Introduction
Renal cell cancer (RCC) is the twelfth most common
cancer worldwide; in 2012, it accounted for about 338,000 new cases
(1). A comprehensive
characterization of the molecular alterations associated with RCC
was recently carried out by the TCGA Kidney consortium (KIRC)
(2). Notably, they described only
limited associations between gene mutations and clinical or
survival parameters in RCC patients. Thus, it was questionable
whether the translation of genetic mutations or variant information
would be useful for future personalized diagnoses, prognoses, or
therapy predictions. This question was consistent with earlier
genetic studies, which demonstrated inconsistent results concerning
associations with clinical pathology or patient survival (3). In contrast, the KIRC data, like many
previous studies, reported that clear cell (cc)RCC typically showed
features of epigenetic alterations, such as hypermethylation of
gene promoters and concurrent loss of gene function, due to
transcriptional silencing. Indeed, it has been consistently
observed, in a substantial number of genes, that DNA
hypermethylation occurs with high frequency, and the degree of
hypermethylation is correlated with clinical and pathological
features of patients. In addition, the tumor suppressors known as
RAS-associated domain family 1 (RASSF1A) and secreted
frizzled-related protein 1 (SFRP1) were each found in 30% to
>70% of tumors. These tumor suppressors were associated with
both clinical pathological features and outcomes (4–7). In
RCC, a significant incidence of epigenetic alterations has been
found in the COL1A1, IGFBP1, EDNRB and KRT19 genes,
in cell lines and primary tumors (8,9). A
recent report demonstrated tumor-specific hypermethylation in RCC,
which was found to be correlated with adverse pathology and was
associated with overall survival among patients undergoing targeted
therapy (10).
In view of the fact that a multitude of
hypermethylated genes (11) have
been described and that the KIRC data provide a large number of new
candidate methylated loci for ccRCC, the question arises, how can
we identify the most relevant genes? Based on traditional genetic
approaches, we might assume that the most promising candidates for
future translational purposes would be genes that are associated
with functional losses, high alteration frequencies in tumors, and
altered clinical behaviors in patients. Here, we focused on the
clinical relevance of DNA methylation in the neural epidermal
growth factor-like 1 (NELL1) gene. NELL1 is a multimeric
extracellular glycoprotein, originally identified in neural tissues
(12). The gene is located on
chromosome 11p15.1, and it is involved in osteogenic
differentiation in mammals and bone regeneration in defects of the
calvarium (13,14).
NELL1 expression has been detected in normal
embryonic kidney, brain and prostate, and in bladder cancer,
lymphomas and different cancer cell lines (15–18). A
recent study revealed that NELL1 protein expression was reduced in
RCC tissues (19). Considering that
promoter hypermethylation and diminished mRNA expression were
detected in RCC cell lines, transcriptional silencing was assumed
to explain low NELL1 expression in RCC (19).
No studies have described NELL1 promoter
methylation in RCC tissues or its correlation to
clinicopathological parameters or clinical outcome. Here, we
investigated NELL1 promoter methylation in cancerous and
adjacent non-cancerous tissues derived from patients with RCC and a
subset of patients with ccRCC and in 18 malignant and benign renal
cancer cell lines. Results were confirmed in silico with the
KIRC data set.
Materials and methods
Tissue specimens
Ninety-eight tumor tissue samples were obtained from
kidney surgeries carried out between 2001 and 2005 at Eberhard
Karls University, Tübingen. Tissue sampling, storage, and
processing were performed as described previously (20). Patient characteristics are provided
in Table I. Pairs of tissue samples
from tumors (TU) and adjacent tumor-free tissue (adN) were obtained
from 63 patients.
 | Table I.Clinicopathological parameters of the
discovery and evaluation cohort. |
Table I.
Clinicopathological parameters of the
discovery and evaluation cohort.
|
| Hannover
cohort | TCGA cohort |
|---|
| Total cases | 98 | 284 |
| Sex |
|
Female | 35 | 96 |
|
Male | 63 | 188 |
| Age (years) |
|
Median | 64 | 61 |
|
Minimum | 35 | 26 |
|
Maximum | 91 | 90 |
|
T-classification |
|
pT1 | 10 | 15 |
|
pT1a | 26 | 72 |
|
pT1b | 17 | 46 |
|
pT2 | 7 | 32 |
|
pT3 | 3 | 3 |
|
pT3a | 9 | 65 |
|
pT3b | 22 | 38 |
|
pT3c | 1 | NA |
|
pT4 | 1 | 8 |
| Lymph node
status |
| N0 | 85 | 127 |
| N1 | 13 | 9 |
| Nx | NA | 148 |
| Distant
metastasis |
| M0 | 79 | 232 |
| M1 | 19 | 52 |
|
Differentiation |
| G1 | 22 | 5 |
|
G1-2 | 14 | NA |
| G2 | 48 | 117 |
|
G2-3 | 6 | NA |
| G3 | 8 | 112 |
| G4 | NA | 48 |
| Gx | NA | 2 |
| Status of
disease |
|
Localizeda | 51 | NA |
|
Advancedb | 46 | NA |
| Paired tissue
samplesc | 63 | 160 |
The Ethics Committee of the institution of Eberhard
Karls University approved the study (vote no. 128/2003V and
1213–2011) and informed consent was obtained from each patient. Two
pathologists evaluated all specimens, with respect to tumor stage,
grade and histological subtypes. Tumor stages were assessed
according to the UICC 2002 issue of the TNM system, and nuclear
grading was based on the Fuhrman grading system (21). Histological subtypes were assessed
according to the consensus classification of RCC. Localized RCC was
defined as pT ≤2, N0/M0; advanced tumors were defined as pT ≥3
and/or N1/M1. Low-stage tumors were defined as the group of pT1 and
pT2 tumors; high stage tumors were defined as the group of pT3 and
T4 tumors. Follow-up data were available for 24 patients. The
duration of the follow-up was calculated from the date of surgery
to the date of recurrence, progression, death, or the last
follow-up.
