Introduction
Liver cancer, including hepatocellular carcinoma
(HCC) and cholangiocarcinoma in adults, and hepatoblastoma mainly
in children, is one of the most common malignant tumors and is the
most frequent cause of cancer-associated death worldwide (1). Much progress has been made in liver
cancer research and current options for the treatment of the early
stage liver cancer include radio frequency ablation, hepatic
resection and transplantation (2,3).
Unfortunately, >50% of HCC cases are diagnosed at an
intermediate or advanced tumor stage (4). Oral administration of sorafenib, a
small molecule multikinase inhibitor, is currently the only
treatment that substantially prolongs survival in patients with
advanced HCC, making sorafenib the standard treatment for advanced
stage HCC. Although sorafenib treatment improves survival among
patients with advanced HCC, toxicity and unsatisfactory antitumor
effects remain unsolved issues with this drug (5,6).
Although novel molecular targeting agents including regorafenib
have been approved, none of the inhibitors have demonstrated
satisfactory results. Therefore, there remains a need for improved
therapeutics for liver cancer.
Thioredoxin (Trx) and thioredoxin reductase (TrxR)
provide a coupled redox system, which serves key roles in
maintaining redox reactions in biosynthetic pathways and
controlling redox homeostasis in cells. Trx, a redox regulatory
protein, can be oxidized by abundant reactive oxygen species (ROS).
TrxR reactivates Trx by reduction, providing a circuit for
sequential turnover in multiple oxidation/reduction cycles
(7).
It has been suggested that TrxR serves important
roles in diverse physiological and pathological processes,
including apoptosis, cancer, chronic inflammation, autoimmune
diseases and neurodegenerative disorders (7). In previous years, several studies have
reported that Trx or TrxR are overexpressed in tumors including
lung, breast, colorectal, pancreatic, hepatocellular, gastric,
myeloma, non-Hodgkin lymphoma and acute lymphocytic leukemia
(8,9). TrxR contributes to tumor growth
through the hypoxia inducible factor-1 pathway and is essential to
maintain tumor phenotypes and metastasis (10,11).
Accumulating evidence indicates that TrxR/Trx is an important
modulator of tumor development. Therefore, targeting TrxR/Trx is a
promising strategy for cancer treatment (8,12).
Due to its involvement in pathological processes,
inhibiting TrxR is an important clinical goal. Recently, several
studies have described inhibitors of TrxR, for example, gold (I)
N-heterocyclic carbene complexes, that exhibit clear and strong
anti-proliferative effects on a wide range of tumor cells (13). Generally, TrxR inhibitors display
antitumor effects by promoting the apoptosis of tumor cells
(8,9,14).
TrxR expression has been demonstrated to be associated with liver
cancer (15,16); however, whether TrxR can serve as a
therapeutic target for liver cancer has yet to be determined.
In the present study, whether blocking TrxR using
the inhibitor chloro(triethylphosphine)gold(I) (AA1), could inhibit
the growth of liver cancer cells in vitro and in vivo
was examined. Targeting TrxR resulted in the production of ROS, the
inhibition of autophagy and the induction of lesions in the
mitochondrial membranes in liver cancer cells. Additionally, AA1
induced a profound antitumor immune response in the tumor immune
microenvironment.
Materials and methods
Cell culture and reagents
Human liver cancer cell line HepG2 (17), the C57BL/6-derived hepatoma cell
line Hepa1-6, MDA-MB231, Hela, B16, K562, HL-60, A549, H7402, PLC
and HepG2.2.15 cells were purchased from the Cell Bank of Type
Culture Collection of the Chinese Academy of Sciences (Shanghai,
China). Cells were cultured in Dulbecco's modified Eagle's medium
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented
with 10% fetal bovine serum (Sijiqing; Zhejiang Tianhang
Biotechnology Co., Ltd., Hangzhou, China) at 37°C with 5%
CO2. AA1, a potent TrxR inhibitor, was kindly provided
by Professor Minyong Li at the School of Pharmaceutical Sciences,
Shandong University (Shandong, China) (18). Cisplatin was obtained from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany), and was dissolved
in 0.9% NACl at a final concentration of 100 mg/ml.
Animal models
6-week-old male nude mice (n=6, 19–21 g) and C57BL/6
mice (n=7, 21–23 g) were purchased from Beijing HFK Bioscience Co.,
Ltd., (Beijing, China) and maintained under specific pathogen-free
conditions (temperature 25°C, relative humidity 45%) with a 12-h
light-dark cycle. A total of 2×106 Hepa1-6 cells in 200
µl PBS were injected into nude mice or C57BL/6 mice and seven days
later, AA1 (2 mg/kg) or PBS as a control was intraperitoneally
injected into tumor-bearing mice every two days for seven total
injections. Cisplatin (3 mg/kg) was as a positive control in
tumor-bearing nude mice. Tumor volumes were determined by measuring
length (l) and width (w) at the indicated time points. At the end
of treatment, the mice were sacrificed and the tumors removed and
used for ROS and tumor immune environment evaluation. All animals
were kept in standard laboratory conditions and provided with food
and water ad libitum. The animal experiments were approved
by the Research Ethics Committee of Shandong University.
TrxR activity analysis by
3-carboxy-4-nitrophenyl disulfide (DNTB) assay
HepG2 or Hepa1-6 cells were incubated with different
concentrations of AA1 for 48 h. Cells were harvested and cell
lysates were prepared with a RIPA lysis buffer (Beyotime Institute
of Biotechnology, Shanghai, China) in the presence of protease
inhibitors. Protein concentration of the supernatant was determined
using the Bradford reagent. A total of 25 µg of extract were
incubated with 1 mM NADPH and 2 mM selenocystine (SC) in Tris EDTA
buffer [50 mM Tris-HCl and 2 mM EDTA, (pH 7.5)] in a total volume
of 100 µl. Samples of protein only, protein and NADPH, and protein
and SC were used as controls. The reaction mixture was placed into
96-well microplates and monitored at 30 sec intervals over a 20 min
period, and the absorbance was measured at 412 nm (BioRad
Laboratories, Inc., Hercules, CA, USA) (9).
Cell viability assay
HepG2 and Hepa1-6 cells (4×103) were
seeded in 96-well plates, then HepG2 were treated with AA1 (1.25,
2.5, 5 and 10 µM), and Hep1-6 cells were treated with AA1 (0.3,
0.6, 1.25, 2.5 and 5 µM) at the indicated time points (24 and 48
h), then MTT assays performed to measure the inhibitory effects of
AA1 on the proliferation of liver cancer cell. At last, DMSO was
used to dissolve the purple formazan, and the absorbance of the
plate was measured at 490 nm.
Cell cycle analysis and apoptosis
assay
HepG2 cells (1.5×105) were seeded in
12-well plates. The following day, cells were treated with AA1 at
the indicated concentrations (2.5, 5 and 10 µM). Following 24 h,
cells were harvested, washed twice with ice-cold PBS and fixed in
70% ethanol at 4°C overnight. Cells were then washed once with
ice-cold PBS and re-suspended in 1 ml of staining reagent
containing 50 mg/ml propidium iodide (PI) and 100 mg/ml RNase.
Cells were incubated for 30 min in the dark, then cell cycle
analysis was performed using a flow cytometer (BD FACS Calibur; BD
Biosciences, Franklin Lakes, NJ, USA) (19). To determine rates of apoptosis,
cells underwent the same treatment and analysis as for cell cycle
analysis but were stained with Annexin V-fluorescein isothiocyanate
(FITC)/PI (eBioscience; Thermo Fisher Scientific, Inc.), according
to the manufacturer's protocol. The data were analyzed using WinMDI
version 2.8 software (The Scripps Research Institute, La Jolla, CA,
USA).
Evaluation of mitochondrial membrane
potential (MMP)
HepG2 and Hepa1-6 cells (1×105) were
seeded in 60 mm cell culture dishes. Then, HepG2 cells were treated
with 2.5 and 5 µM, Hep1-6 cells were treated with 1.25 and 2.5 µM,
48 h later, cells were harvested and washed twice with ice-cold
PBS, then incubated with JC-1 (10 µg/ml; cat. no. C2006; Beyotime
Institute of Biotechnology) in the dark for 15 min at 37°C. Cells
were washed three times with ice-cold PBS and analyzed by
fluorescence microscopy (Olympus Corporation, Tokyo, Japan) using
emission wavelengths of 590 and 525 nm.
