Introduction
Colorectal cancer (CRC) represents the most commonly
diagnosed tumor worldwide, and the mortality rate of CRC is higher
in developed areas than in developing areas (1). Despite the current treatment
strategies including surgery and radio-chemotherapy resulting in an
improved treatment outcome, the median survival time of patients
with malignant CRC is only 20–24 months (2). Currently, mounting evidence indicates
that CRC is an epidemiologically heterogeneous disease and the
molecular mechanisms of CRC are not fully clarified (3). Therefore, identification of novel
molecular biomarkers involved in mediating the pathogenesis of CRC
and predicting the prognosis of CRC patients is of primary
concern.
MicroRNAs (miRNA), a class of single-stranded
noncoding RNAs, play a critical role in degrading target mRNAs
through forming hybrids with the 3′-untranslated region sequences.
Several studies have revealed that miRNAs serve as oncogenes or
tumor suppressors in CRC carcinogenesis (4). miR-296, mapped to the human 20q13.32
gene locus, is characterized as a critical mediator which is
involved in the biological processes of cell growth, angiogenesis
and apoptosis (5,6). Previously, miR-296 has been revealed
to contribute to the development of various malignant tumors, such
as ovarian, prostate, lung and breast cancers (7–10). It
is noteworthy that Kunte et al (11) demonstrated that miR-296 is
profoundly downregulated at the early stages of colon tumors and
this may play a critical role in the pathological process of CRC
neoplasia (11). However, its
expression pattern, clinical significance, and biological functions
in CRC remain unclear.
Arrestin β1 (ARRB1), a mainstay of the β-arrestin
family, has been revealed to serve as a multifunctional adaptor
contributing to the mediation of several signaling pathways which
are involved in cancer cellular progression (12–14).
Emerging evidence demonstrates that ARRB1 may interact with histone
acetyl-transferase, facilitating its recruitment to target
histones, with consequent increased chromatin acetylation and
transcription activation (13,15,16).
Although ARRB1 functions as a tumor oncogene that is associated
with aggressive progression in various malignant cancers (17,18),
its biological significance in CRC has not been extensively
elucidated.
The present study verified that miR-296 was
significantly decreased in CRC patients. Upregulation of miR-296 in
CRC cells resulted in decreased cell proliferation and increased
cell apoptosis. Importantly, ARRB1 was confirmed as a novel target
of miR-296, and it was elucidated that the miR-296-ARRB1-RAC-α
serine/threonine-protein kinase (AKT) axis contributes to
regulation of tumor growth and cell apoptosis of SW480 and HCT-116
cells.
Materials and methods
Tissue samples
CRC specimens were collected from 108 patients (60
male patients and 48 female patients; mean age, 53.32±11.32 years;
median age, 55 years) with CRC at Huizhou Municipal Central
Hospital (Huizhou, China) from January 2008 to December 2014. CRC
tissue use in the present study was approved by the Specialty
Committee on Ethics of Biomedicine Research (PJ2008-012-03),
Huizhou Municipal Central Hospital. In this study, human tissue
acquisition and use complied with the National Regulations on the
Use of Clinical Samples in China, and was performed in accordance
with the ethical standards from the Declaration of Helsinki. All
patients gave written informed consent. In the present study,
subjects' rights and interests were fully protected. There was no
potential risk to the subjects. The board of ethics committee
agreed to the study work as planned. The follow-up was carried out
in all patients, with survival time being censored in July 2014.
The follow-up procedure occurred every 6 months by telephone and
the final follow-up for patients occurred in January 2014. The
selection criteria were as follows: i) The subject was diagnosed
with CRC and had no history of other tumors; ii) complete clinical
data was available for the subject, including age, sex, clinical
manifestations and mean tumor diameter; iii) patients receiving
chemotherapy or radiotherapy prior to surgery were excluded. The
clinical characteristics of patients with CRC are presented in
Table I.
 | Table I.Association between miR-296 expression
and clinicopathological variables of colorectal cancer
patients. |
Table I.
Association between miR-296 expression
and clinicopathological variables of colorectal cancer
patients.
|
|
| MicroRNA-296
expression |
|
|---|
|
|
|
|
|
|---|
| Characteristics | Value | High | Low | P-value |
|---|
| No. of patients | 108 | 50 | 58 |
|
| Age, years |
|
|
| 0.15 |
|
<50 | 64 | 30 | 34 |
|
|
≥50 | 44 | 20 | 24 |
|
| Sex |
|
|
| 0.14 |
|
Male | 60 | 28 | 32 |
|
|
Female | 48 | 22 | 26 |
|
| MTD, cm |
|
|
| 0.18 |
| <5
cm | 77 | 35 | 42 |
|
| ≥5
cm | 31 | 15 | 16 |
|
| Tumor location |
|
|
| 0.11 |
|
Rectum | 46 | 20 | 26 |
|
|
Colon | 62 | 30 | 32 |
|
| Distant
metastasis |
|
|
| 0.02 |
|
Present | 41 | 24 | 17 |
|
|
Absent | 67 | 26 | 41 |
|
| Lymph node
metastasis |
|
|
|
<0.01 |
|
Present | 31 | 6 | 25 |
|
|
Absent | 77 | 54 | 23 |
|
| TNM stage |
|
|
|
<0.01 |
| II | 43 | 31 | 12 |
|
|
III/IV | 65 | 19 | 46 |
|
Cell culture
Human CRC cell lines (HT-29, SW620, LoVo, SW480 and
HCT-116) and the normal colon cell line NCM 460 were obtained from
the American Type Culture Collection (Manassas, VA, USA) and
cultured in Dulbecco's modified Eagle's medium (DMEM,
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) supplemented with
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), 100 U/ml penicillin and 100 µg/ml streptomycin
at 37°C in a humidified atmosphere containing 5%
CO2.
miRNAs and transfection
The miR-296 mimic, the miR-NC and the miR-296
inhibitor were purchased from Invitrogen (Thermo Fisher Scientific,
Inc.). Sequences were as follows: miR-296 mimics, sense
5′-AGGGCCCCCCCUCAAUCCUGU-3′, antisense 5′-ACAGGAUUGAGGGGGGGCCCU-3′;
miR-NC, sense 5′-UUCUCCGAGGACGUCUGACUGUTT-3′, antisense
5′-ACGUGACACGUUCGGAGGGTT-3′; miR-296 inhibitor,
5′-ACAGGCCGGACAAGUCGAAUG. The ARRB1 coding sequence was cloned into
a pCDNA3.1 vector (Invitrogen; Thermo Fisher Scientific, Inc.),
whereas a blank vector was used as a negative control. Short
interfering RNA (siRNA) against ARRB1 (ARRB1 siRNA) (sense,
5′-CCGACUCACAGUCCAUCAATT-3′; antisense, 5′-UUGAGGAUCUACGTACGGTT-3′)
and negative control with siRNA-negative control [green fluorescent
protein (GFP)-siRNA] (sense, 5′-ACGCCAUUGGUAUCGUUTT-3′; antisense,
5′-AAGACUAUTUCAAUGGUCCTT-3′) were synthesized and purified by
Shanghai GenePharma Co., Ltd. (Shanghai, China). Cells were
transfected with the miRNA mimics, siRNA and pCDNA3.1-ARRB1 using
Lipofectamine® 3000 Reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. For
transfection, 2 µg miR-296 mimic, miR-NC, miR-296 inhibitor,
pcDNA3.1, pcDNA3.1-ARRB1, ARRB1 siRNA or GFP-siRNA vectors were
diluted in 200 µl Opti-MEM (Thermo Fisher Scientific, Inc.) and
were incubated at room temperature for 5 min. Subsequently, the
diluted vectors and Lipofectamine 3000 were combined and incubated
for a further 20 min at room temperature, prior to their addition
to cells (4×105/well) seeded in 6-well plates. After
transfection for 6 h, the culture medium was replaced with
RPMI-1640 medium containing 10% FBS (HyClone; GE Healthcare Life
Sciences, Logan, UT, USA), followed by incubation for a further 24
h at 37°C and 5% CO2. At 48 h post-transfection, the
cells were used in subsequent experiments. ARRB1 was predicted as a
direct downstream target of miR-296 by Target Scan 7.2 database
(http://www.targetscan.org).