Primary and cancer cell lines
Prostate cancer (LN-cap, DU-145), urothelial cancer
(CLS439, HB-CLS2, HB-CLS1, 5637), and RCC (ACHN, A498, 786-O,
RCC-GS, RCC-HS, RCC-MF) cell lines were purchased from Lonza
(Basel, Switzerland). Primary normal cell lines (RPTEC, primary
renal proximal tubular epithelial cells) were acquired from Cell
Lines Services (CLS, Eppelheim, Germany). All cells were cultured
according to the manufacturer's protocol and as described
previously (20,22).
DNA extraction and bisulfite
conversion
DNA extraction, bisulfite conversion, and control of
tumor cell content were performed as reported previously (22). Total DNA was extracted from 20
serial frozen sections (20 µm) from each tumoral and normal
specimen after proteinase K digestion, with the standard
phenol/chloroform extraction method. Each 1 µg of extracted DNA was
then subject to bisulfite conversion, as described previously, and
aliquots were stored at −20°C (5).
Additionally, two serial sections of each tissue sample were
stained with hematoxylin-eosin and re-evaluated by
pathologists.
Bisulfite-pyrosequencing for NELL1
promoter methylation analysis
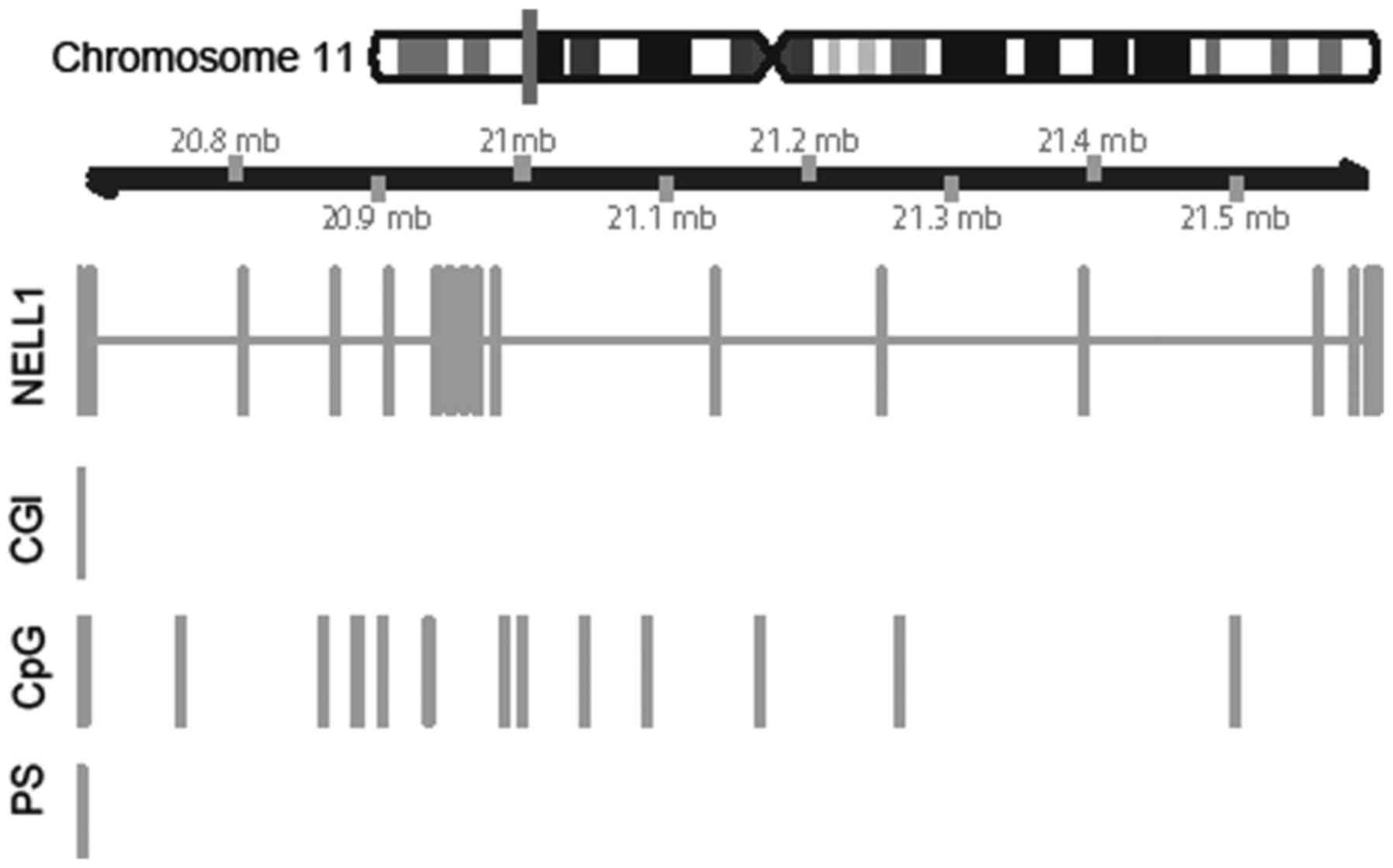
We performed methylation analyses of ten
NELL1 CpG sites (base positions: 20,587, ~591, ~596, ~604,
~611, ~615, ~618, ~626, ~634, ~636, ~691) within the CpG island
(CGI), which extended over the 5′UTR and exon 1 of the NELL1
gene (Fig. 1, ‘CGI’ and ‘Pyro’).
Analyses were performed with bisulfite-pyrosequencing, according to
the manufacturer's protocol (Qiagen, Hilden, Germany) and as
described previously (6,23). We used a two-step PCR protocol. For
the first step, the primers were: Forward
(5′-GGGATGAATTGTGGTAATT-3′) and reverse
(5′-GGGACACCGCTGATCGTTTACTACRAAAATCTAAACTACAAA-3′). For the second
step, the primers were: Forward (5′-GGAAGTAAAGAGGGTAATATTG-3′) and
reverse (Biotin-5′-GGGACACCGCTGATCGTTTA-3′). Measurements were
performed with the PyroMark Q24 pyrosequencing system (Qiagen,
Hilden, Germany).
