Introduction
Glioblastoma (GBM) is the most common primary
malignant brain tumor (1),
representing 45.2% of malignant tumors and 15.6% of all primary
brain tumors. GBM is characterized by rapid proliferation, invasion
into the surrounding normal tissue and vascularization, making it
highly aggressive and deadly. At present, the standard treatment
for newly diagnosed GBM is surgical resection, followed by adjuvant
radiotherapy and chemotherapy; however, the prognosis of GBM
patients is very poor, with an average survival rate of only 15
months (2)U.S. Therefore, it is
urgent and critical to identify alternative therapeutic approaches,
and more importantly, to explore the molecular mechanisms
underlying GBM initiation and progression.
Arsenic resistance protein 2 (Ars2) is a gene
product that was first isolated from a hamster cell line and was
found to be resistant to sodium arsenite (2). Ars2 contains several domains: an
amino-terminal arginine-rich domain, a central RNA binding domain,
and a zinc finger domain, which are all common in RNA-binding
proteins (3). Ars2 is a highly
conserved gene, which is highly conserved in plants and yeast
(4,5). In recent years, many studies have
suggested that Ars2 plays an important role in embryonic
development (5–7) and in the biosynthesis of microRNAs
(8,9); furthermore, it binds to the promoter
of Sox2, a positive regulatory transcription factor in neural stem
cells (10). The Ars2 gene is
necessary for early embryonic development (7,11), and
the absence of the Ars2 protein leads to excessive apoptosis in
early embryos (5). Ars2 can also be
incorporated into the CBP80 and Drosha complexes in the nuclear CBC
(12), where it participates in the
cutting and maturation of primary miRNAs (13). This incorporation improves the
accuracy of the cutting of some miRNAs, including miR-21, let-7 and
miR-155 (12). When the expression
of Ars2 is downregulated, the processing of pri-miRNA was found to
be clearly diminished, and the levels of miRNA were decreased
(14–16). In recent years, it has been found
that Ars2 is highly expressed in some tumors and that it acts on
miR-21 to participate in tumor regulation (17). Some reports have indicated that Ars2
may play a key role in liver cancer and cholangiocarcinoma
(17,18). However, there is little research on
Ars2 in tumors, and its mechanism remains unclear. In the present
study, we investigated the effects of Ars2 on cell proliferation in
glioma growth.
Materials and methods
Cell culture
The human glioblastoma cell lines A172, LN-229, U251
and U87MG, and the human normal brain astrocyte cell line HEB were
grown in Dulbecco's modified Eagle's medium (DMEM) supplemented
with 10% fetal bovine serum (FBS) plus 1% penicillin and
streptomycin (P/S). A172, LN-229 and U87MG cell lines were obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA), U251 was purchased from the China Academia Sinica Cell
Repository (Shanghai, China), and HEB was a generous gift from Dr
Juan Tan (Southwest Hospital, Army Medical University, Chongqing,
China). The identification of cell genetic quality of the cell
lines LN-229 and U87MG (HTB-14) was performed using STR profiling
by Wuhan Genecreate Biological Engineering Co., Ltd., China. The
lentiviral packaging cell line 293FT was cultured in DMEM
containing 10% FBS, 0.1 mM MEM non-essential amino acids, 1 mM MEM
sodium pyruvate, 4 mM L-glutamine, 1% P/S, and 0.5 mg/ml G418. All
cells were cultured at 37°C in a humidified incubator with 5%
CO2. All the growth media, FBS and supplemental reagents
were obtained from Invitrogen/Life Technologies (Thermo Fisher
Scientific, Inc., Waltham, MA, USA).
Lentiviral constructs and
infection
The lentiviral constructs pLK0.1-puro-GFPsh and
pLK0.1-puro-Ars2sh were used in the knockdown studies. First, the
lentiviral constructs were transfected into 293FT packaging cells
using Invitrogen® Lipofectamine® 2000 reagent
(Thermo Fisher Scientific, Inc.). Next, the virus-containing
supernatant was harvested and tittered and then used to infect the
target cells with 4 µg/ml Polybrene (Santa Cruz Biotechnology,
Inc., Dallas, TX, USA). After the final round of infection, the
target cells were cultured in the presence of 2 µg/ml puromycin
(Life Technologies; Thermo Fisher Scientific, Inc.) for 3 days.
Finally, the drug-resistant cells were pooled.
Real-time qPCR assay
Human glioblastoma cells were harvested and lysed
with Trizol (Invitrogen®; Thermo Fisher Scientific,
Inc.) to purify the total RNA, which was then reverse transcribed
into cDNA using M-MLV (Promega Corp., Madison, WI, USA). The mRNA
transcript levels of Ars2, NLK, PRKX, GNG12, PAK2, TAOK2, AKT2 and
RAC2 were determined by real-time qPCR, using the SYBR®
Green PCR Master Mix (Takara). The real-time qPCR assay was
performed in triplicate and carried out using the OneStep Plus7500
Real-Time PCR system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) under the following conditions: 95°C for 10 min, followed by
40 cycles of 5 sec at 95°C and 30 sec at 60°C. All of the
individual values were normalized to the GAPDH control. Relative
mRNA expression levels were calculated by using the ΔΔCq method
(19). Sequences of qRT-PCR primers
were designed as follows: GAPDH-F: 5′-AACGGATTTGGTCGTATTGGG-3′ and
GAPDH-R: 5′-CCTGGAAGATGGTGATGGGAT-3′; Ars2-F:
5′-CACCATGTCCTGCCTATCCAG-3′ and Ars2-R:
5′-CGAAACCACTCCTCATCTTTGTG-3′; NLK-F: 5′-CGCAAAAATGATGGCGGCTTA-3′
and NLK-R: 5′-CCCAGGGTTTAACATGGCTG-3′; PRKX-F:
5′-CTGGACGTGGCATGACGAG-3′ and PRKX-R: 5′-TCGATGGCACAGATGATCTCT-3′;
GNG12-F: 5′-AGCAAGCACCAACAATATAGCC-3′ and GNG12-R:
5′-AGTAGGACATGAGGTCCGCT-3′; PAK2-F: 5′-CACCCGCAGTAGTGACAGAG-3′ and
PAK2-R: 5′-GGGTCAATTACAGACCGTGTG-3′; TAOK2-F:
5′-GGACTTTGGTTCTGCGTCCAT-3′ and TAOK2-R:
5′-TCGATGCAGGTTATCCCCAAG-3′; AKT2-F: 5′-GGTGCAGAGATTGTCTCGGC-3′ and
AKT2-R: 5′-GCCCGGCCATAGTCATTGTC-3′; RAC2-F:
5′-TCTGCTTCTCCCTCGTCAG-3′ and RAC2-R:
5′-TCACCGAGTCAATCTCCTTGG-3′.
