion induced
by TGF-β1.
COX-2 upregulation is essential for
TGF-β1-induced EMT
COX-2 has been reported to be a mediator of
TGF-β1-induced EMT (24). However,
whether it plays a key role in EMT induced by TGF-β1 in TCC has not
been clarified. As presented in Fig. 3A
and B, TGF-β1 increased COX-2 expression, upregulated the
expression of N-cadherin, Vimmm entin, β-catenin and ZEB1 and
downregulated expression of E-cadherin. To further explore the role
of COX-2 in TGF-β1-induced EMT, COX-2 expression was knocked down
using specific siRNA. As presented in Fig. 3C, when COX-2 expression was
silenced, N-cadherin, Vimentin, β-catenin and ZEB1 expression was
decreased, with E-cadherin expression increased. In addition, the
mesenchymal morphology induced by TGF-β1 was reversed by COX-2
knockdown (Fig. 3D). These results
indicated that COX-2 expression is essential for TGF-β1-induced
EMT.
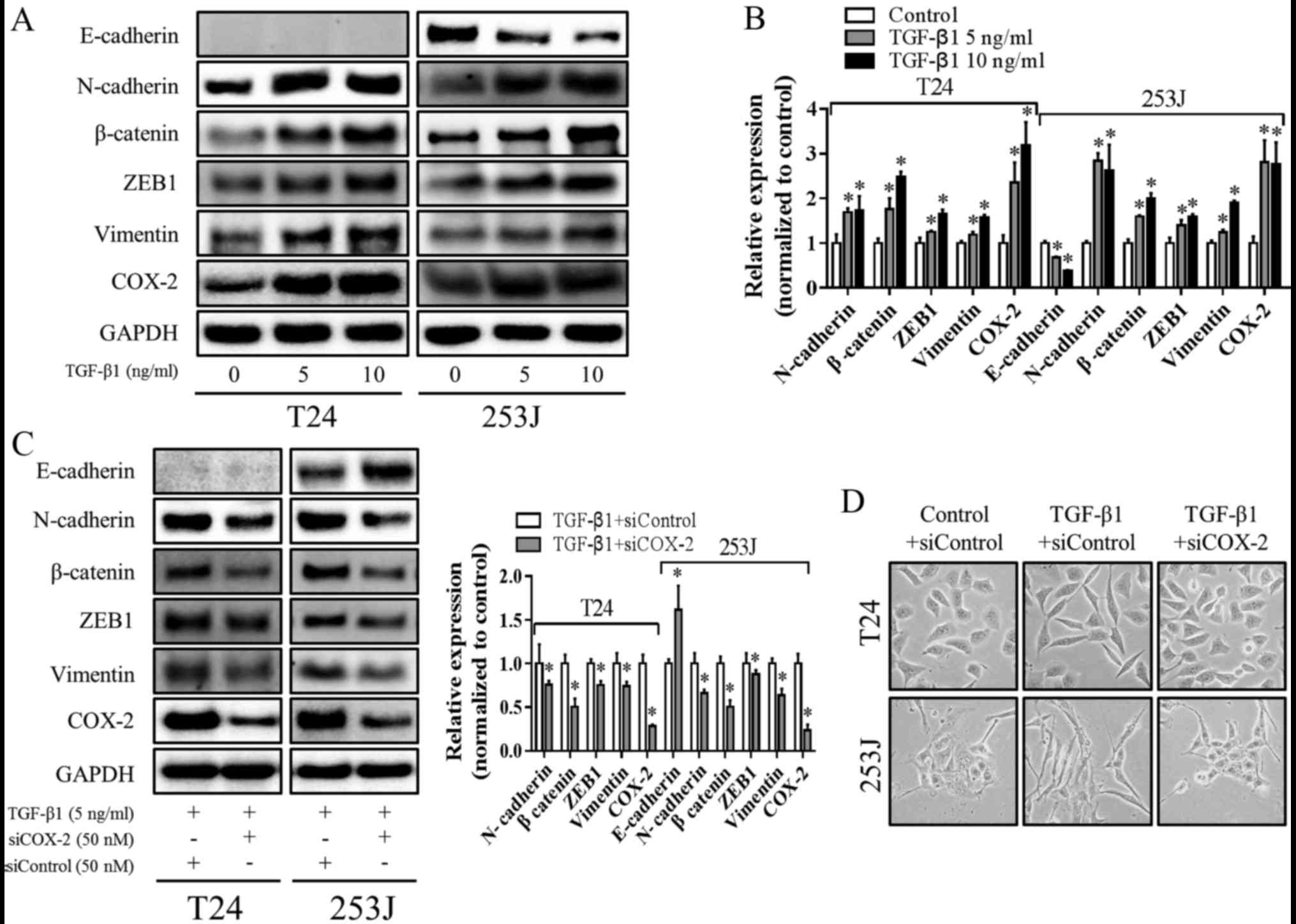 | Figure 3.COX-2 is essential for TGF-β1-induced
EMT. (A) Cells were treated with TGF-β1 at 5 ng/ml or 10 ng/ml for
48 h, and the expression of E-cadherin, N-cadherin, Vimentin,
β-catenin, ZEB1 and COX-2 were determined using western-blotting
and (B) statistically analyzed using densitometry. *P<0.05 vs.
control. (C) Cells were transfected with control siRNA (siControl)
or siCOX-2 for 24 h and then treated with 5 ng/ml TGF-β1 for
additional 48 h, and the levels of E-cadherin, N-cadherin,
Vimentin, β-catenin, ZEB1 and COX-2 were determined using western