Cell proliferation assay
3-(4, 5-dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide (MTT) assay was used to assess cell
viability. Cells were seeded in 96-well plates and were incubated
with 0.2 mg/ml MTT for 4 h at 37°C. After removal of the medium,
the formazan crystals produced by live cells were dissolved in 150
µl dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA) and the
absorbance of each sample was read at a wavelength of 570 nm using
an Ultra multi-functional microplate reader (Tecan US, Inc.,
Morrisville, NC, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Tumor specimens were frozen at −75°C to use. Total
RNA, including miRNA, was extracted from cells/tissues with
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), and RNA was reverse transcribed into cDNA using a qPCR RT
kit (FSQ-101; Toyobo Life Science, Osaka, Japan), according to the
manufacturer's protocol. cDNA was amplified by Platinum SYBR Green
qPCR SuperMix-UDG (Invitrogen; Thermo Fisher Scientific, Inc.).
RT-qPCR was performed according to the manufacturer's protocol. The
PCR conditions were 10 min at 95°C, 1 min at 55°C, 40 cycles of 15
sec at 95°C, followed by 30 sec at 55°C. For mRNA analysis, RT-qPCR
was performed using the Platinum SYBR Green qPCR SuperMix-UDG
(Qiagen GmbH, Hilden, Germany) using the 7500 Fast Real-time PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
relative fold expression of the target gene was normalized to
β-actin and was calculated according to the 2−∆∆Cq
method (19). The PCR primer
sequences for ARRB1 (forward, 5′-CCTGGATGTCTTGGGTCTG-3′ and
reverse, 5′-TGATGGGTTCTCCGTGGTA-3′) were as previously described
(20). The primer sequences for
β-actin were as follows: Forward, 5′-ACCAACTGGGACGACATGGAGAAA-3′
and reverse, 5′-TAGCACAGCCTGGATAGCAACGTA-3′.
Northern blot analysis
Northern blot analysis was performed as described
previously (21). Antisense RNA
probes were 3′-end-labeled with 32P-γATP by T4 polynucleotide
kinase; miR-296 antisense probe sequence
5′-CCCUUCCAGAGGGCCCCCCCUCAAUCCUGUUGUGCCUA-3′ and U6 spliceosomal
RNA antisense probe sequence 5′-GCAGGGGCCATGCTAATCTTCTCTGTATCG-3′
were used.
Luciferase reporter assay
Cells were seeded in 24-well plates and incubated
for 24 h prior to transfection. The ARRB1-3′untranslated region
(UTR)-WT (wild-type) or ARRB1-3′UTR-MT (mutant) were transfected
with a miR-296 mimic or miR-NC into cells. For the luciferase
reporter assay, 3×104 cells were seeded into 48-well
plates overnight, followed by transfection with miR-296 (50
pmol/ml), Renilla luciferase pRL-TK vector (100 ng/ml;
Promega Corporation, Madison, WI, USA) and pGL3 plasmid with WT/MT
3′UTR of ARRB1 gene using Lipofectamine® 2000. After
transfection for 24 h, the luciferase activity was determined by
the Dual Luciferase Reporter Assay System (Promega Corporation).
Renilla luciferase activities were normalized to firefly
luciferase activities.
Apoptosis assay
Cell apoptosis was assessed with ApoScreen Annexin V
Apoptosis kit (Bender MedSystems, GmbH, Vienna, Austria). After
being washed twice with PBS, cell samples were collected and
fluorescein isothiocyanate added. All cells were analyzed by flow
cytometry (FACScan; BD Biosciences, Franklin Lakes, NJ, USA).
Analyses were performed using CellQuest software version 5.0 (BD
Biosciences). Experiments were conducted triple times.
Protein extraction and western
blotting
Western blot analysis was conducted according to
protocols described in a previous study (22). The protein concentration of cell
lysates was quantified using a bicinchoninic acid kit (Beyotime
Institute of Biotechnology, Haimen, China), and 50 µg protein for
each sample was separated by SDS-PAGE on 10% gels and was then
transferred to a polyvinylidene fluoride (PVDF) membrane (EMD
Millipore, Billerica, MA, USA). The membranes were blocked in 5%
non-fat milk diluted with Tris-buffered saline-Tween (TBST) (10 mM
Tris-HCl, pH 7.5; 150 mM NaCl; 0.1% Tween-20) at room temperature
for 1 h and were incubated overnight at 4°C with primary
antibodies. Primary antibodies used were against ARRB1 (catalog no.
sc-377015; 1:500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
AKT (catalog no. GTX110613; 1:600; GeneTex, Inc., Irvine, CA, USA),
B cell lymphoma (Bcl)-2 (catalog no. sc-20067; 1:500; Santa Cruz
Biotechnology, Inc.), signal transducer and activator of
transcription 3 (STAT3) (catalog no. ab-11935; 1:500; Abcam,
Cambridge, MA, USA), Bcl-2-associated X protein (Bax) (catalog no.
sc-23959; 1:500; Santa Cruz Biotechnology, Inc.), Bcl-2 homologous
antagonist/killer (Bak) (catalog no. sc-7873; 1:500; Santa Cruz
Biotechnology, Inc.), NADPH oxidase activator (Noxa) (catalog no.
14766; 1:1,000; Cell Signaling Technology Inc., Danvers, MA, USA),
p53 upregulated modulator of apoptosis (PUMA) (catalog no. 24633;
1:1,000; Cell Signaling Technology, Inc.) and β-actin (catalog no.
ab8227; 1:1,000; Abcam). The membranes were incubated with
horseradish peroxidase-conjugated goat anti-rabbit (catalog no.
ab6721; 1:1,000; Abcam) or rabbit anti-mouse immunoglobulin G
secondary antibodies (catalog no. ab6728; 1:1,000; Abcam) for 2 h
at room temperature. The proteins were visualized using Enhanced
Chemiluminescence Plus reagents (Amersham; GE Healthcare, Chicago,
IL, USA), and band density was measured using ImageJ software
(National Institutes of Health, Bethesda, MD, USA), and the values
were normalized to the densitometric values of β-actin in each
sample.
Transmission electron microscopy
(TEM)
Cultured cells were fixed for 2 h at 4°C with 2.5%
glutaraldehyde in 0.2 M cacodylate buffer (pH 7.3), and post-fixed
in 1% osmium tetroxide in 0.45 M cacodylate buffer (pH 7.3). The
fixed material was dehydrated in a graded series of alcohol and
embedded in Epon 812. For light microscopy, semi-thin sections were
stained with Toluidine Blue and observed under a Reichert
microscope equipped with Nomarski optics. Images were captured
using a 12-bit charge-coupled device camera. For TEM, ultra-thin
sections were classically contrasted with Ultracut S and were
stained with uranyl acetate and lead citrate for 30 and 5 min at
room temperature, respectively. The stained sections were observed
under a JEOL transmission electron microscope (JEOL, Ltd., Tokyo,
Japan) at ×1200 magnification.
Immunohistochemistry
Human tissues were fixed in 4% formalin for 4 h at
room temperature, and were then embedded in paraffin and cut into
3-µm sections. The sections were then deparaffinized in xylol for
20 min at 60°C and rehydrated in a graded ethanol series (75%
alcohol for 1.5 h, 95% alcohol for 1.5 h, 95% alcohol for 1 h,
anhydrous ethanol for 2.5 h). Antigen retrieval was performed by
heating the samples in a microwave for 20 min in 1 mM EDTA buffer
(pH 8.0). Thereafter, endogenous peroxidase activity was quenched
by 30 min incubation in 3% methanolic hydrogen peroxide solution at
room temperature. The sections were incubated in nonimmune serum
(Invitrogen; Thermo Fisher Scientific, Inc.) for 30 min and were
then incubated overnight at 4°C in ARRB1 primary monoclonal
antibody (cat. no., ab31868; 1:500; Abcam). After washing in TBST,
the immunolabeled sections were incubated with a horseradish
peroxidase-conjugated secondary antibody (catalog no. AS09 602;
1:500; Agrisera AB, Vännäs, Sweden) for 20 min at room temperature,
and then with peroxidase-conjugated complex (Dako, Glostrup,
Denmark) for 20 min. Finally, the sections were visualized with
3,3′-diaminobenzidine and counterstained with hematoxylin. To
ensure the specificity of the immunostaining, negative controls
were prepared by replacing the primary antibody with non-immune
serum. Two independent pathologists examined five random fields (1
field = 0.159 mm2 at ×100 magnification; Leica DMRX;
Leica Microsystems, Inc., Buffalo Grove, IL, USA) in each sample
and scored each sample without knowledge of patient outcome
(double-blinded).