Statistical analysis
Statistical analyses of NELL1 methylation
levels in paired TU and adN tissue samples were performed with the
two-sided paired t-test. Tumor subgroups were assessed with
univariate logistic regression analysis and dichotomization, as
specified. Visual comparisons of subgroups were performed with
notched boxplots, which show the median, the 25% quartiles, and the
estimated confidence intervals (notches). Univariate Cox regression
analyses were performed to examine recurrence-free survival (RFS).
Hazard ratios (HR), 95% confidence intervals (95% CI), and P-values
were calculated. Survival was evaluated with the Kaplan-Meier
analysis. All analyses were performed with R version 3.2. The
‘maxRank’ package was used to calculate the optimum cutoff for the
survival analysis (24). Therefore,
the corresponding value of 15% is a result of a mathematical
calculation in R using the Package ‘OptimalCutpoint’.
P-values <0.05 were considered to indicate
statistical significance. In silico validation was carried
out with the subset of level 3 data from the KIRC data set,
annotated for NELL1. Distributions of the methylation
differences obtained for each pair of TU and corresponding adN
samples were calculated for each annotated CpG site. We used
quality criteria for statistical information on the sites.
Consequently, only CpG sites that showed approximately normal or
bimodal density distributions were considered for further
evaluation. We excluded CpG sites characterized by methylation
differences that exhibited more or less discrete and constant
values, in the paired tissue group comparisons.
Results
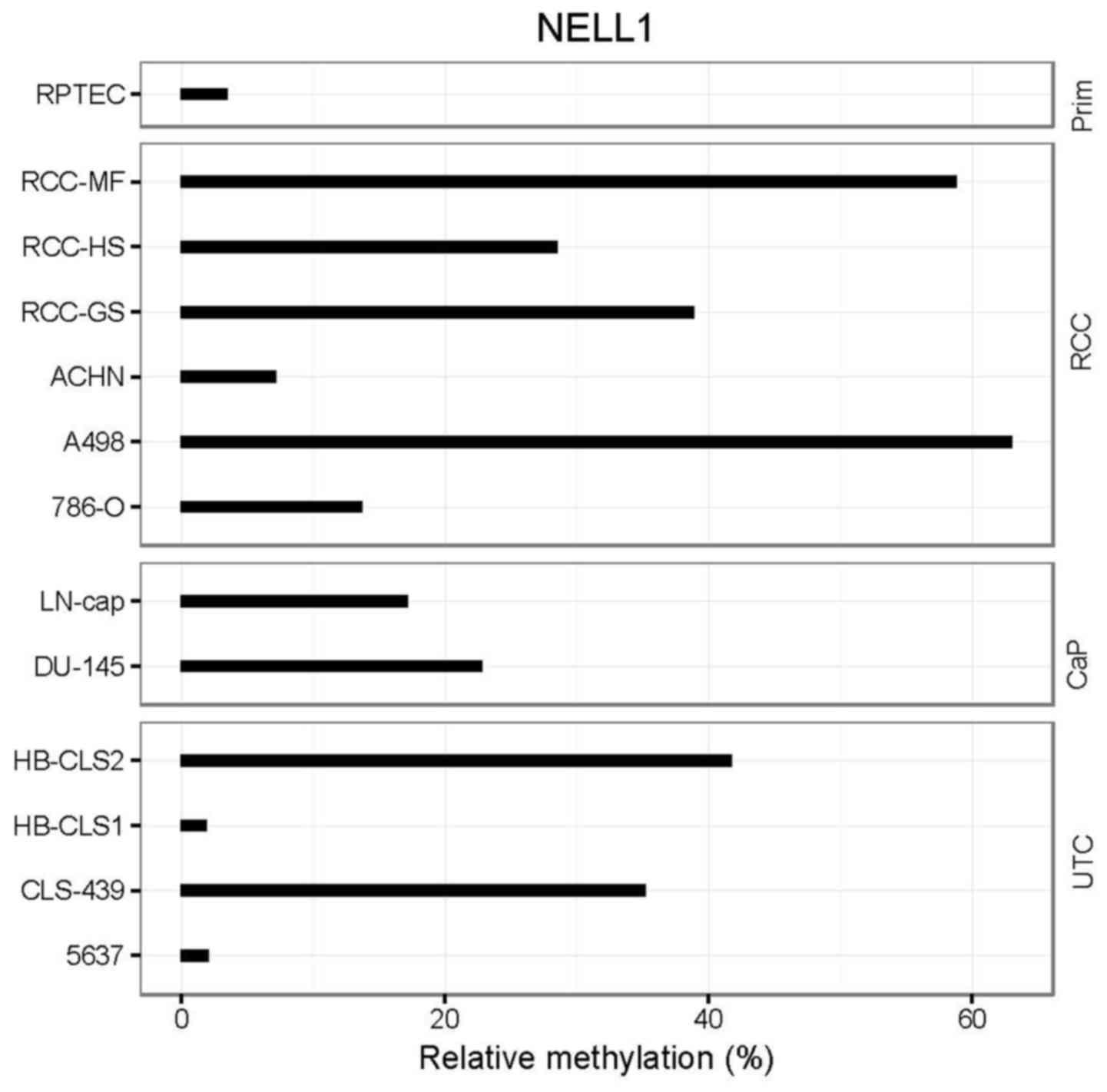
NELL1 methylation in tumor cell line
models
Twelve cell lines that represented urological cancer
models and one normal tissue cell model (RPTECs) were analyzed for
NELL1 methylation. RPTECs demonstrated low relative
methylation values (~3%) and a large number of kidney tumor cell
lines, including A498, RCC-HS, and RCC-GS, displayed high relative
methylation (30–60%; Fig. 2). Two
kidney cancer cell lines (786-O and ACHN) displayed medium relative
methylation (10–20% maximum). The prostate cancer cell lines,
LN-cap and DU-145, exhibited elevated methylation, and two
(HB-CLS2, CLS-439) out of four urothelial cancer cell lines
displayed high methylation (~40%, Fig.