blotting and statistically analyzed using densitometry. GAPDH was
used as internal control. *P<0.05 vs. siControl. (D) Cells were
transfected with siControl or siCOX-2 for 24 h and then treated
with/without 5 ng/ml TGF-β1 for another 48 h, representative images
of cell morphology (magnification, ×200) were captured using a
microscope. COX-2, prostaglandin-endoperoxide synthase 2; TGF-β1,
transforming growth factor-β1; EMT, epithelial-mesenchymal
transition; ZEB1, zinc finger E-box binding homeobox 1; si, small
interfering RNA. |
Silibinin inhibition of TGF-β1-induced
EMT is associated with COX-2 downregulation
As COX-2 was shown to play a pivotal role in
TGF-β1-induced EMT, the present study next investigated the
potential effects of silibinin on COX-2. As presented in Fig. 4A, in the presence of TGF-β1,
expression of COX-2, N-Cadherin, Vimentin, β-catenin and ZEB1 was
downregulated by silibinin, and E-Cadherin expression was
upregulated. To further confirm the role of COX-2, cells were
treated with silibinin with/without COX-2-knockdown in the presence
of TGF-β1. As presented in Fig. 4B,
silibinin or siCOX-2 decreased the expression of COX-2, N-cadherin,
Vimentin, β-catenin and ZEB1, and increased the expression of
E-Cadherin. When COX-2 knockdown was combined with silibinin,
COX-2, N-cadherin, Vimentin, β-catenin and ZEB1 expression levels
were decreased further, while E-cadherin expression was increased
further. These data suggested that inhibition of TGF-β1-induced EMT
by silibinin is associated with COX-2 downregulation.