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA,
USA). Experimental data are presented as the mean ± standard
deviation from triplicate experiments. A Student's t-test was used
to analyze differences between two groups. A one-way analysis of
variance and Tukey's post hoc test was performed to detect
statistical differences among multiple groups. Pearson's
χ2 tests were used for analysis of the association
between clinicopathological features and miR-296 expression in CRC
patients. The correlation between miR-296 and ARRB1 expression was
examined by Spearman's correlation coefficient. Overall survival
(OS) curves were calculated using the Kaplan-Meier method and were
compared using the log-rank test. Possible prognostic factors were
analyzed by univariate analysis for their potential association
with OS. Factors with a P<0.05 in the univariate analysis were
included in the multivariate analysis, in order to analyze their
potential association with OS using Cox regression with the
backward stepwise method. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-296 is frequently downregulated in
CRC specimens
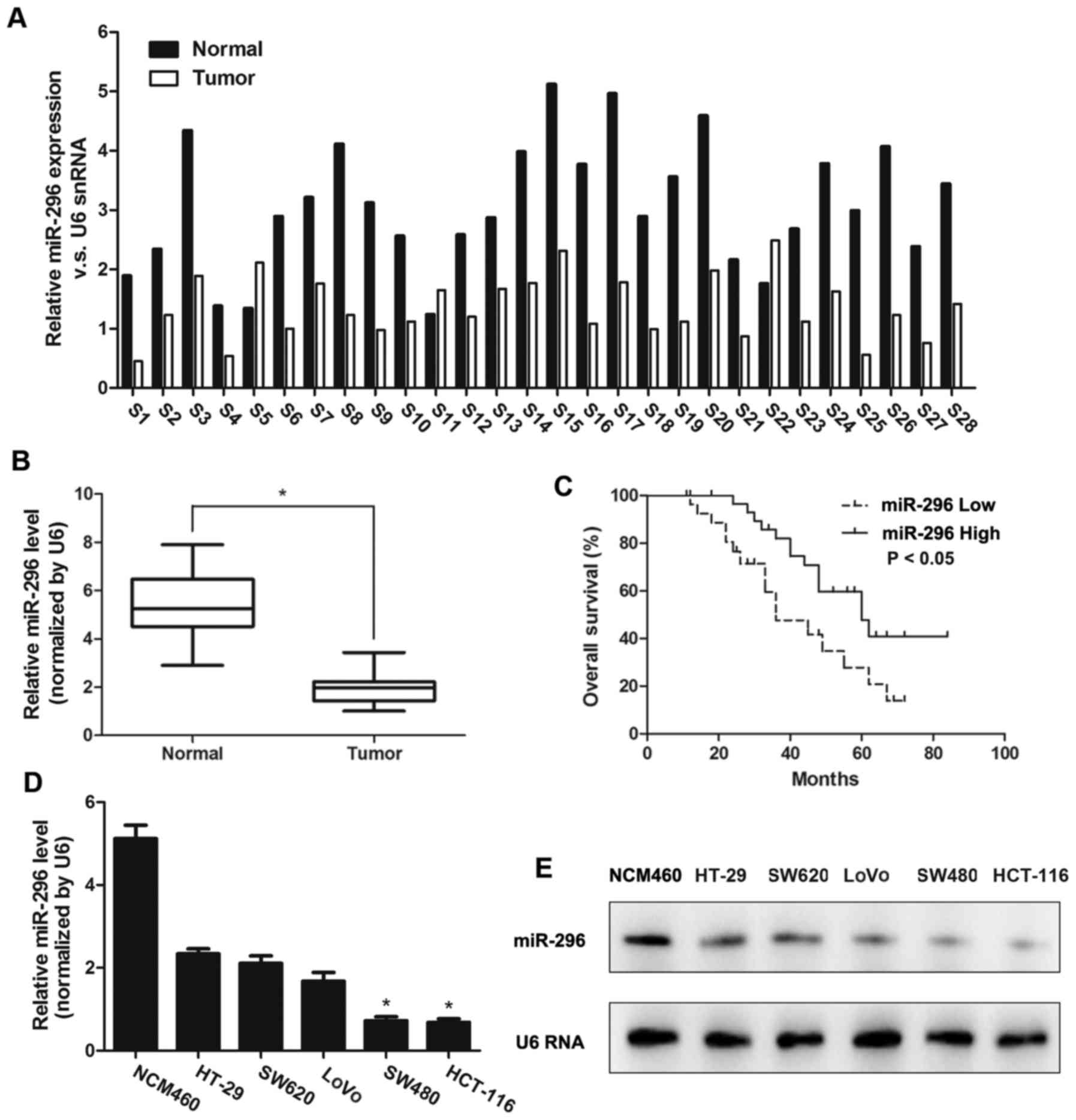
The present study first used RT-qPCR to measure the
miR-296 expression in 28 CRC tissues and corresponding adjacent
normal specimens. It was demonstrated that miR-296 downregulation
was detected in 25/28 (89.28%) CRC tumors (Fig. 1A), which indicated that the decrease
of miR-296 is a frequent event in CRC. The RT-qPCR data revealed
that miR-296 expression in the tumors was approximately 2.8-fold
lower compared with adjacent normal tissues (4.98±1.02 vs.
1.99±0.54; P<0.05; Fig. 1B). The
mean miR-296 expression level of CRC tissues was 4.98, which was
utilized to divide CRC patients into either a low expression group
(<4.98, n=58) or a high expression group (≥4.98, n=50). The
present study then investigated the association between miR-296
expression and clinicopathological parameters (Table I). The expression level of miR-296
was inversely associated with tumor node metastasis (TNM) stage
(P<0.01), lymph node metastasis (P<0.01) and distant
metastasis (P=0.02). No significant association was identified
between miR-296 expression and other clinical parameters.
Univariate survival analysis revealed a significant
association between expression of miR-296, TNM stage (P=0.032),
distant metastasis (P=0.006), lymph node metastasis (P=0.021) and
recurrence-free survival rate (Table
II), while no significant association was observed between
recurrence-free survival rate and sex, age, tumor size, or tumor
location. Multivariate analysis using the Cox proportional hazards
model for all variables included in the univariate analysis further
revealed that expression of miR-296 (P=0.008), TNM stage (P=0.021),
distant metastasis (P=0.007), and lymph node metastasis (P=0.002)
at diagnosis were independent prognostic factors for patients with
CRC (Table III).
| Table II.Univariate analysis of factors
associated with survival of colorectal cancer patients. |
Table II.
Univariate analysis of factors
associated with survival of colorectal cancer patients.
|
| Survival |
|---|
|
|
|
|---|
| Variable | HR | 95% CI | P-value |
|---|
| Sex (male vs.
female) | 0.758 | 0.397–1.076 | 0.417 |
| Age (≥50
vs.<50) | 0.812 | 0.276–1.374 | 0.398 |
| MTD (≥5 cm vs.
<5 cm) | 0.541 | 0.322–1.179 | 0.743 |
| MicroRNA-296 (low
vs. high) | 1.843 | 1.031–2.458 | 0.012a |
| Tumor location | 0.742 | 0.512–1.592 | 0.732 |
| TNM stage | 0.965 | 0.651–1.868 | 0.032a |
| Lymph node
metastasis | 0.713 | 0.251–0.964 | 0.021a |
| Distant
metastasis | 0.433 | 0.113–0.879 | 0.006a |
| Table III.Multivariate analyses of factors
associated with survival of colorectal patients. |
Table III.
