Introduction
Multidrug resistance (MDR) is a major impediment to
effective cancer chemotherapy and the treatment of infectious
diseases (1). The development of
drug resistance remains a dominant obstacle toward curative cancer
treatment. MDR can be induced by numerous mechanisms, including
reduced drug uptake, altered cell cycle checkpoints and cell cycle
arrest, altered drug targets, increased drug efflux by drug
transporters, or sequestration of anticancer drugs in lysosomes,
intracellular organelles and intercellular vesicles (2–12).
Overexpression of various ATP-dependent efflux pumps, particularly
ATP binding cassette (ABC) subfamily B member 1, which encodes
multidrug resistance protein 1 (MDR1, also known as
P-glycoprotein), is a well-known resistance mechanism against
several chemotherapeutic agents; for example, taxanes,
anthracyclines, vinca alkaloids and epipodopyllotoxins. This has
been demonstrated in previous studies (13), both in tumor cell lines and in
numerous tumor types, including solid tumors and hematological
malignancies (13).
In tumor cells, MDR1 pumps out anticancer drugs,
such as doxorubicin, taxanes, anthracyclines, vinca alkaloids and
epipodopyllotoxins, resulting in decreased intracellular drug
concentrations (14,15). MDR1-mediated drug resistance can be
effectively overcome by either blocking the function of MDR1 or by
inhibiting its expression (16). In
addition, other mechanisms underlying drug resistance have been
described, including activation of p53, deletion or inactivation of
the pro-apoptotic gene caspase 3, increased expression of the
proto-oncogene B-cell lymphoma 2 (Bcl-2), suppression of the
apoptosis pathway and increased protein expression of ATP
transporters, all of which can promote tumor cell resistance to
chemotherapeutic drugs (17–21).
Apoptosis involves a series of morphological and
biochemical events, including DNA degradation, cell shrinkage,
protein cleavage, protein cross-linking, phosphatidylserine
exposure, increased mitochondrial membrane permeability and cell
membrane deformities (22). There
are two major apoptotic pathways: The extracellular pathway, which
is mediated by death receptors, and the intracellular pathway,
which is also known as mitochondrial apoptosis. Although these two
pathways function separately, they are interconnected and
eventually converge on the same target (23). The process of apoptosis is closely
regulated by cells through a series of mitochondrial pathway
molecules, particularly the Bcl-2 family (24). Apoptosis is a normal form of cell
death that is closely associated with biological development, organ
formation and physical balance. The most prominent feature of tumor
cells is the ability to escape control by the host and to avoid
cell apoptosis.
The ubiquitin-proteasome pathway (UPP) is an
important pathway that selectively degrades proteins in
vivo; the majority of eukaryotic protein degradation is
accomplished in this manner (25).
This pathway also serves an important role in the regulation of
apoptosis. It has previously been reported that proteasome
inhibitors can induce cellular apoptosis in various tumor cells by
blocking the ubiquitination pathway; these findings provide a
direct and powerful means for studying the role of the UPP in
apoptosis (25). In addition, the
proteasome inhibitor, MG132, induces nuclear damage, reduces
mitochondrial transmembrane potential, releases cytochrome
c, induces the formation of reactive oxygen species (ROS),
activates caspase-8, and affects Bcl-2 family proteins to affect
MDR. Therefore, proteasome inhibitors are closely associated with
drug resistance in cancer therapy (26–28).
Natural products are garnering interest in the field
of cancer therapy. Some compounds extracted from fruits,
vegetables, oilseeds and herbs have been reported to modulate the
activity of MDR1 (29–31). Levistolide A is a natural compound
extracted from the rhizome of Angelicae sinensis (Oliv.)
(32). It has been used clinically
in China to treat gynecological symptoms, anemia, chronic
bronchitis, asthma, rheumatism and other diseases (33). With regards to antitumor therapies,
the most important effects of Angelicae sinensis are that it
enhances the efficacy and reduces the normal cell toxicity of
chemotherapeutic drugs (34–36).
Our previous study screened and identified levistolide A as an
active compound capable of inhibiting tumor cell proliferation
(37). The present study aimed to
reveal the mechanisms underlying the synergistic effects of
levistolide A towards doxorubicin-induced apoptosis of k562/dox
cells.
Materials and methods
Reagents
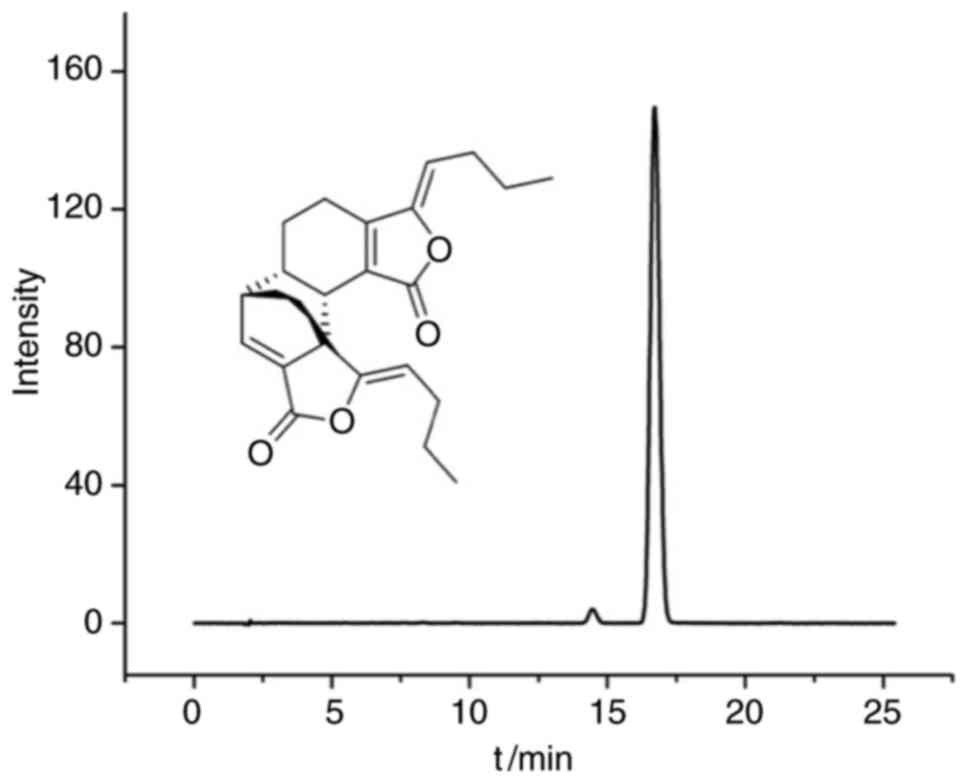
Levistolide A (Fig.
1) was obtained from Shanghai Suo Laibao Biotechnology Co.,
Ltd. (Shanghai, China). Doxorubicin was obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Antibodies against
the apoptosis regulators Bcl-2 (cat. no. 4223) and Bcl-2-associated
X protein (Bax; cat. no. 5023) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Primary antibodies against
MDR1 (cat. no. sc-8313) were obtained from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA), against breast cancer
resistance protein [BCRP (ERP2099); cat. no. ab108312) were
purchased from Abcam (Cambridge, MA, USA) and GAPDH (cat. no.
AB-P-R001) were obtained from Hangzhou Goodhere Biotechnology Co.,
Ltd. (Hangzhou, China) MG132, dimethyl sulfoxide (DMSO) and MTT
were purchased from Sigma-Aldrich; Merck KGaA. RPMI-1640 medium was
obtained from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA,
USA).
For all experiments, stock solutions of the drugs
were dissolved in DMSO. The final concentrations of the drugs were
obtained by diluting the stock solutions in RPMI-1640 culture
medium. The final concentration of DMSO was always <0.2% in
order to avoid cell toxicity.
Cell culture
Doxorubicin-induced MDR erythroleukemic k562/dox
cells and parental k562 cells were obtained from the Institute of
Biochemistry and Cell Biology, Chinese Academy of Sciences
(Shanghai, China). To maintain doxorubicin resistance, k562/dox
cells were cultured with 1.0 µM doxorubicin. Cells were maintained
in RPMI-1640 medium supplemented with 10% (v/v) heat-inactivated
fetal bovine serum (HyClone; GE Healthcare Life Sciences, Logan,
UT, USA) at 37°C under a humidified atmosphere containing 5%
CO2.
Cytotoxicity assays
Cell viability was determined using the MTT assay.
k562 and k562/dox cells were seeded into 96-well plates
(5×103 cells/well) and were then treated with various
concentrations of doxorubicin (with or without levistolide A) for
48 h at 37°C. In addition, k562/dox cells were incubated with 0, 2,
4, 8 and 10 µM MG132 for 24 h at 37°C. After discarding the medium,
the MTT solution (500 µg/ml) was added and cells were incubated for
4 h at 37°C. The formazan was solubilized with DMSO and relative
cell viability was measured at 490 nm with a microplate reader
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Growth inhibition
in response to various concentrations of doxorubicin and
levistolide A was calculated using GraphPad Prism 7.0 (GraphPad
Software, Inc., La Jolla, CA, USA).