Determination of ROS production
HepG2 and Hepa1-6 cells (1.5×105) were
seeded in 60-mm cell culture dishes. Following treatment with AA1
for 6 h, cells were incubated with 10 µM 2′,7′-dichlorofluorescein
diacetate (DCFH-DA; cat. no. D6883; Sigma-Aldrich; Merck KGaA) for
15 min in the dark, washed three times with ice-cold PBS and the
ROS levels analyzed by fluorescence microscopy (Olympus
Corporation) (20).
Nuclear staining
HepG2 and Hepa1-6 cells (1.5×105) were
seeded in 60-mm cell culture dishes. Following treatment for 12 h,
cells were harvested and washed three times with cold PBS,
following which cells were fixed with 4% polyoxymethylene for 10
min at room temperature. Then, cells were rinsed with PBS, and
incubated with 1 µM DAPI (cat. no. C1002; Beyotime Institute of
Biotechnology) for 10 min at 37°C in dark. Then, the cells were
imaged using a fluorescent microscope (CK30-F200; Olympus
Corporation).
Reverse transcription-quantitative
polymerase chain reaction analysis
Total RNA was extracted from cells using TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) and used to synthesize
cDNA using M-MLV Reverse Transcriptase (Invitrogen; Thermo Fisher
scientific, Inc.) in accordance with the manufacturer's protocol.
The mRNA level of Bcl-2 was determined by Reverse
transcription-quantitative PCR (RT-qPCR) with SYBR Green Master Mix
(Roche Diagnostics, Indianapolis, IN, USA) using an iCycleriQ
real-time PCR system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). qPCR amplification was conducted for 35 cycles, each cycle
consisting of denaturation (95°C for 5 sec), annealing (55°C for 20
sec) with a single fluorescence measurement taken at the end of the
annealing step, and extension (72°C for 20 sec). The ∆∆Cq method
was applied for gene number determination (21). The expression of human Bcl-2 was
normalized to GAPDH levels. Primer sequences for quantitative
real-time PCR were as follows: GAPDH (forward primer,
5′-TGCACCACCAACTGCTTAGC-3′; reverse primer,
5′-GGCATGGACTGTGGTCATGAG-3′) and Bcl-2 (forward primer,
5′-CTGAGTACCTGAACCGGCA-3′; reverse primer,
5′-GAGAAATCAAACAGAGGCCG-3′).
Western blotting
Total protein was extracted from cells lysed with a
RIPA lysis buffer (Beyotime Institute of Biotechnology) and the
concentrations were determined with bicinchoninic acid methods.
Then, proteins (30 µg/lane) were separated by SDS-PAGE on a 10%
polyacrylamide gel and transferred onto polyvinylidene difluoride
membranes membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked with TBS buffer containing 5% non-fat milk
for 1 h at room temperature. Then the protein were incubated with
specific antibodies (1:1,000): Phosphorylated (p)-38 (cat. no.
4511; Cell Signaling Technology, Inc., Danvers, MA, USA), P38 (cat.
no. 8690; Cell Signaling Technology, Inc.), p-mitogen-activated
protein kinase 8 (p-JNK; cat. no. 4668S; Cell Signaling Technology,
Inc.), JNK (cat. no. 3708S; Cell Signaling Technology, Inc.),
p-extracellular signal regulated kinase (p-ERK; cat. no. 14227S;
Cell Signaling Technology, Inc.), ERK (cat. no. 4348S; Cell
Signaling Technology, Inc.), p-mammalian target of rapamycin
(p-mTOR; cat. no. ab109268; Abcam, Cambridge, UK), mTOR (cat. no.
ab32028; Abcam), p62 (cat. no. ab109012; Abcam), light chain-3
(LC3B; cat. no. 2275S), Bcl-2 (cat. no. ab32124; Abcam) and β-actin
(cat. no. 3700; Cell Signaling Technology, Inc.). The membranes
were then washed and incubated with goat anti-rabbit (cat. no.
A0208; Beyotime Institute of Biotechnology) or goat anti-mouse
(cat. no. A0216; Beyotime Institute of Biotechnology) horseradish
peroxidase-conjugated secondary antibody (1:10,000) for 1 h. After
being washed, the membranes were developed using enhanced
chemiluminescence (ECL; EMD Millipore). Densitometric analysis of
blots was performed in ImageJ (version 1.41o; National Institutes
of Health, Bethesda, MD, USA).
Autophagy flux assays
HepG2 cells were seeded in a 12-well plate
(1.5×105) and infected with Ad-mCherry-green
fluorescence protein (GFP)-LC3B (Beyotime Institute of
Biotechnology) (Multiplicity of infection 15) for 48 h, then
preincubated for 2 h with or without 1 mg/ml N-acetylcysteine
(NAC). This was followed by treatment with 10 µM AA1 for 6 h. Cells
were fixed with 4% paraformaldehyde for 24 h at 4°C and
microphotographs of mCherry-GFP-LC3 fluorescence were acquired by
fluorescence microscopy (Olympus Corporation).
Flow cytometry
Infiltrating mononuclear cells were isolated from
the whole tumor tissue. Flow cytometric analysis was performed
using BD FACSCalibur and FACSAria III instruments. Antibodies used
in this study included FITC-labeled anti-Natural killer (NK)1.1
(cat. no. 11-5941-85) and anti-cluster of differentiation (CD)4
(cat. no. 11-0041-82); phycoerythrin (PE)-labeled anti-mouse-CD69
(cat. no. 12-0691-83); PE-cyanine 5.5-labeled anti-mouse CD3e (cat.
no. 45-0031-82) and PE-labeled anti-CD8 (cat. no. 12-0081-82).
Antibodies were obtained from eBioscience; Thermo Fisher
Scientific, Inc. (22). The cells
were blocked with 10% normal rat serum (prepared in our lab) for 30
min, and stained with a saturating amount of the antibodies for 1 h
at 4°C, then washed with PBS for three times. Cells were acquired
on a FACSCalibur flow cytometer (BD Bioscience) and analyzed using
WinMDI version 2.8 software (The Scripps Research Institute, San
Diego, CA, USA).
Statistical analyses
All values are presented as the mean ± standard
error of the mean of three independent experiments. Statistical
analysis was performed using a paired Student's t-test or
one-way analysis of variance followed by Tukey's post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Targeting TrxR inhibits liver cancer
cell growth
To evaluate the feasibility of targeting TrxR in
liver cancer, TrxR activity was first examined using a DNTB assay.
Among various cancer cell lines including MDA-MB231 (breast
cancer), Hela (cervical cancer), B16 (melanoma cells), K562 and
HL-60 (leukemia cell), A549 (lung cancer), HepG2, H7402, PLC and
HepG2.2.15 cells (liver cancer), liver cancer cell lines, including
HepG2 and H7402, were demonstrated to exhibit a high level of TrxR
activity (data not shown). Subsequently, the effects of the TrxR
inhibitor AA1 on the growth of HepG2 cells and the mouse hepatoma
cell line Hepa1-6 in vitro was analyzed using an MTT assay.