Multivariate analyses of factors
associated with survival of colorectal patients.
| Variable | HR | 95% CI | P-value |
|---|
| MicroRNA-296 (low
vs. high) | 1.765 | 1.239–3.581 | 0.008a |
| TNM stage | 1.132 | 0.542–1.783 | 0.021a |
| Lymph node
metastasis | 2.349 | 0.396–3.486 | 0.002a |
| Distant
metastasis | 2.132 | 0.987–3.783 | 0.007a |
Next, the present study investigated the prognostic
value of miR-296 between high and low expression groups using the
Kaplan-Meier survival method. These results demonstrated that the
patients with high expression levels of miR-296 exhibited a
significantly prolonged overall survival (OS) compared with those
with low expression levels of miR-296 (P<0.05; Fig. 1C).
The present study further measured the miR-296
expression in one normal colon cell line NCM 460 and five CRC cell
lines, including HT-29, SW620, LoVo, SW480 and HCT-116 by RT-qPCR
analysis (Fig. 1D) and Northern
blotting (Fig. 1E). SW480 and
HCT-116 cells revealed a significantly lower expression level of
miR-296 compared with HT-29, SW620, LoVo and NCM 460 cells.
Therefore, SW480 and HCT-116 cells were selected for use in
subsequent experiments.
Overexpression of miR-296 inhibits
cell survival and promotes cell apoptosis of CRC cells in
vitro
To investigate the antitumor effects of miR-296 in
CRC cells, the present study upregulated the miR-296 expression
level by treating with miR-296 mimics. As presented in Fig. 2A, transfection of SW480 and HCT-116
cells with miR-296 mimics resulted in a significant increase in
miR-296 expression. Consistent with the previously demonstrated
critical role of miR-296 in cell survival, miR-296
mimics-transfected SW480 and HCT-116 cells (overexpressing miR-296)
revealed significantly lower survival rates compared with miR-NC
transfected cells as evidenced by MTT assay (Fig. 2B). In addition, upregulation of
miR-296 expression in SW480 and HCT-116 cell lines enhanced CRC
cell apoptosis as measured by flow cytometry assays (Fig. 2C). Furthermore, TEM analysis
revealed classical apoptotic changes in SW480 and HCT-116 cells
transfected with miR-296 mimics, such as nuclear condensation and
fragmentation and cell blebbing morphological alterations (Fig. 2D). These data strongly suggested
that miR-296 suppressed CRC cell survival through enhancing cell
apoptosis.
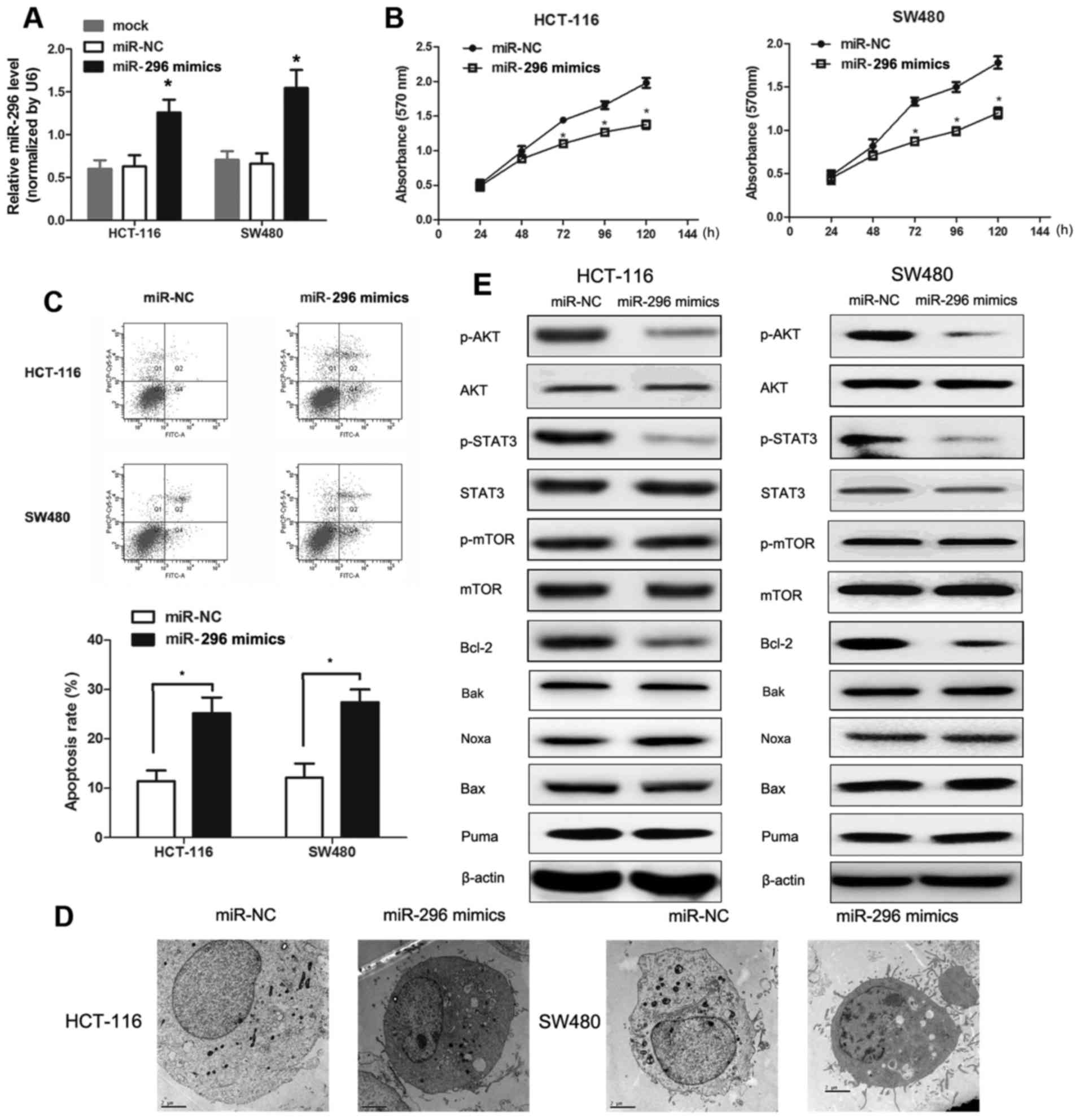 | Figure 2.Effects of miR-296 on cell
proliferation, cell apoptosis and the expression of associated
molecules in SW480 and HCT-116 cell lines. (A) Reverse
transcription-quantitative polymerase chain reaction analysis
demonstrated that miR-296 expression was significantly enhanced in
SW480 and HCT-116 cells transfected with miR-296 mimics compared to
cells transfected with miR-NC. *P<0.05 vs. miR-NC-transfected
cells. (B) Effect of miR-296 upregulation on SW480 and HCT-116 cell
proliferation as measured by MTT assay. *P<0.05 vs.
miR-NC-transfected cells. (C) Effect of miR-296 upregulation on
SW480 and HCT-116 cell apoptosis rates. The number of apoptotic
cells was significantly higher in SW480 and HCT-116 cells
overexpressing miR-296 as measured by propidium iodide staining and
flow cytometry. *P<0.05. (D) Morphological analysis of SW480 and
HCT-116 cell apoptosis induced by miR-296 mimics. The morphology of
colorectal cancer cells were imaged by TEM. Magnification in
transmission electron microscopy analysis was ×6,000. (E) Altered
AKT signaling and apoptosis are associated with overexpression of
miR-296. Detection of p-AKT, AKT, p-STAT3, STAT3 and
apoptosis-associated proteins in SW480 and HCT-116 cells was
performed by western blotting. miR, microRNA; NC, negative control;
p, phosphorylated; AKT, RAC-α serine/threonine-protein kinase;
STAT3, signal transducer and activator of transcription 3; mTOR,
serine/threonine-protein kinase mTOR; Bcl-2, B cell lymphoma 2;
Bak, Bcl-2 homologous antagonist/killer; Bax, Bcl-2 associated X,
apoptosis regulator; Noxa, NADPH oxidase activator; Puma, p53
upregulated modulator of apoptosis. |
miR-296-induced apoptosis is
associated with dephosphorylation of AKT
The AKT signaling pathway plays an important role in
controlling CRC cellular processes, such as apoptosis and
proliferation. Signal transducer and activator of transcription 3
(STAT3) has been reported to mediate cell survival and apoptosis in
CRC cells through the AKT signaling pathway. To determine whether
AKT pathway activation is involved in miR-296-induced apoptosis,
the expression and activities of AKT signaling pathway molecules
were examined in cellular apoptosis pathways. Western blot analysis
revealed that AKT phosphorylation and STAT3 phosphorylation were
significantly lower in SW480 and HCT-116 cells expressing the
miR-296 mimics compared with cells transfected with miR-NC, while
no changes in the expression levels of total AKT, total STAT3,
p-serine/threonine-protein kinase mTOR (mTOR) and total mTOR were
observed (Fig. 2E). Moreover, a
significant decrease in the protein expression level of
anti-apoptotic protein Bcl-2 in SW480 and HCT-116 cells
overexpressing miR-296 was observed, whereas the protein levels of
the apoptotic proteins Bax, Bak, Noxa and PUMA did not appear to
differ between the two groups (Fig.
