Introduction
α-Smooth muscle actin (α-SMA) is encoded by the
ACTA2 gene and contributes to tumor cell migration and invasion
through the suppression of E-cadherin (1–3). The
abnormal induction of α-SMA expression was found to be associated
with shorter disease-free survival in lung, colorectal and
pancreatic cancers (4–6). Recently, we reported that breast
cancer patients with high α-SMA and HER2 levels had a poorer
prognosis than patients with low α-SMA and HER2 levels (2). The level of α-SMA expression is
significantly increased during epithelial-mesenchymal transition
(EMT) and is regulated by various stimuli such as IL-1β, IL-6 and
TGF-β1 (7,8).
The TP53 gene encoding p53 is the single most
frequently inactivated gene in human cancers, with somatic missense
mutations being present in approximately 50% of all invasive tumors
(9). These mutation frequencies of
TP53 are lower in breast cancers than in other solid tumors
(10). The level of TP53 protein
expression is stabilized and activated in response to a wide
variety of cellular stresses such as DNA damage, ultraviolet
irradiation, hypoxia and nucleotide depletion (11–13).
TP53 works as a transcriptional activator and regulates cell cycle
arrest, senescence, apoptosis, DNA replication and repair through
the expression of its downstream target genes such as p21, Bax,
NOXA and p53R2 (14,15).
In the present study, the clinical significance of
α-SMA expression was investigated in estrogen receptor-positive
[(ER(+)] breast cancer patients and the effect of TP53 on α-SMA
expression was evaluated in tamoxifen-resistant breast cancer
cells. In this study, it was found that α-SMA expression is
associated with the survival of ER(+) breast cancer patients. In
addition, we observed that wild-type TP53 upregulates α-SMA
expression in tamoxifen-resistant breast cancer cells.
Materials and methods
Reagents
Dulbecco's modified Eagle's medium (DMEM) was
purchased from Thermo Fisher Scientific, Inc. (Hemel Hempstead,
UK). Fetal bovine serum (FBS) was purchased from HyClone/GE
Healthcare Life Sciences (Logan, UT, USA). Phenol red-free DMEM,
penicillin (100 U/ml), and 100 mg/ml streptomycin were purchased
from Life Technologies (Rockville, MD, USA). 4-Hydroxytamoxifen
(4-OHT) was purchased from Sigma-Aldrich;Merck KGaA (Darmstadt,
Germany). Proteome Profiler Human Phospho-Kinase Antibody Array
Kits were purchased from R&D Systems (Minneapolis, MN, USA).
Nutlin3 (TP53 activator, Nut3) and pifithrin-α (TP53 inhibitor,
PFT-α) were purchased from Selleck Chemicals (Houston, TX, USA).
Anti-p53 (1:1,000 dilution; cat. no. sc-126) and mouse monoclonal
anti-β-actin (1:1,000 dilution; cat. no. sc-47778) antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Anti-α-SMA (1:20,000 dilution; cat. no. ab124964) antibody
was purchased from Abcam (Cambridge, UK). ECL Western Blotting
Detection Reagents (West-Q Chemiluminescent Substrate Plus kit)
were obtained from GenDepot (Barker, TX, USA).
Cell culture
Tamoxifen-sensitive and -resistant MCF-7 (TP53
wild-type), MDA-MB-231 (TP53-mutant R280K), and Hs578T (TP53-mutant
V157F) human breast cancer cells were grown in a humidified
atmosphere of 95% air and 5% CO2 at 37°C in DMEM
supplemented with 10% FBS, 2 mM glutamine, 100 IU/ml penicillin and
100 µg/ml streptomycin. Each cell line was maintained in culture
medium without FBS for 24 h before the experiments.
Establishment of tamoxifen-resistant
MCF-7 breast cancer cells
Tamoxifen-sensitive (TamS) and -resistant (TamR)
breast cancer cell lines were kindly provided by Professor Keon
Wook Kang (Seoul National University, Seoul, Korea). The TamR cells
were established using methodology as previously reported (16,17).