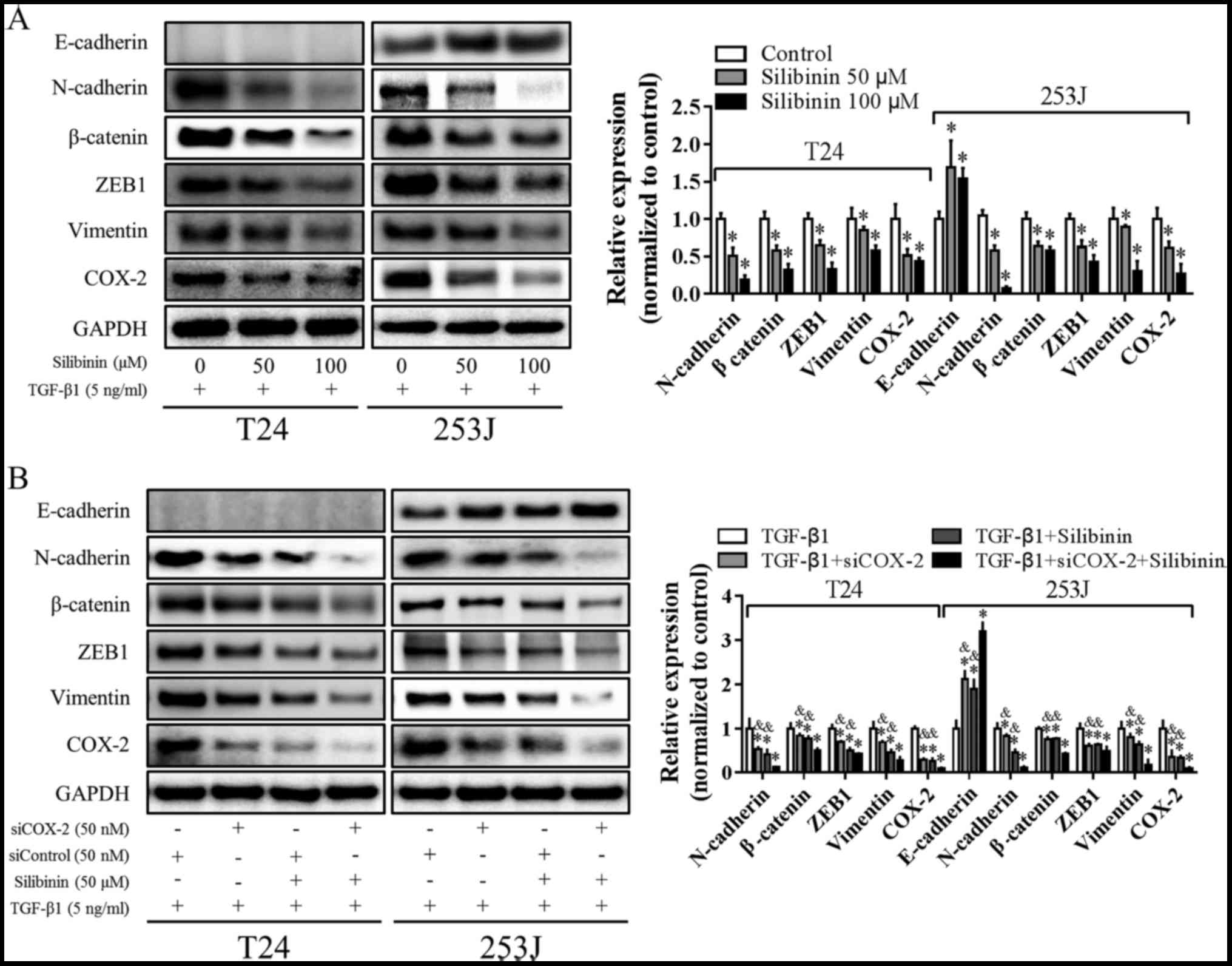 | Figure 4.Silibinin inhibits TGF-β1-induced EMT
and downregulates COX-2 expression. (A) Cells were treated with
silibinin (0, 50 and 100 µM) in the presence of 5 ng/ml TGF-β1 for
48 h; E-cadherin, N-cadherin, Vimentin, β-catenin, ZEB1 and COX-2
expression levels were determined using western blotting and
statistically analyzed using densitometry. *P<0.05 vs. control.
(B) Cells were transfected with siControl or siCOX-2 for 24 h and
then treated with/without 50 µM silibinin in the presence of 5
ng/ml TGF-β1 for an additional 48 h. E-cadherin, N-cadherin,
Vimentin, β-catenin, ZEB1 and COX-2 were determined using western
blotting and statistically analyzed. GAPDH was used as an internal
control. *P<0.05 vs. TGF-β1 treatment; &P<0.05
vs. triple treatment group. TGF-β1, transforming growth factor β1;
EMT, epithelial-mesenchymal transition; COX-2,
prostaglandin-endoperoxide synthase 2; ZEB1, zinc finger E-box
binding homeobox 1; si, small interfering RNA. |
Discussion
TGF-β1-induced cell migration and invasion via EMT
induction has been reported in various cancer models, including
bladder cancer; however, whether this promotion of metastasis and
EMT induction can be modulated by silibinin is unclear. The present
study confirmed that TGF-β1 promoted cell migration and invasion
via inducing EMT in TCC cells. Additionally, silibinin treatment
attenuated the migration and invasion induced by TGF-β1 via EMT
suppression.