2E). These results suggested that AKT, STAT3 and Bcl-2 are
critical downstream effectors of miR296-induced apoptosis and
suppression of proliferation in SW480 and HCT-116 cells.
Upregulation of miR-296 expression in CRC cells may inhibit the
activity of the AKT-STAT3 signaling pathway, which ultimately
results in promoting CRC cell apoptosis by suppressing levels of
Bcl-2, an important anti-apoptotic protein.
ARRB1 is a direct target of
miR-296
Since ARRB1 was predicted as a direct downstream
target of miR-296 using the TargetScan 7.2 database (http://www.targetscan.org) (Fig. 3A), the present study next validated
whether miR-296 regulates ARRB1 expression by using a luciferase
reporter gene assay. As presented in Fig. 3B, overexpression of miR-296 in SW480
cells significantly suppressed the luciferase activity of the WT
3′-UTR of ARRB1, whereas no change in the luciferase activity of
the Mut 3′-UTR of ARRB1was observed. RT-qPCR revealed that miR-296
mimics resulted in a significant increase in miR-296 expression and
miR-296 inhibitor caused a significant decrease in miR-296
expression in SW480 cells (Fig.
3C). In addition, upregulation of miR-296 expression
substantially decreased endogenous ARRB1 mRNA and protein levels in
SW480 cells expressing miR-296 mimics compared with cells
expressing only miR-NC, detected using RT-qPCR and western blot
analysis (Fig. 3D and E). In
contrast to SW480 cells overexpressing miR-296, SW480 cells
underexpressing miR-296 showed enhanced ARRB1 mRNA and protein
expression levels compared with the control (Fig. 3D and E). Furthermore, the
association between miR-296 level and ARRB1 expression in CRC
tissues was investigated, to confirm that ARRB1 was a novel target
of miR-296. Immunohistochemical (ICH) staining indicated that ARRB1
expression in CRC with high expression level of miR-296 was
significantly lower than in CRC with low miR-296 expression
(P<0.05; Fig. 3F). RT-qPCR
analysis revealed that ARRB1 mRNA expression was lower in the
miR-296 high-expressing tumors compared with miR-296 low-expressing
tumors, consistent with results from ICH (P<0.05; Fig. 3G). As depicted in Fig. 3H, an inverse correlation was
observed between ARRB1 and miR-296 expression levels in human CRC
tissues (r= −0.352, P=0.0055). Therefore, the aforementioned
results suggested that miR-296 reduced ARRB1 expression by
targeting its 3′-UTR and ARRB1 is a direct downstream target of
miR-296 in CRC.
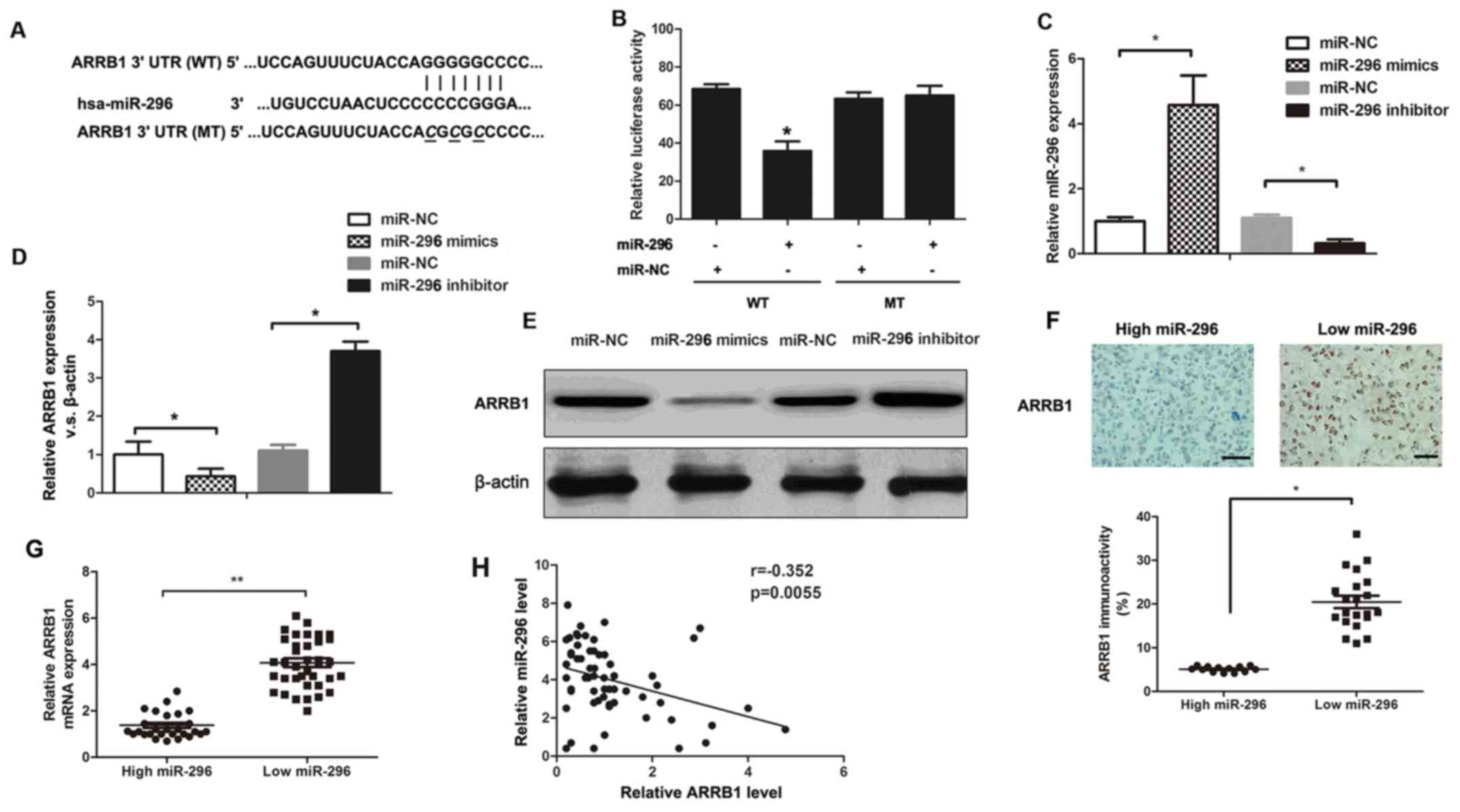 | Figure 3.ARRB1 is a novel target of miR-296.