Western blotting
Human glioblastoma cells or xenograft tumors were
harvested and washed once with ice-cold PBS. Cell pellets or tumor
tissues were suspended in SDS sample buffer, boiled for 10 min and
then centrifuged at 10,000 × g for 10 min. Fifty micrograms of the
protein samples were separated using 10% SDS-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to a PVDF membrane (EMD
Millipore Corp., Billerica, MA, USA). Next, the PVDF membrane was
probed with both primary and secondary antibodies and finally
visualized by enhanced chemiluminescence (ECL) (Beyotime Institute
of Biotechnology, Haimen, China). The primary antibodies were as
follows: Rabbit anti-human Ars2 (dilution 1:2,000; cat. no.
ab192999; Abcam, Cambridge, UK), mouse anti-human α-tubulin
(dilution 1:2,000; cat. no. B-5-1-2; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), rabbit anti-human CDK2 (dilution 1:1,000; cat.
no. 2546S; Cell Signaling Technology, Inc., Danvers, MA, USA),
rabbit anti-human CDK4 (dilution 1:1,000; cat. no. 12790S; Cell
Signaling Technology), rabbit anti-human cyclin D1 (dilution
1:1,000; cat. no. 2922S; Cell Signaling Technology), rabbit
anti-human cyclin E2 (dilution 1:1,000; cat. no. ab40890; Abcam),
rabbit anti-human p21 (dilution 1:1,000; cat. no. ab109199; Abcam),
rabbit anti-human PAK2 (dilution 1:1,000; cat. no. 2608S; Cell
Signaling Technology), rabbit anti-human ERK1/2 (dilution 1:500;
cat. no. 4695S; Cell Signaling Technology), rabbit anti-human
phospho-ERK1/2 (Thr202/Tyr204) (dilution 1:2,000; cat. no. 4376S;
Cell Signaling Technology), rabbit anti-human MEK1/2 (dilution
1:1,000; cat. no. 9122S; Cell Signaling Technology), and rabbit
anti-human phospho-MEK1/2 (Ser217/221) (1:500; cat. no. 9121S; Cell
Signaling Technology). Horseradish peroxidase-conjugated goat
anti-mouse and goat anti-rabbit IgG (1:20,000; KPL, Inc.,
Gaithersburg, MD, USA) were used as secondary antibodies.
Histology and
immunohistochemistry
The tumor tissue was embedded in paraffin blocks,
sectioned at a thickness of 4 µm and stained with hematoxylin and
eosin (H&E). For the immunohistochemical examination, the
sections were deparaffinized, rehydrated, and then treated with 10
mmol/l citrate buffer (pH 6.0) at 95°C for antigen retrieval, and
subsequently washed in PBS. The endogenous peroxidase activity was
quenched with 0.6% H2O2 in methanol, and the
sections were blocked with normal goat serum. They were then
incubated sequentially with primary antibodies, rabbit anti-human
Ars2 (dilution 1:100; cat. no. NBP2-15473; Novus Biologicals,
Littleton, CO, USA), mouse anti-Ki-67 (dilution 1:100; clone no.
550609; BD Pharmingen; BD Biosciences, San Jose CA, USA), and
secondary antibodies, biotinylated goat anti-rabbit, goat
anti-mouse IgG, and the ABC reagent (Vector Laboratories, Inc.,
Burlingame, CA, USA). The immunostaining was visualized with
3,3′-diaminobenzidine (Sigma-Aldrich; Merck KGaA). The sections
were then counterstained with hematoxylin before being examined
using a Nikon 80i light microscope (Nikon, Tokyo, Japan). Nuclear
immunostaining was considered as positive for protein accumulation.
The immunohistochemistry staining score was identified as 0 (no
detectable immunostaining), 1 (few nuclei), 2 (up to 10% of
nuclei), 3 (10–50% nuclei), and 4 (>50% nuclei) (20–22).
Cell growth and proliferation
assays
U87MG and LN-229 cells were seeded and cultured in
6-well culture plates at a concentration of 1×105
cells/well. For the cell growth assays, the cells were harvested
and counted daily for 7 days using a hemocytometer, and cell growth
was monitored using trypan blue dye analysis. For the cell
proliferation assays, U87MG and LN-229 cells were plated in 96-well
culture plates. The cell proliferation was determined by MTT
(Sigma-Aldrich; Merck KGaA) analysis. Briefly, 10 µl of MTT was
added into 100 µl of medium in each well, and the cells were
incubated at 37°C for 2 h. Next, 100 µl of DMSO was added to each
well to dissolve MTT, and the plate was shaken for 20 min on the
table concentrator. The absorbance was measured at a wavelength of
560 nm using a microplate reader (Model 550; Bio-Rad
Laboratories).
BrdU staining
U87MG and LN-229 cells were seeded and cultured in
24-well culture plates at a concentration of 2×103
cells/well. The cells were then incubated with 10 µg/ml BrdU
(Sigma-Aldrich; Merck KGaA) for 1 h, washed with phosphate-buffered
saline (PBS), and fixed in 4% paraformaldehyde (PFA) for 20 min.
Subsequently, the cells were pretreated with 1 mol/l HCl and
blocked with 10% goat serum for 1 h, followed by staining with a
monoclonal rat primary antibody against BrdU (dilution 1:200; cat.
no. ab6326; Abcam) at 4°C overnight. They were then incubated with
the Alexa FluorR® 594 goat anti-rat IgG secondary
antibody (H+L; Invitrogen; Thermo Fisher Scientific, Inc.) for 2 h.
DAPI (300 nM) was used for nuclear staining, and the percentage of
BrdU-positive cells was calculated from at least 10 microscopic
fields (Olympus CKX41; Olympus Corp., Tokyo, Japan).
Soft agar clonogenic assay
U87MG cells were mixed in 0.3% Noble agar in DMEM
supplemented with 10% FBS and plated at 1,500 cells/well into
6-well plates containing a solidified bottom layer (0.6% Noble agar
in the same growth medium). After 21 days, colonies were stained
with 5 mg/ml MTT and photographed using an Olympus CKX41 inverted
miscroscope (Olympus Corp.).
Tumor xenograft assay
Six female NOD/SCID mice (4 weeks of age, 18 g of
average weight) were used in the xenograft assay, and they were
maintained under specific pathogen-free (SPF) conditions in the
animal facility of Southwest University (Chongqing, China). In
generally, all mice were housed in the SPF room with temperature
20–26°C, and the air cleanliness was ten thousand grade (≥0.5 µm
particles ≤ ten thousand/cubic foot). The animal illumination and
working illumination were controlled in 15–20 Lx and 150–300 Lx
respectively, and the alternation time of light and shade was 12/12
h. The SPF mice fodder was purchased from Chongqing TengXin
Biotechnology Co., Ltd. (Chongqing, China). The drinking water of
mice was sterilized by high pressure steam (121.3°C, 25 min) before
use. For each mouse, both flanks were injected subcutaneously with
1×106 U87MG cells suspended in 200 µl of serum-free
DMEM. One week after tumor cell injection, tumor growth was
measured using calipers, and tumor volume was calculated using the
formula 4/3пr3, where ‘r’ is the radius of the tumor.
The xenograft tumors were removed and weighed after three weeks of
growth. The tumors were washed with ice-cold PBS and prepared for
the following histological, immunohistochemical and western blot
assays.
Flow cytometry
For cell cycle analysis, 1×106 cells were
harvested and washed twice with ice-cold PBS, fixed with 70%
ethanol, stained with propidium iodide (PI) (BD Biosciences), and
incubated with RNaseA for 30 min at room temperature. For the cell
apoptosis analysis, adherent and floating cells were pooled,
collected by centrifugation at 211 × g for 5 min, and washed once
with ice-cold PBS. Apoptotic cells were determined using the
Annexin V-fluorescein isothiocyanate (FITC) kit (Sigma-Aldrich;
Merck KGaA) according to the manufacturer's instructions. Finally,
the samples were analyzed by flow cytometry using a FACS C6 (BD
Biosciences), and the data were analyzed with BD CellQuest Pro
software (BD Biosciences).