Briefly, MCF-7 cells were washed with PBS, and the culture medium
was replaced with phenol red-free DMEM containing 10%
charcoal-stripped steroid-depleted FBS and 0.1 µM 4-OHT. The cells
were continuously exposed to this treatment regimen for two weeks,
and the 4-OHT concentration was increased gradually up to 3 µM over
a 9-month period. Initially, cell growth rates were depressed.
However, after exposure to the medium for 9 months, the rate of
cell growth increased gradually, indicating the establishment of
tamoxifen-resistant cells.
Analysis of a public database
Expression data were downloaded from a public
database [Kaplan-Meier plotter database (http://kmplot.com/breast)] (18). The clinical value of ACTA2 mRNA
expression was analyzed by Kaplan-Meier survival plots in 209 ER(+)
breast cancer patients with tamoxifen treatment (GSE2034) (19). The hazard ratios with 95% confidence
intervals and log-rank P-values were calculated.
Human Phospho-Kinase Antibody
Array
TamS and TamR cells were seeded at 1×106
cells/plate in two separate 100-mm dishes. Protein lysates from
TamS and TamR cells were prepared using the supplied buffers and
300 µg of protein was hybridized to each array from the Proteome
Profiler Human Phospho-Kinase Antibody Array (R&D Systems,
Minneapolis, MN, USA). Further steps were performed based on the
manufacturer's protocol.
Soft agar colony formation assay
TamR breast cancer cells were seeded at a density of
5×104 cells/well in 6-well plates in growth medium
containing 0.7% agar (1.5 ml/well). The cells were seeded on top of
a layer of growth medium containing 1.4% agar (2 ml/well). Next,
growth medium (500 µl) with 10% FBS was added on top of the agar.
In addition, 3 µM 4-OHT was added on top of the agar for some of
the plates. The cells were plated and cultured in a 37°C incubator
for two weeks. After two weeks, the viable colonies were stained
with 0.01% crystal violet and observed using an Olympus CK40
inverted microscope at ×10 magnification (Olympus Corp., Tokyo,
Japan).
Flow cytometric analysis (FACS)
As in a previous study, we performed an apoptosis
assay using the Annexin V-Fluorescein Isothiocyanate (FITC)
Apoptosis Kit-I (BD Pharmingen; BD Biosciences, San Diego, CA, USA)
(3). Briefly, cells
(1×106 cells/ml) were collected and washed twice with
PBS, and then resuspended in 500 µl of staining solution containing
5 µl FITC-conjugated Annexin V and propidium iodide (PI). After
incubation for 15 min at room temperature (RT) in the dark, the
cells were immediately analyzed on a flow cytometer. Apoptotic
cells were double-stained with Annexin V and PI and then they were
analyzed using the FACS Vantage system (Becton-Dickinson, San
Diego, CA, USA). The percentage of cells undergoing apoptosis was
determined.
Western blotting
Cell lysates were prepared to detect α-SMA, TP53,
and β-actin expression. Equal amounts of protein (50 µg) were
boiled for 5 min in Laemmli sample buffer, and then electrophoresed
on 10% sodium dodecyl-sulfate polyacrylamide (SDS-PAGE) gels.
Separated proteins were transferred to polyvinylidene fluoride
(PVDF) membranes, and the membranes were blocked with 10% skim milk
in Tris-buffered saline (TBS) containing 0.01% Tween-20 (TBS/T) for
15 min. Blots were washed three times in TBS/T and then incubated
overnight with antibodies against α-SMA, TP53 and β-actin in TBS/T
buffer at 4°C. The blots were then washed three times in TBS/T and
subsequently incubated with secondary HRP-conjugated antibodies
(1:2,000 dilution; cat. nos. sc-2004 and sc-2005; Santa Cruz
Biotechnology) in TBS/T buffer. After 1 h of incubation at room
temperature (RT), the blots were washed three times in TBS/T and
positive immunoreactive proteins were detected using the West-Q
Chemiluminescent Substrate Plus kit.