(A) The putative miR-296 binding sequences in ARRB1 3′-UTR. (B) A
significant decrease in the relative luciferase activity was
observed following co-transfection of ARRB1-3′-UTR with the miR-296
mimics *P<0.05 vs. miR-NC-transfected cells. (C) RT-qPCR
analysis of miR-296 expression levels after the introduction of
mimic control, miR-296 mimic, inhibitor control, or miR-296
inhibitor to sw480 cells. *P<0.05. (D) RT-qPCR and (E) western
blotting analysis of ARRB1 protein levels following the
introduction of mimic control, miR-296 mimic, inhibitor control, or
miR-296 inhibitor to SW480 cells. *P<0.05. (F) Representative
immunostaining showed negative expression of ARRB1 in miR-296
high-expressing CRC tissue and positive expression of ARRB1 in
miR-296 low-expressing CRC tissue. The expression of ARRB1 protein
in miR-296 high-expressing tumors was significantly lower than that
in miR-296 low-expressing tumors. Scale bar, 10 µm. *P<0.05. (G)
The expression of ARRB1 mRNA in miR-296 high-expressing tumors was
significantly lower than that in miR-296 low-expressing tumors.
*P<0.01. (H) A significant inverse correlation between the mRNA
levels of miR-296 and ARRB1 was observed in CRC tissues. P=0.0055.
miR, microRNA; NC, negative control; UTR, untranslated region; CRC,
colorectal cancer; WT, wild-type; MT, mutated; ARRB1, arrestin
β1. |
ARRB1 functions as an oncogene in
CRC
The present study examined ARRB1 mRNA and protein
expression levels in HT-29, SW620, LoVo, SW480 and HCT-116 cell
lines by RT-qPCR and western blot analysis (Fig. 4A and B). As predicted, SW480 and
HCT-116 cells exhibited significantly increased ARRB1 expression
levels compared with HT-29, SW620, and LoVo cells, at both the mRNA
and protein level. Therefore, SW480 and HCT-116 cells were selected
for subsequent in vitro experiments. Western blot analysis
revealed that both ARRB1 mRNA and protein expression levels were
significantly inhibited by siRNA targeting ARRB1 (Fig. 4C and D). An MTT assay was next used
to investigate the contribution of ARRB1 to SW480 and HCT-116 cell
proliferation. It was observed that downregulation of ARRB1 led to
a significant reduction in survival of SW480 and HCT-116 cells
(Fig. 4E). In addition, flow
cytometry analysis revealed that SW480 and HCT-116 cells that
underexpressed ARRB1 exhibited a significantly increased rate of
apoptosis relative to cells expressing the GFP-siRNA vector
(Fig. 4F). These results clearly
indicated that ARRB1 functions as a critical oncogene through
enhancing cell survival and suppressing cell apoptosis in
vitro.
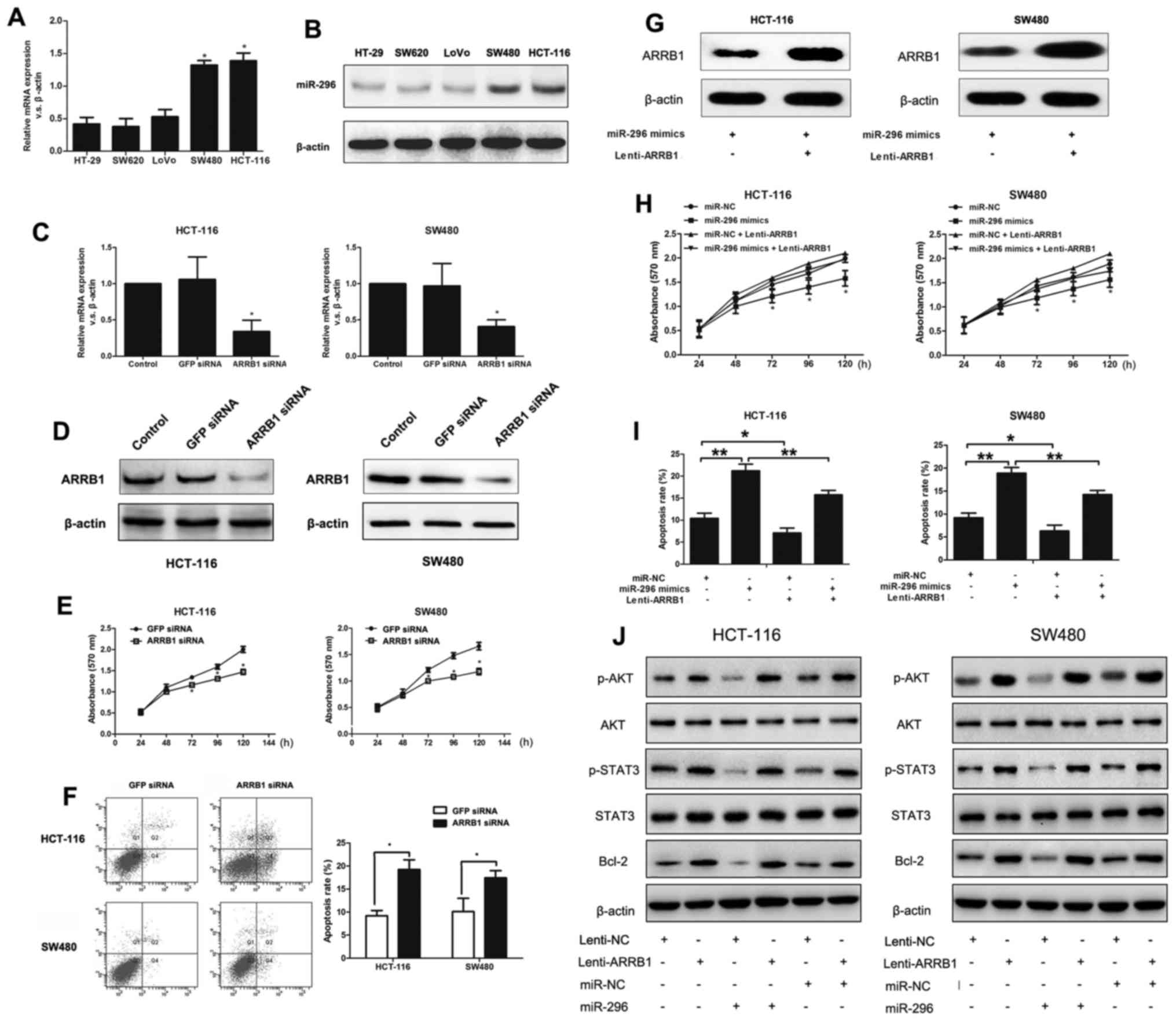 | Figure 4.Overexpression of ARRB1 partially
rescues miR-296-regulated cell growth and apoptosis in SW480 and
HCT-116 cells. (A) mRNA levels of ARRB1 measured in five CRC cell
lines by RT-qPCR. *P<0.05 vs. HT-29, SW620 and LoVo cells. (B)
Expression of ARRB1 protein in five CRC cell lines measured by
western blotting. (C) The mRNA levels of ARRB1 expression were
suppressed in SW480 and HCT-116 cells after transfection of ARRB1
siRNA for 72 h, detected by RT-qPCR. *P<0.05 vs.
GFP-siRNA-transfected cells. (D) Western blot analysis showing the
results of ARRB1 expression in HCT-116 and SW480 cells after
ARRB1-siRNA transfection. (E) Effect of ARRB1 underexpression on
SW480 and HCT-116 cell proliferation, as measured by MTT assay.
*P<0.05 vs. GFP-siRNA-transfected cells. (F) Effect of ARRB1
underexpression on SW480 and HCT-116 cell apoptosis rates. The
number of apoptotic cells was significantly higher in SW480 and
HCT-116 cells underexpressing ARRB1, as measured by apoptosis
assay. *P<0.05. (G) Western blot analysis of CRC cells
co-transfected with miR-296 mimics and Lenti-ARRB1. (H) MTT assay
of SW480 and HCT-116 cells co-transfected with miR-296 mimics and
Lenti-ARRB1 or the control. *P<0.05 vs. SW480 and HCT-116 cells
co-transfected with miR-296 mimics and Lenti-ARRB1. (I) Apoptosis
assay of SW480 and HCT-116 cells co-transfected with miR-296 mimics
and Lenti-ARRB1 or the control. *P<0.05, **P<0.01. (J) ARRB1
overexpression blocks the effect of miR-296 on dephosphorylation of
AKT signaling without affecting total AKT and STAT3 expression.
