Introduction
Ovarian cancer is the one of the most lethal
gynecological cancer types. Approximately 85–90% of ovarian
malignancies manifest as epithelial ovarian cancers (1). Ovarian cancer is a heterogeneous
disease, which has been divided into two major subtypes based on
histopathological, molecular and genetic criteria (2). Type I tumors comprise low-grade
serous, clear cell, endometrioid and mucinous carcinomas, whereas
Type II tumors comprise high-grade serous carcinomas,
undifferentiated carcinomas and carcinosarcomas. Ovarian cancer
remains a highly lethal malignancy, primarily as a result of
inability to detect the cancer at an early, and thus more
treatable, stage (3). Studies have
demonstrated that ovarian cancer development is associated with the
accumulation of certain gene mutations, as well as alterations in
epigenetic modifications, which are associated with downstream
activation of signaling pathways (4,5).
Low-grade serous, clear cell, endometrioid and mucinous carcinomas
are often associated with mutations in KRAS, BRAF, AT-rich
interaction domain 1A (ARID1A), phosphatidylinositol 3-kinase
catalytic subunit α (PIK3CA), CTNNB1 and ERBB2 but only very rarely
in the tumor-suppressor gene tumor protein 53 (TP53) (6–9).
Patients with high-grade serous ovarian cancer present with highly
unstable genomes and harbor TP53 mutations in ~95% of cases
(10). Although numerous studies
have attempted to elucidate the general mechanisms of
carcinogenesis in ovarian cancer, the molecular mechanisms, both
genetic and epigenetic, that underlie ovarian tumorigenesis are not
clearly understood.
MicroRNAs (miRNAs or miRs) are non-coding RNAs,
19–22 nucleotides in length, which are usually involved in the
post-transcriptional downregulation of gene expression via base
pairing with the 3′-untranslated region (3′-UTR) of target mRNAs.
MiRNAs have been demonstrated to regulate both the translatability
and turnover of numerous mRNAs involved in various biological
processes, and hence miRNAs often serve as oncogenes or tumor
suppressors (11). Mounting
evidence indicates that miRNAs help define a particular cancer
phenotype via modulating the expression of genes and signaling
pathways that are often involved in tumorigenesis (12). The signaling pathways involved in
tumorigenesis that are frequently activated in human cancer thus
represent attractive targets for therapies based on small
molecules. Therefore, defining the networks of genetic and
epigenetic interactions that govern tumorigenesis in any particular
cancer type could inform molecular strategies for effective
treatments across cancer subtypes.
The aim of the current study was to investigate
whether epigenetic drivers could cause genetic alterations that
typically lead to ovarian cancer. Next-generation sequencing data
from our previous study (13) was
analyzed to identify potential miRNAs that, upon dysregulation,
could significantly impact both growth factor signaling pathways
and the rate of genetic mutations that often lead to ovarian
cancer.
Materials and methods
Bioinformatics analysis
Target prediction tools TargetScan (version 7.1;
http://www.targetscan.org/vert_71/)
and miRanda (http://www.mirbase.org/) were used to
search potential target miRNAs for PIK3CA, ARID1A, ETS1 and
DNA-dependent protein kinase catalytic subunit (PRKDC).
Cell lines and culture
The ovarian cancer cell lines TOV21G and TOV112D
were obtained from the American Type Culture Collection (Manassas,
VA, USA). All cell lines were grown in a 1:1 mixture of complete
Medium 199 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and MCDB 105 (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
supplemented with 15% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin (Gibco; Thermo
Fisher Scientific, Inc.). All cells were cultured in a humidified
incubator at 37°C with 5% CO2. The procedure used to
isolate stromal cells from an ectopic endometriotic implant has
been described previously (14,15).
Ovarian endometrial tissue from 3 patients of reproductive age
(aged 21 to 42 years) who underwent laparoscopic surgery was
obtained from the Department of Obstetrics and Gynecology,
Kaohsiung Medical University Hospital (Kaohsiung, Taiwan). For each
case, a final diagnosis of endometriosis was made according to the
revised American Society of Reproductive Medicine classification
(1997) in female patients that had been surgically resected between
June 2014 and April 2016 (16). All
samples were histologically confirmed by pathologists. All patients
had regular menstrual cycles and had not received any hormone
treatment within the 6 months prior to gynecological surgery.
The tissue samples were enzymatically dissociated
using collagenase II, then stromal cells were separated from
epithelial glands by wire sieves. The procedure used to isolate
stromal cells from an ectopic endometriotic implant has been
described previously (14,15). Filtered stromal cells were then
cultured in Dulbecco's Modified Eagle's Medium Nutrient Mixture F12
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS.
All cells were cultured in a humidified incubator at 37°C with 5%
CO2. The study protocol was approved by the Ethics
Committee of the Institutional Review Board of the Kaohsiung
Medical University Hospital (approval no. KMUH-IRB-20140031), and
informed written consent was obtained from each patient.
Serum collection
Serum samples were obtained from patients who
underwent surgery with indications of gynecological disease at the
Department of Obstetrics and Gynecology of Kaohsiung Medical
University Hospital. A total of 9 subjects aged 21 to 55 years
participated in the present study, including 3 healthy controls, 3
patients with endometriosis, and 3 patients with ovarian cancer (1
endometrioid and 2 clear cell carcinoma). In each case of
endometriosis-associated ovarian cancer (EAOC), a final diagnosis
of endometriosis-associated clear cell cancer, endometrioid cancer
or ovarian cancer was made according to FIGO and WHO criteria in
patients who were surgically resected between June 2014 and April
2016 (17). Each diagnosis of
endometriosis, either by laparoscopy or laparotomy, followed the
classification of the revised American Society of Reproductive
Medicine, 1997 (16). The patients
with endometriosis and EAOC were histologically confirmed by
examination of a laparoscopy. All patients had regular menstrual
cycles and had not received any hormone treatment in the 6 months
prior to gynecological surgery. Healthy controls were diagnosed as
having other benign diseases, including urinary incontinence,
pelvic organ prolapse or ovarian hemorrhagic cyst. Exclusion
criteria included post-menopausal status, endometrial cancer,
hyperplasia, endometrial polyps, infectious diseases, adenomyosis,
inflammatory diseases, malignancy, autoimmune disease or
cardiovascular disease.
The study protocol was approved by the Institutional
Review Board of Kaohsiung Medical University Hospital (approval no.