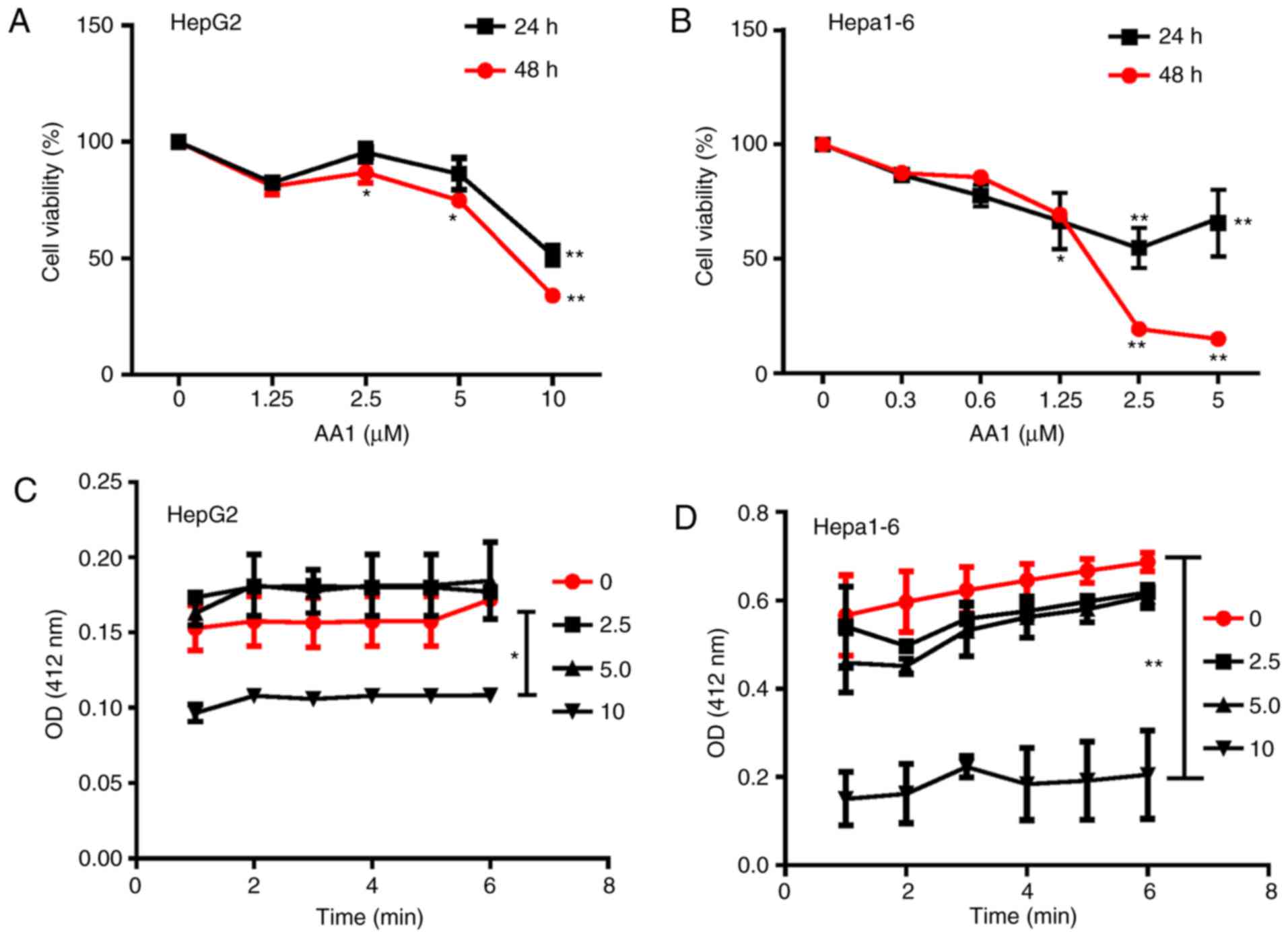
As demonstrated in Fig. 1A and B,
treatment with AA1 for 24 or 48 h inhibited the proliferation of
HepG2 and Hepa1-6 cells significantly at ≥2.5 µM (P<0.05). In
addition, it was observed that TrxR activity was inhibited in HepG2
and Hepa1-6 cells treated with a range of AA1 concentrations and
significant inhibition occurred at 10 µM (P<0.05; Fig. 1C and D). These results demonstrated
that targeting TrxR can inhibit the growth of liver cancer cells
in vitro.
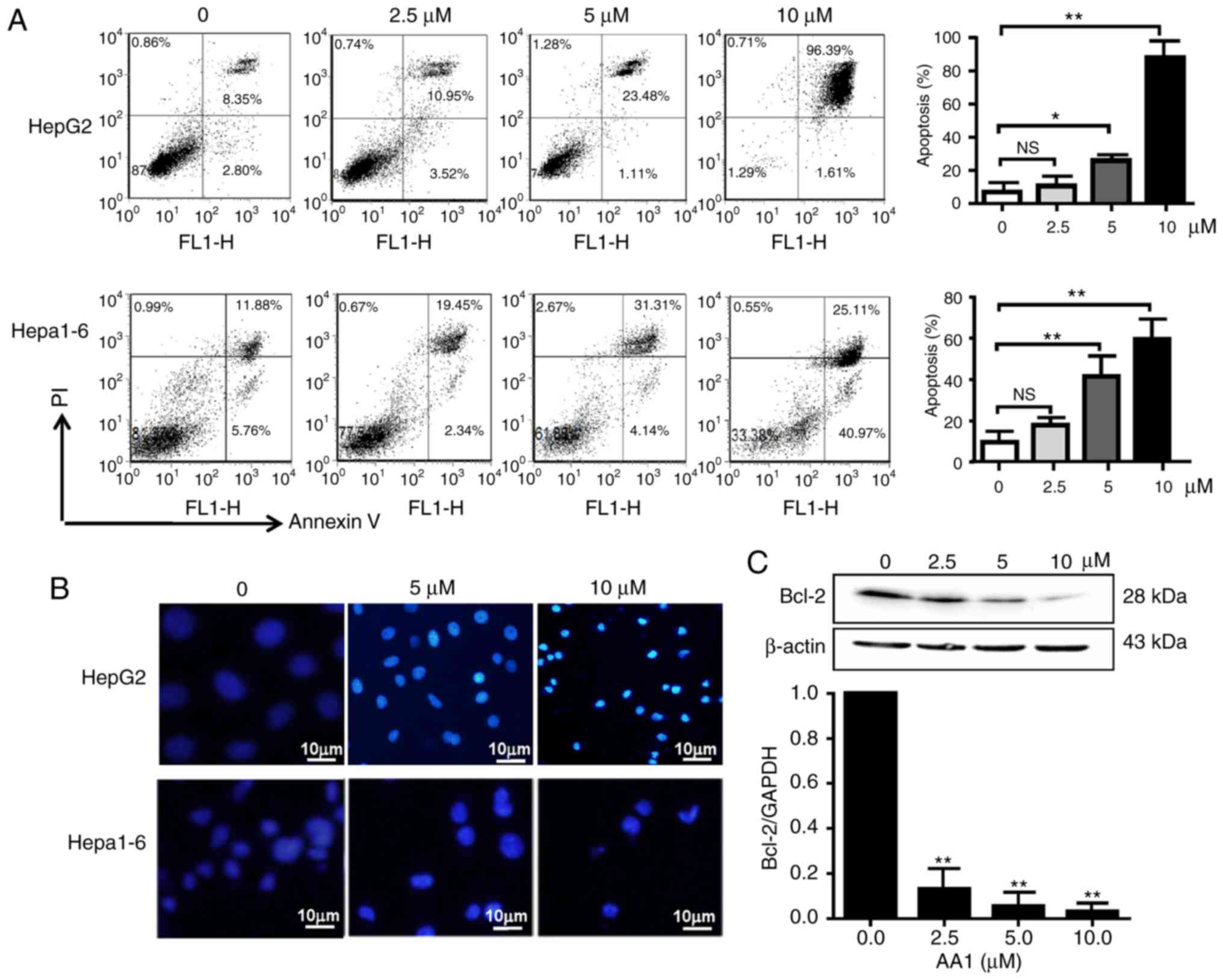
Targeting TrxR induces apoptosis in
liver cancer cells in a ROS-dependent manner
Cell cycle analysis demonstrated that TrxR
inhibition by AA1 has no effect on the cell cycle progression of
liver cancer cells (data not shown). Therefore, the pro-apoptotic
effects of AA1 in liver cancer cell lines were examined using an
Annexin V/PI staining assay. As demonstrated in Fig. 2A, HepG2 and Hepa1-6 cells underwent
dose-dependent apoptotic cell death following treatment with AA1
for 12 h, with a significant increase in apoptosis was demonstrated
at ≥5 µM (P<0.05). Cell nuclear morphology was also examined as
an indicator of apoptosis. As demonstrated in Fig. 2B, compared with the untreated cells,
which remained dim, HepG2 cells treated with the TrxR inhibitor
exhibited a bright blue appearance and the nucleus appears to be
condensed and fragmented. Examination of expression of the
anti-apoptotic gene Bcl-2 by qPCR and western blotting demonstrated
that inhibition of TrxR significantly decreased expression in a
dose-dependent manner in HepG2 cells (P<0.01; Fig. 2C), a similar trend of Bcl-2 mRNA
inhibition was also observed in Hep1-6 cells after AA1 treatment
(data not shown).