The nature of the interaction (whether it was
synergistic, additive or antagonistic) between doxorubicin and
levistolide A, as a function of their concentrations and cell
growth inhibition, was assessed using the combination index (CI)
method (38). For the combination
of two compounds: CI=D1/Dx1 + D2/Dx2 +
D1×D2/Dx1xDx2; where D1 and D2 are the
concentrations required to observe the × % effect when K562/dox
cells were cotreated with doxorubicin and levistolide A;
Dx1 and Dx2 are the concentrations required
to observe the × effect with each of the two drugs alone. The CI
helps to identify synergistic (CI<1), additive (CI=1) or
antagonistic (CI>1) interactions.
Cellular doxorubicin accumulation
assay
The k562/dox and k562 cells were cultured in 6-well
plates for 24 h to allow attachment. Subsequently, the cells were
incubated with DMSO alone (control), with 1.2 µM doxorubicin, or
with doxorubicin plus 50 or 100 µM levistolide A for 24 h. After
washing twice with PBS, the cells were treated with 400 µl PBS and
were gently mixed before being analyzed using a FACSort flow
cytometer with CellQuest Pro Ver. 6.0 software (both BD
Biosciences, Franklin Lakes, NJ, USA).
Cell apoptosis
The k562/dox cells (1×105 cells/ml) were
seeded into 6-well plates. Once they reached 80% confluence, cells
were treated with 1.2 µM doxorubicin; 0, 10 or 100 µM levistolide
A; or with both drugs for 24 h. Apoptosis was detected using the
Annexin V-fluorescein isothiocyanate/propidium iodide (PI) double
staining method. The cells were washed twice with ice-cold PBS,
resuspended in PBS (100 µl) and were incubated with Annexin V and
PI labeling solution (5 µl) at room temperature for 15 min.
Subsequently, the cells were gently vortexed and incubated for 15
min at room temperature in the dark. Following the addition of 400
µl 1X binding buffer, the percentage of apoptotic cells was
measured within 1 h using the BD Accuri™ C6 Plus Flow Cytometer (BD
Biosciences).
ROS assay
The k562/dox cells (1×105 cells/ml) were
seeded into 6-well plates. Once they reached 80% confluence, cells
were treated with 1.2 µM doxorubicin; 0, 10 or 100 µM levistolide
A; or with both drugs for 12 h. ROS were detected using the ROS
Assay kit (cat. no. 50101ES01; Shanghai Yi San Biotechnology Co.,
Ltd., Shanghai, China), according to the manufacturer's
protocol.
JC-1 mitochondrial membrane potential
assay
The k562/dox cells (1×105 cells/ml) were
seeded into 6-well plates. Once they reached 80% confluence, cells
were treated with 1.2 µM doxorubicin; 0, 10 or 100 µM levistolide
A; or with both drugs for 24 h. The mitochondrial membrane
potential was analyzed using the JC-1 Mitochondrial Membrane
Potential kit (cat. no. 40706ES60; Shanghai Yi San Biotechnology
Co., Ltd.), according to the manufacturer's protocol.
Caspase 3 colorimetric assay
The k562/dox cells (1×105 cells/ml) were
seeded into 6-well plates. Once they reached 80% confluence, cells
were treated with 1.2 µM doxorubicin; 0, 50 or 100 µM levistolide
A; or with both drugs for 24 h. Caspase 3 levels were detected
using the caspase 3 colorimetric assay kit (cat. no. BC3830;
Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China), according to the manufacturer's protocol.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was used to evaluate MDR1 mRNA expression
following treatment with levistolide A (0, 0.01, 0.1, 1, 10 and 100
µM) for 24 h. Total RNA was extracted from k562/dox cells using the
E.Z.N.A.® Total RNA kit I (Omega Bio-tek, Inc.,
Norcross, GA, USA), according to the manufacturer's protocol, and
cDNA was synthesized using reverse transcriptase (PrimeScript™ RT
reagent kit; cat. no. RR047A; Takara Bio, Inc., Otsu, Japan),
according to the manufacturer's protocol. RT-qPCR
(SYBR®-Green JumpStart™ Taq ReadyMix™; cat. no.
S5193; Sigma-Aldrich; Merck KGaA) amplification was performed using
a TP-810 Thermal Cycler system (Takara Bio, Inc.). The primer
sequences were as follows: MDR1, forward
5′-GCAGCTGGAAGACAAATACACAAA-3′, and reverse
5′-CCCCAACATCGT-GCACATC-3′; and GAPDH, forward
5′-TCTGCAGGAGACAAGACCTG-3′ and reverse 5′-GACTTGAGGAGCTGGAGAGG-3′.
The PCR protocol was as follows: One cycle of denaturation at 95°C
for 2 min; 40 cycles of denaturation at 95°C for 15 sec, annealing
at 60°C for 30 sec and extension at 72°C for 30 sec, followed by a
final extension step at 4°C for 1 min. Relative quantification
(ΔCq) (39) was used to determine
relative mRNA expression levels, with GAPDH as the internal
reference.
Western blot analysis
Following incubation with various concentrations of
levistolide A, doxorubicin, or both drugs for 24 h; or incubation
with various concentrations of levistolide A combined with MG132 (2
µM), k562/dox cells or k562 cells were washed twice with cold PBS
and lysed with lysis buffer (Xi'an Hat Biotechnology Co., Ltd.,
Xi'an, China). Proteins were extracted using the total protein
extraction kit (Xi'an Hat Biotechnology Co., Ltd.); the cells were
treated with lysis buffer and proteins were extracted from the
lysates using the protein extraction kit. Lysates were centrifuged
at 3,600 × g for 20 min at 4°C. The bicinchoninic acid protein
assay kit (Thermo Fisher Scientific, Inc.) was used to quantify
protein concentrations. Proteins were denatured by mixing with an
equal volume of loading buffer (100 µl loading buffer and 4 µl
mercaptoethanol) and boiling at 100°C for 5 min. An aliquot (30 µg
protein) of the supernatants was run on a 10% SDS polyacrylamide
gel. Proteins were then transferred onto polyvinylidene fluoride
membranes, which were blocked with 5% non-fat milk in Tris-buffered
saline plus 0.1% Tween-20 for 2 h at 25°C. Proteins were detected
using the following antibodies at 4°C for 24 h: Anti-GAPDH
(1:1,000), anti-MDR1 (1:1,000), anti-Bcl-2 (1:1,000), anti-Bax
(1:1,000) and anti-BCRP (1:1,000). The blots were washed and were
then incubated with their respective secondary antibodies
(1:20,000; cat. no. ANM02-4; Zhuangzhi Biotechnology Co., Ltd.,
Xi'an, China) at 25°C for 2 h. Proteins were visualized using an
enhanced chemiluminescence detection reagent and GAPDH was used as
a reference to estimate relative protein levels. ImageJ k1.45
(National Institutes of Health, Bethesda, MD, USA) was used to
semi-quantify protein expression.
Statistical analysis
Data are expressed as the means ± standard
deviation. Differences between control and experimental values were
assessed using one-way analysis of variance followed by
Bonferroni's multiple comparisons test (GraphPad 7.0 Software;
GraphPad Software, Inc., La Jolla, CA, USA.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Antiproliferative effects of
levistolide A on k562 and k562/dox cells
The effects of levistolide A and doxorubicin on k562
and k562/dox proliferation after 48 h were assessed using the MTT
assay (Fig. 2A-D). Levistolide A
exhibited a mild, dose-dependent inhibitory effect on the survival
of k562 (Fig. 2A) and k562/dox
(Fig. 2C) cells at 100 µM; however,
cell viability was still >80% at this concentration. The half
maximal inhibitory concentration (IC50) of doxorubicin
in k562 and k562/dox cells was 2.86±0.64 and 102.56±2.89 µM,
respectively (Table I). The
IC50 of levistolide A in k562/dox cells was 200.5±18.66
µM (Fig. 2E). Subsequently, the
present study examined whether levistolide A modified the
sensitivity of k562 and k562/dox cells to doxorubicin.
Co-incubation with ≥10 µM levistolide A markedly increased the
response of k562/dox cells to doxorubicin (Fig. 2D). Conversely, the synergistic
effect of levistolide A plus doxorubicin was absent in k562 cells
(Fig. 2B). The interaction between
levistolide A and doxorubicin was analyzed using the CI method, as
shown in Fig. 2F. In k562/dox
cells, levistolide A and doxorubicin exhibited a synergistic effect
when the doxorubicin concentration was >0.3 µM, the levistolide
A concentration was >10 µM, and the combined inhibition was
>5% (Fig. 2D). Conversely,
treatment of k562/dox (Fig. 2A) and
k562 (Fig. 2C) cells with each of
these two compounds individually produced lower antiproliferative
effects. These results indicated that levistolide A could increase
the antiproliferative effects of doxorubicin in k562/dox cells
(Fig. 2D), but not in k562 cells
(Fig. 2B).