KMUHIRB-G(I)-20150046), and written informed consent was obtained
from each participant. Whole blood was drawn in EDTA-coated tubes
(BD Biosciences, Franklin Lakes, NJ, USA) and centrifuged at 2,200
× g for 10 min at room temperature. The resultant sera were
aliquoted into Eppendorf tubes and stored at −80°C for use in
reverse transcription (RT) and quantitative polymerase chain
reaction (qPCR) studies.
RNA extraction, cDNA synthesis and
qPCR
RNA was extracted from patient sera using bulk
reagents from the MasterPure Complete DNA and RNA Purification kit
(Epicentre; Illumina, Inc., San Diego, CA, USA) or purified from
cultured cells using TRIzol reagent (Thermo Fisher Scientific,
Inc.). RT was performed using the Deoxy + HiSpec RT kit (cat.
FYT501-100R; Yeastern Biotech Co., Ltd., Taipei, Taiwan) and the
TaqMan MicroRNA Reverse Transcription kit (cat. 4366596; Applied
Biosystems; Thermo Fisher Scientific, Inc.). Then, qPCR was
performed using the ABI 7900 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). mRNA was amplified
using the Eztime Fast Real-Time PCR Premix (2X, SYBR-Green, ROX;
Yeastern Biotech Co., Ltd.), and 18S mRNA served as the internal
control. The levels of miRNAs were measured using specific primers
to miR-381 (000571), miR-203 (000507) and RNU6 (001093; Applied
Biosystems; Thermo Fisher Scientific, Inc.). RNU6 served as the
internal control. The reaction mixture included 2 µl cDNA, 5 µl
TaqMan Universal PCR Master Mix II (2X; cat. no. 4440042; Applied
Biosystems; Thermo Fisher Scientific, Inc.), 1 µl specific primers
and 2.5 µl RNase-free water for a final reaction volume of 10 µl.
The thermocycling conditions were initiated by uracil-N glycosylase
activation at 50°C for 2 min and initial denaturation at 95°C for
10 min, followed by 40 cycles at 95°C for 15 sec, and annealing at
60°C for 60 sec. Expression levels were normalized to that of an
endogenous control. Relative miRNA and mRNA levels were determined
using the 2−∆∆Cq method (18). The qPCR primers for HOXD10, 18S,
PIK3CA, PRKDC, ETS1 and ARID1A were synthesized by Genemessenger
Scientific Co., Ltd. (Kaohsiung, Taiwan), and their sequences are
listed in Table I.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene name | Sequence |
|---|
| 18S | F:
5′-CATGGCCGTTCTTAGTTGGT-3′ |
|
| R:
5′-CGCTGAGCCAGTCAGTGTAG-3′ |
| PIK3CA | F:
5′-TTCTTACATTTTCGTAAGTGTTACTCA-3′ |
|
| R:
5′-CGAAGGTATTGGTTTAGACAGAAA-3′ |
| PRKDC | F:
5′-TGCTTTTCCATGAGCTATTACA-3′ |
|
| R:
5′-TGGCAACTTAACGTGTTTGA-3′ |
| ETS1 | F:
5′-GAACGAATTTGGGAACATGC-3′ |
|
| R:
5′-CTTCCCTTCATCCACCTCCT-3′ |
| ARID1A | F:
5′-CCAGCAGAACTCTCACGACC-3′ |
|
| R:
5′-CTGAGCGAAGGACGAAGACG-3′ |
| RNU6B |
CGCAAGGATGACACGCAAATTCGTGAAGCGTTCCATATTTTT |
| miR-381 |
UAUACAAGGGCAAGCUCUCUGU |
| miR-203 |
GUGAAAUGUUUAGGACCACUAG |
| MiR-381 ChIP (−565
to −574) | F:
5′-GTGTGCTCATCAGCGCTTTTT-3′ |
|
| R:
5′-ACAGCTCCCACGGCTCAT-3′ |
| MiR-381 ChIP (−682
to −691) | F:
5′-CGGAGCACTGGCTCTGTCTA-3′ |
|
| R:
5′-GGACTGCGAGCTGGATCAT-3′ |
Transient transfection of cells
TOV21G and TOV112D cells were transiently
transfected with 45 nM of miRNA mimics, inhibitors, and 1 µg of
short hairpin RNAs (shRNAs) or other plasmids was performed using
Turbofect Transfection Reagent (Thermo Fisher Scientific, Inc.).
The miRIDIAN miRNA mimic (miR-381 mimics, cat. no.
C-300690-03-0010) and mimic negative control (cat. no.
CN-001000-01-05) were purchased from GE Healthcare Dharmacon, Inc.
(Lafayette, CO, USA). The mirVana miRNA inhibitor (miR-381
inhibitor, cat. no. MH10242) and inhibitor negative control (cat.
no. 4464078) were obtained from Ambion (Thermo Fisher Scientific,
Inc.). The shRNAs included shRNA-homeobox D10 (HOXD10)#1 (cat. no.
TRCN0000274091), shRNA-HOXD10#2 (cat. no. TRCN0000274093),
shRNA-HOXD10#3 (cat. no. TRCN0000274028) and control shRNA (cat.
no. TRCN0000274091; National RNAi Core Facility at the Institute of
Molecular Biology, Academia Sinica, Taipei, Taiwan). pDNA-HOXD10
was purchased from Addgene, Inc. (Cambridge, MA, USA). Following 24
h of transfection, the cells were subjected to assays measuring
mRNA and protein expression, cell viability, migration and colony
formation.
Cell viability assay
Cell viability was measured using a Cell Counting
Kit-8 (CCK-8) assay (Honeywell Fluka; Thermo Fisher Scientific,
Inc.). Briefly, ovarian cancer cell lines were seeded into 96-well
plates at a density of 5×103 cells/well in a total
volume of 100 µl medium with 15% (v/v) FBS, transfected with miRNA
mimics, inhibitors or scrambled-sequence controls, and incubated at
37°C in an atmosphere of 5% CO2. The following day, 90
µl culture medium and 10 µl CCK-8 reagent were added to each well.
Cell viability was assayed after 2 h using an enzyme-linked
immunosorbent assay reader (Multiskan EX; Thermo Fisher Scientific,
Inc.) at 450 nm (reference, 650 nm). In each case, three
independent experiments were performed.