Inhibitor treatment
The MEK inhibitor, trametinib, was purchased from
MCE® MedChemExpress (Monmouth Junction, NJ, USA). The
vector encoding human Ars2 was constructed by PCR-based
amplification and subsequently cloned into the
pCDH-CMV-MCS-EF1-copGFP vector to generate the recombinant plasmid.
U87MG cells were prepared to 70–80% confluence in 6-well plates,
and were transiently transfected with plasmids using
Invitrogen® Lipofectamine® 2000 reagent
(Thermo Fisher Scientific, Inc.). At 48 h after transfection, U87MG
cells were harvested and plated in 96-well culture plates. Then
cells were treated with 2 nM (23,24)
trametinib, and the cell proliferation was determined by MTT
assay.
Patient tumor tissues
All patient tissues used in the experiment were
purchased as a tissue microarray from Alenabio Company (Item no:
BS17015a, BS17016a; http://www.alenabio.com). The brain tumor microarray
chip BS17015a included 38 cases of astrocytoma, 14 cases of
glioblastoma, 6 cases of oligodendrocytoma, 1 case of anaplastic
ependymoma, 1 case of medulloblastoma, and 3 cases of peripheral
brain tissue, one point of each sample. And the glioma tissue
microarray chip BS17016a included 35 cases of glioma with different
pathological stage and 5 cases of normal peripheral tissue, taking
two points for each sample.
Quantification and statistical
analysis
Each value was confirmed using at least three
independent experiments. Quantitative data are expressed as the
mean ± SD. GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA,
USA) was applied for statistical analysis. A two-tailed Student's
t-test was performed for paired samples. One- or two-way analysis
of variance, followed by Dunnett's or Bonferroni's multiple
comparison were performed for comparing multiple groups. P<0.05
was considered to indicate a statistically significant result. All
calculations were performed using the SPSS software package 14.0
(SPSS, Inc., Chicago, IL, USA).
Results
Ars2 is commonly expressed in
glioblastoma
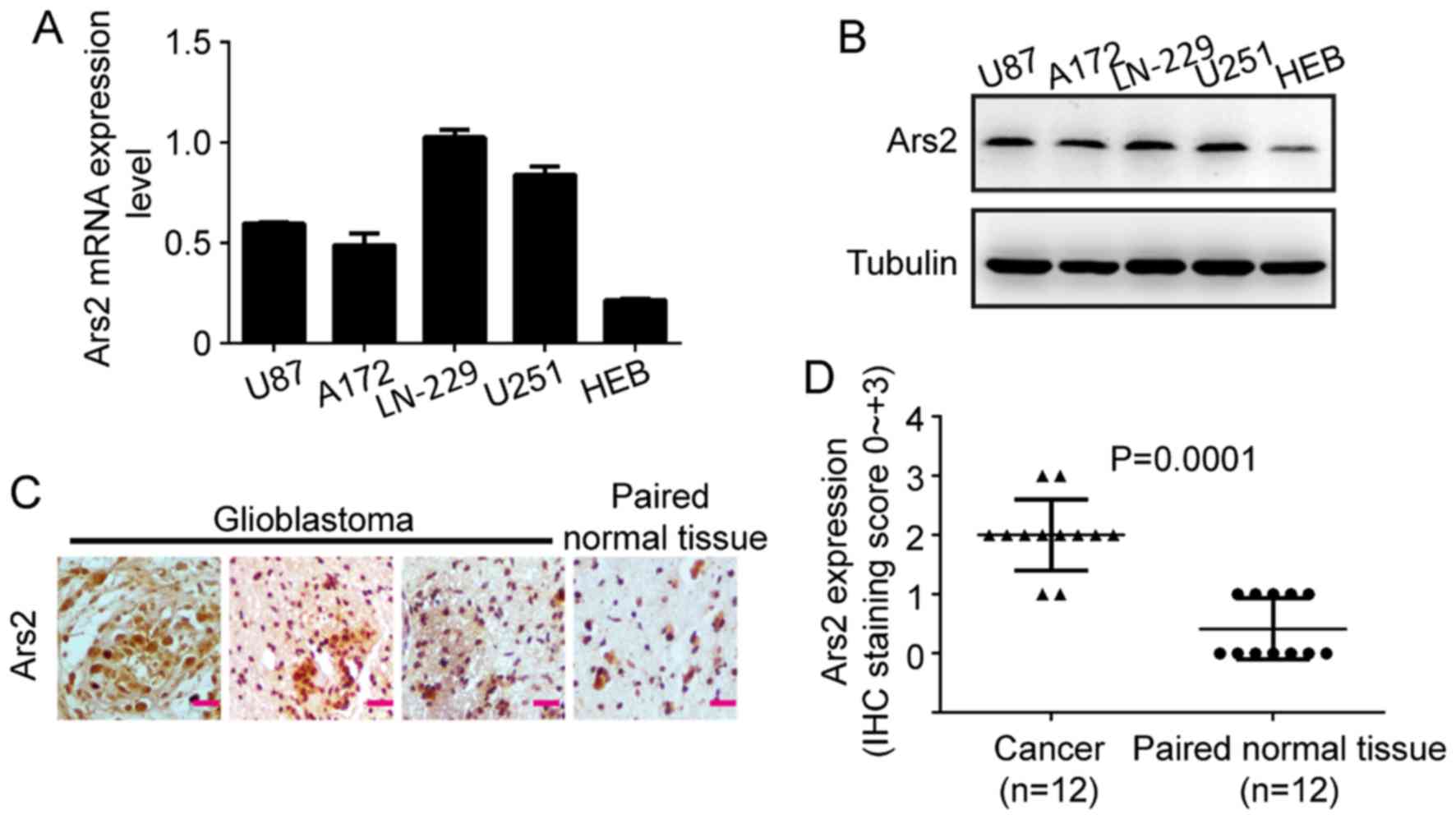
To investigate whether Ars2 is associated with
glioblastoma cell proliferation, we first examined the expression
levels of Ars2 in various human glioblastoma cell lines and tumor
tissues. The results of qRT-PCR and western blot analyses showed
that Ars2 was commonly expressed in four glioblastoma cell lines
(Fig. 1A and B), such as A172,
LN-229, U87MG and U251. In addition, the results revealed that Ars2
was expressed at a higher level in tumor cells than that in HEB, a
normal human brain astrocyte cell line. The results of the
immunohistochemical assay in 12 paired samples showed that Ars2 was
highly expressed in human glioblastoma tissues and was lower in
adjacent normal brain tissues (Fig. 1C
and D). Therefore, we proposed that Ars2 may have an important
role in glioblastoma.