EMT is a dynamic process and is an initial step in
metastasis whereby localized cancer cells become aggressive and
migrate to a distant site. During this process, cells lose
cuboidal-like epithelial morphology and lose expression of
epithelial proteins, such as E-cadherin. Additionally, cells gain
an elongated spindle-like mesenchymal morphology, with increased
expression of mesenchymal proteins, including N-cadherin (25). Several signaling pathways have been
demonstrated to induce EMT, including TGF-β, fibroblast growth
factor, epidermal growth factor, hepatocyte growth factor,
Wnt/β-catenin and Notch signaling (26). Other crucial regulators, including
hedgehog, nuclear factor-κB and activating transcription factor 2
have also been implicated in EMT. However, the signaling pathways
or regulators that are involved in EMT induction may vary, as the
process is tumor tissue- and cell type-dependent (27–29).
Notably, TGF-β1 regulates the function of transcriptional
regulators, including Snail1, Slug and Twist, by modulating their
expression or altering their binding patterns via canonical or
non-canonical signaling pathways, resulting in decreased E-cadherin
expression and increased expression of N-cadherin, Vimentin and
metalloproteinases in epithelial cells (30–32).
Crosstalk between inflammation and EMT signaling is
a current topic of research interest. EMT induced by inflammatory
factors, such as interleukin-6 and tumor necrosis factor-α in the
tumor microenvironment have been studied in various tumor models
(6,33–36).
Previously, it was reported that COX-2, a crucial inflammation
mediator, is a link between inflammation signaling and EMT in
prostate cancer, and that its inactivation represses the expression
of genes involved in EMT (37).
COX-2 has also been reported to be highly expressed in bladder TCC
and positively associated with tumor grade (38). However, whether COX-2 plays a role
in TGF-β1-induced EMT is unclear. The present study, to the best of
the author's knowledge, was the first to demonstrate that COX-2 is
a key mediator of EMT induced by TGF-β1 in TCC. COX-2-knockdown
inhibited TGF-β1-induced expression of EMT-associated genes and
cell morphology transformation. However, how COX-2 mediates
TGF-β1-induced EMT in TCC is unclear, and further experiments are
required to unmask the underlying mechanisms.
Silibinin has been demonstrated to be a potent EMT
suppressor in numerous studies in vitro and in vivo
(39), but whether TGF-β1-induced
EMT can be inhibited by silibinin is unknown. The present study
demonstrated that TGF-β1-induced EMT was suppressed by silibinin,
and that COX-2 downregulation was involved in the underlying
mechanism. Inhibition of COX-2 by silibinin has also been reported
in a previous study that investigated the anti-inflammatory
potential of the compound (7). The
present study focused on the role of COX-2 in TGF-β1-induced EMT.
Silencing of COX-2 in TCC cells together with silibinin treatment
resulted in further decrease of COX-2 expression, and increased the
inhibition of EMT. This confirmed the vital role of COX-2 in
regulating silibinin-induced anti-metastatic effects. However, how
COX-2 downregulation by silibinin contributes to TGF-β1-induced EMT
suppression is unclear and further investigation is required.
The present study demonstrated that silibinin at 50
µM treatment for 48 h had no impact on cell growth. However, a
previous study reported that silibinin (10 µM) significantly
suppresses the proliferation of bladder cancer T24 cells (40). Previous studies, in addition to the
results of the present study, have revealed that a 50 µM dose of
silibinin did not significantly affect cell proliferation after 48
h treatment, including in bladder cancer, prostate cancer, renal
cancer and breast cancer (13,16,17,41–44).