Western blot analysis also indicated that miR-296 suppressed Bcl-2
protein expression, via regulation of ARRB1. Western blot analyses
were performed in triplicate, and representative results are shown.
miR, microRNA; NC, negative control; CRC, colorectal cancer; ARRB1,
arrestin β1; si, small interfering; GFP, green fluorescent protein;
p, phosphorylated; AKT, RAC-α serine/threonine-protein kinase;
STAT3, signal transducer and activator of transcription 3; Bcl-2, B
cell lymphoma-2. |
miR-296 inhibits cell growth and
promotes cell apoptosis through suppressing ARRB1-mediated AKT
activation
To figure out whether ARRB1 is a critical mediator
in miR-296-regulated cell survival and apoptosis, full-length ARRB1
was transfected into SW480 and HCT-116 cells overexpressing
miR-296. It was verified that miR-296 mimics significantly
decreased ARRB1 protein levels and this effect could be in part
alleviated by ARRB1 transfection in CRC cells, detected by western
blot analysis (Fig. 4G). The MTT
and apoptosis assays revealed that upregulation of ARRB1 in SW480
and HCT-116 cells transfected with miR-296 mimics reversed the
decreased growth and enhanced apoptosis that was observed in cells
transfected only with miR-296 mimics (Fig. 4H and I). These results implied that
miR-296 suppresses cell growth and induces cell apoptosis by an
ARRB1-dependent mechanism.
The association between miR-296 and ARRB1 in SW480
and HCT-116 cells prompted investigation of the activation of the
AKT signaling pathway under altering ARRB1 expression levels.
Following enhancement of the expression of ARRB1 in SW480 and
HCT-116 cells by using Lenti-ARRB1, it was observed that ARRB1
activated the AKT signaling pathway by increasing the
phosphorylation levels of AKT and STAT3 without affecting the total
AKT and STAT3 expression. The expression levels of Bcl-2 increased
in CRC cells transfected with Lenti-ARRB1. Notably, overexpression
of ARRB1 significantly abrogated the decrease in the
phosphorylation of AKT and phosphorylation of STAT3 in cells
overexpressing miR-296 (Fig. 4J).
In addition, ARRB1 rescued cells from the inhibitory effect of
miR-296 on Bcl-2 expression (Fig.
4J). These results demonstrated that miR-296-induced apoptosis
in CRC may be associated with the inactivation of AKT/STAT3
signaling pathway, which is regulated by ARRB1.
Discussion
Previous evidence has indicated that miR-296 serves
as a tumor suppressor in a variety of malignant cancer cells.
Previously, Shivapurkar et al (23) demonstrated that underexpression of
miR-296 in serum predicts chemotherapy resistance in patients
receiving systemic chemotherapy for metastatic colon cancer.
Furthermore, it was reported that miR-296 inhibits CRC metastasis
by mediating S100A4, which highlights the potential of miR-296 as
an important target against CRC metastasis (24). These results prompted speculation
that miR-296 may serve as a tumor suppressor for CRC cells. The
present study revealed that miR-296 was underexpressed in 89.28% of
the CRC patients. Kaplan-Meier analysis revealed that lower miR-296
expression was positively associated with poor prognosis. The
results identified miR-296 as a promising predictive biomarker,
which may aid in the optimization of treatment stratification.
Notably, it was demonstrated that SW480 and HCT-116 cells revealed
significantly lower expression levels of miR-296 compared with
HT-29, SW620, and LoVo cells. It has been reported that miR-296
directly targets protein kinase C α to suppress focal adhesion
kinase-Ras-c-Myc signaling, thus stimulating its own expression in
a feedback loop in lung adenocarcinoma (25). Deng et al (26) demonstrated that suppression of MK2
expression downregulates Ras/Braf/Erk/Mek/c-Myc axis and promotes
cytoplasmic translocation of c-Myc, which activates miR-296
expression by a feedback loop (26). ATCC information suggests that both
SW480 and HCT-116 cells have mutations in the Ras proto-oncogene,
while the other three lines (HT-29, SW620, and LoVo cells) have no
mutations in the Ras proto-oncogene. Perhaps these altered Ras
mutations may contribute to the lower expression level of miR-296.
However, this issue requires further studies in the future.
Over the past decades, a wide range of studies have
demonstrated that the AKT signaling pathway is associated with
multiple cellular functions. Accumulating evidence suggests that
the AKT signaling pathway regulates various cellular biological
processes including cell apoptosis, proliferation, angiogenesis,
and invasion, all of which are involved in CRC development. It is
noteworthy that miR-296 targets AKT2 in pancreatic cancer and
functions as a potential tumor suppressor (27). Therefore, it is reasonable to
speculate that miR-296 may mediate several important signaling
pathways, particularly the AKT signaling pathway, and ultimately
contributes to tumorigenesis. ARRB1 serves as a ubiquitous,
multifunctional scaffolding protein, and it connects the activated
receptors with diverse signaling pathways within the cell (12,17).
ARRB1 may have different effects on cellular processes in different
signaling pathways. The repressed protein ARRB1 indirectly
regulates transcription factors involved in DNA damage processing
and apoptosis through binding to regulators such as IκBα and MDM2
(28). Furthermore, it was reported
that ARRB1 mediates prolonged phosphorylation of AKT in
glioblastoma cells induced by NK1R activation (29). Given the importance of ARRB1 in the
AKT signaling pathway, the present study examined the expression of
ARRB1 after miR-296 transfection, to clarify the mechanisms of
apoptosis in CRC cells. It was indicated that miR-296 enhanced CRC
cell apoptosis by inhibiting ARRB1 expression, a novel downstream
target of miR-296, which serves to reduce AKT signaling pathway
activity, leading to increased apoptosis. However, the in
vivo functions and molecular mechanism of miR-296 and ARRB1 in
CRC remain to be fully elucidated, and further studies are required
in the future.
Apoptosis is an inborn process that allows the cells
to systematically inactivate, destroy their own components and
ultimately results in cell death. Bcl-2 is a proto-oncogene that
suppresses apoptosis and has been indicated in the pathogenesis of
various tumor types including CRC (30). In addition, CRC with high Bcl-2
expression levels exhibits lower rates of spontaneous apoptosis
compared with CRC with low Bcl-2 expression (31). Aberrant activation of the Bcl-2 gene
may be an early event in colorectal carcinogenesis that can
suppress apoptosis in vivo (31). Interestingly, miR-296 is also
suppressed at the premalignant stages of colon carcinogenesis
(11). The present study indicated
that miR-296 regulates Bcl-2 expression in CRC cells, induced by
AKT pathway inactivation. The lack of Bcl-2 activation in miR-296
transfected CRC cells was at least partly responsible for the
enhanced cell apoptosis. Moreover, it was demonstrated that
overexpression of miR-296 in CRC resulted in a decrease in Bcl-2
expression, whereas the level of Bax, or PUMA remained unchanged,
implying a functional interaction between miR-296 and the
Bcl-2-depedent apoptosis pathways. Although there is no direct
interaction of miR-296 with Bcl-2, it is reasonable to speculate
that miR-296 plays a critical role in the Bcl-2 pathway. Additional
studies are essential to fully describe the miR-296-associated
Bcl-2 pathway in the future.
In conclusion, the results of the present study
indicated that ARRB1 is a direct target of miR-296. In addition,
miR-296 resulted in the promotion of cell apoptosis and the
suppression of cell growth by regulating the ARRB1-AKT-mediated
signaling pathway. The results add to the accumulating evidence
that miR-296 plays a critical role in CRC tumorigenesis and
targeting miR-296 may represent an ideal gene-targeting strategy
for CRC treatment.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZZ and CC designed and performed the experiments;
YX, ZZ, and XZ contributed to the data analysis; CC enrolled
patients and measured the RNA levels in the clinical samples; ZZ
initiated the study and wrote the manuscript; and all authors have
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The use of human tissues was approved by the
Specialty Committee on Ethics of Biomedicine Research (PJ2008-
012-03), Huizhou Municipal Central Hospital, and patient consent
was obtained.
Patient consent for publication
Written informed consent was obtained.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang J, Song YX, Ma B, Wang JJ, Sun JX,
Chen XW, Zhao JH, Yang YC and Wang ZN: Regulatory roles of
non-coding RNAs in colorectal cancer. Int J Mol Sci.