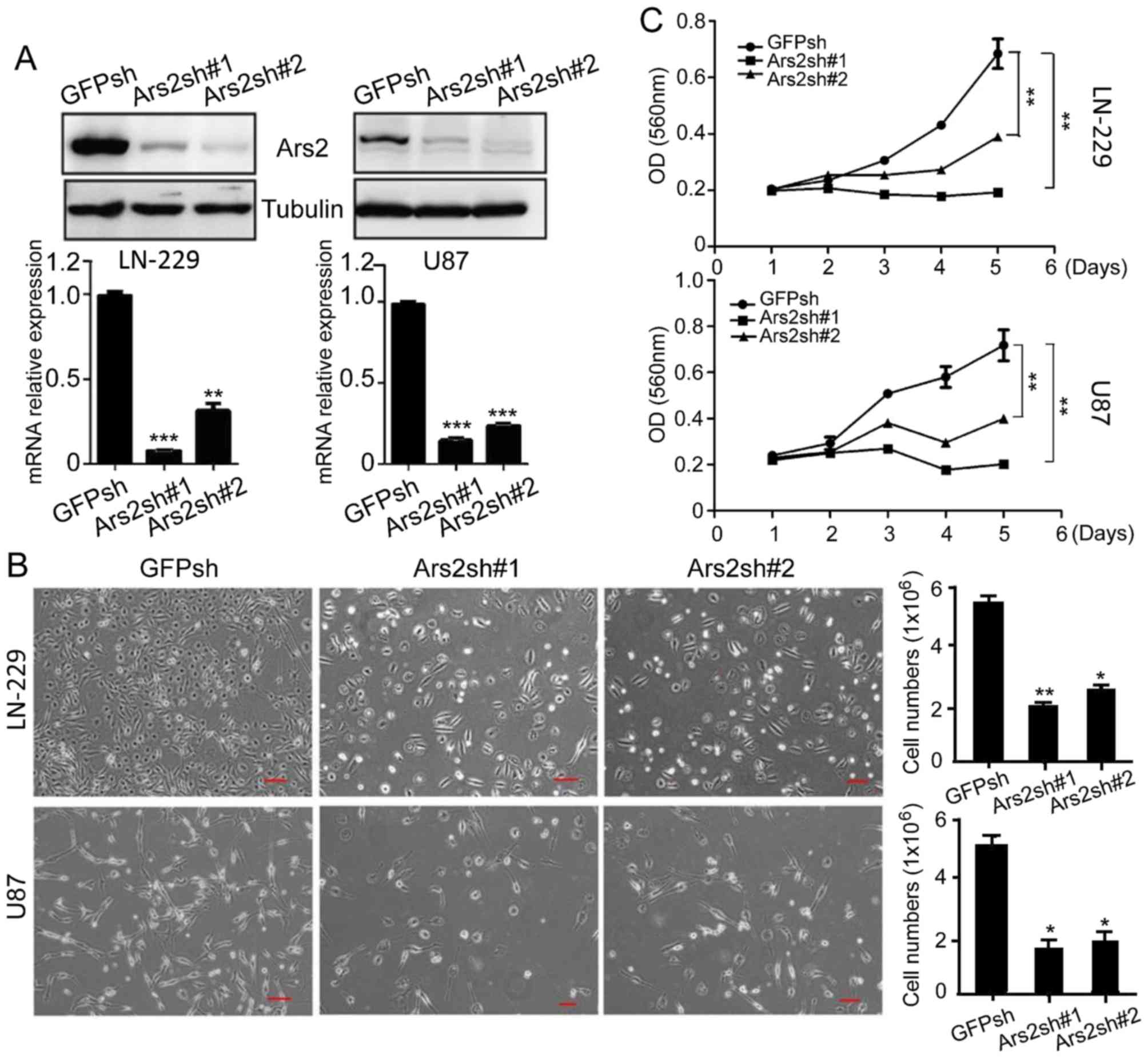
Knockdown of Ars2 inhibits the
proliferation and self-renewal of glioblastoma cells
To confirm the connection between Ars2 and
glioblastoma cell proliferation, we performed downregulation
analysis of Ars2 in glioblastoma cells. We knocked down Ars2 in
LN-229 and U87MG cells, with GFPsh as a control (Fig. 2A). The results of the cell counting
and MTT analyses verified that downregulation of Ars2 significantly
inhibited the proliferation of LN-229 and U87MG cells (Fig. 2B and C). Moreover, we performed BrdU
staining to confirm the cell proliferation status (25), and the results showed that the
percentage of BrdU-positive cells was significantly decreased after
Ars2 knockdown (Fig. 3A and B).
Next, we checked for a functional role of Ars2 in maintaining the
self-renewal ability of U87MG cells. The results showed that
U87MG-Ars2-knockdown cells gave rise to tiny and scant colonies in
soft agar, and these cells formed small spheres, when compared to
the control (Fig. 3C and D). These
data revealed that downregulation of Ars2 significantly inhibited
the proliferation and self-renewal ability of glioblastoma cells,
indicating that the Ars2 gene may be a key factor in glioblastoma
growth.
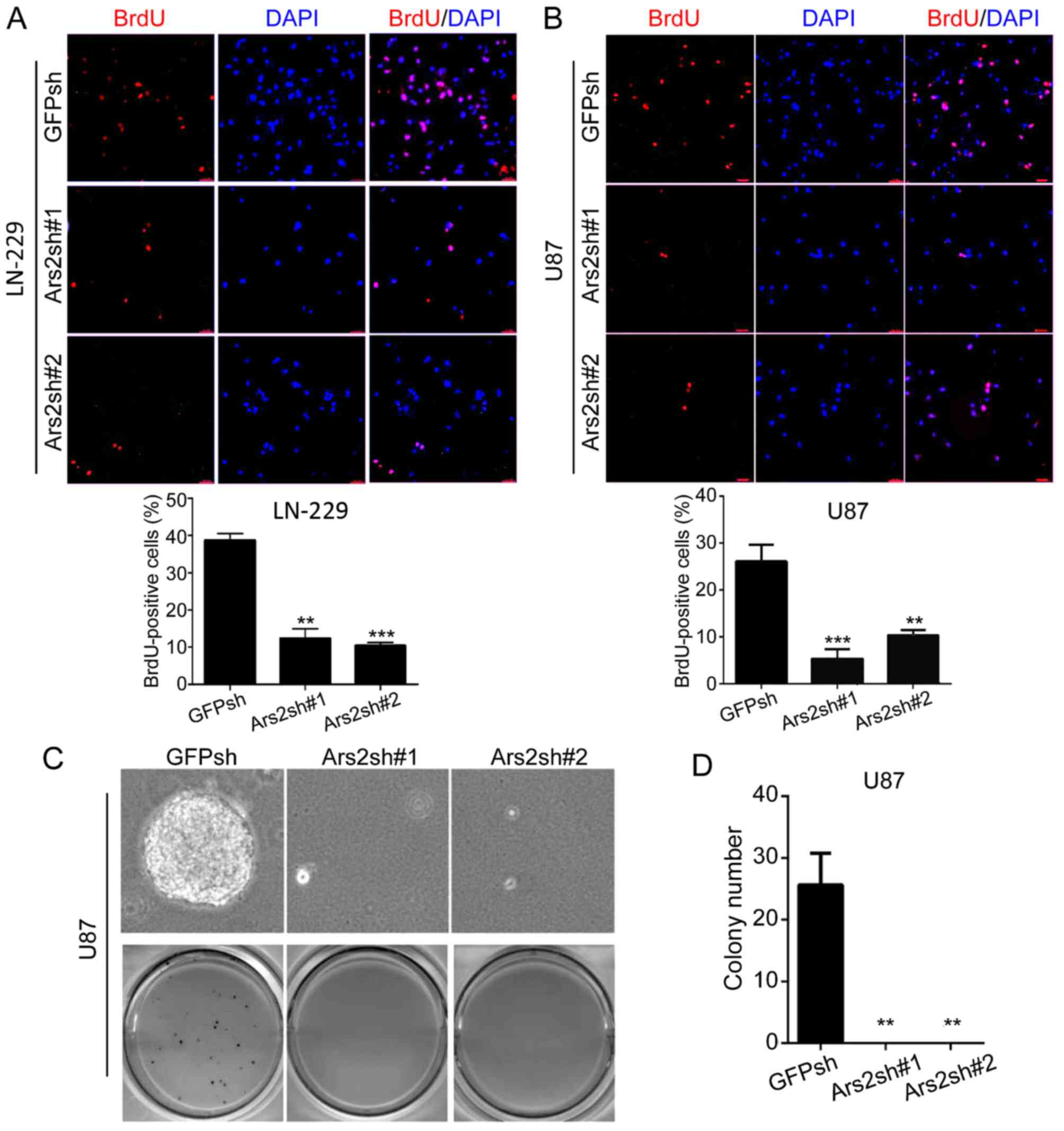 | Figure 3.Downregulation of Ars2 inhibits BrdU
incorporation and the self-renewal ability of glioblastoma cells.