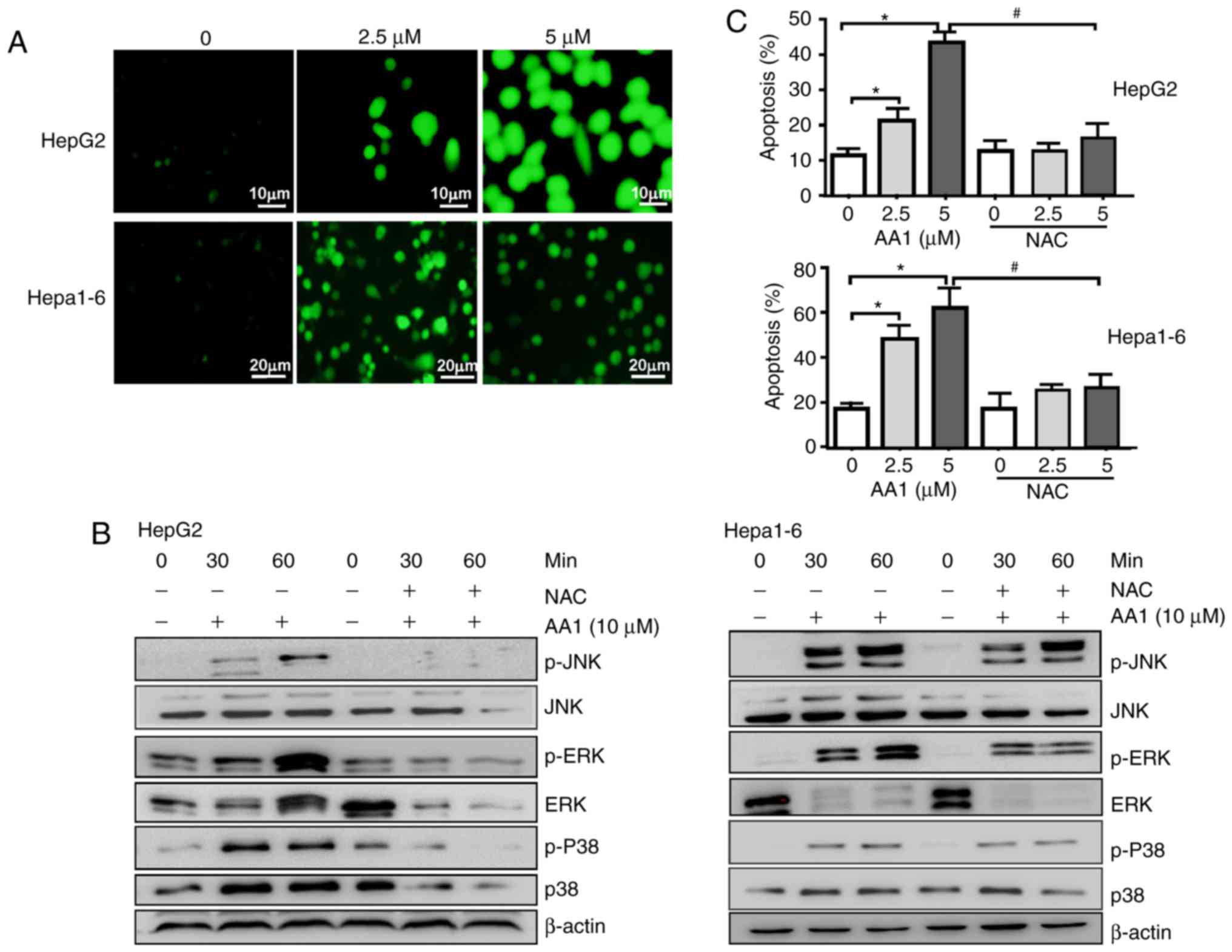
It was reported that TrxR inhibition promotes
apoptosis in a ROS-dependent manner in pancreatic and lung cancer
cells (9,23). Therefore, whether TrxR
inhibition-induced apoptosis of liver cancer cells was associated
with an increase in ROS levels was examined. Using the
redox-sensitive fluorescent probe DCFH-DA, TrxR inhibition by AA1
was demonstrated to cause a dose-dependent increase in ROS levels
in HepG2 and Hepa1-6 cells (Fig.
3A). The p38-mitogen associated protein kinase (MAPK) signaling
pathway is activated by ROS production, which has previously been
suggested to promote cell apoptosis (24). As demonstrated in Fig. 3B, TrxR inhibition by AA1 increases
the levels of p-p38, p-JNK and p-ERK in a time-dependent manner;
notably, the ROS inhibitor NAC almost completely abolished p38,
JNK, ERK activation induced by AA1 in HepG2 and Hepa1-6 cells.
Furthermore, AA1-induced pro-apoptotic effects in HepG2 and Hepa1-6
cells are almost abolished by pre-treatment with NAC (Fig. 3C). These results indicated that
AA1-induced ROS activate the MAPK signal pathway and promote
apoptosis in liver cancer cells.
Targeting TrxR inhibits autophagy of
liver cancer cells in a ROS-independent way
As previously described, ROS induce autophagy which
then contributes to a reduction of ROS levels. Autophagy serves
important roles in cell survival as well as in the regulation of
cell death, especially in apoptosis-signaling pathways. To
investigate whether the activation of autophagy contributes to the
effects of AA1, the activation of autophagy in AA1-treated HepG2
and Hepa1-6 cells was examined. LC3 expression was demonstrated to
decease significantly in cells treated with AA1 for 6 h
(P<0.05); however, NAC treatment does not reverse autophagosome
inhibition resulting from AA1 treatment (Fig. 4A). The conversion of LC3-I to LC3-II
was monitored using western blot analysis. Consistently, the ratio
of LC3-II to LC3-I in HepG2 and Hepa1-6 cells treated with AA1
decreased significantly compared with the control group
(P<0.05). This effect is also observed following NAC
pretreatment (P<0.05; Fig.
4B).
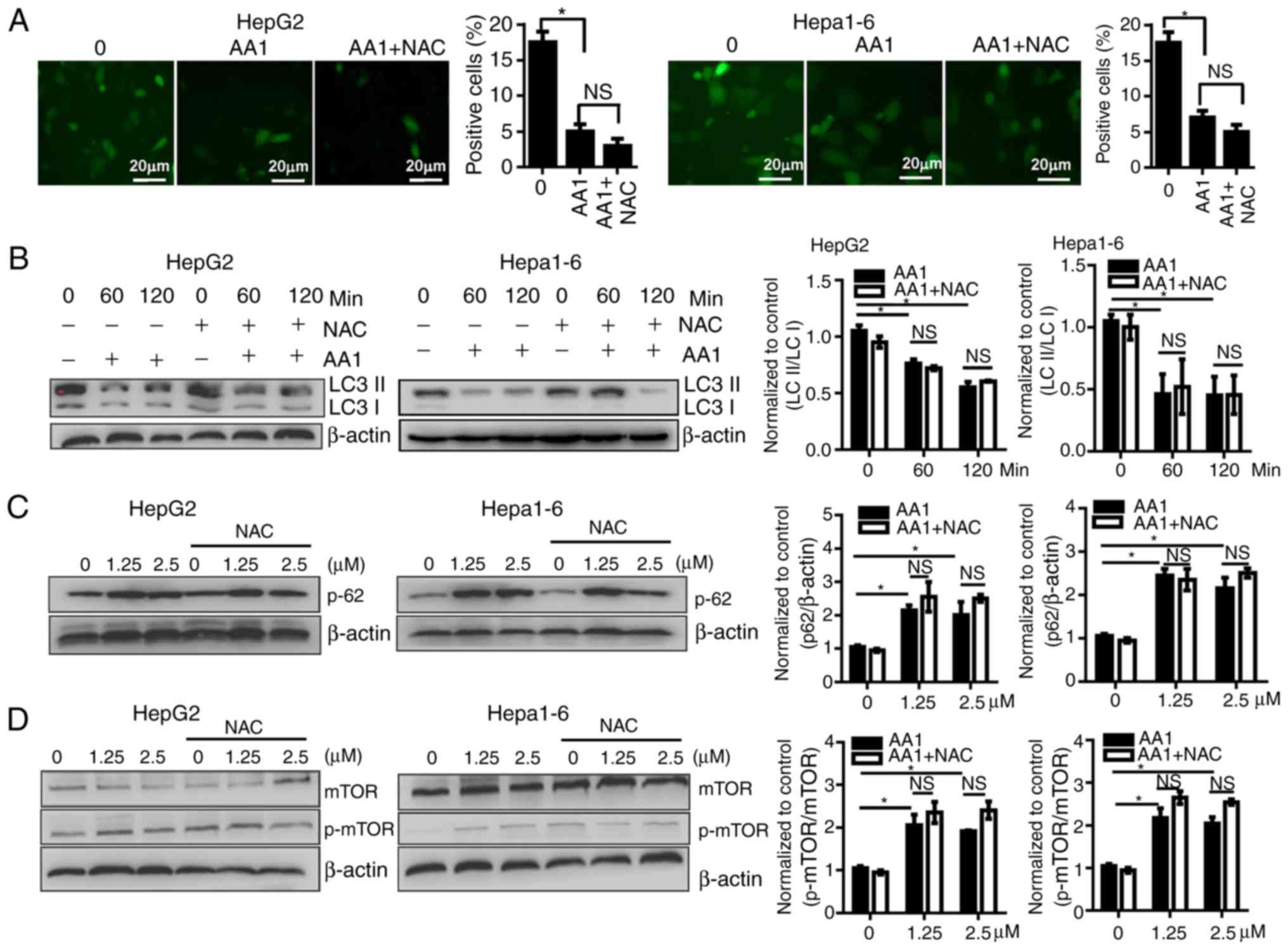 | Figure 4.Targeting TrxR inhibits autophagy of
liver cancer cells in a reactive oxygen species-independent manner.