2).
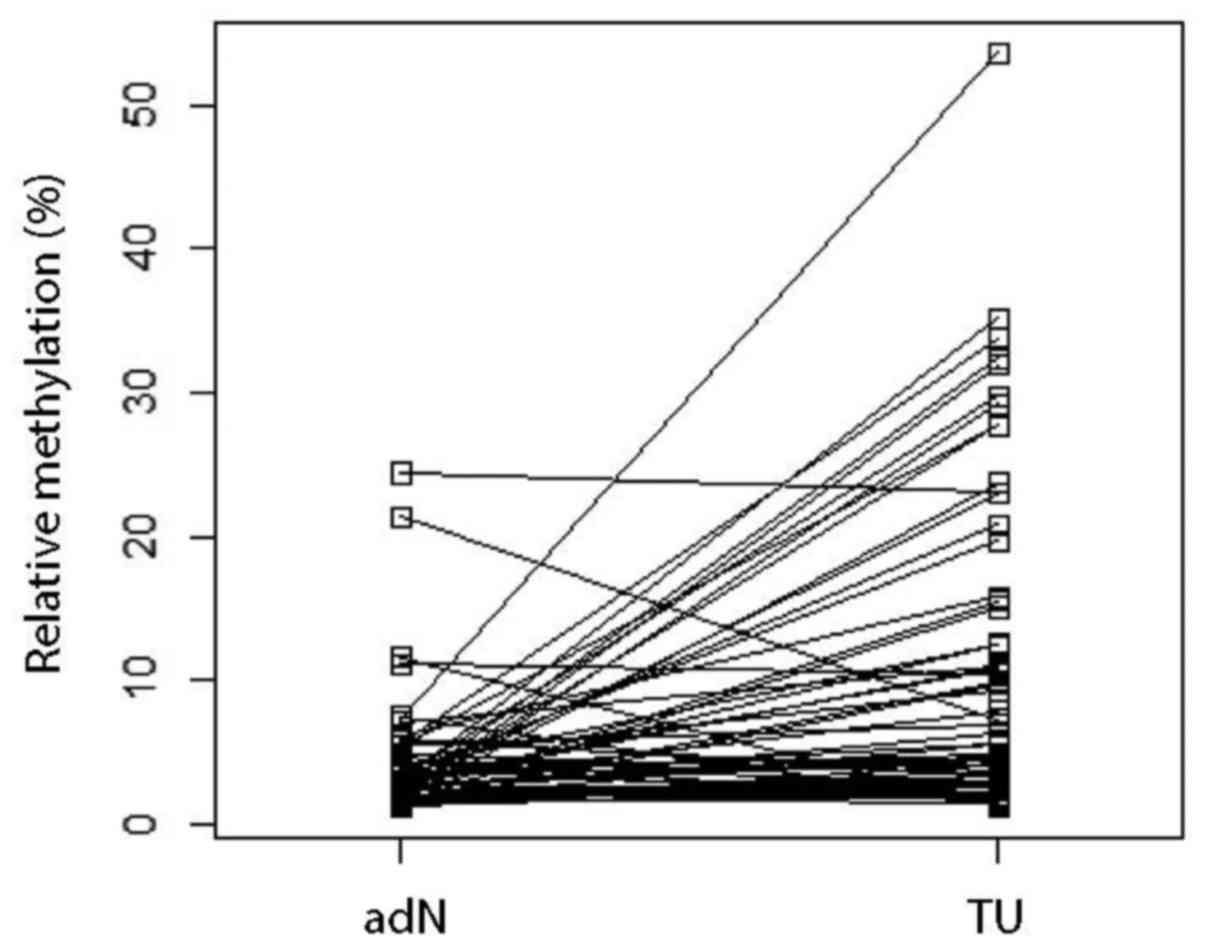
NELL1 DNA hypermethylation in paired
tumor and adjacent normal renal tissue samples
To investigate whether NELL1 showed
tumor-specific DNA hypermethylation, we compared TU paired with adN
tissue samples. We found that TU tissues displayed a significantly
higher mean methylation level (6.8%) compared to paired adN tissues
(paired t-test, P=1.64e−5, Fig. 3).