(A and B) Glioblastoma cells with GFP knockdown or Ars2 knockdown
were plated at 2×103 cells per well in 24-well culture
plates. After cells were adherent, a 1-h incubation with BrdU was
performed, and the immunofluorescence was detected by anti-BrdU
antibody. Scale bar, 20 µm. Finally, the ratio of BrdU-positive
cells was calculated. (C) U87MG cells with GFP knockdown or Ars2
knockdown were plated at 1×103 cells per well in 6-well
culture plates. After 14 to 21 days of culture, soft agar colonies
grew from cells with GFP knockdown or Ars2 knockdown. As shown,
cells with Ars2 knockdown were observed to give rise to tiny and
scant colonies in soft agar. Colonies >0.5 mm or those
containing >50 cells were recorded. For all the data in A, B and
D, each value represents the average obtained from three
independent experiments and the error bars show the SD. Statistical
analyses were performed using two-tailed Student's t-tests,
***P<0.001, **P<0.01. Ars2, arsenic resistance protein 2. |
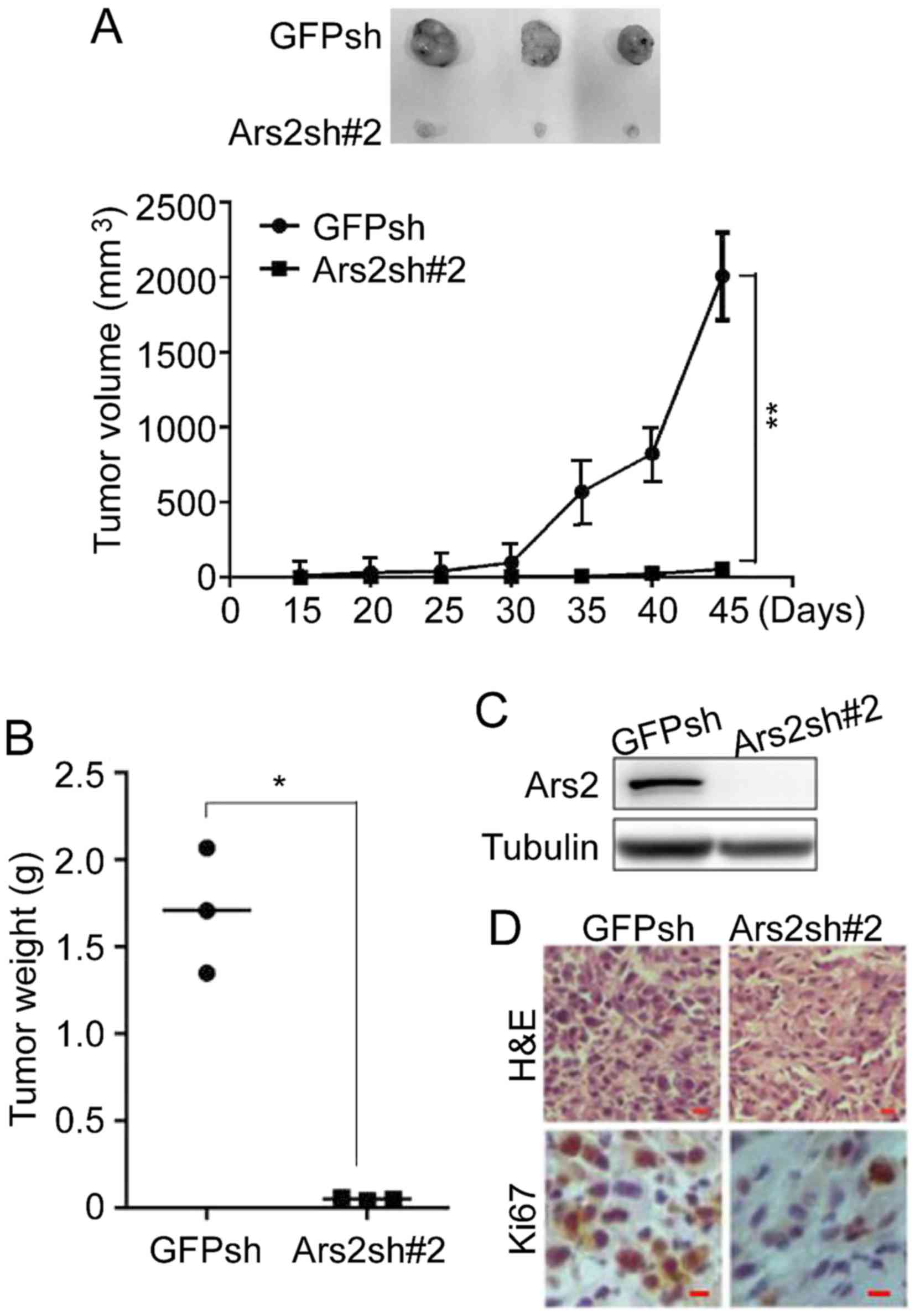
Knockdown of Ars2 inhibits
tumorigenicity of glioblastoma cells
Next, the xenograft tumor growth assay was performed
to test for a role of Ars2 in the regulation of the tumorigenicity
of glioblastoma cells. U87MG-Ars2sh cells were injected into
NOD/SCID mice to form xenograft tumors, and U87MG-GFPsh cells were
used as control. As shown in Fig.
4A, the volume of the xenograft tumors in the Ars2sh group was
much lower than that calculated in the GFPsh group, indicating that
downregulation of Ars2 inhibited the tumorigenicity of
neuroblastoma cells. Furthermore, the tumor weights of the Ars2sh
group were much lower than in the GFPsh group (Fig. 4B). Furthermore, to investigate the
cell proliferation status of the xenograft tumors, the tumors were
removed and then checked by immunoblot and immunohistochemical
analyses. The results shown in Fig. 4C
and D demonstrated that downregulation of Ars2 significantly
decreased the number of Ki67-positive cells, indicating that the
cell proliferation in xenograft tumors was inhibited by Ars2
knockdown. All of the above data indicated that Ars2 knockdown
inhibited the tumorigenicity of glioblastoma cells.