HepG2 or Hepa1-6 cells were infected with Ad-mCherry-green
fluorescent protein-LC3B for 48 h and following pretreatment with
NAC for 2 h or no treatment, cells were then treated with 10 µM AA1
for 6 h. (Aa) Autophagosomes were imaged by fluorescence microscopy
(scale bar 20 µm), quantitative analysis of LC3 expression was
presented in (Ab). (B) LC3-II and LC3-I levels were examined by
western blotting following treatment with AA1 (10 µM), AA1+NAC
pretreated, or NAC alone for different time points in Hepa1-6 and
HepG2 cells. (C) The levels of p62 protein were detected following
treatment with an increasing concentration (0–2.5 µM) of AA1 for 3
h in Hepa1-6 and HepG2 cells with or without NAC pretreatment. (D)
Activation of mTOR in HepG2 and Hepa1-6 cells treated as described
above was detected by western blotting. The western blots are
presented on the left and quantitative analysis is presented in the
right panel. Experiments were run in triplicate. *P<0.05 vs. the
untreated group (0 µM). mTOR, mammalian target of rapamycin; LC3,
light chain 3; NAC, N-acetylcysteine; AA1,
chloro(triethylphosphine)gold(I); TrxR, thioredoxin reductase; NS,
not significant. |
The p62 protein has previously been demonstrated to
be preferentially degraded by autophagy, so levels of p62 reflect
alterations in autophagy (25). As
the levels of p62 were examined by western blotting, it was noticed
that the levels of p62 increased noticeably in 1.25 or 5 µM
AA1-treated HepG2 and Hepa1-6 cells, and that NAC does not
influence the expression of p62 (Fig.
4C). These data further support that AA1 inhibition of
autophagy is not dependent on ROS.
Activity of the mTOR is associated with the status
of autophagy (26). TrxR inhibition
with increasing concentrations of AA1 was demonstrated to
significantly induce phosphorylation of mTOR in Hepa1-6 and HepG2
cells (P<0.05) and NAC pretreatment does not prevent AA1-induced
mTOR activation (Fig. 4D).
Together, these results indicate that targeting TrxR inhibits
autophagy in liver cancer cells in an ROS-independent manner.
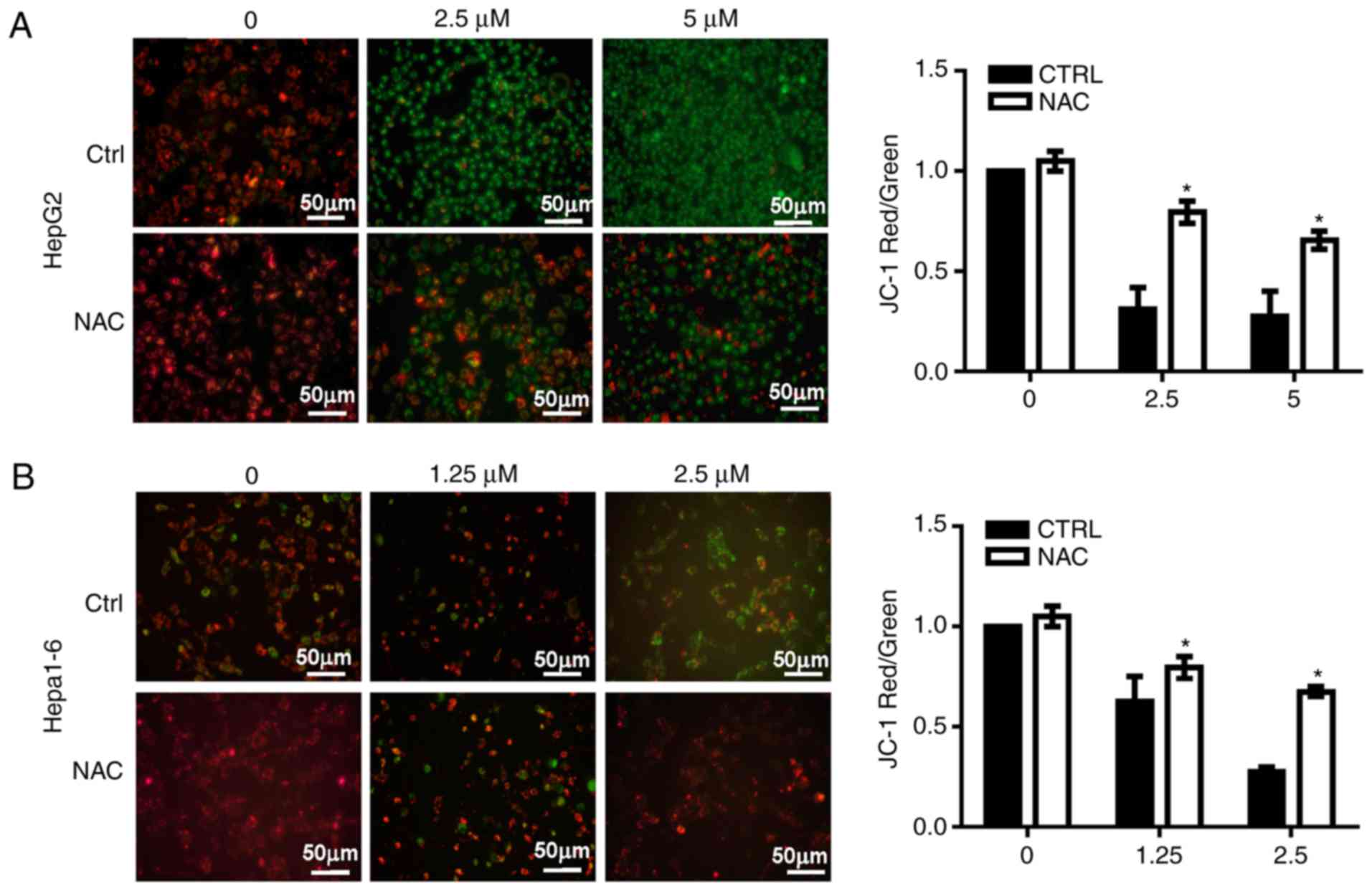
Inhibition of TrxR induces
mitochondrial membrane lesions in liver cancer cells
Since increases in ROS may occur due to
dysfunctional mitochondria (9), the
MMP was analyzed in HepG2 and Hepa1-6 cells treated with AA1 using
JC-1 dye. An increase in green fluorescence and a decrease of red
fluorescence was noted, indicating a decline in the MMP in
AA1-treated cells without NAC. NAC significantly suppressed the
inhibition of MMP induced by AA1 in HepG2 (P<0.05; Fig. 5A) and Hepa1-6 cells at 2.5 µM
(P<0.05; Fig. 5B). These
observations provide a clear indication that TrxR inhibition
induces lesions in the mitochondrial membrane in liver cancer
cells.