Colony formation assay
Ovarian cancer cells transduced with the miR-381
mimic, miR-381 inhibitor, or scrambled-sequence controls were
plated in triplicate into 6-well plates (500 cells/well), cultured
in a humidified incubator at 37°C with 5% CO2 for 2
weeks, then fixed with 4% paraformaldehyde and stained with 0.1%
crystal violet at room temperature for 1 h. Colonies were counted
by visual inspection. Digital images of the colonies were obtained
using a camera. Colonies were counted using ImageJ software
(version 1.47; National Institutes of Health, Bethesda, MD, USA).
Triplicate wells were measured for each treatment group.
In vitro migration assay
A total of 5×104 cells were seeded into
the upper chamber of each Transwell insert (8 µm pore size) in a
24-well plate in serum-free medium. In the lower chambers, medium
containing 15% FBS was added as a chemoattractant. Following
incubation for 18 h at 37°C, non-migrated cells in the upper
chamber were removed with cotton swabs. The migrated cells on the
lower chamber surface were fixed with 4% paraformaldehyde for 15
min and stained with 0.1% crystal violet at room temperature for
15–20 min. The number of migrated cells was calculated by counting
three different fields of view under a phase-contrast microscope.
In each case, three independent experiments were performed.
Western blotting
Proteins were extracted using
radioimmunoprecipitation lysis buffer (EMD Millipore, Billerica,
MA, USA) containing several protease and phosphatase inhibitors
(Roche Applied Science, Madison, WI, USA). Protein concentration
was determined with the Bio-Rad DC Protein Assay kit from Bio-Rad
Laboratories, Inc. (Hercules, CA, USA). Cell lysates were analyzed
by western blotting. Proteins (30 µg) were resolved on a 10%
BIS-TRIS SDS gel) at 100 V and transferred onto nitrocellulose
membranes at 90 V for 1.5 h. Non-specific binding was blocked by
incubating the membranes in 5% nonfat dry milk in PBS for 1 h.
Then, the membranes were incubated with primary antibodies
overnight at 4°C. Primary antibodies were as follows: Mouse
monoclonal anti-HOXD10 (cat. no. TA800777; 1:500; OriGene
Technologies, Inc., Rockville, MD, USA) or rabbit monoclonal
anti-PIK3CA (cat. no. TA302178; 1:1,000; OriGene Technologies,
Inc.). Expression of β-actin was used as a loading control and was
detected with monoclonal anti-actin (cat. no. A5316; 1:5,000,
Sigma-Aldrich; Merck KGaA). The secondary antibodies used were
horseradish peroxidase-conjugated goat anti-mouse IgG or
anti-rabbit IgG (Santa Cruz Biotechnology, Inc., Dallas, TX, USA).
Secondary antibodies were incubated at room temperature for 1 h.
Enhanced chemiluminescence reagents (EMD Millipore) were used for
immunodetection.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assays were performed using the Chromatin