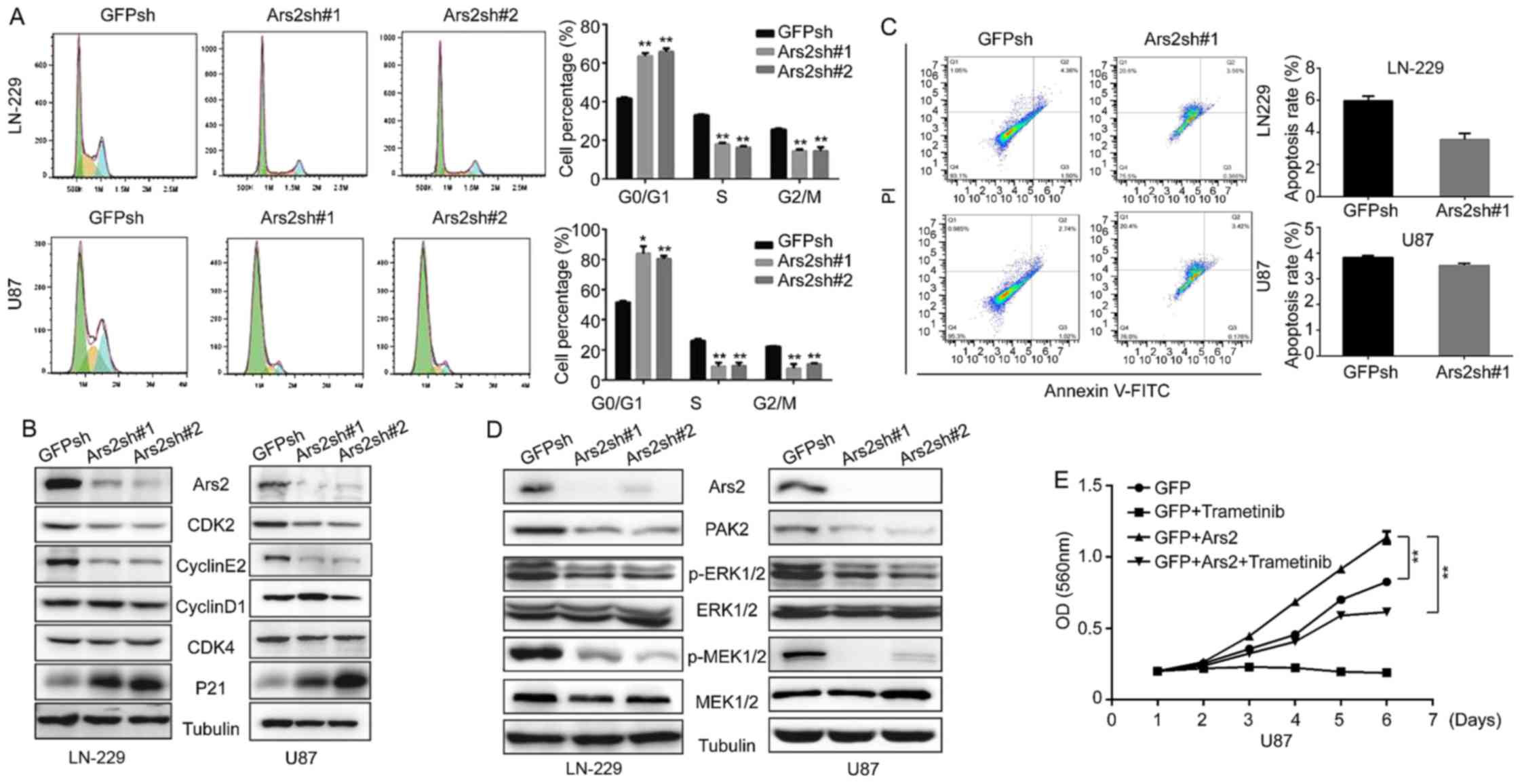
Ars2 is involved in MAPK pathway
activation and is associated with the glioblastoma cell cycle
Since Ars2 knockdown inhibits the proliferation and
tumorigenicity of glioblastoma cells, we used flow cytometric
detection to test how Ars2 influences glioblastoma cell
proliferation. As shown in Fig. 5A,
downregulation of Ars2 in glioblastoma cells induced cell cycle
arrest in the G1 phase; furthermore, the number of S phase cells
was clearly decreased after Ars2 knockdown. We next analyzed the
expression of cell cycle-related proteins. The result of the
western blot analysis verified that downregulation of Ars2
significantly decreased the expression of CDK2 and cyclin E2
(Fig. 5B), which are required for
the transition from G1 to S phase in the cell cycle. There was also
a marked upregulation of P21 expression (Fig. 5B). This protein is known as
cyclin-dependent kinase inhibitor 1 and is primarily associated
with inhibition of CDK2 (26–28).
These results confirmed that knockdown of Ars2 inhibited cell cycle
progression.
In addition, we assessed cell apoptosis after
knockdown of Ars2. As shown in Fig.
5C, the result revealed that there was no significant
difference between the Ars2 knockdown group and the control group.
However, we found that the MAPK/ERK pathway was inhibited after
Ars2 knockdown (Fig. 5D). It is
known that activation of the MAPK/ERK pathway is necessary and
sufficient to promote production of the cyclin E/CDK2 complex,
which allows progression from G1-phase to S-phase in most mammalian
cells (29–31). The results in Fig. 5D show that the phosphorylation and
activation of MEK and ERK were both inhibited by Ars2 knockdown. To
further confirm that the effect of Ars2 knockdown on cell
proliferation was dependent on the MAPK/ERK pathway, we next
overexpressed Ars2 in MAPK inhibitor-treated U87MG cells.
Overexpression of Ars2 restored cell proliferation in the MAPK
inhibitor-treated cells (Fig. 5E),
suggesting that the reduced cell proliferation induced by knockdown
of Ars2 is due to the inhibition of the MAPK/ERK pathway. Our
findings provide new insight into the mechanism of how Ars2
regulates cell cycle entry and cell proliferation by showing that
Ars2 knockdown leads to inhibition of the MAPK/ERK pathway,
ultimately resulting in the suppression of cell cycle progression
and cell proliferation.
Discussion
Ars2 is a highly conservative gene located on the
long arm of human chromosome 7 (32). It is reported that Ars2 plays an
important role in the biosynthesis of microRNAs, and loss of Ars2
leads to decreased levels of a variety of microRNAs, including
miR-21, miR-155 and let-7 (12,13).
Some researchers have observed that the expression of Ars2 in
cholangiocarcinoma tissues is significantly higher than that in
paracancerous tissues (18), and
that the expression of Ars2 is positively correlated to the
expression of miR-21, but negatively correlated with the expression
of PTEN and PDCD4 (17). After
interfering with Ars2 function, the proliferation and invasion of
cholangiocarcinoma cells were weakened, and the formation of mature
miR-21 molecules was inhibited (17). However, the role of Ars2 in
glioblastoma and its molecular mechanism have not yet been
reported. Our present results provide evidence that Ars2 regulates
glioblastoma proliferation and the cell cycle.