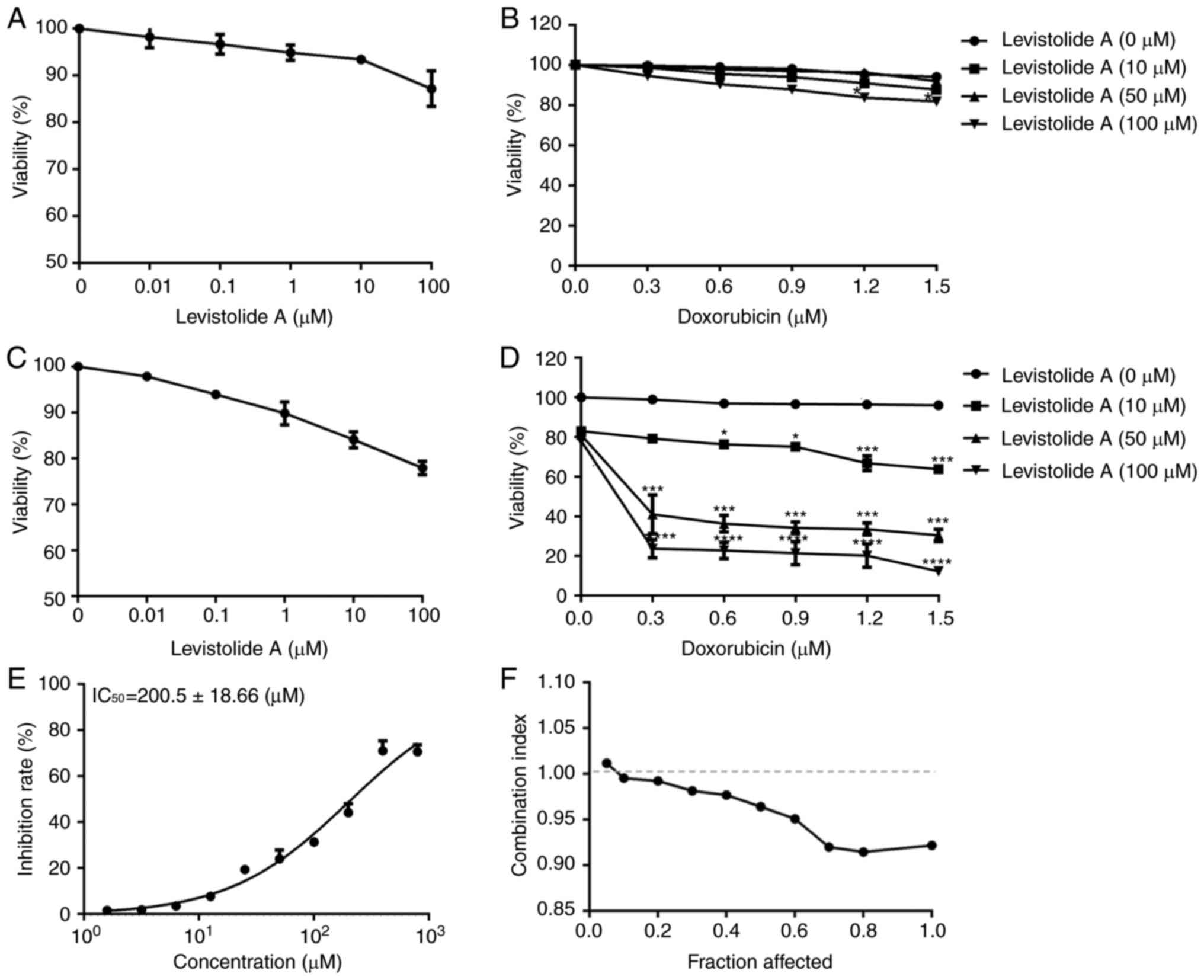 | Figure 2.Effects of levistolide A on k562 and
k562/dox cell viability. (A) Following treatment with various
concentrations of levistolide A for 48 h, the viability of k562
cells was evaluated with MTT assays. (B) After k562 cells were
treated with 0, 0.3, 0.6, 0.9, 1.2 or 1.5 µM doxorubicin together
with 0, 10, 50 or 100 µM levistolide A for 48 h, cell viability was
determined with MTT assays. (C) Following treatment with various
concentrations of levistolide A in for 48 h, the viability of
k562/dox cells was evaluated with MTT assays. (D) After k562/dox
cells were treated with 0, 0.3, 0.6, 0.9, 1.2 or 1.5 µM doxorubicin
together with 0, 10, 50, or 100 µM levistolide A for 48 h, cell
viability was determined with MTT assays. (E) Following treatment
with various concentrations of levistolide A for 48 h, the
IC50 in k562/dox cells was evaluated with MTT assays.
(F) Combination index of levistolide A combined with doxorubicin in
k562/dox cells. One-way analysis of variance (Bonferroni's multiple
comparisons test) was used to determine statistical significance.
Data are presented as the means ± standard deviation, n=3.
*P<0.05 vs. doxorubicin (0 µM); ***P<0.001 vs. doxorubicin (0
µM); ****P<0.0001 vs. doxorubicin (0 µM). |
 | Table I.Determination of multidrug resistance
according to the sensitivity of k562 and k562/dox cells to
doxorubicin. |
Table I.
Determination of multidrug resistance
according to the sensitivity of k562 and k562/dox cells to
doxorubicin.
|
| IC50
(µM) |
|
|---|
|
|
|
|
|---|
| Treatment | k562 | k562/dox | Fold increase in
resistance |
|---|
| Doxorubicin | 2.86±0.64 |
102.56±2.89a | 35.86 |
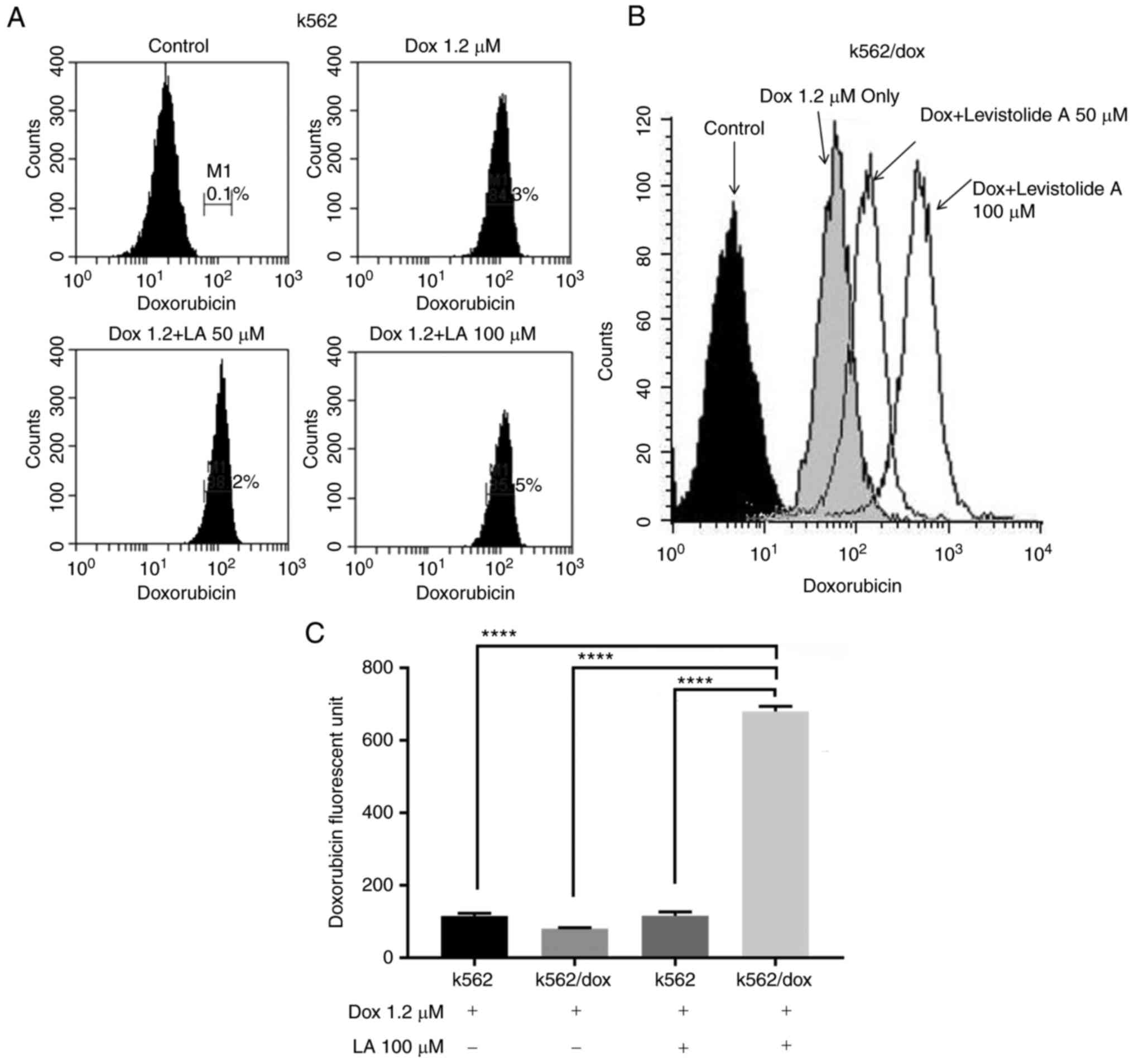
Levistolide A promotes
doxorubicin-induced apoptosis by increasing its intracellular
concentration
The present study conducted fluorescence-activated
cell sorting analyses to investigate whether levistolide A
increased the sensitivity of k562/dox cells to doxorubicin by
increasing its intracellular accumulation (Fig. 3). The experiments revealed that the
fluorescence intensity of intracellular doxorubicin was increased
in a dose-dependent manner in k562/dox cells, indicating that
levistolide A promoted the intracellular accumulation of
doxorubicin (Fig. 3B and C).