Immunoprecipitation Assay kit (Upstate Biotechnology, Inc., Lake
Placid, NY, USA). The ALGGEN-PROMO prediction software (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3)
was used to identify potential transcription factor binding sites
within the miR-381 promoter (19).
Chromatin was incubated with anti-HOXD10 (cat. no. TA800777; 1:500;
OriGene Technologies, Inc.) or goat anti-mouse IgG (control, cat.
no. TA130004; 1:500: OriGene Technologies, Inc.). PCR was conducted
with primers complementary to the miR-381 promoter region. The
primer sequences are presented in Table
I. RT semi-quantitative PCR was performed to amplify the
transcription factor binding sequence of miR-381. RT-PCR analysis
of relative expression was performed with the one-step Emerald Amp
GT PCR Master mix (Takara Bio, Inc., Otsu, Japan). Thermocycling
was performed under the following conditions: 94°C for 5 min; 35
cycles of 94°C for 30 sec, 58°C for 30 sec and 72°C for 30 sec. PCR
products were analyzed by electrophoresis with 2% agarose gels and
visualized by UV light following staining with EtB‘Out’ Nucleic
Acid Staining Solution (cat. no. FYD007-200P; Yeastern Biotech Co.,
Ltd).
Statistical analysis
Data are presented as the mean ± standard deviation
from at least three independent experiments by GraphPad Prism 7
(GraphPad Software, Inc., La Jolla, CA, USA). For the in
vitro cell migration assays, groups were compared using the
Mann-Whitney U test. For all other results, one-way analysis of
variance followed by Tukey's honest significant difference test was
used to analyze differences among multiple groups. Student's t-test
was used to analyze differences between two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Analysis of differentially expressed
miRNAs and prediction of target genes
In our previous work, the genetic alterations that
are commonly observed in endometriosis-associated ovarian cancers
were characterized; the genes PIK3CA, ARID1A, ETS1 and PRKDC were
identified to be the most frequently mutated genes in patient
samples (13). Further examination
revealed potential interactions between miRNAs and the mutated
mRNAs in ovarian tumors. To characterize the mechanisms by which
genetic alterations promote ovarian cancer, TargetScan (version
7.1) and miRanda were used to predict potential upstream regulators
of the mutated gene (20). The two
most commonly mutated genes encoded mRNAs are regulated by two
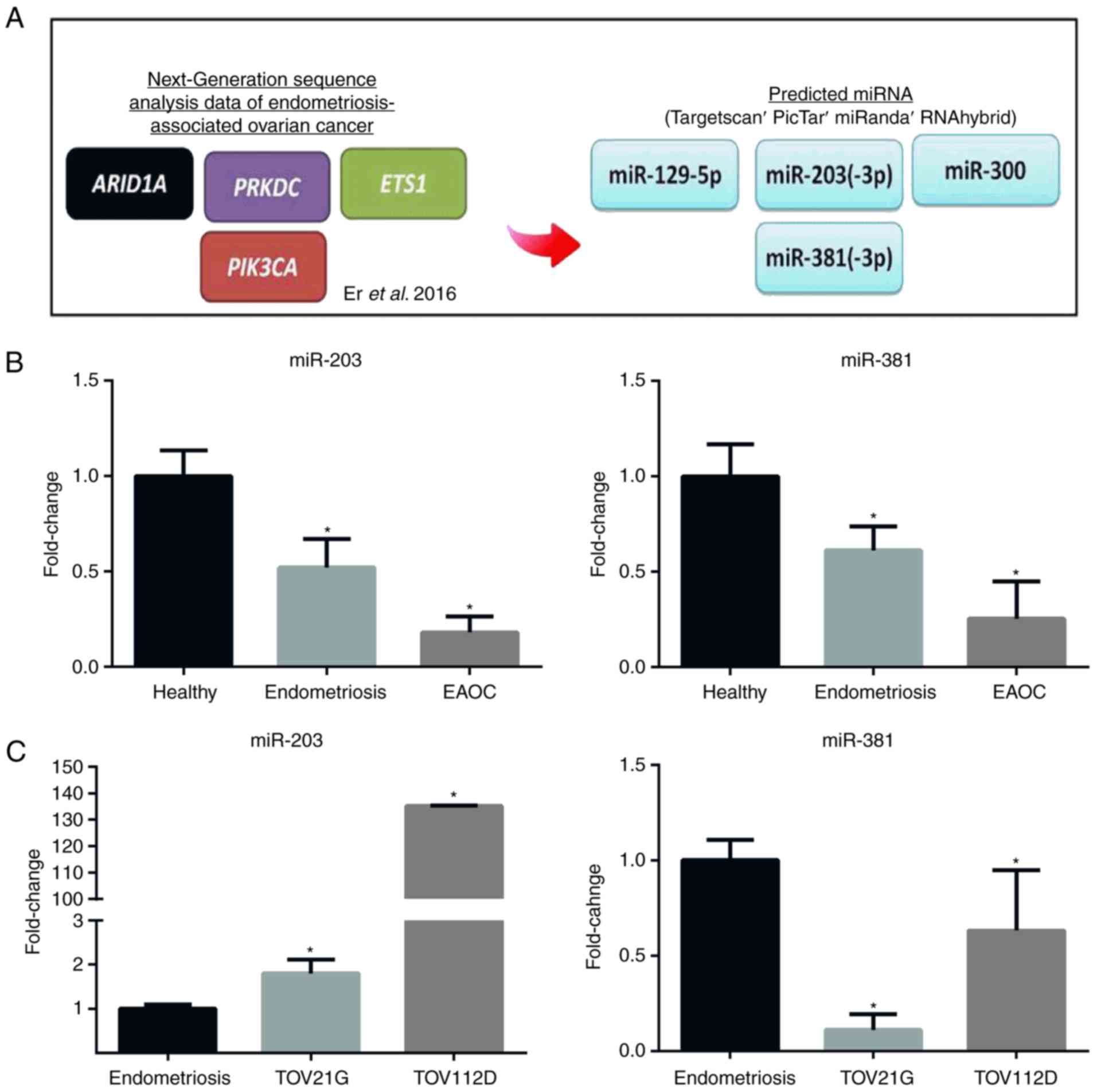
miRNAs, namely miR-203 and miR-381 (Fig. 1A). Therefore, in the present study,
the expression level of miR-203 and miR-381 was determined in tumor
samples from patients with ovarian cancer and in ovarian cancer
cell lines. The results revealed that miR-381 and miR-203 were
expressed at a significantly lower level in the sera of patients
with ovarian cancer compared with healthy patients and