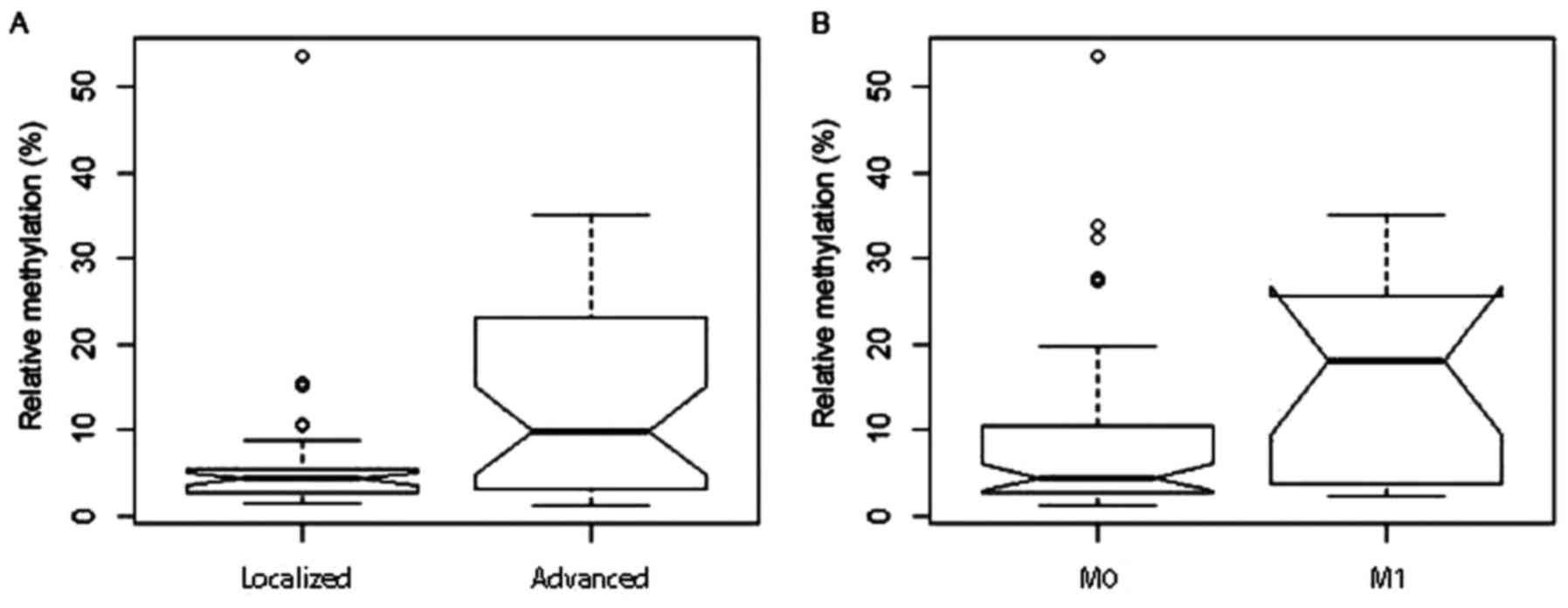
NELL1 DNA methylation and
clinicopathological parameters
To investigate whether the clinicopathological
parameters of the patients might be associated with altered
NELL1 DNA methylation, patients were grouped according to
different clinicopathological parameters, and then subjected to
univariate logistic regression analyses, following the
corresponding dichotomization. We found that methylation was
significantly increased in the group with advanced tumors (mean
methylation 12.6%) compared to those with localized disease [6.1%;
P=0.002; odds ratio (OR)=1.08; Fig.
4A]. Furthermore, the presence of distant metastases in
patients was associated with a higher mean methylation (15.8%)
compared to patients without clinically detectable distant
metastasis (7.8%, P=0.004; OR: 1.07; Fig. 4B). Low- and high-stage tumor groups
had mean methylation values of 7.5 and 11.8%, respectively
(P=0.044; OR: 1.05). No significant difference in mean methylation
was observed between groups that exhibited low and high
histological tumor grades (Table
II).
| Table II.Overview of NELL1 DNA
methylation analyses in primary RCC tissue samples, and statistical
comparison with clinicopathological parameters. |
Table II.
Overview of NELL1 DNA
methylation analyses in primary RCC tissue samples, and statistical
comparison with clinicopathological parameters.
| NELL1
methylation | Mean relative
methylation (%) |
P-valued | Odds ratio | 95% confidence
interval |
|---|
|
Localized/advanceda | 6.07 | 12.62 | 0.002 | 1.08 | 1.03–1.15 |
| Low/high
gradeb | 8.60 | 13.63 | 0.096 | 1.04 | 0.99–1.09 |
| Distant metastasis
(M0 vs. M1) | 7.75 | 15.85 | 0.004 | 1.07 | 1.02–1.12 |
| Low/high
stagec | 7.48 | 11.76 | 0.044 | 1.05 | 1.00–1.09 |
NELL1 promoter methylation and
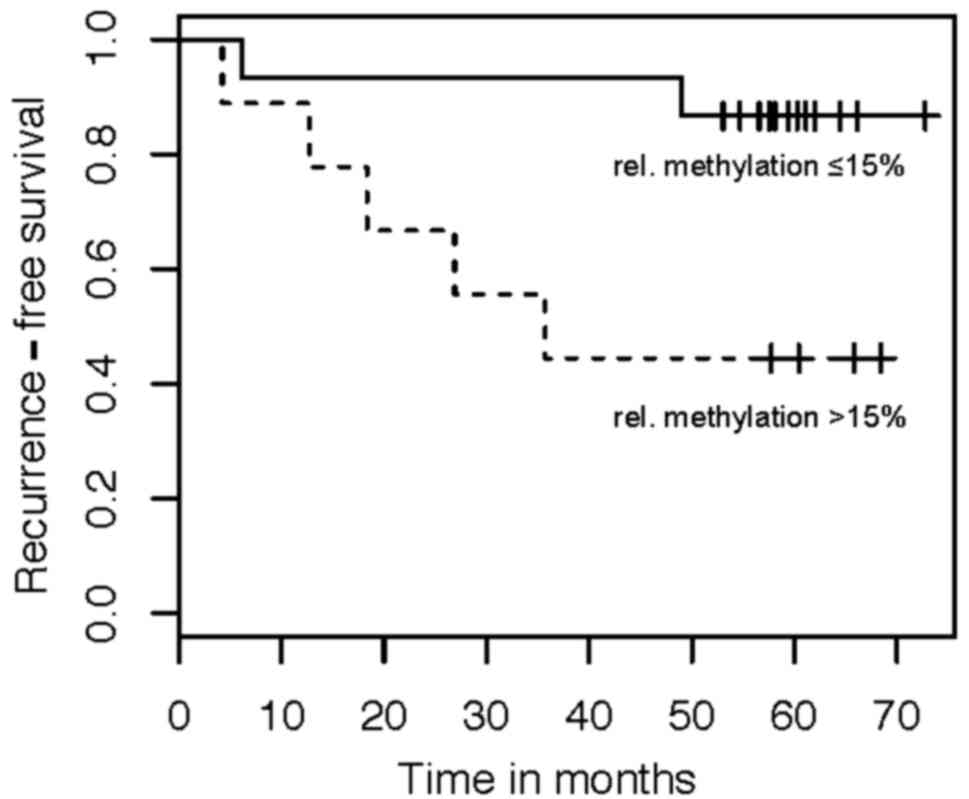
recurrence-free survival
Kaplan Meier and Cox regression analyses were
performed to assess the relationship between NELL1 DNA
methylation and the RFS of patients. We dichotomized patients
according to the degree of DNA methylation, based on a cut-off
value of 15% relative methylation. The Kaplan Meier analysis
indicated that high NELL1 methylation was associated with a
worse RFS (Fig. 5). The univariate
Cox regression analysis identified NELL1 methylation as a
significant risk factor for RFS (HR: 4.15, 95% CI: 1.10–15.6;
P=0.035).