Conversely, levistolide A had little effect on the intracellular
accumulation of doxorubicin in k562 cells (Fig. 3A and C). In addition, levistolide A
increased the doxorubicin effective concentration in k562/dox cells
(Fig. 2D). Therefore, combined
treatment of k562/dox cells with levistolide A and doxorubicin
significantly increased their response to doxorubicin.
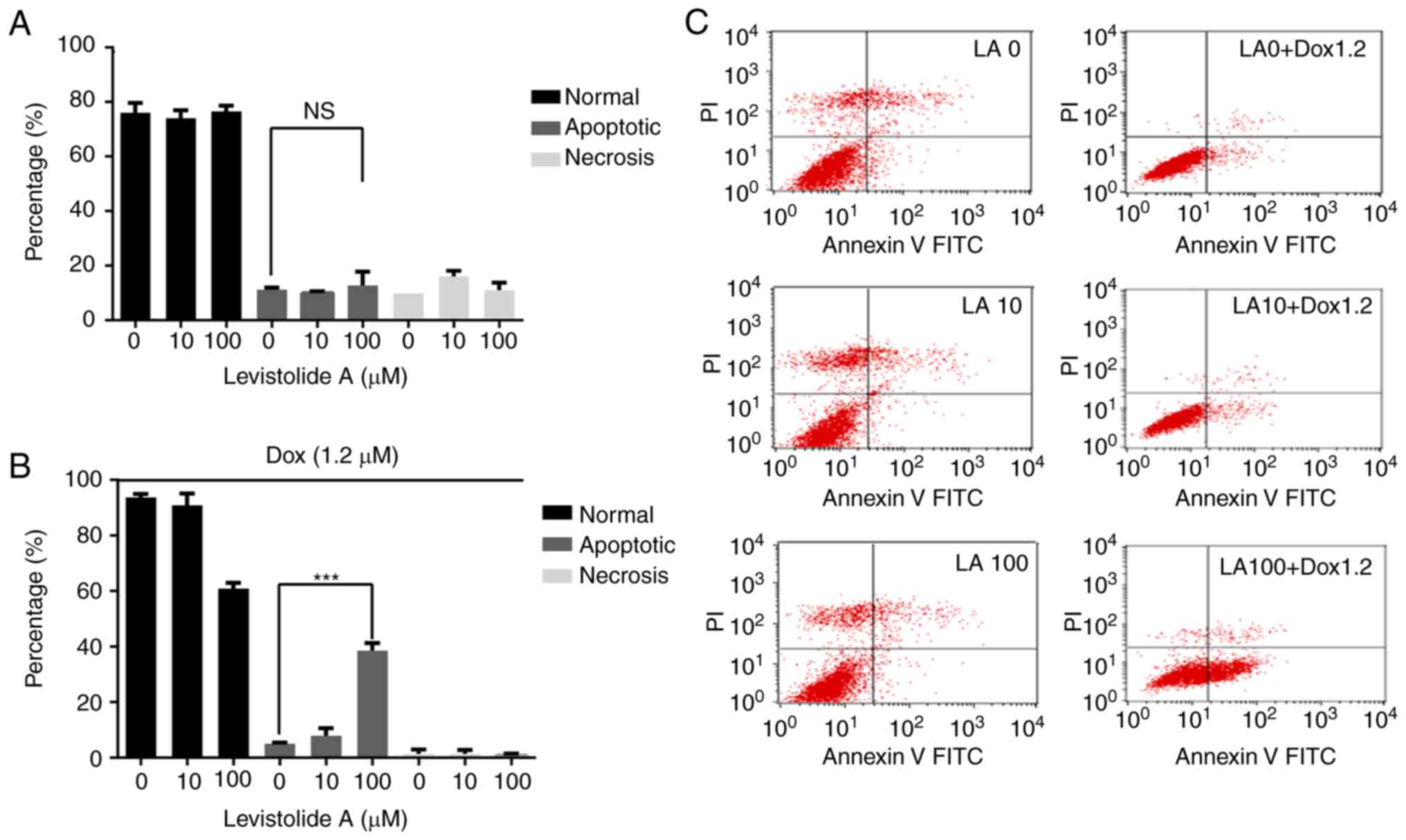
K562/dox cells were treated with various
concentrations of levistolide A alone or combined with 1.2 µM
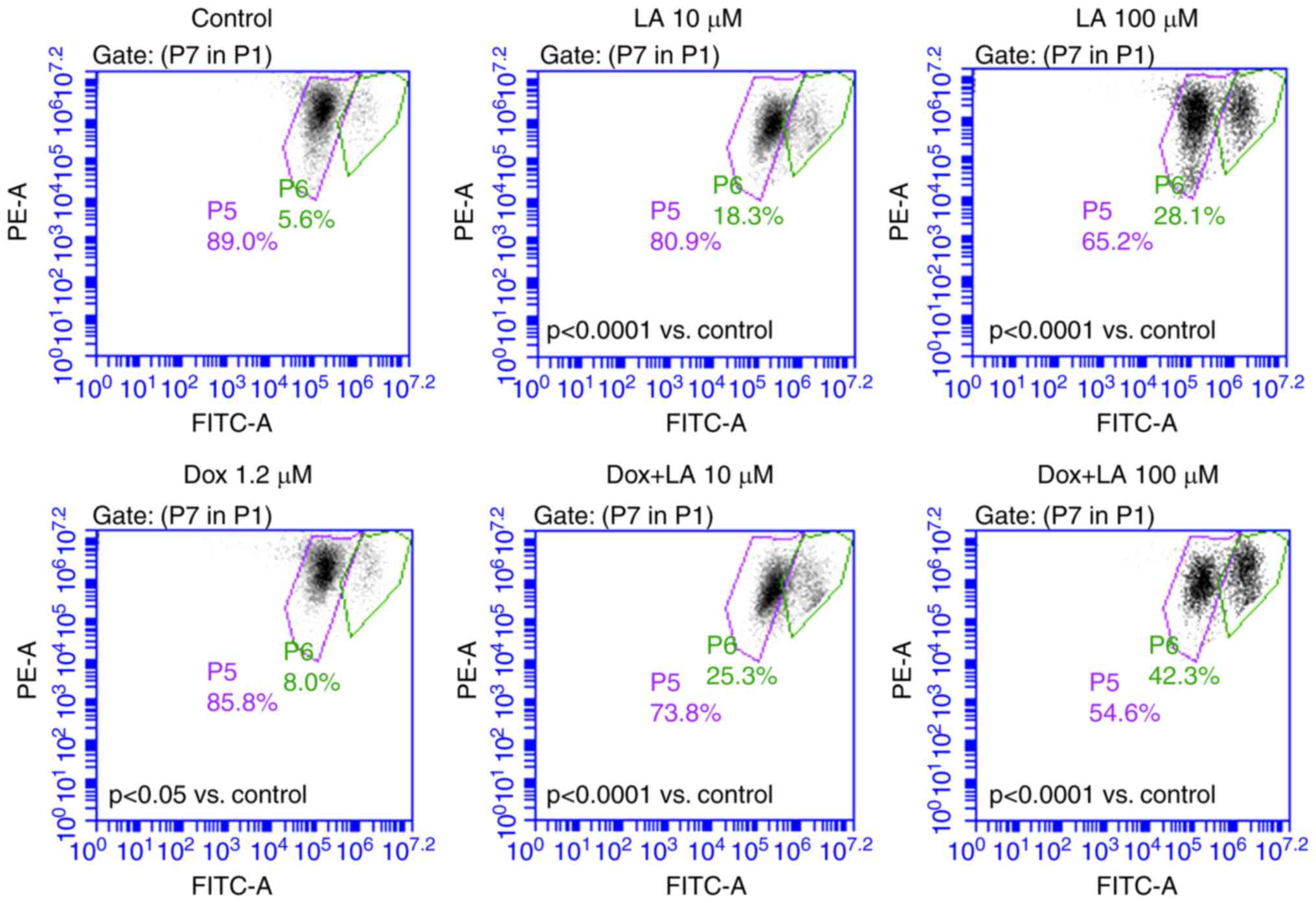
doxorubicin, and apoptosis was analyzed by flow cytometry (Fig. 4). The percentage of apoptotic cells
was significantly increased in the combined treatment group
(Fig. 4B). The results revealed
that levistolide A combined with doxorubicin increased the
percentage of apoptotic cells in a dose-dependent manner (Fig. 4).
The present study also investigated whether
increased expression of ROS could explain the synergistic effects
of levistolide A and doxorubicin (Fig.