endometriosis patients, and in ovarian cancer cell lines compared
with controls (Fig. 1B and C).
Subsequent studies focused on miR-381 because it was the most
consistently downregulated miRNA in patient sera (Fig. 1B) and ovarian cancer cell lines
(Fig. 1C).
Identification of potential miRNA-mRNA
interactions specific to ovarian cancer
To address whether miR-381 affects the cellular
levels of mRNAs encoding PIK3CA, ARID1A, EST1 and PRKDC, ovarian
cancer cell lines were transfected with an miRNA mimic or miRNA
inhibitor specific for these individual mRNAs and then mRNA
expression was assessed. The PIK3CA mRNA level was significantly
reduced in ovarian cancer cell lines transfected with miR-381
mimics compared with a non-specific control mimic; by contrast,
PIK3CA mRNA level was significantly increased following
transfection with an miR-381 inhibitor compared with a control
non-specific inhibitor (Fig. 2A and
B). However, the cellular levels of ARID1A, ETS1 and PRKDC
mRNAs did not respond consistently to transfection with various
miRNA mimics and inhibitors (Fig. 2A
and B). Next, the effect of various concentrations of miR-381
mimics or inhibitors on the cellular level of PIK3CA mRNA was
examined in the various transiently transfected ovarian cancer cell
lines (Fig. 2C). The results
indicated that 45 nM of miR-381 mimic or inhibitor affected the
cellular level of PIK3CA mRNA and protein (Fig. 2C and D), suggesting that miR-381
effectively suppresses the cellular level of both PIK3CA mRNA and
protein.
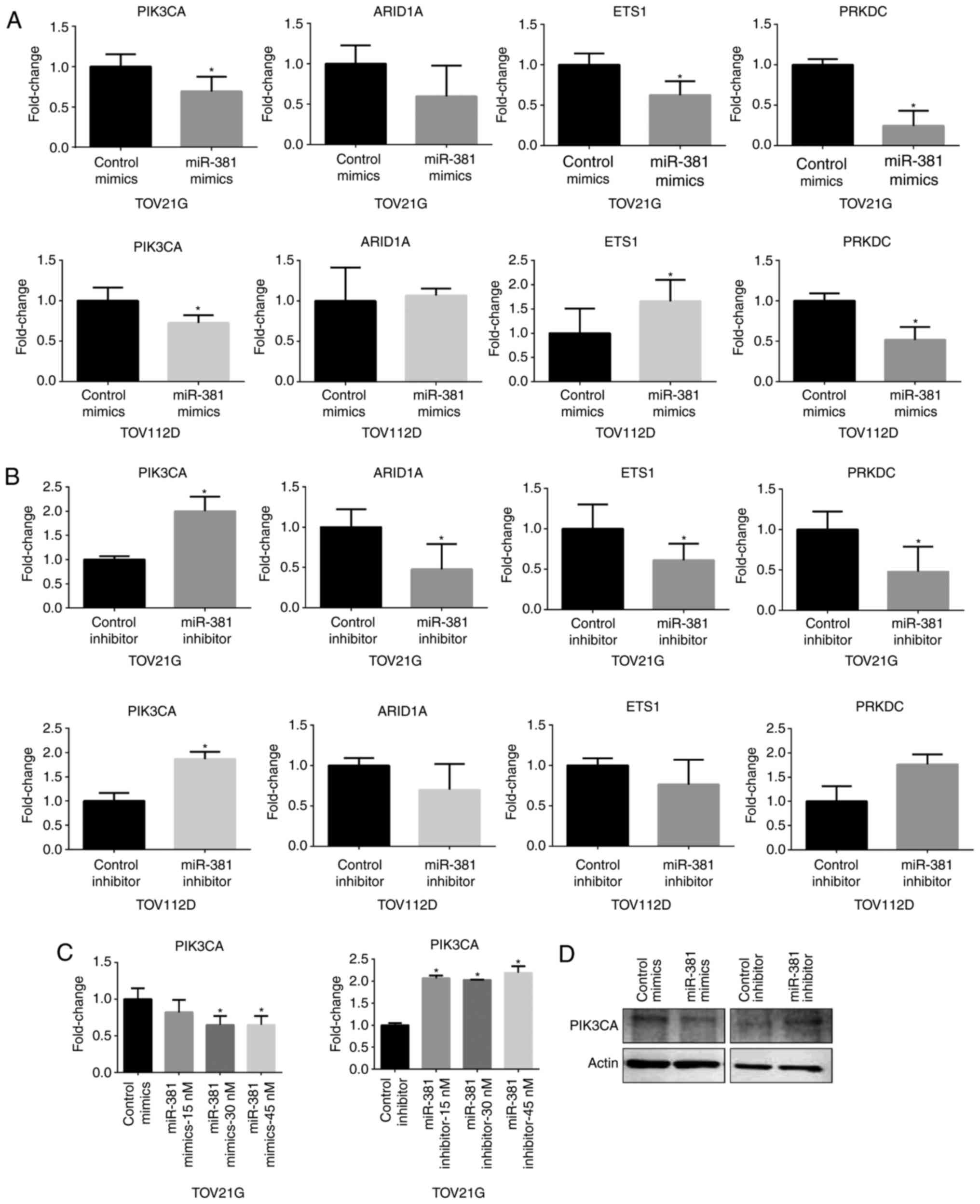 | Figure 2.MiR-381 affects the expression of
PIK3CA. (A) qPCR analysis of PIK3CA, ARID1A, ETS1 and PRKDC
following transfection of cells with miR-381 mimics. (B) qPCR
analysis of PIK3CA, ARID1A, ETS1 and PRKDC following transfection
of cells with miR-381 inhibitors. (C) qPCR analysis of PIK3CA
following transfection of cells with miR-381 mimics or inhibitor at
different doses. (D) Western blot analyses of PIK3CA levels
following treatment of TOV112D ovarian cancer cells with miR-381
mimics or miR-381 inhibitors. Actin was used a loading control.
Data are presented as the mean ± standard deviation from three
independent experiments. qPCR, quantitative polymerase chain
reaction; miR, microRNA; PIK3CA, phosphatidylinositol 3-kinase
catalytic subunit α, PRKDC, DNA-dependent protein kinase catalytic
subunit; ARID1A, AT-rich interaction domain 1A. Data are presented
as the mean ± standard deviation from three independent
experiments. *P<0.05 vs. control group. |
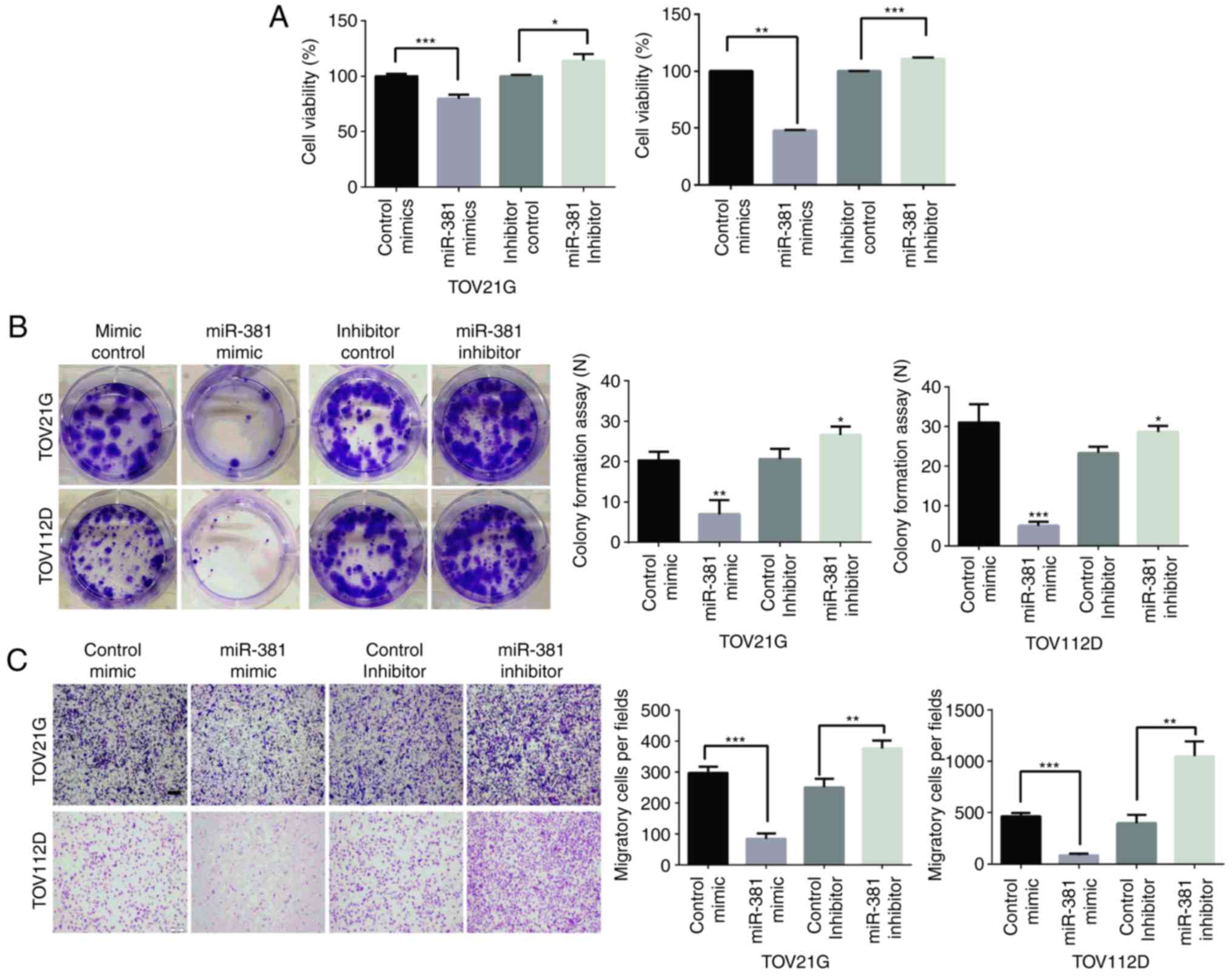
Experimental upregulation of miR-381
suppresses cell proliferation, cell migration and colony
formation
To investigate how miR-381 suppression promotes
ovarian cancer progression, cell proliferation and migration
assessed following targeted knockdown or overexpression of miR-381.