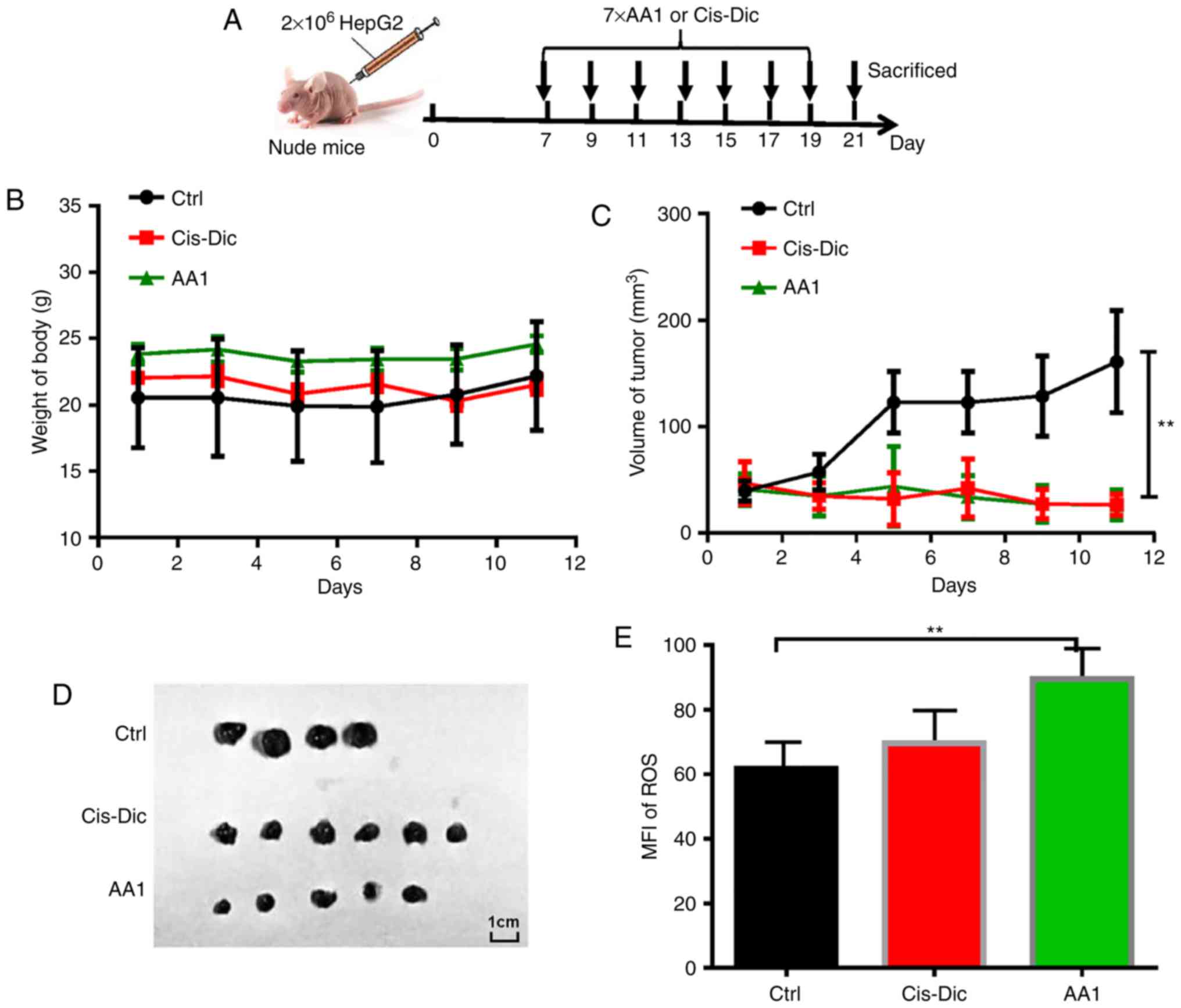
Targeting TrxR inhibits tumor growth
in HepG2 tumor-bearing nude mice
To investigate the effects of targeting TrxR on
liver cancer growth in vivo, AA1 was administered to nude
mice subcutaneously injected with 2×106 HepG2 cells. A
widely used anticancer drug cis-diamine dichloroplatinum
(Cis-Dic) was used for comparison (Fig. 6A). The body weights of AA1-and
Cis-Dic-treated groups were not significantly different from
the untreated control group (Fig.
6B). The size of the tumors in the AA1- and
Cis-Dic-treated mice were significantly smaller compared
with the control mice, with the inhibitory effects of AA1 similar
to Cis-Dic (P<0.01; Fig.
6C and 6D). Furthermore, as
demonstrated in Fig. 6E, AA1
treatment significantly increases the expression of ROS in HepG2
cells isolated from tumor-bearing nude mice (P<0.01). These data
suggest that TrxR inhibition efficiently inhibits the growth of
liver cancer in vivo.
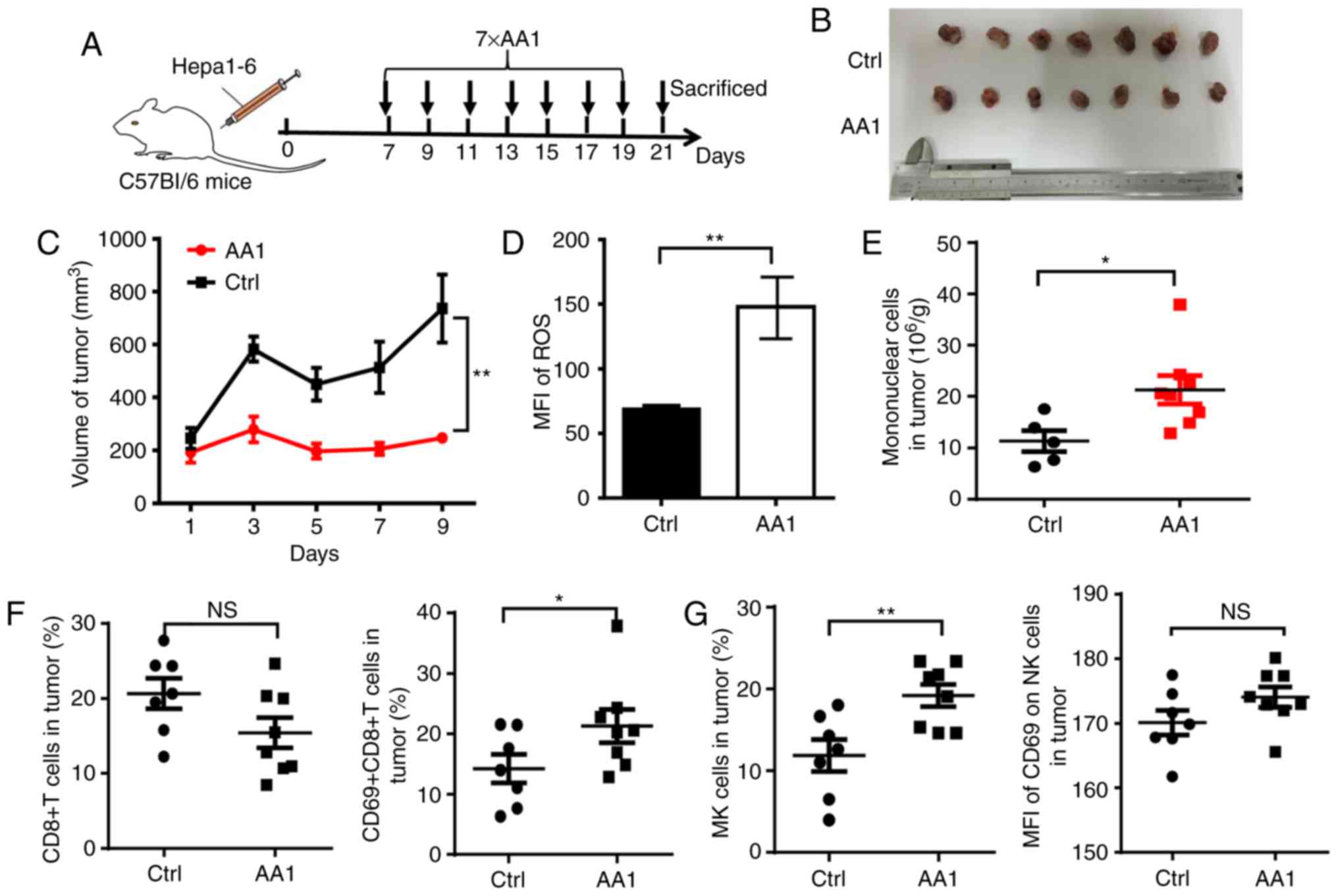
Targeting TrxR improves the tumor
microenvironment
To identify whether TrxR inhibition affects the
tumor microenvironment, another mouse model was adopted with an
intact immune system. A total of 2×106 Hepa1-6 cells
were inoculated into C57BL/6 mice and beginning seven days
post-inoculation, AA1 or a solvent control were administered every
two days for a total of seven injections (Fig. 7A). Similar to what was observed in
HepG2 tumor-bearing nude mice, the growth of liver cancer was
significantly suppressed by AA1 treatment as compared with the
control group (P<0.01; Fig. 7B
and 7C) and is accompanied by a
significant increase in ROS levels (P<0.01; Fig. 7D). Next mononuclear cells were
isolated from tumor tissues and the phenotypes of immune cells were
analyzed. Compared with the control mice, a significantly increased
infiltration of mononuclear cells was observed in tumor tissues
from mice treated with AA1 (P<0.05; Fig. 7E). Since it is well known that
CD8+ T and natural killer (NK) cells serve key roles in
antitumor immunity, the frequency and activation of these cells was
next evaluated by fluorescence-activated cell sorting. As
demonstrated in Fig. 7F, although
the percentage of CD8+ T cells is not significantly
affected by AA1 treatment, the expression of the activation marker
CD69 is significantly increased in CD8+ T cells from
AA1-treated mice (P<0.05). In addition, the frequency and CD69
expression levels of NK cells increased in tumor tissues from
AA1-treated mice, while the CD69 upregulation was non-significant.