5). The k562/dox cells were treated with 0, 10 or 100 µM
levistolide A, either alone or combined with 1.2 µM doxorubicin for
12 h. The results revealed that levistolide A dose-dependently
promoted doxorubicin-induced apoptosis of k562/dox cells via
increasing ROS levels. Furthermore, analysis of the mitochondrial
potential using JC-1 staining indicated that levistolide A
synergistically enhanced doxorubicin-induced cell death (Fig. 6). The k562/dox cells were treated
with 0, 10 or 100 µM levistolide A, either alone or combined with
1.2 µM doxorubicin for 24 h. The present study revealed that
levistolide A dose-dependently promoted doxorubicin-induced
apoptosis of k562/dox cells by reducing mitochondrial membrane
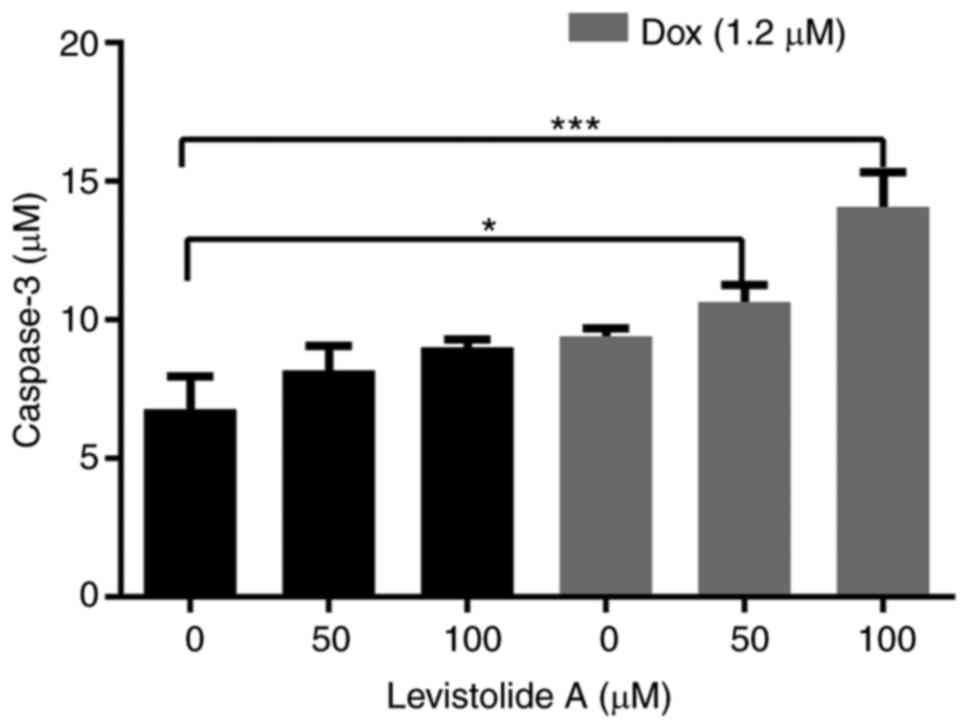
potential. In addition, increased expression levels of caspase 3
were detected in the combined treatment group (Fig. 7). Western blotting was also
performed to analyze the expression levels of apoptotic markers,
including Bax and Bcl-2, in k562/dox cells treated with levistolide
A alone or combined with doxorubicin for 24 h (Fig. 8A). Levistolide A alone (50 and 100
µM) induced a slight dose-dependent decrease in Bcl-2 (Fig. 8Ac) compared with in control,
untreated cells. However, levistolide A (50 and 100 µM) combined
with doxorubicin (1.2 µM) resulted in a significant (P<0.001)
dose-dependent reduction in Bcl-2 expression (Fig. 8Ac), when compared with untreated,
control cells. Treatment of k562 cells with levistolide A alone or
in combination with doxorubicin for 24 h revealed that levistolide
A alone induced a slight dose-dependent decrease in Bcl-2 when
compared with control, untreated cells; levistolide A (50 and 100
µM) combined with doxorubicin (1.2 µM) resulted in a similar
downregulation in Bcl-2 (Fig. 8Bc),
thus indicating that levistolide A does not enhance
doxorubicin-induced apoptosis of k562 cells. On the basis of these
results, it may be suggested that levistolide A markedly enhanced
the doxorubicin-induced mitochondrial apoptotic cascade in k562/dox
cells.
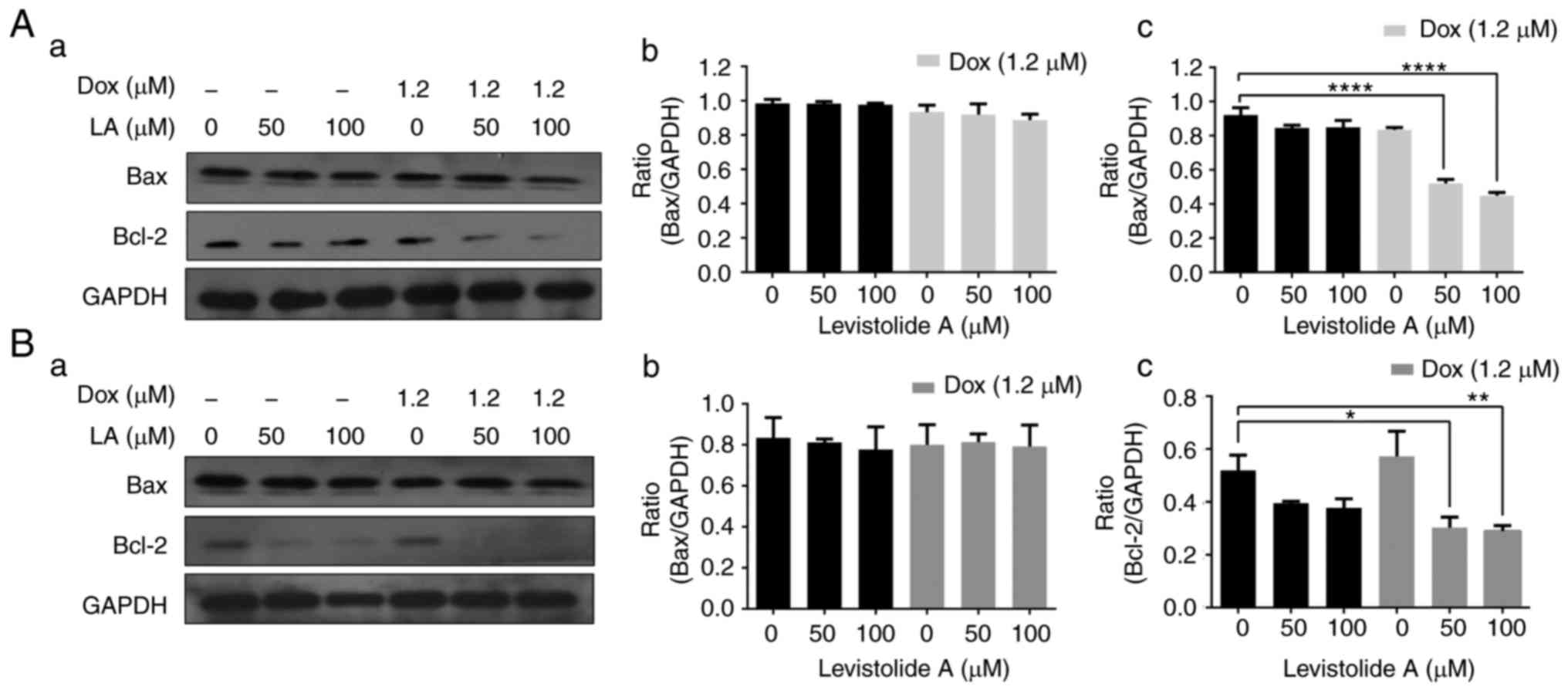 | Figure 8.Levistolide A enhances
doxorubicin-induced k562/dox cell apoptosis by decreasing the
levels of Bcl-2. (Aa) Protein expression levels of Bcl-2 and Bax
were detected in k562/dox cells by western blotting. Cells were
treated with various concentrations of levistolide A (0, 50 and 100
µM) alone or combined with 1.2 µM doxorubicin for 24 h. (Ab) Bax
protein expression in k562/dox cells was semi-quantified by
densitometric analysis, and was normalized to GAPDH protein. (Ac)
Bcl-2 protein expression in k562/dox cells was semi-quantified by