Cell proliferation was evaluated by CCK-8 and colony formation
assays. The downregulation of miR-381 significantly increased cell
proliferation, and inhibition of miR-381 activity significantly
decreased proliferation (Fig. 3A).
Similarly, in a colony formation assay, which is an in vitro
cell survival assay based on the ability of a single cell to grow
into a colony (21), clonogenic
survival decreased following transfection of cells with miR-381
mimics, whereas survival was enhanced in miR-381
inhibitor-transfected cells (Fig.
3B). Furthermore, cell migration was evaluated by Transwell
assays, in which overexpression of miR-381 significantly decreased
migration, whereas an miR-381 inhibitor significantly increased
migration (Fig. 3C). These results
indicated that miR-381 suppresses the growth and migration of
ovarian cancer cells.
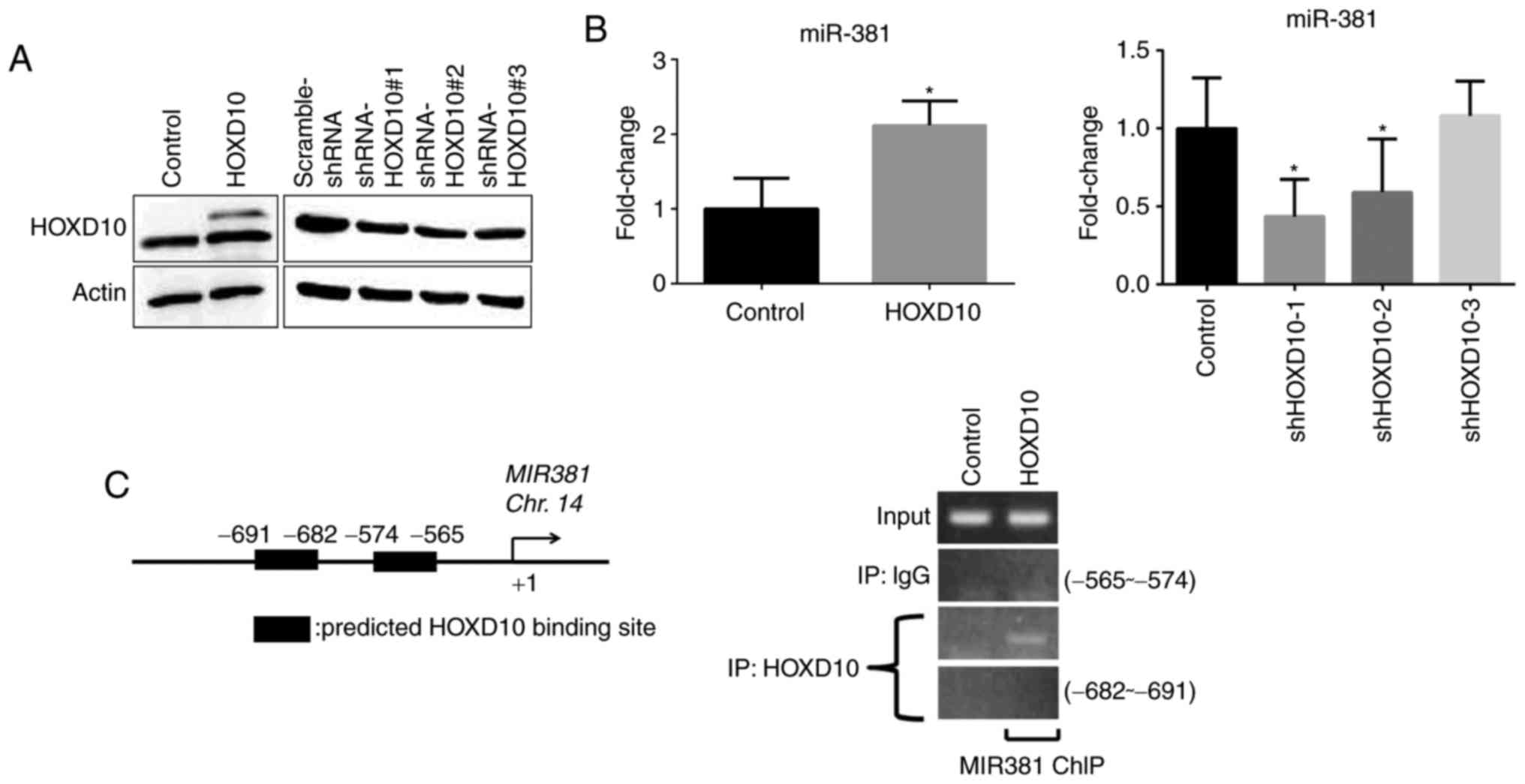
HOXD10 drives the expression of
miR-381
The current results indicated that miR-381 is an
upstream regulator of PIK3CA in ovarian cancer cells. Previous
studies have identified that dysregulation of the
phosphatidylinositol 3-kinase (PI3K) pathway, which is commonly
observed in human tumors, drives tumorigenesis; therefore, the PI3K
pathway is an appealing therapeutic target (22). Hence, the upstream regulators of
miR-381 with respect to the progression of ovarian cancer were
investigated. The ALGGEN-PROMO prediction program (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3)
was used to identify potential transcription factor binding sites
within the miR-381 promoter (19,21,23).
HOXD10 is a tumor-suppressor gene that suppresses tumor invasion
and hence could potentially modulate the cellular level of miR-381
(24). To determine whether miR-381
level is regulated by HOXD10, ovarian cancer cells were transfected
with an overexpression plasmid encoding HOXD10. Subsequently,
HOXD10 was efficiently overexpressed (Fig. 4A), resulting in a significantly
increased cellular level of miR-381 expression (Fig. 4B). Conversely, HOXD10 knockdown
resulted in a decreased level of miR-381 in cells (Fig. 4B). Furthermore, it was investigated
whether HOXD10 modulates the cellular level of miR-381. The
ALGGEN-PROMO program was used to predict the promoter region for
miR-381. ChIP assays revealed that HOXD10 positively regulated
transcription of the miR-381 gene by binding to the region −565 to
−574 bp upstream of the miR-381 coding sequence (Fig. 4C).