(Fig. 7G). These data suggested
that targeting TrxR can stimulate the activation of the immune
response, enhancing antitumor efficiency.
Discussion
In the present study, it was demonstrated that
targeting TrxR inhibits the growth of liver cancer cells by
inducing apoptosis through a ROS-dependent pathway.
Mechanistically, TrxR inhibition results in the production of ROS,
which activates the MAPK signaling pathway. Additionally, lesions
in the mitochondrial membranes in liver cancer are induced by
targeting TrxR and are accompanied by the inhibition of the protein
kinase B (Akt)/mTOR pathway and autophagy. The results of xenograft
experiments in nude mice were highly consistent with in
vitro studies. Notably, TrxR inhibition improves the tumor
immune microenvironment in mouse model.
The Trx/TrxR system is a major protein disulfide
reduction system in mammalian cells. TrxR is required to convert
oxidized Trx into its functional reductive form, which can scavenge
ROS and improves cell viability under oxidative stress (27). TrxR, is overexpressed in a number of
tumor cells including liver cancer, pancreatic cancer and lung
cancer, and has emerged as a novel target for cancer treatment
(9,23). To date, a number of synthetic and
natural therapeutic compounds that exhibit anticancer properties
have been classified as TrxR inhibitors. The results of the present
study demonstrated that TrxR inhibition inhibits the growth of
liver cancer in vitro and in vivo. Although no
disruption to the cell cycle of HepG2 cells is observed following
treatment with the TrxR inhibitor AA1, the number of apoptotic
cells was increased. Further investigation demonstrated that the
levels of ROS increased with TrxR inhibition and mediated
AA1-induced liver cancer cell apoptosis, in conjunction with
downregulation of Bcl-2. This is consistent with previous
observations demonstrating that the accumulation of ROS can result
in DNA damage and apoptosis (24,28).
Trx is a physiological inhibitor of mitogen-activated protein
kinase kinase kinase 5 (ASK1) that interacts directly and therefore
disrupts ASK1-p38-MAPK-dependent apoptosis (9,29,30).
Given that this interaction only occurs with Trx in its reduced
form, AA1 likely reduces the binding of ASK1 to Trx. As a result,
free ASK1 leads to phosphorylation of p38-MAPK, which is detected
in liver cancer cells, as well as the activation of JNK and
ERK.
The phosphoinositol 3 kinase/Akt/mTOR signaling
pathway and the Ras/Raf1/ERK1/2 pathway are known to regulate
autophagy in cellular responses (31,32).
In the present study, the activation of mTOR signaling was
demonstrated and the conversion of LC3-I to LC3-II was decreased by
AA1 treatment, which is consistent with observations of other types
of cancer treated with TrxR inhibitors (33,34).
Targeting TrxR induces the inhibition of autophagy independently of
ROS. This may mean that AA1 directly inhibits autophagy, or
autophagy inhibition occurs prior to ROS accumulation. These
results demonstrate the impact of the redox environment on
autophagy-apoptosis interplay in various cancer cells.
Mitochondria serve an important role in the
regulation of apoptosis and the mitochondria-mediated apoptosis
pathway is accompanied by MMP depolarization, followed by
pro-apoptotic molecule release from the mitochondria into the
cytosol (35). In the present
study, JC-1 was used as a fluorescent probe, which selectively
enters into mitochondria and undergoes a reversible color
alteration from green to red as the membrane potential increases.
TrxR inhibition was observed to decrease MMP in liver cancer cells
treated with AA1, demonstrating an additional mechanism
contributing to TrxR inhibition-induced apoptosis in liver cancer
cells.
A previous study demonstrated that
CD4+CD25+Foxp3+ regulatory T cells
(Tregs) are enriched in the tumor microenvironments of melanoma
patient and demonstrate a positive correlation with Trx levels,
while CD8+ T cell responses were not evaluated (36). In the present study, mononuclear
cells were observed to accumulate in the tumor tissue following AA1
treatment of C57BL/6 Hepa1-6 tumor-bearing mice and infiltrating
CD8+ T, and NK cells are activated. The present study
hypothesized that AA1 treatment may inhibit the function of Tregs,
then the inhibition of CD8+T cells and NK cells was
reversed in tumor environment. Importantly, in the present study
and another previous study hepatocellular damage following TrxR
inhibition was not observed (16).
Although AA1 demonstrates interesting anticancer effects in liver
cancer, more work needs to be done to better understand the
underlying immunological mechanisms.
In conclusion, the results of the present study
indicate that a disruption in the redox balance generated by
targeting TrxR can efficiently inhibit the growth of liver cancer
in vitro and in vivo. TrxR inhibition induces
apoptotic cell death through ROS and mitochondrial dysfunction. In
addition, blockage of autophagy increases the sensitivity of liver
cancer to TrxR inhibition. Notably, TrxR inhibition results in a
potent immune response in a mouse model. Together, the results of
the present study indicate that TrxR is a potential antitumor
target for liver cancer chemotherapy.
Acknowledgements
The authors would like to thank Professor Minyong Li
(School of Pharmaceutical Sciences, Shandong University) for
providing the potent TrxR inhibitor, AA1.
Funding
The present study was supported by the Shandong
Provincial Key Research and Development Program (grant no.
2017GSF18159), and the Shandong Provincial Natural Science
Foundation (grant no. ZR2017BH029), the Natural Science Foundation
of China (grant no. 81373222) and the Fundamental Research Fund of
Shandong University (grant no. 2017JC004).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
QH and JZ designed the study. HL and GW performed
the experiments. HL and QH analyzed the data. QH wrote the
manuscript. All authors have read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the Research
Ethics Committee of Shandong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nault JC, Sutter O, Nahon P, Ganne-Carrié
N and Séror O: Percutaneous treatment of hepatocellular carcinoma:
State of the art and innovations. J Hepatol. Oct 13–2017.(Epub
ahead of print).
|
|
2
|
Kim E, Kim D, Lee JS, Yoe J, Park J, Kim
CJ, Jeong D, Kim S and Lee Y: Capicua suppresses hepatocellular
carcinoma progression by controlling ETV4-MMP1 axis. Hepatology.