densitometric analysis, and was normalized to GAPDH protein. (Ba)
Protein expression levels of Bcl-2 and Bax were detected in k562
cells by western blotting. Cells were treated with various
concentrations of levistolide A (0, 50 and 100 µM) alone or
combined with 1.2 µM doxorubicin for 24 h. (Bb) Bax protein
expression in k562 cells was semi-quantified by densitometric
analysis, and was normalized to GAPDH protein. (Bc) Bcl-2 protein
expression in k562 cells was semi-quantified by densitometric
analysis, and was normalized to GAPDH protein. One-way analysis of
variance (Bonferroni's multiple comparisons test) was used to
determine statistical significance. Data are presented as the means
± standard deviation, n=3. *P<0.05, **P<0.01, ****P<0.0001
vs. levistolide A (0 µM)-treated cells. Bax, Bcl-2-associated X
protein; Bcl-2, B-cell lymphoma 2. |
Levistolide A downregulates MDR1
expression through the UPP
MDR1 has been reported to be overexpressed in
k562/dox cells (40) and absent in
k562 cells; this finding was confirmed in the present study
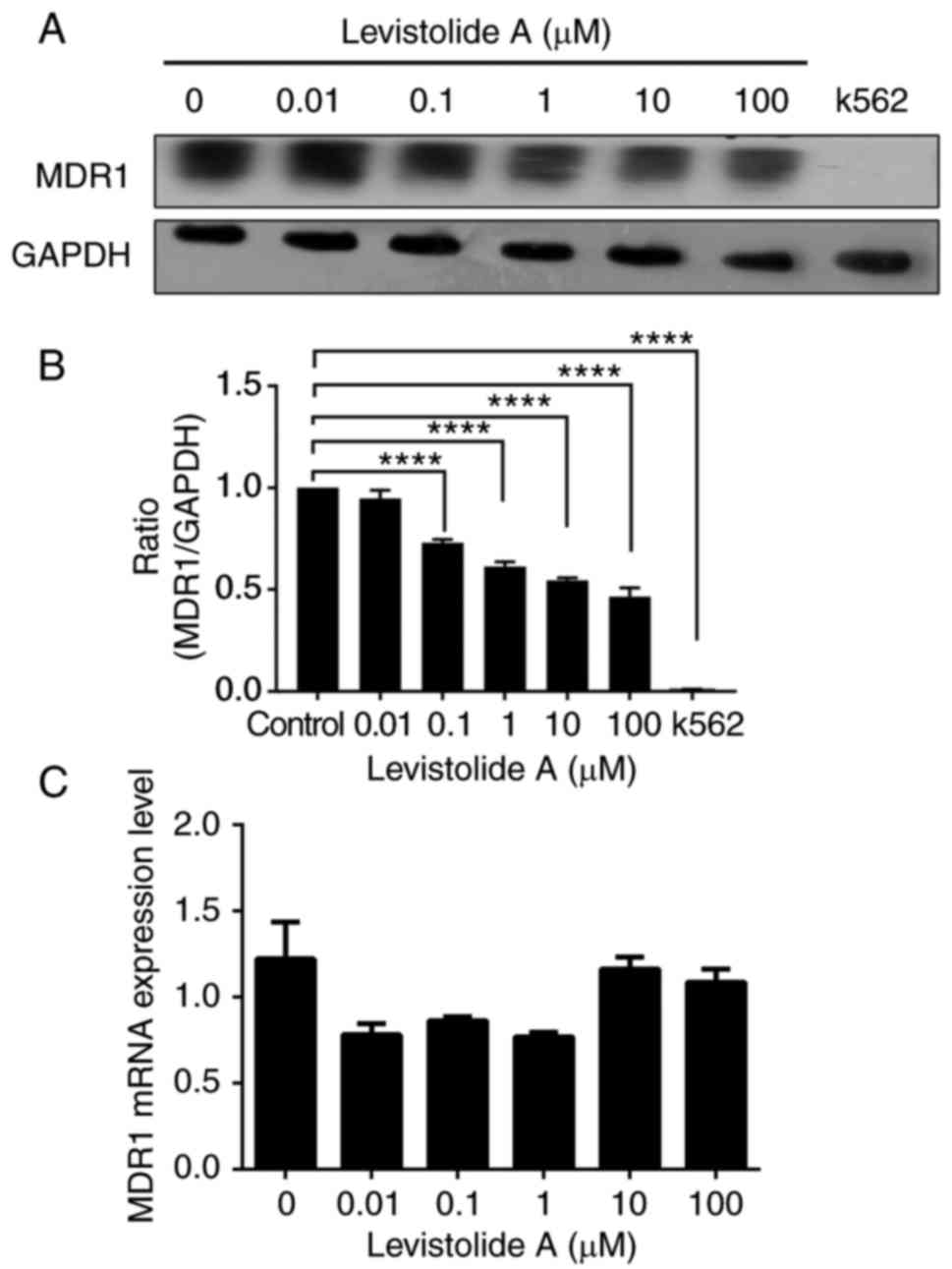
(Fig. 9A and B). Notably,
levistolide A could downregulate MDR1 expression in k562/dox cells
(Fig. 9A and B) without affecting
MDR1 mRNA expression (Fig. 9C),
thus suggesting that it acted via a post-translational pathway.
Therefore, k562/dox cells were treated with MG132, a potent and
cell-permeable proteasome inhibitor, at a low, non-toxic
concentration (2 µM); the results revealed that MG132 exhibited a
dose-dependent inhibitory effect on the survival of k562/dox cells
(Fig. 10A). The results indicated
that MG132 may attenuate levistolide A-induced downregulation
(Fig. 9A) of MDR1 protein levels
(Fig. 10B). Based on these
results, it may be hypothesized that levistolide A induces MDR1
degradation via the UPP.
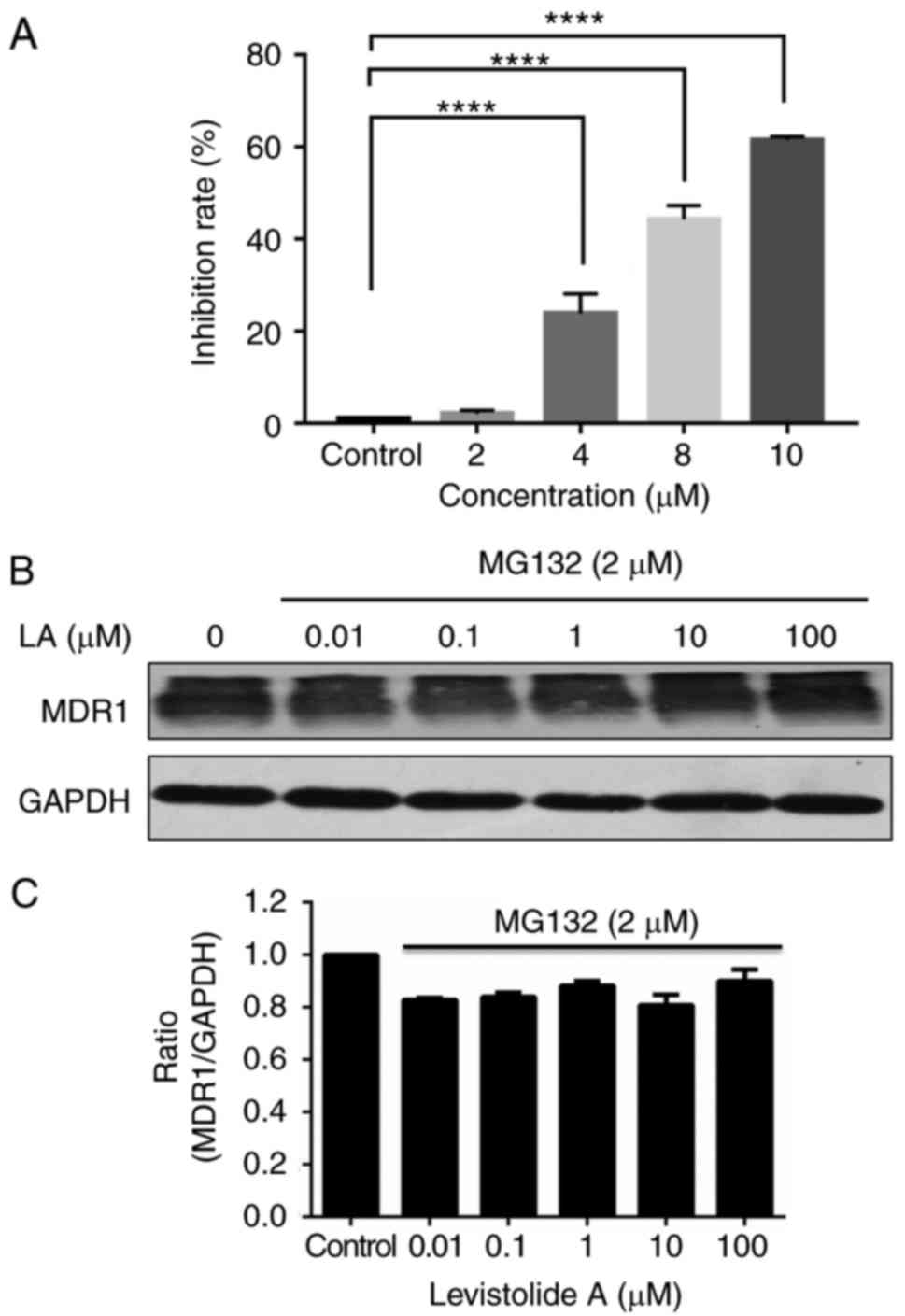 | Figure 10.Levistolide A inhibits the protein
expression levels of MDR1 in k562/dox cells via the
ubiquitin-proteasome pathway. (A) MG132 exhibited a dose-dependent
inhibitory effect on the survival of k562/dox cells; the k562/dox
cells were incubated with 0, 2, 4, 8 and 10 µM MG132 for 24 h. (B)
The k562/dox cells were incubated with 2 µM MG132 together with 0,
0.01, 0.1, 1, 10 or 100 µM levistolide A for 24 h, and MDR1 protein
expression was detected by western blotting. (C) MDR1 protein
expression in k562/dox cells was semi-quantified by densitometric
analysis, and was normalized to GAPDH protein. One-way analysis of
variance (Bonferroni's multiple comparisons test) was used to
determine statistical significance. Data are presented as the means
± standard deviation of three independent experiments.