Specifically, in the study published in Carcinogenesis in
2004, silibinin (50 µM) treatment showed no effect on T24 cell
proliferation as determined by cell growth assay and flow cytometry
analysis, which was consistent with the results of the present
study (43). One possibility was
that the batches of silibinin from the supplier used in the present
study were different from those used in the aforementioned
study.
In conclusion, the findings of the present study
demonstrated that TGF-β1 promoted TCC migration and invasion via
induction of EMT and upregulation of COX-2. Silibinin inhibited
TGF-β1-induced metastasis via inhibition of EMT, which was
associated with COX-2 downregulation. The study broadens
understanding of TGF-β1- induced EMT and the anti-metastasis
capacity of silibinin, and may indicate future treatment strategies
for metastatic TCC.
Acknowledgements
Not applicable.
Funding
The present study was partly supported by the
National Natural Science Foundation of China (grant nos. NSFC
81672538 to JZ, NSFC 81672557 and 81372279 to PG, NSFC 81602495 to
ZM and NSFC 81302227 to YC).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FL, YS, PG, LC, DH and JZ conceived and designed the
experiments; FL, YS, JJ, CY, XT, BJ, KW, ZM, YC and XW performed
the experiments; FL, YS, DH and JZ analyzed the data; FL, YS, DH
and JZ wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
National Cancer Institute: Bladder Cancer
Treatment (PDQ®): Health Professional Version. National
Cancer Institute (US); Bethesda, MD: 2002, https://www.ncbi.nlm.nih.gov/books/NBK65962/
|
|
2
|
Packiam VT, Johnson SC and Steinberg GD:
Non-muscle-invasive bladder cancer: Intravesical treatments beyond
Bacille Calmette-Guérin. Cancer. 123:390–400. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Krishna SR and Konety BR: Current concepts
in the management of muscle invasive bladder cancer. Indian J Surg
Oncol. 8:74–81. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jeon HM and Lee J: MET: Roles in
epithelial-mesenchymal transition and cancer stemness. Ann Transl
Med. 5:52017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pradella D, Naro C, Sette C and Ghigna C:
EMT and stemness: Flexible processes tuned by alternative splicing
in development and cancer progression. Mol Cancer. 16:82017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lopez-Novoa JM and Nieto MA: Inflammation
and EMT: An alliance towards organ fibrosis and cancer progression.
EMBO Mol Med. 1:303–314. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tyagi A, Agarwal C, Dwyer-Nield LD, Singh
RP, Malkinson AM and Agarwal R: Silibinin modulates TNF-α and IFN-γ
mediated signaling to regulate COX2 and iNOS expression in
tumorigenic mouse lung epithelial LM2 cells. Mol Carcinog.
51:832–842. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fabregat I, Fernando J, Mainez J and
Sancho P: TGF-beta signaling in cancer treatment. Curr Pharm Des.
20:2934–2947. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Helmy A, Hammam OA, El Lithy TR and El
Deen Wishahi MM: The role of TGF-beta-1 protein and TGF-beta-R-1
receptor in immune escape mechanism in bladder cancer. MedGenMed.
9:342007.PubMed/NCBI
|
|
10
|
Levy L and Hill CS: Alterations in
components of the TGF-beta superfamily signaling pathways in human
cancer. Cytokine Growth Factor Rev. 17:41–58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bayram D, Çetin ES, Kara M, Özgöçmen M and
Candan IA: The apoptotic effects of silibinin on MDA-MB-231 and
MCF-7 human breast carcinoma cells. Hum Exp Toxicol. 36:573–586.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gu J, Tang SJ, Tan SY, Wu Q, Zhang X, Liu
CX, Gao XS, Yuan BD, Han LJ, Gao AP, et al: An open-label,
randomized and multi-center clinical trial to evaluate the efficacy
of Silibinin in preventing drug-induced liver injury. Int J Clin
Exp Med. 8:4320–4327. 2015.PubMed/NCBI
|
|
13
|
Li F, Ma Z, Guan Z, Chen Y, Wu K, Guo P,
Wang X, He D and Zeng J: Autophagy induction by silibinin
positively contributes to its anti-metastatic capacity via
AMPK/mTOR pathway in renal cell carcinoma. Int J Mol Sci.