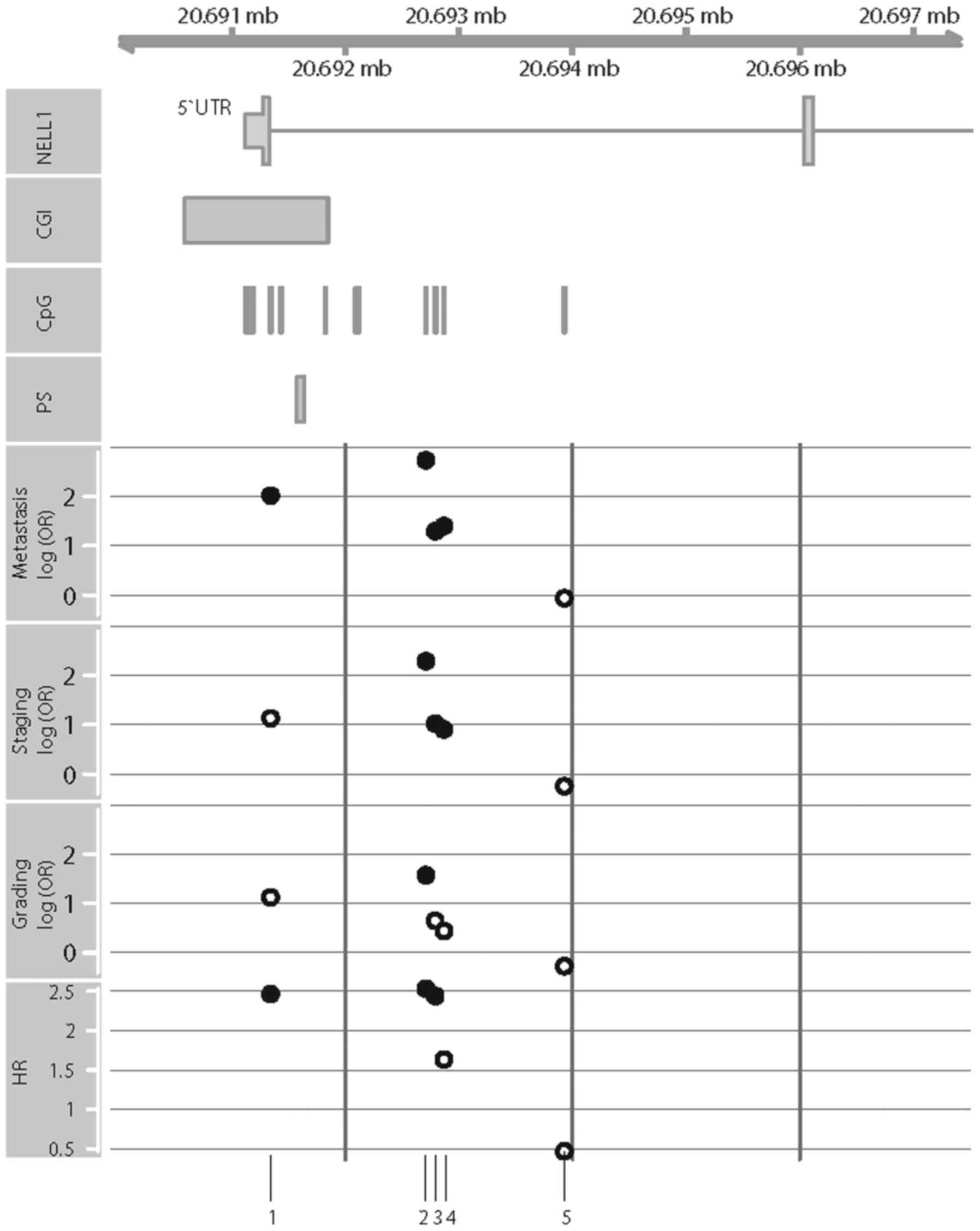
In silico validation with KIRC
data
We assessed the KIRC data set for associations
between tumor-specific hypermethylation, clinicopathological
parameters and survival, in an independent patient cohort. Out of
15 annotated CpG sites selected in a pre-analysis quality check
(see Materials and methods), five showed hypomethylation (paired
t-test, P<0.05, adjusted with Bonferroni-Hochberg for multiple
testing) and six showed hypermethylation (P<0.05). We observed
significant associations between the methylation state and the
state of metastasis in seven out of 15 CpG sites (univariate
logistic regression: P=1.8×10−2 to 7.3×10−7,
OR: 12-546). Four of these CpG sites were located within or
adjacent to the CGIs presented in Fig.
6. Notably, the corresponding CpG sites were also associated
with high-stage tumors, high-grade tumors, and the RFS of patients
(Fig. 6).