****P<0.0001 vs. control cells. MDR, multidrug resistance
protein 1. |
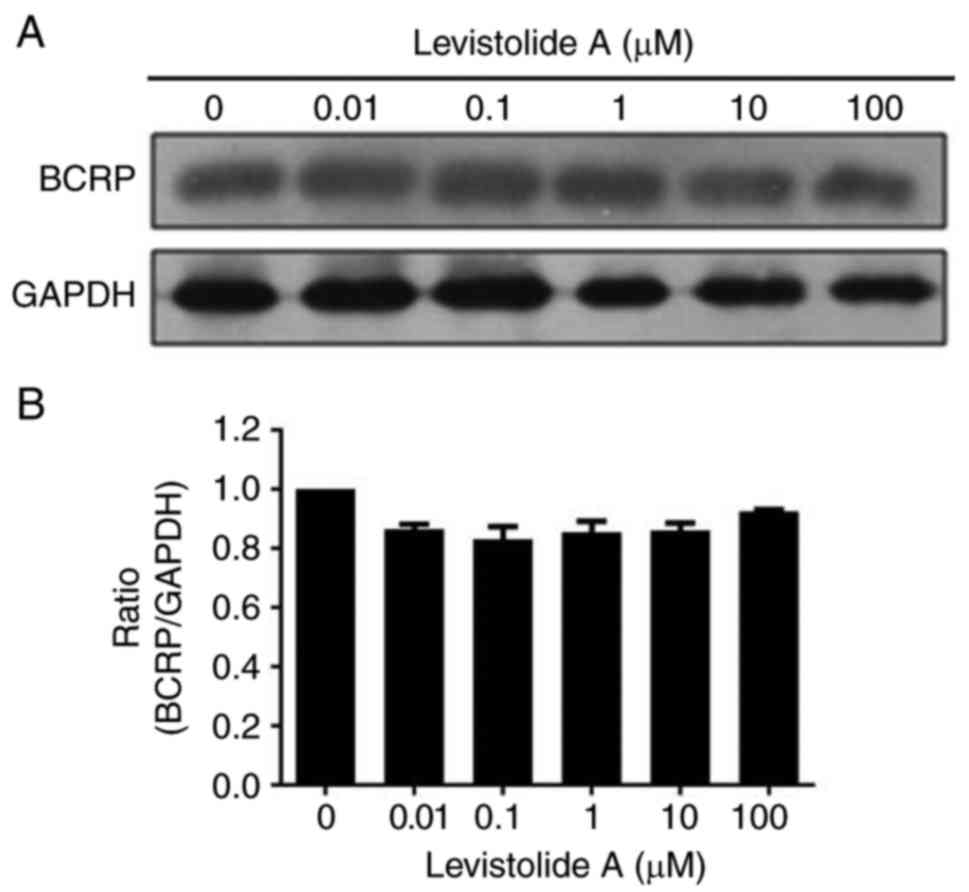
Western blot analysis revealed that BCRP protein
expression was not significantly altered following treatment with
levistolide A (Fig. 11).
Therefore, it may be hypothesized that the effects of levistolide A
on k562/dox cells specifically involves attenuation of MDR via the
UPP, rather than BCRP.
Discussion
Levistolide A is a natural compound extracted from
the rhizome of Angelicae sinensis (Oliv.). According to
Traditional Chinese Medicine, the roots of Angelicae
sinensis are used to tonify circulation and improve blood
stasis. It has also been used clinically for the treatment of
gynecological symptoms, anemia, chronic bronchitis, asthma,
rheumatism and other diseases (33). Angelicae is often included in
vegetable-rich diets due to its beneficial chemopreventive,
antitumor, antioxidant and antimutagenic effects (41).
The IC50 of levistolide A in k562/dox
cells was 200.5±18.66 µM, whereas the IC50 of
doxorubicin in k562 and k562/dox cells was 2.86±0.64 and
102.56±2.89 µM, respectively. Individually, and at low
concentrations (≤100 µM), levistolide A and doxorubicin exhibited
little effects on k562 and k562/dox cell proliferation and
apoptosis. However, levistolide A increased the cytotoxic effects
of doxorubicin against k562/dox cells compared with its effect on
k562 cells. In addition, levistolide A promoted the intracellular
accumulation of doxorubicin in k562/dox cells when compared with
k562 cells, thus suggesting that the MDR1 pump was inhibited by
levistolide A. Compared with in k562/dox cells, k562 cells
exhibited lower sensitivity to the combination of levistolide A and
doxorubicin. The process of apoptosis is closely regulated by a
series of molecules associated with the Bcl-2 family, which are
particularly linked to mitochondrial pathways (24). The present study revealed that
levistolide A promoted doxorubicin-induced apoptosis of k562/dox
cells via increased ROS, reduced mitochondrial membrane potential,
decreased Bcl-2 levels and increased caspase 3 expression.
Treatment with levistolide A alone had only a mild effect on ROS,
JC-1 mitochondrial membrane potential, Bcl-2 and caspase 3 levels
when compared with control, untreated cells. The combination of the
two drugs had little effect on Bax, thus suggesting that apoptosis
was Bax-independent. In k562 cells, levistolide A alone or combined
with doxorubicin induced a significant dose-dependent decrease in
Bcl-2 expression, indicating that levistolide A induced apoptosis
of k562 cells, whereas the pro-apoptotic effect of doxorubicin was
weaker in k562/dox cells. These findings indicated that levistolide
A promoted doxorubicin-induced cell apoptosis via mitochondrial
pathways and by increasing the intracellular concentration of
doxorubicin.
MDR1 is a member of the ABC transporter superfamily.
Various ABC transporters, such as MDR1 and BCRP, have overlapping
substrates (42). MDR1 has been
reported to be overexpressed in k562/dox cells (40), and this was confirmed in the present
study. Notably, this study revealed that levistolide A effectively
suppressed MDR1 protein expression; however, MDR1 mRNA expression
was not significantly altered in cells treated with levistolide A.
Therefore, it may be hypothesized that the decrease in MDR1 levels
is a result of post-transcriptional regulation. The majority of
eukaryotic protein degradation is accomplished by the UPP, and
MG132 is an effective and reversible aldehyde peptide-specific
proteasome inhibitor. Therefore, this study used MG132 to validate
the hypothesis that levistolide A downregulates the expression of
MDR1 through the UPP pathway. It was revealed that MG132 (2 µM)
could reverse the levistolide A-induced reduction in MDR1
expression, thus indicating that levistolide A may negatively
regulate MDR1 expression via the UPP. In addition, the protein
expression levels of BCRP were not significantly altered following
treatment with levistolide A. Therefore, the effects of levistolide
A may be mediated by downregulation of MDR1 expression, and not
BCRP.
Fei et al reported that levistolide A
modulates MDR1 function and can overcome MDR1-mediated MDR in
Bcap37/MDR1 cells. In addition, levistolide A inhibits
3,3′-diethyloxacarbocyanine iodide efflux from Bcap37/MDR1 cells
without affecting MDR1 expression (32). In the present study, the levistolide
A-mediated reversal of doxorubicin resistance in k562/dox cells
occurred via modulation of MDR1 expression.
In conclusion, the present findings suggested that
levistolide A synergistically increased the cytotoxicity of
doxorubicin and promoted doxorubicin-induced apoptosis of k562/dox
cells. This effect involved the apoptotic cascade, which was
regulated by the mitochondrial system, and downregulation of MDR1
expression. Therefore, levistolide A may be a promising drug
candidate for the treatment of doxorubicin-resistant cancer by
targeting MDR1.
Acknowledgements
Not applicable.
Funding
The present study was financially supported by the
National Natural Science Foundation of China (grant nos. 81101725,
81202495 and 81230079), the Fundamental Research Funds for the
Central University (grant no. xjj2016125), the Natural Science
Foundation of Shaanxi Province (grant no. 2017JM8035), and the
Hospital Fund of the First Affiliated Hospital of Xi'an Jiaotong
University (grant no. 2016QN-12).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YD performed the research, analyzed the data and
wrote the paper. WN and TZ performed the research and wrote the
paper. JW performed statistical analysis. JC, HC and RW performed
cell culture. HA designed the study, performed the research,
analyzed the data and wrote the paper. All authors read and
approved the final manuscript.
Ethical approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang YL: Tiny mutation triggers drug
resistance for patients with one type of leukemia. The University
of Chicago Medicine Communications. http://www.uchospitals.edu/news/2014/20140528-leukemia.html
|
|
2
|
Chai S, To KK and Lin G: Circumvention of
mult-drug resistance of cancer cells by Chinese herbal medicines.