16:8415–8429. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma Z, Liu W, Zeng J, Zhou J, Guo P, Xie H,
Yang Z, Zheng L, Xu S, Wang X, et al: Silibinin induces apoptosis
through inhibition of the mTOR-GLI1-BCL2 pathway in renal cell
carcinoma. Oncol Rep. 34:2461–2468. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sozen H, Celik OI, Cetin ES, Yilmaz N,
Aksozek A, Topal Y, Cigerci IH and Beydilli H: Evaluation of the
protective effect of silibinin in rats with liver damage caused by
itraconazole. Cell Biochem Biophys. 71:1215–1223. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu K, Zeng J, Li L, Fan J, Zhang D, Xue Y,
Zhu G, Yang L, Wang X and He D: Silibinin reverses
epithelial-to-mesenchymal transition in metastatic prostate cancer
cells by targeting transcription factors. Oncol Rep. 23:1545–1552.
2010.PubMed/NCBI
|
|
17
|
Zeng J, Sun Y, Wu K, Li L, Zhang G, Yang
Z, Wang Z, Zhang D, Xue Y, Chen Y, et al: Chemopreventive and
chemotherapeutic effects of intravesical silibinin against bladder
cancer by acting on mitochondria. Mol Cancer Ther. 10:104–116.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singh RP, Tyagi A, Sharma G, Mohan S and
Agarwal R: Oral silibinin inhibits in vivo human bladder tumor
xenograft growth involving down-regulation of survivin. Clin Cancer
Res. 14:300–308. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu K, Ning Z, Zeng J, Fan J, Zhou J, Zhang
T, Zhang L, Chen Y, Gao Y, Wang B, et al: Silibinin inhibits
β-catenin/ZEB1 signaling and suppresses bladder cancer metastasis
via dual-blocking epithelial-mesenchymal transition and stemness.
Cell Signal. 25:2625–2633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chandrasekaran S, Marshall JR, Messing JA,
Hsu JW and King MR: TRAIL-mediated apoptosis in breast cancer cells
cultured as 3D spheroids. Plos One. 9:e1114872014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brito RB, Malta CS, Souza DM, Matheus LH,
Matos YS, Silva CS, Ferreira JM, Nunes VS, França CM and Dellê H:
1-Methyl-D-tryptophan potentiates TGF-β-induced
epithelial-mesenchymal transition in T24 human bladder cancer
cells. PLoS One. 10:e01348582015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu CL, Ho JY, Chou SC and Yu DS: MiR-429
reverses epithelial-mesenchymal transition by restoring E-cadherin
expression in bladder cancer. Oncotarget. 7:26593–26603.
2016.PubMed/NCBI
|
|
24
|
Xian X, Huang L, Zhang B, Wu C, Cui J and
Wang Z: WIN 55,212-2 inhibits the epithelial mesenchymal transition
of gastric cancer cells via COX-2 signals. Cell Physiol Biochem.
39:2149–2157. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen T, You Y, Jiang H and Wang ZZ:
Epithelial-mesenchymal transition (EMT): A biological process in
the development, stem cell differentiation and tumorigenesis. J
Cell Physiol. 232:3261–3272. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kong D, Li Y, Wang Z and Sarkar FH: Cancer
stem cells and epithelial-to-mesenchymal transition
(EMT)-phenotypic cells: Are they cousins or twins? Cancers (Basel).
3:716–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vlahopoulos SA, Logotheti S, Mikas D,
Giarika A, Gorgoulis V and Zoumpourlis V: The role of ATF-2 in
oncogenesis. Bioessays. 30:314–327. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huber MA, Beug H and Wirth T:
Epithelial-mesenchymal transition: NF-kappaB takes center stage.