67:2287–2301. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shen Q, Eun JW, Lee K, Kim HS, Yang HD,
Kim SY, Lee EK, Kim T, Kang K, Kim S, et al: BANF1, PLOD3, SF3B4 as
early-stage cancer decision markers and drivers of hepatocellular
carcinoma. Hepatology. 67:1360–1377. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Finn RS, Zhu AX, Farah W, Almasri J, Zaiem
F, Prokop LJ, Murad MH and Mohammed K: Therapies for advanced stage
hepatocellular carcinoma with macrovascular invasion or metastatic
disease: A systematic review and meta-analysis. Hepatology.
67:422–435. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dietrich P, Koch A, Fritz V, Hartmann A,
Bosserhoff AK and Hellerbrand C: Wild type Kirsten rat sarcoma is a
novel microRNA-622-regulated therapeutic target for hepatocellular
carcinoma and contributes to sorafenib resistance. Gut.
67:1328–1341. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ray EM and Sanoff HK: Optimal therapy for
patients with hepatocellular carcinoma and resistance or
intolerance to sorafenib: Challenges and solutions. J Hepatocell
Carcinoma. 4:131–138. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saccoccia F, Angelucci F, Boumis G,
Carotti D, Desiato G, Miele AE and Bellelli A: Thioredoxin
reductase and its inhibitors. Curr Protein Pept Sci. 15:621–646.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Li X, Han X, Liu R and Fang J:
Targeting the thioredoxin system for cancer therapy. Trends
Pharmacol Sci. 38:794–808. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheng X, Holenya P, Can S, Alborzinia H,
Rubbiani R, Ott I and Wölfl S: A TrxR inhibiting gold(I) NHC
complex induces apoptosis through ASK1-p38-MAPK signaling in
pancreatic cancer cells. Mol Cancer. 13:2212014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hellfritsch J, Kirsch J, Schneider M,
Fluege T, Wortmann M, Frijhoff J, Dagnell M, Fey T, Esposito I,
Kölle P, et al: Knockout of mitochondrial thioredoxin reductase
stabilizes prolyl hydroxylase 2 and inhibits tumor growth and
tumor-derived angiogenesis. Antioxid Redox Signal. 22:938–950.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harris IS, Treloar AE, Inoue S, Sasaki M,
Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA,
et al: Glutathione and thioredoxin antioxidant pathways synergize
to drive cancer initiation and progression. Cancer Cell.
27:211–222. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Benhar M, Shytaj IL, Stamler JS and
Savarino A: Dual targeting of the thioredoxin and glutathione
systems in cancer and HIV. J Clin Invest. 126:1630–1639. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang D, Xu Z, Yuan J, Zhao YX, Qiao ZY,
Gao YJ, Yu GA, Li J and Wang H: Synthesis and molecular recognition
studies on small-molecule inhibitors for thioredoxin reductase. J
Med Chem. 57:8132–8139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rubbiani R, Kitanovic I, Alborzinia H, Can
S, Kitanovic A, Onambele LA, Stefanopoulou M, Geldmacher Y,
Sheldrick WS, Wolber G, et al: Benzimidazol-2-ylidene gold(I)
complexes are thioredoxin reductase inhibitors with multiple
antitumor properties. J Med Chem. 53:8608–8618. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li C, Peng Y, Mao B and Qian K:
Thioredoxin reductase: A novel, independent prognostic marker in
patients with hepatocellular carcinoma. Oncotarget. 6:17792–17804.
2015.PubMed/NCBI
|
|
16
|
Zheng X, Ma W, Sun R, Yin H, Lin F, Liu Y,
Xu W and Zeng H: Butaselen prevents hepatocarcinogenesis and
progression through inhibiting thioredoxin reductase activity.
Redox Biol. 14:237–249. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
López-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: Hep G2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar
|
|
18
|
Sutton BM, McGusty E, Walz DT and
DiMartino MJ: Oral gold. Antiarthritic properties of
alkylphosphinegold coordination complexes. J Med Chem.
15:1095–1098. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Y, Zheng B, Han Q, Zhang C, Tian Z
and Zhang J: Targeting blockage of STAT3 inhibits hepatitis B
virus-related hepatocellular carcinoma. Cancer Biol Ther.
17:449–456. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen W, Zou P, Zhao Z, Weng Q, Chen X,
Ying S, Ye Q, Wang Z, Ji J and Liang G: Selective killing of
gastric cancer cells by a small molecule via targeting TrxR1 and
ROS-mediated ER stress activation. Oncotarget. 7:16593–16609.
2016.PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang Y, Han Q, Hou Z, Zhang C, Tian Z and
Zhang J: Exosomes mediate hepatitis B virus (HBV) transmission and
NK-cell dysfunction. Cell Mol Immunol. 14:465–475. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Arambula JF, McCall R, Sidoran KJ, Magda
D, Mitchell NA, Bielawski CW, Lynch VM, Sessler JL and Arumugam K:
Targeting antioxidant pathways with ferrocenylated N-heterocyclic
carbene supported gold(I) complexes in A549 lung cancer cells. Chem
Sci. 7:1245–1256. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gan P, Gao Z, Zhao X and Qi G: Surfactin
inducing mitochondria-dependent ROS to activate MAPKs, NF-κB and
inflammasomes in macrophages for adjuvant activity. Sci Rep.
6:393032016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Din FV, Valanciute A, Houde VP, Zibrova D,
Green KA, Sakamoto K, Alessi DR and Dunlop MG: Aspirin inhibits
mTOR signaling, activates AMP-activated protein kinase, and induces
autophagy in colorectal cancer cells. Gastroenterology.
142(1504–1515): e3. 2012.PubMed/NCBI
|
|
27
|
Jin R, Gao Y, Zhang S, Teng F, Xu X, Aili
A, Wang Y, Sun X, Pang X, Ge Q and Zhang Y: Trx1/TrxR1 system
regulates post-selected DP thymocytes survival by modulating
ASK1-JNK/p38 MAPK activities. Immunol Cell Biol. 93:744–752. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang M, Harashima N, Moritani T, Huang W
and Harada M: The roles of ROS and caspases in TRAIL-induced
apoptosis and necroptosis in human pancreatic cancer cells. PLoS
One. 10:e01273862015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tsuchiya A, Kaku Y, Nakano T and Nishizaki
T: Diarachidonoylphosphoethanolamine induces apoptosis of malignant
pleural mesothelioma cells through a Trx/ASK1/p38 MAPK pathway. J
Pharmacol Sci. 129:160–168. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang X, Qin J, Zou J, Lv Z, Tan B, Shi J,
Zhao Y, Ren H, Liu M, Qian M and Du B: Extracellular ADP
facilitates monocyte recruitment in bacterial infection via ERK
signaling. Cell Mol Immunol. 15:58–73. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yokoi K, Kobayashi A, Motoyama H, Kitazawa
M, Shimizu A, Notake T, Yokoyama T, Matsumura T, Takeoka M and
Miyagawa SI: Survival pathway of cholangiocarcinoma via AKT/mTOR
signaling to escape RAF/MEK/ERK pathway inhibition by sorafenib.
Oncol Rep. 39:843–850. 2018.PubMed/NCBI
|
|
32
|
An H and Harper JW: Systematic analysis of
ribophagy in human cells reveals bystander flux during selective
autophagy. Nat Cell Biol. 20:135–143. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nagakannan P and Eftekharpour E:
Differential redox sensitivity of cathepsin B and L holds the key
to autophagy-apoptosis interplay after Thioredoxin reductase
inhibition in nutritionally stressed SH-SY5Y cells. Free Radic Biol
Med. 108:819–831. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin YX, Gao YJ, Wang Y, Qiao ZY, Fan G,
Qiao SL, Zhang RX, Wang L and Wang H: pH-sensitive polymeric
nanoparticles with gold(I) compound payloads synergistically induce
cancer cell death through modulation of autophagy. Mol Pharm.
12:2869–2878. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang SY, Chien CC, Hseu RS, Huang VYJ,
Chiang SY, Huang CJ, Chen SK, Tsai RY, Lin HT and Cheng YC:
Ganoderma microsporum immunomodulatory protein induces apoptosis
and potentiates mitomycin C-induced apoptosis in urinary bladder
urothelial carcinoma cells. J Cell Biochem. 119:4592–4606. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang X, Dong H, Li Q, Li Y and Hong A:
Thioredoxin induces Tregs to generate an immunotolerant tumor
microenvironment in metastatic melanoma. Oncoimmunology.
4:e10274712015. View Article : Google Scholar : PubMed/NCBI
|