Discussion
Emerging evidence has demonstrated that several
miRNAs are mutated in human cancers, resulting in their
dysregulation. These miRNAs may thus act as tumor suppressors or
oncogenes. MiRNAs serve as important regulators of gene expression
in various human cancer types and hold promise for the development
of cancer diagnoses or therapies (25,26).
Several studies have investigated the association between miRNAs
and the various phenotypes observed in ovarian tumors (27–29).
However, the associations between mutations in miRNAs and/or
gene-coding regions and cancer risk have not been fully elucidated.
The present study was an extension of our previous work, in which
next-generation sequencing was used to identify the majority of
endometriosis-associated somatic mutations associated with ovarian
cancer, including mutations in ARID1A, PIK3CA, ETS1 and PRKDC,
among others (13). Based on the
results of that previous study, in the present study it was
identified that miR-381 is an upstream regulator of PIK3CA using an
miR prediction program and the regulation and role of miR-381 in
ovarian cancer cells was explored. MiR-381 was observed to be
downregulated in ovarian cancer samples and clear cell and
endometrioid cancer cells. Exploration of the effect of miR-381 on
ovarian cancer cells confirmed that miR-381 inhibits cell migration
and proliferation.
MiR-381 has been identified to be involved in a
variety of tumorigenic processes, including cell proliferation,
apoptosis, migration and invasion, complicating the regulatory
network in human cancer. For instance, miR-381 has been identified
to be diversely deregulated in several cancer types, including
colorectal and breast cancers, and this deregulation could affect
epithelial-mesenchymal transition as well as proliferation,
invasion, and migration of cancer cells via targeting TWIST and
CXCR4 mRNAs (30,31). In addition, Xia et al
(32) reported the downregulation
of miR-381 in ovarian epithelial cancer tissue. The current
findings are consistent with these previous studies in that miR-381
was identified to act as a tumor suppressor in various cancer
types.
Chemotherapy is the principal treatment for ovarian
cancer; however, the acquisition of chemoresistance is a major
problem with respect to patient long-term survival. Previous
studies have demonstrated that the level of miR-381 correlates with
the development of drug resistance following chemotherapy (33,34).
Consistent with this, the PI3K-AKT pathway and its components
(principally PIK3CA), which target miR-381, are often mutated, and
dysregulation of this pathway or its components increases the
chemoresistance observed in ovarian cancer (35–37).
Therefore, it is proposed that the deregulation of miR-381 may have
consequential effects on the PI3K-AKT pathway and thus contribute
to chemoresistance. Additionally, it was identified in the current
study HOXD10 is a novel transcription factor for miR-381. HOXD10
drives increased transcription of miR-381, perhaps leading to the
downregulation of PIK3CA. Previous work has demonstrated that
HOXD10 is frequently downregulated in other cancer types, including
breast cancer (38), ovarian cancer
(24), bladder cancer (39) and cholangiocellular carcinoma
(40). Ectopic expression of HOXD10
significantly blocks tumor cell migration and invasiveness,
indicating that HOXD10 may serve as a tumor suppressor (38,41). A
previous study revealed that HOXD10 is a direct target of miR-10b,
which promotes cell invasion through the RhoC/AKT signaling pathway
(24).
In addition to the aforementioned results, a role
for HOXD10 has been established in angiogenesis and in modulating
cell motility in numerous cancer types (24,39,42,43).
Similar to the results reported previously, the current results
establish a functional connection between HOXD10, miR-381 and
PIK3CA.
In summary, the current results demonstrate that
miR-381 is significantly downregulated in ovarian cancer cells and
that miR-381 overexpression may inhibit the migration and
proliferation ability of ovarian cancer cells in vitro.
Additionally, expression of the miR-381 gene is directly regulated
by HOXD10, and HOXD10 may mediate the effect of miR-381 on ovarian
cancer cell migration and proliferation. The current results
suggest that miR-381 functions as a tumor suppressor through
targeting PIK3CA in ovarian cancer.
Acknowledgements
Not applicable.
Funding
This study was supported by the Kaohsiung Medical
University Hospital Research Fund (grant no. KMUH106-6R38),
Ministry of Science and Technology, Taiwan (grant nos. MOST
105-2314-B-037-052-MY3 and MOST 106-2320-B-037-019) and National
Sun Yat-Sen University-KMU Joint Research Project (grant no.
NSYSUKMU107-P031).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
CYH and EMT designed the experiments. CYH and EMT
conducted the experiments and wrote the manuscript. THH, TKE, CCT
and HSC provided the research materials and analyzed the data. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research.
Ethics approval and consent to
participate
The study protocol was approved by the Institutional
Review Board of Kaohsiung Medical University Hospital (approval no.
KMUHIRB-G(I)-20150046), and written informed consent was obtained
from each participant.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cho KR and Shih Ie M: Ovarian cancer. Annu
Rev Pathol. 4:287–313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kurman RJ and Shih Ie M: The origin and
pathogenesis of epithelial ovarian cancer: A proposed unifying
theory. Am J Surg Pathol. 34:433–443. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Koshiyama M, Matsumura N and Konishi I:
Recent concepts of ovarian carcinogenesis: Type I and type II.
Biomed Res Int. 2014:9342612014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shen H and Laird PW: Interplay between the
cancer genome and epigenome. Cell. 153:38–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berdasco M and Esteller M: Aberrant
epigenetic landscape in cancer: How cellular identity goes awry.
Dev Cell. 19:698–711. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shih IeM and Kurman RJ: Ovarian
tumorigenesis: A proposed model based on morphological and
molecular genetic analysis. Am J Pathol. 164:1511–1518. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jones S, Wang TL, Shih IeM, Mao TL,
Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B, et
al: Frequent mutations of chromatin remodeling gene ARID1A in
ovarian clear cell carcinoma. Science. 330:228–231. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Campbell IG, Russell SE, Choong DY,
Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB and
Phillips WA: Mutation of the PIK3CA gene in ovarian and breast
cancer. Cancer Res. 64:7678–7681. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y,
Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et
al: ARID1A mutations in endometriosis-associated ovarian
carcinomas. N Engl J Med. 363:1532–1543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ahmed AA, Etemadmoghadam D, Temple J,
Lynch AG, Riad M, Sharma R, Stewart C, Fereday S, Caldas C, Defazio
A, et al: Driver mutations in TP53 are ubiquitous in high grade
serous carcinoma of the ovary. J Pathol. 221:49–56. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Er TK, Su YF, Wu CC, Chen CC, Wang J,
Hsieh TH, Herreros-Villanueva M, Chen WT, Chen YT, Liu TC, et al:
Targeted next-generation sequencing for molecular diagnosis of
endometriosis-associated ovarian cancer. J Mol Med. 94:835–847.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kao AP, Wang KH, Chang CC, Lee JN, Long
CY, Chen HS, Tsai CF, Hsieh TH and Tsai EM: Comparative study of
human eutopic and ectopic endometrial mesenchymal stem cells and
the development of an in vivo endometriotic invasion model. Fertil
Steril. 95:1308–1315.e1. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ryan IP, Schriock ED and Taylor RN:
Isolation, characterization, and comparison of human endometrial
and endometriosis cells in vitro. J Clin Endocrinol Metab.