The basic process of cellular life is the cell
cycle, and infinite progression through the cell cycle allows cells
to proliferate. In the course of continuous evolution, cells
establish a series of regulatory mechanisms to ensure that the cell
cycle proceeds in an orderly manner to allow cells to maintain
normal tissue structure and function (33). When the proliferation of cells is
not controlled by the body and there is infinite proliferation,
cancer is formed (34). The cell
cycle is regulated by the activities of cyclin-CDK complexes
(35), and abnormalities of cyclin
and CDKs, or the absence of CDK inhibitors, can cause cell cycle
disorders, leading to out-of-control cell proliferation (34). In response to extracellular signals,
such as growth factors, cyclin D is the first cyclin produced in
the cell cycle and it binds to existing CDK4 to form the active
cyclin D-CDK4 complex (36,37). The cyclin D-CDK4 complex in turn
phosphorylates the retinoblastoma susceptibility protein (Rb), and
the hyperphosphorylated Rb separates from the E2F/DP1/Rb complex to
activate E2F (38). Activation of
E2F results in transcriptional activation of certain key factors
such as cyclin E, cyclin A, DNA polymerase, among others (36). Cyclin E binds to CDK2, forming the
cyclin E-CDK2 complex, which pushes the cell from the G1 to S phase
(39). In the present study, our
findings showed that Ars2 knockdown dramatically suppressed
expression of cyclin E and CDK2, and upregulated P21 expression.
This may be the major reason that caused the cell cycle arrest in
the G1 phase, which then induced the inhibition of cell
proliferation and tumorigenicity of glioblastoma cells.
In the last few decades, a number of studies have
illustrated that the most basic feature of cancer cells is that
they can maintain unlimited proliferation. It was found that 40% of
human melanomas contain activating mutations that can affect the
structure of the B-Raf protein, consequently affecting the basic
signaling function of the Raf mitogen-activated protein kinase
(MAPK) pathway, thus affecting cell proliferation (40). It is known that the MAPK/ERK pathway
plays an important role in promoting cell growth and proliferation
in many mammalian cells (31). In
general, the presence of extracellular growth signals trigger the
activation of canonical receptor tyrosine kinases and subsequent
activation of the small GTPase Ras (31), which then leads to a series of
phosphorylation events downstream in the MAPK cascade
(Raf-MEK-ERK), ultimately resulting in the phosphorylation and
activation of MAPK/ERK. The activation of MAPK/ERK kinase activity
causes the phosphorylation of its many downstream targets,
including the cyclin D-CDK4 complex (4), which contributes to the
hyperphosphorylation of Rb and the subsequent activation of E2F
(40), promoting the expression of
its targets cyclin E and CDK2, finally allowing cells to progress
from G1 to S-phase (4,31,40).
Thus, we investigated the MAPK/ERK pathway in our study. We found
that the MAPK/ERK pathway is crucial for the regulation of
glioblastoma cell proliferation by Ars2. The activation and
phosphorylation of MEK and ERK were both inhibited by Ars2
knockdown. Moreover, overexpression of Ars2 restored cell
proliferation of glioblastoma cells treated by the MAPK inhibitor.
Our results provide evidence of the regulation of cell
proliferation by Ars2 through the MAPK/ERK pathway.
In summary, in our research, we verified an
important role of Ars2 in glioblastoma cell proliferation and
tumorigenicity. Our results confirmed that Ars2 knockdown led to
the suppression of proliferation, self-renewal and tumorigenicity
of glioblastoma cells. Additionally, Ars2 knockdown induced the
downregulation of MEK1/2 and ERK1/2 phosphorylation, leading to
inhibition of the MAPK/ERK pathway. This effect then led to
decreased cyclin E2 and CDK2 expression, finally causing G1 arrest
in the glioblastoma cells that ultimately repressed cell
proliferation. In accordance with these results, we confirmed that
Ars2 serves as an important mediator in regulating glioblastoma
cell proliferation and maintaining stemness, and we suggest that
Ars2 may be a potential therapeutic target for glioblastoma
treatment. Moreover, Ars2 was reported to play important roles in
microRNA biosynthesis (8,9,12), and
consequently many microRNAs were found to affect cancer development
in recent years (41–44). It should be important to ascertain
the association among Ars2, microRNAs and cancer. Therefore, we
will focus on the regulatory mechanism of Ars2 linked with
microRNAs in cancer, and explore the potential correlation of Ars2,
microRNAs, the MAPK pathway and cancer cell proliferation in future
research.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81502574, 31501100
and 81672502), the Chongqing Research Program of Basic Research and
Frontier Technology (grant no. cstc2017jcyjAX0304), the Fundamental
Research Funds for the Central Universities (grant no. SWU118033),
the Chongqing University Innovation Team Building Special Fund
(grant no. CXTDX201601010), and the Natural Science Research
Projects of Chongqing Three Gorges Medical College (grant no.
2016×mpxz04).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
XXK and YP carried out the most of the experiments.
XXK wrote the paper, and YP helped to edit the manuscript. KC, DZ
and FW participated in the cell experiments and animal experiments.
SZ and JM helped to do statistical analysis and the flow cytometry
analysis. XH and GZ participated in immunoblot assays and
immunofluorescence staining. HC conceived, designed the study and
helped to revise the manuscript. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
This study was conducted in accordance with approved
guidelines. The animal study protocol was preapproved by the
Institutional Animal Care and Use Committee of Southwest
University. All efforts were made to minimize animal suffering. The
patient tissue microarray used in the experiment was obtained from
Alenabio Company. All the patients provided written informed
consent to participate. Tissue analysis was also approved by the
Ethics Committee of the Southwest University.
Patient consent for publication
Not applicable.
Competing interests
The authors disclose no potential conflicts of
interest.
References
|
1
|
Ostrom QT, Gittleman H, Farah P, Ondracek
A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 15
Suppl 2:ii1–56. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rossman TG and Wang Z: Expression cloning
for arsenite-resistance resulted in isolation of tumor-suppressor
fau cDNA: Possible involvement of the ubiquitin system in arsenic
carcinogenesis. Carcinogenesis. 20:311–316. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gruber JJ, Olejniczak SH, Yong J, La Rocca
G, Dreyfuss G and Thompson CB: Ars2 promotes proper
replication-dependent histone mRNA 3′ end formation. Mol Cell.
45:87–98. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wilson MD, Riemer C, Martindale DW,
Schnupf P, Boright AP, Cheung TL, Hardy DM, Schwartz S, Scherer SW,
Tsui LC, et al: Comparative analysis of the gene-dense ACHE/TFR2
region on human chromosome 7q22 with the orthologous region on
mouse chromosome 5. Nucleic Acids Res. 29:1352–1365. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wilson MD, Wang D, Wagner R, Breyssens H,
Gertsenstein M, Lobe C, Lu X, Nagy A, Burke RD, Koop BF and Howard
PL: ARS2 is a conserved eukaryotic gene essential for early
mammalian development. Mol Cell Biol. 28:1503–1514. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prigge MJ and Wagner DR: The arabidopsis
serrate gene encodes a zinc-finger protein required for normal
shoot development. Plant Cell. 13:1263–1279. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Amsterdam A, Nissen RM, Sun Z, Swindell
EC, Farrington S and Hopkins N: Identification of 315 genes
essential for early zebrafish development. Proc Natl Acad Sci USA.
101:12792–12797. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grigg SP, Canales C, Hay A and Tsiantis M:
SERRATE coordinates shoot meristem function and leaf axial
patterning in Arabidopsis. Nature. 437:1022–1026. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang L, Liu Z, Lu F, Dong A and Huang H:
SERRATE is a novel nuclear regulator in primary microRNA processing
in Arabidopsis. Plant J. 47:841–850. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Andreu-Agullo C, Maurin T, Thompson CB and
Lai EC: Ars2 maintains neural stem-cell identity through direct
transcriptional activation of Sox2. Nature. 481:195–198. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Golling G, Amsterdam A, Sun Z, Antonelli
M, Maldonado E, Chen W, Burgess S, Haldi M, Artzt K, Farrington S,
et al: Insertional mutagenesis in zebrafish rapidly identifies
genes essential for early vertebrate development. Nat Genet.