Chin Med. 5:262010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goler-Baron V and Assaraf YG: Structure
and function of ABCG2-rich extracellular vesicles mediating
multidrug resistance. PLoS One. 6:e160072011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goler-Baron V, Sladkevich I and Assaraf
YG: Inhibition of the PI3K-Akt signaling pathway disrupts
ABCG2-rich extracellular vesicles and overcomes multidrug
resistance in breast cancer cells. Biochem Pharmacol. 83:1340–1348.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gonen N and Assaraf YG: Antifolates in
cancer therapy: Structure, activity and mechanisms of drug
resistance. Drug Resist Updat. 15:183–210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ifergan I, Scheffer GL and Assaraf YG:
Novel extracellular vesicles mediate an ABCG2-dependent anticancer
drug sequestration and resistance. Cancer Res. 65:10952–10958.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lebedeva IV, Goler-Baron V and Assaraf YG:
Overcoming multidrug resistance via photo destruction of ABCG2-rich
extracellular vesicles sequestering photosensitive
chemotherapeutics. PLoS One. 7:e354872012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Livney YD and Assaraf YG: Rationally
designed nanovehicles to overcome cancer chemoresistance. Adv Drug
Deliv Rev. 65:1716–1730. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Raz S, Sheban D, Gonen N, Stark M, Berman
B and Assaraf YG: Severe hypoxia induces complete antifolate
resistance in carcinoma cells due to cell cycle arrest. Cell Death
Dis. 5:e10672014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhitomirsky B and Assaraf YG: Lysosomal
sequestration of hydrophobic weak base chemotherapeutics triggers
lysosomal biogenesis and lysosome-dependent cancer multidrug
resistance. Oncotarget. 6:1143–1156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhitomirsky B and Assaraf YG: Lysosomes as
mediators of drug resistance in cancer. Drug Resist Updat.
24:23–33. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou X, Li D, Wang X, Zhang B, Zhu H and
Zhao J: Galectin-1 is overexpressed CD133+ human lung
adenocarcinoma cells and promotes their growth and invasiveness.
Oncotarget. 6:3111–3122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Genovese I, Ilari A, Assaraf YG, Fazi F
and Colotti G: Not only P-glycoprotein: Amplification of the
ABCB1-containing chromosome region 7q21 confers multidrug
resistance upon cancer cells by coordinated overexpression of an
assortment of resistance related proteins. Drug Resist Updat.
32:23–46. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Glavinas H, Krajcsi P, Cserepes J and
Sarkadi B: The role of ABC transporters in drug resistance,
metabolism and toxicity. Curr Drug Deliv. 1:27–42. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Varma MV, Ashokraj Y, Dey CS and
Panchagnula R: P-glycoprotein inhibitors and their screening: A
perspective from bioavailability enhancement. Pharmacol Res.
48:347–359. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gottesman MM, Fojo T and Bates SE:
Multidrug resistance in cancer: Role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hipfner DR, Deeley RG and Cole SP:
Structural, mechanistic and clinical aspects of MRP1. Biochim
Biophys Acta. 146:1359–1376. 1999.
|
|
18
|
Ozvegy C, Litman T, Szakács G, Nagy Z,
Bates S, Váradi A and Sarkadi B: Functional characterization of the
human multidrug transporter, ABCG2, expressed in insect cells.
Biochem Biophys Res Commun. 285:111–117. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roy S, Kenny E, Kennedy S, Larkin A,
Ballot J, Perez De Villarreal M, Grown J and O'Driscoll L:
MDR1/P-glycoprotein and MRP-1mRNA and protein expression in
non-small cell lung cancer. Anticancer Res. 27:1325–1330.
2007.PubMed/NCBI
|
|
20
|
Sparrebppm A, Danesi R, Ando Y, Chan J and
Figg WD: Phamacogenomics of ABC transporters and its role in cancer
chemotherapy. Drug Resist Updat. 6:71–84. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sanchez C, Mendoza P, Contreras HR,
Vergara J, McCubrey JA, Huidobro C and Castellón EA: Expression of
multidrug resistance proteins in prostate cancer is related with
cell sensitivity to chemotherapeutic drugs. Prostate. 69:1448–1459.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Igney FH and Krammer PH: Death and
anti-death: Tumour resistance to apoptosis. Nat Rev Cancer.
2:277–288. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shore GC and Nguyen M: Bcl-2 proteins and
apoptosis: Choose your partner. Cell. 135:1004–1006. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Glickman MH and Ciechanover A: The
ubiquitin-proteasome protcolytic pathway: Destruction for the sake
of construction. Physiol Rev. 82:373–428. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee CS, Han ES, Ha YS and Bang H:
Differential effect of calmodulin antagonists on MG132-induced
mitochondrial dysfunction and cell death in PC12 cells. Brain Res
Bull. 67:225–234. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bang JH, Han ES, Lim L and Lee CS:
Differential response of MG132 cytotoxicity against small cell lung
cancer cells to change in cellular GSH contents. Biochem Pharmacol.
68:659–666. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan XB, Yang DS, Gao X, Feng J, Shi ZL and
Ye Z: Caspase-8 dependent osteosarcoma cell apoptosis induced by
proteasome inhibitor MG132. Cell Biol Int. 31:1136–1143. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chung SY, Sung MK, Kim NH, Jang JO, Go EJ
and Lee HJ: Inhibition of Pglycoprotein by natural products in
human breast cancer cells. Arch Pharm Res. 28:823–828. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mothana RA, Lindequist U, Gruenert R and
Bednarski PJ: Studies of the in vitro anticancer, antimicrobial and
antioxidant potentials of selected Yemeni medicinal plants from the
island Soqotra. BMC Complement Altern Med. 9:72009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Patanasethanont D, Nagai J, Matsuura C,
Fukui K, Sutthanut K, Sripanidkulchai BO, Yumoto R and Takano M:
Modulation of function of multidrug resistance associated-proteins
by Kaempferia parviflora extracts and their components. Eur
J Pharmacol. 566:67–74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen F, Wang T, Wang J, Wang ZQ and Qian
M: Levistolide A overcomes P-glycoprotein-mediated drug resistance
in human breast carcinoma cells. Acta Pharmacol Sin. 29:458–464.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang JC, Zhang JN, Wu YT and Li ZX: Effect
of the water extract and ethanol extract from traditional Chinese
medicines Angelicae sinensis (Oliv.) Diels, Ligusticum
chuanxiong Hort. and Rheum palmatum L on rat liver
cytochrome P450 activity. Phytother Res. 20:1046–1051. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lian N, Liu JB and Yan QM: Effect of
jiawei donggui buxue decoction on the survival time of mice with
cancer. Chin J Clin Rehabil. 8:5736–5737. 2004.
|
|
35
|
Lian N, Liu YM and Liu Y: Effect on
modified Danggui Buxue decoction to curative effect of tumor
treated by chemotherapy. J Chengdu Univ TCM. 26:pp. 18–19. 2003,
http://kns.cnki.net/KCMS/detail/detail.aspx?dbcode=CJFQ&dbname=CJFD2003&filename=CDZY200303006&v=MzI0MTNJOUZZb1I4ZVgx
THV4WVM3RGgxVDNxVHJXTTFGckNVUkxLZVp1ZG1Ge
URrVjc3SkppblJkN0c0SHRMTXI=.
|
|
36
|
Li BH and Lian N: Clinical observation on
synergia and attenuation of Jiawei Danggui buxue decoction to
radiotherapy and chemotherapy of tumor: Attachment report of 392
cases. J Cheng Univ TCM. 28:pp. 7–9. 2005, http://kns.cnki.net/KCMS/detail/detail.aspx?dbcode=CJFQ&dbname=CJFD2005&filename=CDZY200502
002&v=MjE2MzZyV00×RnJDVVJMS2VadWRtRnlEbFZMM0pKaW5SZDdHNEh0VE1yWTlGWm9SOGVYMUx1eFlTN0RoMVQzcVQ=.
|
|
37
|
Zhang T, Ding Y, An H, Feng L and Wang S:
Screening anti-tumor compounds from Ligusticum wallichii
using cell membrane chromatography combined with high performance
liquid chromatography and mass spectrometry. J Sep Sci.
38:3247–3253. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jin ZJ: Addition in drug combination
(author's transl). Zhongguo Yao Li Xue Bao. 1:70–76. 1980.(In
Chinese). https://www.ncbi.nlm.nih.gov/pubmed/6461187PubMed/NCBI
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2ΔΔCT method. Methods.
25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu F, Wang Y, Zeng S, Fu X, Wang L and
Cao J: Involvement of annexin A1 in multidrug resistance of
K562/ADR cells identified by the proteomic study. OMICS.
13:467–476. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen D, Shi Y and Tian G: Chuanxiong and
aspirin treatment of transient ischemic attack of clinical
research. J Int Tra Chi West Med. 12:672–674. 1992.
|
|
42
|
Ling V, Kartner N, Sudo T, Siminovitch L
and Riordan JR: Multidrug resistance phenotype in Chinese hamster
ovary cells. Cancer Treat Rep. 67:869–874. 1983.PubMed/NCBI
|