Discussion
NELL1 DNA methylation and epigenetic
silencing of gene expression were previously described in RCC cell
lines, which suggested that epigenetic alterations were potentially
relevant to RCC pathology (19). In
the present study, we aimed to ascertain whether NELL1 DNA
methylation was associated with clinicopathological parameters and
disease outcome in patients with ccRCC. Comparisons between tumors
(TU) and adjacent tumor-free tissue (adN) clearly demonstrated that
tumor-specific hypermethylation occurred in the loci analyzed. An
interrogation of the KIRC data set showed that several CpG sites in
the extended CGI region at exon 1 of the gene also exhibited clear
hypermethylation, which confirmed results from former cell line
experiments and from the present study. Of note, the DNA
methylation observed in cell lines probably reflected both
intra-tumoral and inter-patient variability at each locus
investigated. Indeed, both baseline methylation and age-dependent
methylation show considerable variation among different organ
systems. Thus, some of the observed differences in methylation
reflected the normal biological variations in methylation expected
among different tissues and tissue donors.
Previous studies indicated that the loss of
NELL1 mRNA and concurrent loss of NELL1 protein expression
are associated with altered tumor cell behavior. Those findings
strongly indicated that epigenetic silencing of the NELL1
gene is functionally relevant (19). Therefore, our finding that high
levels of DNA methylation were associated with adverse
clinicopathology in patients supported the hypothesis that the loss
of NELL1 function promotes the development of aggressive
ccRCC.
DNA methylation of NELL1 has also been shown
in other human cancers. For example, a high frequency of
NELL1 methylation was reported in colon cancer, early-stage
esophageal adenocarcinoma (EAC), and gastric cancer (25–27).
Notably, in EAC, NELL1 methylation was significantly
associated with a poor prognosis in patients (25,26).
No previous study had investigated whether NELL1 methylation
might also be correlated with disadvantageous clinical parameters,
such as advanced disease, presence of distant metastases, or worse
survival in RCC. Therefore, our finding that higher methylation was
significantly associated with metastatic and advanced disease in
patients with RCC extended the number of human malignancies that
showed associations between NELL1 methylation and aggressive
tumor biology.
Similarly, our survival analysis indicated that
NELL1 methylation could serve as a prognostic indicator of
RFS in patients. However, due to our small survival cohort, a
multivariate analysis was not feasible; due to this limitation, the
relevance of this part of the clinical evaluation should be
interpreted with caution.
On the other hand, our evaluation of KIRC data
confirmed our finding that NELL1 methylation in the CGI and
adjacent CpG sites were associated with the state of distant
metastasis, the tumor grade, and the tumor stage. Notably, three of
five CpG sites in this region also showed associations with RFS.
However, considering that multivariate significance could not be
achieved in the in silico survival analyses, the association
between NELL1 methylation and patient survival remains to be
confirmed in appropriate study cohorts. Moreover, from a technical
point of view, only a small fraction (roughly estimated at about
1%) of the totally available CpG sites could be considered for
in silico TCGA methylation analyses. Thus, the combined
number of CpG sites was limited to 25 sites in our experimental and
in silico approaches. Hence, most likely, in future, it will
be necessary to extend both the study cohorts and the number of
target CpG sites to determine the relevance of NELL1 DNA
methylation detection for predicting the clinical course of RCC and
for translating these findings into individualized therapy and
follow-up regimes.
In conclusion, our analyses suggested that
NELL1 DNA methylation is a promising candidate prognostic
epigenetic biomarker for more detailed analyses of RCC disease.
This finding provides a potential clinical marker to complement
previously defined functional data (19). Furthermore, our finding that the
prostate and urothelial cancer cell line models demonstrated high
relative methylation levels indicated that NELL1 DNA
methylation might be also relevant to investigations of other
frequent tumor entities. Thus, NELL1 methylation might serve
as a potential target structure for investigations with molecular
therapeutic objectives.
Acknowledgements
We thank Christel Reese for the technical
assistance.
Funding
No funding was received for this study.
Availability of data and materials
All data and materials are available at our local
laboratory (Hannover Medical School, 30625 Hannover, Dept. of
Urology).
Authors contributions
IP wrote the manuscript, prepared the figures, and
participated in the study design. ND carried out the methylation
analyses and participated in the sequence analyses. JH and IP
assembled histopathological, clinicopathological, and survival
data. ND and JH isolated and characterized the tissue samples and
collected the patients' data. WIA re-evaluated the histopathology,
revised the manuscript critically and finally approved the full
version. MAK, CAJvK, HT, and AS contributed to the study design,
revised the manuscript critically and did the final approval of the
version for publication. JS identified the candidate promoter,
conceived the study, developed the study design and analytical
assays, constructed and ran the clinical database, performed the
statistical analyses (together with IP), and participated in the
manuscript preparation. All authors read and approved the final
manuscript and provided consent for publication.
Ethics approval and consent to
participate
The Ethics Committee of the Eberhard Karls
University approved the study (vote no. 128/2003V and 1213–2011)
and informed consent was obtained from each patient.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interest.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Creighton CJ, Morgan M, Gunaratne PH,
Wheeler DA, Gibbs RA, Robertson A, Chu A, Beroukhim R, Cibulskis K,
Signoretti S, et al: Comprehensive molecular characterization of
clear cell renal cell carcinoma. Nature. 499:43–49. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheng L, Zhang S, MacLennan GT,
Lopez-Beltran A and Montironi R: Molecular and cytogenetic insights
into the pathogenesis, classification, differential diagnosis, and
prognosis of renal epithelial neoplasms. Hum Pathol. 40:10–29.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Morris MR, Ricketts C, Gentle D,
Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Latif F and
Maher ER: Identification of candidate tumour suppressor genes
frequently methylated in renal cell carcinoma. Oncogene.
29:2104–2117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peters I, Rehmet K, Wilke N, Kuczyk MA,
Hennenlotter J, Eilers T, Machtens S, Jonas U and Serth J: RASSF1A
promoter methylation and expression analysis in normal and
neoplastic kidney indicates a role in early tumorigenesis. Mol
Cancer. 6:492007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Atschekzei F, Hennenlotter J, Jänisch S,
Großhennig A, Tränkenschuh W, Waalkes S, Peters I, Dörk T,
Merseburger AS, Stenzl A, et al: SFRP1 CpG island methylation locus
is associated with renal cell cancer susceptibility and disease
recurrence. Epigenetics. 7:447–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Onay H, Pehlivan S, Koyuncuoglu M, Kirkali
Z and Ozkinay F: Multigene methylation analysis of conventional
renal cell carcinoma. Urol Int. 83:107–112. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Caceres Ibanez I, Dulaimi E, Hoffman
AM, Al-Saleem T, Uzzo RG and Cairns P: Identification of novel
target genes by an epigenetic reactivation screen of renal cancer.