Cell Cycle. 3:1477–1480. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Katoh Y and Katoh M: Hedgehog signaling,
epithelial-to-mesenchymal transition and miRNA (Ρeview). Int J Mol
Med. 22:271–275. 2008.PubMed/NCBI
|
|
30
|
Ijaz T, Pazdrak K, Kalita M, Konig R,
Choudhary S, Tian B, Boldogh I and Brasier AR: Systems biology
approaches to understanding Epithelial Mesenchymal Transition (EMT)
in mucosal remodeling and signaling in asthma. World Allergy Organ
J. 7:132014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zarzynska JM: Two faces of TGF-beta1 in
breast cancer. Mediators Inflamm. 2014:1417472014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Iwano M: EMT and TGF-beta in renal
fibrosis. Front Biosci (Schol Ed). 2:229–238. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Servais C and Erez N: From sentinel cells
to inflammatory culprits: Cancer-associated fibroblasts in
tumour-related inflammation. J Pathol. 229:198–207. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Raposo TP, Beirão BC, Pang LY, Queiroga FL
and Argyle DJ: Inflammation and cancer: Till death tears them
apart. Vet J. 205:161–174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu H, Ren G, Wang T, Chen Y, Gong C, Bai
Y, Wang B, Qi H, Shen J, Zhu L, et al: Aberrantly expressed Fra-1
by IL-6/STAT3 transactivation promotes colorectal cancer
aggressiveness through epithelial-mesenchymal transition.
Carcinogenesis. 36:459–468. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang L, Jiao M, Wu K, Li L, Zhu G, Wang
X, He D and Wu D: TNF-α induced epithelial mesenchymal transition
increases stemness properties in renal cell carcinoma cells. Int J
Clin Exp Med. 7:4951–4958. 2014.PubMed/NCBI
|
|
37
|
Tong D, Liu Q, Liu G, Xu J, Lan W, Jiang
Y, Xiao H, Zhang D and Jiang J: Metformin inhibits
castration-induced EMT in prostate cancer by repressing
COX2/PGE2/STAT3 axis. Cancer Lett. 389:23–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tabriz HM, Olfati G, Ahmadi SA and
Yusefnia S: Cyclooxygenase-2 expression in urinary bladder
transitional cell carcinoma and its association with
clinicopathological characteristics. Asian Pac J Cancer Prev.
14:4539–4543. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Deep G and Agarwal R: Antimetastatic
efficacy of silibinin: Molecular mechanisms and therapeutic
potential against cancer. Cancer Metastasis Rev. 29:447–463. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Imai-Sumida M, Chiyomaru T, Majid S, Saini
S, Nip H, Dahiya R, Tanaka Y and Yamamura S: Silibinin suppresses
bladder cancer through down-regulation of actin cytoskeleton and
PI3K/Akt signaling pathways. Oncotarget. 8:92032–92042. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gholami M, Moallem SA, Afshar M, Etemad L
and Karimi G: Gestational exposure to silymarin increases
susceptibility of BALB/c mice fetuses to apoptosis. Avicenna J Med
Biotechnol. 9:66–70. 2017.PubMed/NCBI
|
|
42
|
Tyagi AK, Agarwal C, Singh RP, Shroyer KR,
Glode LM and Agarwal R: Silibinin down-regulates survivin protein
and mRNA expression and causes caspases activation and apoptosis in
human bladder transitional-cell papilloma RT4 cells. Biochem
Biophys Res Commun. 312:1178–1184. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tyagi A, Agarwal C, Harrison G, Glode LM
and Agarwal R: Silibinin causes cell cycle arrest and apoptosis in
human bladder transitional cell carcinoma cells by regulating
CDKI-CDK-cyclin cascade, and caspase 3 and PARP cleavages.
Carcinogenesis. 25:1711–1720. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yousefi M, Ghaffari SH, Zekri A, Hassani
S, Alimoghaddam K and Ghavamzadeh A: Silibinin induces apoptosis
and inhibits proliferation of estrogen receptor (ER)-negative
breast carcinoma cells through suppression of nuclear factor kappa
B activation. Arch Iran Med. 17:366–371. 2014.PubMed/NCBI
|