16:19886–19919. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Konda B, Shum H and Rajdev L:
Anti-angiogenic agents in metastatic colorectal cancer. World J
Gastrointest Oncol. 7:71–86. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kocarnik JM, Shiovitz S and Phipps AI:
Molecular phenotypes of colorectal cancer and potential clinical
applications. Gastroenterol Rep (Oxf). 3:269–276. 2015.PubMed/NCBI
|
|
4
|
Farazi TA, Hoell JI, Morozov P and Tuschl
T: MicroRNAs in human cancer. Adv Exp Med Biol. 774:1–20. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang L, Bo X, Zheng Q, Xiao X, Wu L and Li
B: miR-296 inhibits proliferation and induces apoptosis by
targeting FGFR1 in human hepatocellular carcinoma. FEBS Lett.
590:4252–4262. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dinami R, Buemi V, Sestito R, Zappone A,
Ciani Y, Mano M, Petti E, Sacconi A, Blandino G, Giacca M, et al:
Epigenetic silencing of miR-296 and miR-512 ensures hTERT dependent
apoptosis protection and telomere maintenance in basal-type breast
cancer cells. Oncotarget. 8:95674–95691. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee KH, Lin FC, Hsu TI, Lin JT, Guo JH,
Tsai CH, Lee YC, Lee YC, Chen CL, Hsiao M, et al: MicroRNA-296-5p
(miR-296-5p) functions as a tumor suppressor in prostate cancer by
directly targeting Pin1. Biochim Biophys Acta. 1843:2055–2066.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yan W, Chen J, Chen Z and Chen H:
Deregulated miR-296/S100A4 axis promotes tumor invasion by inducing
epithelial-mesenchymal transition in human ovarian cancer. Am J
Cancer Res. 6:260–269. 2016.PubMed/NCBI
|
|
9
|
Luo W, Lin Y, Meng S, Guo Y, Zhang J and
Zhang W: miRNA-296-3p modulates chemosensitivity of lung cancer
cells by targeting CX3CR1. Am J Transl Res. 8:1848–1856.
2016.PubMed/NCBI
|
|
10
|
Savi F, Forno I, Faversani A, Luciani A,
Caldiera S, Gatti S, Foa P, Ricca D, Bulfamante G, Vaira V, et al:
miR-296/Scribble axis is deregulated in human breast cancer and
miR-296 restoration reduces tumour growth in vivo. Clin Sci (Lond).
127:233–242. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kunte DP, DelaCruz M, Wali RK, Menon A, Du
H, Stypula Y, Patel A, Backman V and Roy HK: Dysregulation of
microRNAs in colonic field carcinogenesis: Implications for
screening. PLoS One. 7:e455912012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Witherow DS, Garrison TR, Miller WE and
Lefkowitz RJ: beta-Arrestin inhibits NF-kappaB activity by means of
its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc
Natl Acad Sci USA. 101:8603–8607. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miele E, Po A, Begalli F, Antonucci L,
Mastronuzzi A, Marras CE, Carai A, Cucchi D, Abballe L, Besharat
ZM, et al: β-arrestin1-mediated acetylation of Gli1 regulates
Hedgehog/Gli signaling and modulates self-renewal of SHH
medulloblastoma cancer stem cells. BMC Cancer. 17:4882017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kraemer A, Barjaktarovic Z, Sarioglu H,
Winkler K, Eckardt-Schupp F, Tapio S, Atkinson MJ and Moertl S:
Cell survival following radiation exposure requires miR-525-3p
mediated suppression of ARRB1 and TXN1. PLoS One. 8:e774842013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kang J, Shi Y, Xiang B, Qu B, Su W, Zhu M,
Zhang M, Bao G, Wang F, Zhang X, et al: A nuclear function of
beta-arrestin1 in GPCR signaling: Regulation of histone acetylation
and gene transcription. Cell. 123:833–847. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Parathath SR, Mainwaring LA, Fernandez-L
A, Guldal CG, Nahlé Z and Kenney AM: β-Arrestin-1 links mitogenic
sonic hedgehog signaling to the cell cycle exit machinery in neural
precursors. Cell Cycle. 9:4013–4024. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang Y, Guo Y, Tan S, Ke B, Tao J, Liu H,
Jiang J, Chen J, Chen G and Wu B: β-Arrestin1 enhances
hepatocellular carcinogenesis through inflammation-mediated Akt
signalling. Nat Commun. 6:73692015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zecchini V, Madhu B, Russell R,
Pértega-Gomes N, Warren A, Gaude E, Borlido J, Stark R,
Ireland-Zecchini H, Rao R, et al: Nuclear ARRB1 induces
pseudohypoxia and cellular metabolism reprogramming in prostate
cancer. EMBO J. 33:1365–1382. 2014.PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma H, Wang L, Zhang T, Shen H and Du J:
Loss of β-arrestin1 expression predicts unfavorable prognosis for
non-small cell lung cancer patients. Tumour Biol. 37:1341–1347.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hong L, Han Y, Zhang H, Li M, Gong T, Sun
L, Wu K, Zhao Q and Fan D: The prognostic and chemotherapeutic
value of miR-296 in esophageal squamous cell carcinoma. Ann Surg.
251:1056–1063. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Q, Qian J, Wang J, Luo C, Chen J, Hu
G and Lu Y: Knockdown of RLIP76 expression by RNA interference
inhibits invasion, induces cell cycle arrest, and increases
chemosensitivity to the anticancer drug temozolomide in glioma
cells. J Neurooncol. 112:73–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shivapurkar N, Mikhail S, Navarro R, Bai
W, Marshall J, Hwang J, Pishvaian M, Wellstein A and He AR:
Decrease in blood miR-296 predicts chemotherapy resistance and poor
clinical outcome in patients receiving systemic chemotherapy for
metastatic colon cancer. Int J Colorectal Dis. 28:8872013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He Z, Yu L, Luo S, Li M, Li J, Li Q, Sun Y
and Wang C: miR-296 inhibits the metastasis and
epithelial-mesenchymal transition of colorectal cancer by targeting
S100A4. BMC Cancer. 17:1402017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fu Q, Song X, Liu Z, Deng X, Luo R, Ge C,
Li R, Li Z, Zhao M, Chen Y, et al: miRomics and proteomics reveal a
miR-296-3p/PRKCA/FAK/Ras/c-Myc feedback loop modulated by
HDGF/DDX5/β-catenin complex in lung adenocarcinoma. Clin Cancer
Res. 23:6336–6350. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Deng X, Liu Z, Liu X, Fu Q, Deng T, Lu J,
Liu Y, Liang Z, Jiang Q, Cheng C, et al: miR-296-3p negatively
regulated by nicotine stimulates cytoplasmic translocation of c-Myc
via MK2 to suppress chemotherapy resistance. Mol Ther.
26:1066–1081. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li H, Li J, Shi B and Chen F: MicroRNA-296
targets AKT2 in pancreatic cancer and functions as a potential
tumor suppressor. Mol Med Rep. 16:466–472. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma L and Pei G: Beta-arrestin signaling
and regulation of transcription. J Cell Sci. 120:213–218. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang YX, Li XF, Yuan GQ, Hu H, Song XY,
Li JY, Miao XK, Zhou TX, Yang WL, Zhang XW, et al: β-Arrestin 1 has
an essential role in neurokinin-1 receptor-mediated glioblastoma
cell proliferation and G2/M phase transition. J Biol Chem.
292:8933–8947. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guzińska-Ustymowicz K, Pryczynicz A,
Kemona A and Czyzewska J: Correlation between proliferation
markers: PCNA, Ki-67, MCM-2 and antiapoptotic protein Bcl-2 in
colorectal cancer. Anticancer Res. 29:3049–3052. 2009.PubMed/NCBI
|
|
31
|
Sinicrope FA, Ruan SB, Cleary KR, Stephens
LC, Lee JJ and Levin B: bcl-2 and p53 oncoprotein expression during
colorectal tumorigenesis. Cancer Res. 55:237–241. 1995.PubMed/NCBI
|