78:642–649. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Revised American Society for Reproductive
Medicine classification of endometriosis: 1996. Fertil Steril.
67:817–821. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Prat J: FIGO Committee on Gynecologic
Oncology: FIGO's staging classification for cancer of the ovary,
fallopian tube, and peritoneum: Abridged republication. J Gynecol
Oncol. 26:87–89. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Farre D, Roset R, Huerta M, Adsuara JE,
Roselló L, Albà MM and Messeguer X: Identification of patterns in
biological sequences at the ALGGEN server: PROMO and MALGEN.
Nucleic Acids Res. 31:3651–3653. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li H, Zeng J and Shen K: PI3K/AKT/mTOR
signaling pathway as a therapeutic target for ovarian cancer. Arch
Gynecol Obstet. 290:1067–1078. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ferrara N and Kerbel RS: Angiogenesis as a
therapeutic target. Nature. 438:967–974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakayama I, Shibazaki M, Yashima-Abo A,
Miura F, Sugiyama T, Masuda T and Maesawa C: Loss of HOXD10
expression induced by upregulation of miR-10b accelerates the
migration and invasion activities of ovarian cancer cells. Int J
Oncol. 43:63–71. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K,
Guo J, Zhang Y, Chen J, Guo X, et al: Characterization of microRNAs
in serum: A novel class of biomarkers for diagnosis of cancer and
other diseases. Cell Res. 18:997–1006. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Iorio MV, Visone R, Di Leva G, Donati V,
Petrocca F, Casalini P, Taccioli C, Volinia S, Liu CG, Alder H, et
al: MicroRNA signatures in human ovarian cancer. Cancer Res.
67:8699–8707. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dahiya N and Morin PJ: MicroRNAs in
ovarian carcinomas. Endocr Relat Cancer. 17:F77–F89. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mateescu B, Batista L, Cardon M, Gruosso
T, de Feraudy Y, Mariani O, Nicolas A, Meyniel JP, Cottu P,
Sastre-Garau X and Mechta-Grigoriou F: miR-141 and miR-200a act on
ovarian tumorigenesis by controlling oxidative stress response. Nat
Med. 17:1627–1635. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
He X, Wei Y, Wang Y, Liu L, Wang W and Li
N: MiR-381 functions as a tumor suppressor in colorectal cancer by
targeting Twist1. Onco Targets Ther. 9:1231–1239. 2016.PubMed/NCBI
|
|
31
|
Xue Y, Xu W, Zhao W, Wang W, Zhang D and
Wu P: miR-381 inhibited breast cancer cells proliferation,
epithelial-to-mesenchymal transition and metastasis by targeting
CXCR4. Biomed Pharmacother. 86:426–433. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xia B, Li H, Yang S, Liu T and Lou G:
MiR-381 inhibits epithelial ovarian cancer malignancy via YY1
suppression. Tumour Biol. 37:9157–9167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu Y, Ohms SJ, Li Z, Wang Q, Gong G, Hu Y,
Mao Z, Shannon MF and Fan JY: Changes in the expression of miR-381
and miR-495 are inversely associated with the expression of the
MDR1 gene and development of multi-drug resistance. PLoS One.
8:e820622013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Boren T, Xiong Y, Hakam A, Wenham R, Apte
S, Chan G, Kamath SG, Chen DT, Dressman H and Lancaster JM:
MicroRNAs and their target messenger RNAs associated with ovarian
cancer response to chemotherapy. Gynecol Oncol. 113:249–255. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee S, Choi EJ, Jin C and Kim DH:
Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA
amplification contributes to cisplatin resistance in an ovarian
cancer cell line. Gynecol Oncol. 97:26–34. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mitsuuchi Y, Johnson SW, Selvakumaran M,
Williams SJ, Hamilton TC and Testa JR: The phosphatidylinositol
3-kinase/AKT signal transduction pathway plays a critical role in
the expression of p21WAF1/CIP1/SDI1 induced by cisplatin
and paclitaxel. Cancer Res. 60:5390–5394. 2000.PubMed/NCBI
|
|
37
|
Astanehe A, Arenillas D, Wasserman WW,
Leung PC, Dunn SE, Davies BR, Mills GB and Auersperg N: Mechanisms
underlying p53 regulation of PIK3CA transcription in ovarian
surface epithelium and in ovarian cancer. J Cell Sci. 121:664–674.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Carrio M, Arderiu G, Myers C and Boudreau
NJ: Homeobox D10 induces phenotypic reversion of breast tumor cells
in a three-dimensional culture model. Cancer Res. 65:7177–7185.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xiao H, Li H, Yu G, Xiao W, Hu J, Tang K,
Zeng J, He W, Zeng G, Ye Z and Xu H: MicroRNA-10b promotes
migration and invasion through KLF4 and HOXD10 in human bladder
cancer. Oncol Rep. 31:1832–1838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang H, Zhou J, Mi J, Ma K, Fan Y, Ning J,
Wang C, Wei X, Zhao H and Li E: HOXD10 acts as a tumor-suppressive
factor via inhibition of the RHOC/AKT/MAPK pathway in human
cholangiocellular carcinoma. Oncol Rep. 34:1681–1691. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Reddy SD, Ohshiro K, Rayala SK and Kumar
R: MicroRNA-7, a homeobox D10 target, inhibits p21-activated kinase
1 and regulates its functions. Cancer Res. 68:8195–8200. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Myers C, Charboneau A, Cheung I, Hanks D
and Boudreau N: Sustained expression of homeobox D10 inhibits
angiogenesis. Am J Pathol. 161:2099–2109. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ma L, Teruya-Feldstein J and Weinberg RA:
Tumour invasion and metastasis initiated by microRNA-10b in breast
cancer. Nature. 449:682–688. 2007. View Article : Google Scholar : PubMed/NCBI
|