Cancer Res. 66:5021–5028. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morris MR, Gentle D, Abdulrahman M, Clarke
N, Brown M, Kishida T, Yao M, Teh BT, Latif F and Maher ER:
Functional epigenomics approach to identify methylated candidate
tumour suppressor genes in renal cell carcinoma. Br J Cancer.
98:496–501. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dubrowinskaja N, Gebauer K, Peters I,
Hennenlotter J, Abbas M, Scherer R, Tezval H, Merseburger AS,
Stenzl A, Grünwald V, et al: Neurofilament Heavy polypeptide CpG
island methylation associates with prognosis of renal cell
carcinoma and prediction of antivascular endothelial growth factor
therapy response. Cancer Med. 3:300–309. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Henrique R, Luís AS and Jerónimo C: The
epigenetics of renal cell tumors: From biology to biomarkers. Front
Genet. 3:942012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Watanabe TK, Katagiri T, Suzuki M, Shimizu
F, Fujiwara T, Kanemoto N, Nakamura Y, Hirai Y, Maekawa H and
Takahashi EI: Cloning and characterization of two novel human cDNAs
(NELL1 and NELL2) encoding proteins with six EGF-like repeats.
Genomics. 38:273–276. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ting K, Vastardis H, Mulliken JB, Soo C,
Tieu A, Do H, Kwong E, Bertolami CN, Kawamoto H, Kuroda S, et al:
Human NELL-1 expressed in unilateral coronal synostosis. J Bone
Miner Res. 14:80–89. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aghaloo T, Cowan CM, Chou YF, Zhang X, Lee
H, Miao S, Hong N, Kuroda S, Wu B, Ting K, et al: Nell-1-induced
bone regeneration in calvarial defects. Am J Pathol. 169:903–915.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maeda K, Matsuhashi S, Tabuchi K, Watanabe
T, Katagiri T, Oyasu M, Saito N and Kuroda S: Brain specific human
genes, NELL1 and NELL2, are predominantly expressed in
neuroblastoma and other embryonal neuroepithelial tumors. Neurol
Med Chir. 41:582–589. 2001. View Article : Google Scholar
|
|
16
|
Shah US and Getzenberg RH: Fingerprinting
the diseased prostate: Associations between BPH and prostate
cancer. J Cell Biochem. 91:161–169. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Osman I, Bajorin DF, Sun TT, Zhong H,
Douglas D, Scattergood J, Zheng R, Han M, Marshall KW and Liew CC:
Novel blood biomarkers of human urinary bladder cancer. Clin Cancer
Res. 12:3374–3380. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Slovak ML, Bedell V, Hsu YH, Estrine DB,
Nowak NJ, Delioukina ML, Weiss LM, Smith DD and Forman SJ:
Molecular karyotypes of Hodgkin and Reed-Sternberg cells at disease
onset reveal distinct copy number alterations in chemosensitive
versus refractory Hodgkin lymphoma. Clin Cancer Res. 17:3443–3454.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakamura R, Oyama T, Tajiri R, Mizokami A,
Namiki M, Nakamoto M and Ooi A: Expression and regulatory effects
on cancer cell behavior of NELL1 and NELL2 in human renal cell
carcinoma. Cancer Sci. 106:656–664. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gebauer K, Peters I, Dubrowinskaja N,
Hennenlotter J, Abbas M, Scherer R, Tezval H, Merseburger AS,
Stenzl A, Kuczyk MA, et al: Hsa-mir-124-3 CpG island methylation is
associated with advanced tumours and disease recurrence of patients
with clear cell renal cell carcinoma. Br J Cancer. 108:131–138.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sobin LH and Compton CC: TNM seventh
edition: What's new, what's changed: Communication from the
international union against cancer and the American Joint Committee
on cancer. Cancer. 116:5336–5339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peters I, Eggers H, Atschekzei F,
Hennenlotter J, Waalkes S, Tränkenschuh W, Grosshennig A,
Merseburger AS, Stenzl A, Kuczyk MA, et al: GATA5 CpG island
methylation in renal cell cancer: A potential biomarker for
metastasis and disease progression. BJU Int. 110:E144–E152. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Colella S, Shen L, Baggerly KA, Issa JP
and Krahe R: Sensitive and quantitative universal Pyrosequencing
methylation analysis of CpG sites. Biotechniques. 35:146–150. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Team RDC: R Development Core Team. 2011.
R: A language and environment for statistical computingR Foundation
for Statistical Computing. Vienna, Austria: http://www.R-project.org/
|
|
25
|
Mori Y, Cai K, Cheng Y, Wang S, Paun B,
Hamilton JP, Jin Z, Sato F, Berki AT, Kan T, et al: A genome-wide
search identifies epigenetic silencing of somatostatin,
tachykinin-1, and 5 other genes in colon cancer. Gastroenterology.
131:797–808. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jin Z, Mori Y, Yang J, Sato F, Ito T,
Cheng Y, Paun B, Hamilton JP, Kan T, Olaru A, et al:
Hypermethylation of the nel-like 1 gene is a common and early event
and is associated with poor prognosis in early-stage esophageal
adenocarcinoma. Oncogene. 26:6332–6340. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao C and Zhang Q, Kong D, Wu D, Su C,
Tong J, Chen F and Zhang Q: MALDI-TOF Mass Array analysis of Nell-1
promoter methylation patterns in human gastric cancer. Biomed Res
Int. 2015:1369412015. View Article : Google Scholar : PubMed/NCBI
|