31:135–140. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sabin LR, Zhou R, Gruber JJ, Lukinova N,
Bambina S, Berman A, Lau CK, Thompson CB and Cherry S: Ars2
regulates both miRNA- and siRNA- dependent silencing and suppresses
RNA virus infection in Drosophila. Cell. 138:340–351. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gruber JJ, Zatechka DS, Sabin LR, Yong J,
Lum JJ, Kong M, Zong WX, Zhang Z, Lau CK, Rawlings J, et al: Ars2
links the nuclear cap-binding complex to RNA interference and cell
proliferation. Cell. 138:328–339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Denli AM, Tops BB, Plasterk RH, Ketting RF
and Hannon GJ: Processing of primary microRNAs by the
Microprocessor complex. Nature. 432:231–235. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Han J, Lee Y, Yeom KH, Kim YK, Jin H and
Kim VN: The Drosha-DGCR8 complex in primary microRNA processing.
Genes Dev. 18:3016–3027. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Landthaler M, Yalcin A and Tuschl T: The
human DiGeorge syndrome critical region gene 8 and Its D.
Melanogaster homolog are required for miRNA biogenesis. Curr Biol.
14:2162–2167. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
He Q, Cai L, Shuai L, Li D, Wang C, Liu Y,
Li X, Li Z and Wang S: Ars2 is overexpressed in human
cholangiocarcinomas and its depletion increases PTEN and PDCD4 by
decreasing microRNA-21. Mol Carcinog. 52:286–296. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He Q, Huang Y, Cai L, Zhang S and Zhang C:
Expression and prognostic value of Ars2 in hepatocellular
carcinoma. Int J Clin Oncol. 19:880–888. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Charafe-Jauffret E, Tarpin C, Bardou VJ,
Bertucci F, Ginestier C, Braud AC, Puig B, Geneix J, Hassoun J,
Birnbaum D, et al: Immunophenotypic analysis of inflammatory breast
cancers: identification of an ‘inflammatory signature’. J Pathol.
202:265–273. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shiao YH, Palli D, Caporaso NE, Alvord WG,
Amorosi A, Nesi G, Saieva C, Masala G, Fraumeni JF Jr and Rice JM:
Genetic and immunohistochemical analyses of p53 independently
predict regional metastasis of gastric cancers. Cancer Epidemiol
Biomarkers Prev. 9:631–633. 2000.PubMed/NCBI
|
|
22
|
McDonald JW and Pilgram TK: Nuclear
expression of p53, p21 and cyclin D1 is increased in
bronchioloalveolar carcinoma. Histopathology. 34:439–446. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamaguchi T, Kakefuda R, Tanimoto A,
Watanabe Y and Tajima N: Suppressive effect of an orally active
MEK1/2 inhibitor in two different animal models for rheumatoid
arthritis: a comparison with leflunomide. Inflamm Res. 61:445–454.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yamaguchi T, Kakefuda R, Tajima N, Sowa Y
and Sakai T: Antitumor activities of JTP-74057 (GSK1120212), a
novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro
and in vivo. Int J Oncol. 39:23–31. 2011.PubMed/NCBI
|
|
25
|
Cappella P, Gasparri F, Pulici M and Moll
J: Cell proliferation method: Click chemistry based on BrdU
coupling for multiplex antibody staining. Curr Protoc Cytom. 72:7
34 31–17. 2015.
|
|
26
|
Gartel AL and Radhakrishnan SK: Lost in
transcription: p21 repression, mechanisms, and consequences. Cancer
Res. 65:3980–3985. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Deng C, Zhang P, Harper JW, Elledge SJ and
Leder P: Mice lacking p21CIP1/WAF1 undergo normal development, but
are defective in G1 checkpoint control. Cell. 82:675–684. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD,
Lees E, Harper JW, Elledge SJ and Reed SI: p53-dependent inhibition
of cyclin-dependent kinase activities in human fibroblasts during
radiation-induced G1 arrest. Cell. 76:1013–1023. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Meloche S and Pouyssegur J: The ERK1/2
mitogen-activated protein kinase pathway as a master regulator of
the G1- to S-phase transition. Oncogene. 26:3227–3239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chambard JC, Lefloch R, Pouyssegur J and
Lenormand P: ERK implication in cell cycle regulation. Biochim
Biophys Acta. 1773:1299–1310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yao G, Lee TJ, Mori S, Nevins JR and You
L: A bistable Rb-E2F switch underlies the restriction point. Nat
Cell Biol. 10:476–482. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Waxman S and Anderson KC: History of the
development of arsenic derivatives in cancer therapy. Oncologist. 6
Suppl 2:S3–S10. 2001. View Article : Google Scholar
|
|
33
|
Fu WJ, Li BL, Wang SB, Chen ML, Deng RT,
Ye CQ, Liu L, Fang AJ, Xiong SL, Wen S, et al: Changes of the
tubular markers in type 2 diabetes mellitus with glomerular
hyperfiltration. Diabetes Res Clin Pract. 95:105–109. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fuster D, Torregrosa JV, Setoain X,
Doménech B, Campistol JM, Rubello D and Pons F: Localising imaging
in secondary hyperparathyroidism. Minerva Endocrinol. 33:203–212.
2008.PubMed/NCBI
|
|
35
|
Nigg EA: Cyclin-dependent protein kinases:
Key regulators of the eukaryotic cell cycle. Bioessays. 17:471–480.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lavi O, Ginsberg D and Louzoun Y:
Regulation of modular Cyclin and CDK feedback loops by an E2F
transcription oscillator in the mammalian cell cycle. Math Biosci
Eng. 8:445–461. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Arellano M and Moreno S: Regulation of
CDK/cyclin complexes during the cell cycle. Int J Biochem Cell
Biol. 29:559–573. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hiyama H, Iavarone A and Reeves SA:
Regulation of the cdk inhibitor p21 gene during cell cycle
progression is under the control of the transcription factor E2F.
Oncogene. 16:1513–1523. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Orlando DA, Lin CY, Bernard A, Wang JY,
Socolar JE, Iversen ES, Hartemink AJ and Haase SB: Global control
of cell-cycle transcription by coupled CDK and network oscillators.
Nature. 453:944–947. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Davies MA and Samuels Y: Analysis of the
genome to personalize therapy for melanoma. Oncogene. 29:5545–5555.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Musilova K and Mraz M: MicroRNAs in B-cell
lymphomas: How a complex biology gets more complex. Leukemia.
29:1004–1017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vosa U, Vooder T, Kolde R, Fischer K, Välk
K, Tõnisson N, Roosipuu R, Vilo J, Metspalu A and Annilo T:
Identification of miR-374a as a prognostic marker for survival in
patients with early-stage nonsmall cell lung cancer. Genes
Chromosomes Cancer. 50:812–822. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu H and Mo YY: Targeting miR-205 in
breast cancer. Expert Opin Ther Targets. 13:1439–1448. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View Article : Google Scholar : PubMed/NCBI
|