Real-time PCR
Total RNA was extracted from the cells using
Invitrogen™ TRIzol reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Isolated RNA samples were
then used for RT-PCR. Samples of total RNA (1 µg) were reverse
transcribed into cDNA in 20 µl reaction volumes using a
first-strand cDNA synthesis kit for RT-PCR according to the
manufacturer's instructions (MBI Fermentas, Hanover, MD, USA). Gene
expression levels were quantified by real-time PCR using a SensiMix
SYBR kit (Bioline Ltd., London, UK) and 100 ng of cDNA per
reaction. The primer sequences used for this analysis were as
follows: human TP53 (forward, 5′-GGCCCACTTCACCGTACTAA-3′ and
reverse, 5′-AAGCGAGACCCAGTCTCAAA-3′); human α-SMA (forward,
5′-AGACATCAGGGGGTGATGGT-3′ and reverse,
5′-CATGGCTGGGACATTGAAAG-3′), and GAPDH was used as an internal
control (forward, 5′-ATTGTTGCCATCAATGACCC-3′ and reverse,
5′-AGTAGAGGCAGGGATGATGT-3′). An annealing temperature of 60°C was
used for all the primers. PCR was performed in a standard 384-well
plate format with an ABI 7900HT Real-Time PCR Detection System
(Thermo Fisher Scientific, Inc.). For data analysis, the raw
threshold cycle (CT) value was first normalized
to the housekeeping gene for each sample to obtain a
ΔCT. The normalized ΔCT was
then calibrated to the control cell samples and to obtain the
ΔΔCq values (20).
Adenovirus induction
Adenovirus expressing Lac Z and human TP53
cDNA (Ad-TP53) was a gift from Dr Hyunil Ha (Korean Institute of
Oriental Medicine, Daejeon, Korea). Recombinant adenovirus
expressing human TP53 was reproduced in 293A cells. The expression
of this construct was confirmed by western blot analysis. Each
construct was transfected into Hs578T and BT549 cells for 24 h and
incubated for 24 h in fresh culture media. Ad-TP53-overexpressing
MDA-MB-231 and Hs578T cells were further incubated for 24 h in
serum-free culture media. The cell lysates and culture medium were
then harvested for analysis of α-SMA and TP53 expression.
TP53 siRNA transfection
TP53 siRNA was purchased from Bioneer Corporation
(Daejeon, Korea). A synthetic siRNA against two different TP53
mRNAs was used to inhibit endogenous TP53 gene expression. The
duplex sequences of TGFBRI and TGFBRII siRNA used for this
experiment are as follows: Human #1 TP53 siRNA [sense,
CACUACAACUACAUGUGUA (dTdT) and antisense, UACAUAUGUAGUUGUAGUG
(dTdT)]; and human #2 TP53 siRNA [sense, GAGGUUGGCUCUGACUGUA (dTdT)
and antisense, UACAGUCAGAGCCAACCUC (dTdT)]. It was found that the
optimal siRNA knockdown conditions involved transfecting TamR
cells. Effectene (Qiagen, Inc., Valencia, CA, USA) was used for
transfections with siRNA following protocols provided by the
manufacturer. After the 72 h transfection, the levels of TP53 and
α-SMA mRNA expression were analyzed by real-time PCR.
Statistical analysis
Statistical significance was determined using
Student's t-test. The results are presented as the mean ± SEM. All
the quoted P-values are two-tailed and the differences were
considered statistically significant when the P-value was <0.05.
Statistical analyses were performed using the Microsoft Excel
2016.
Results
α-SMA expression is associated with a
poor prognosis in estrogen receptor-positive breast cancer
patients
In a previous study, we reported that the levels of
mesenchymal marker proteins such as fibronectin and α-SMA were
significantly increased in TamR cells (3). In the present study, we investigated
the clinical significance and the regulatory mechanism of α-SMA in
breast cancer models. We found that the levels of α-SMA mRNA
expression were associated with a poor prognosis in 209 ER(+)
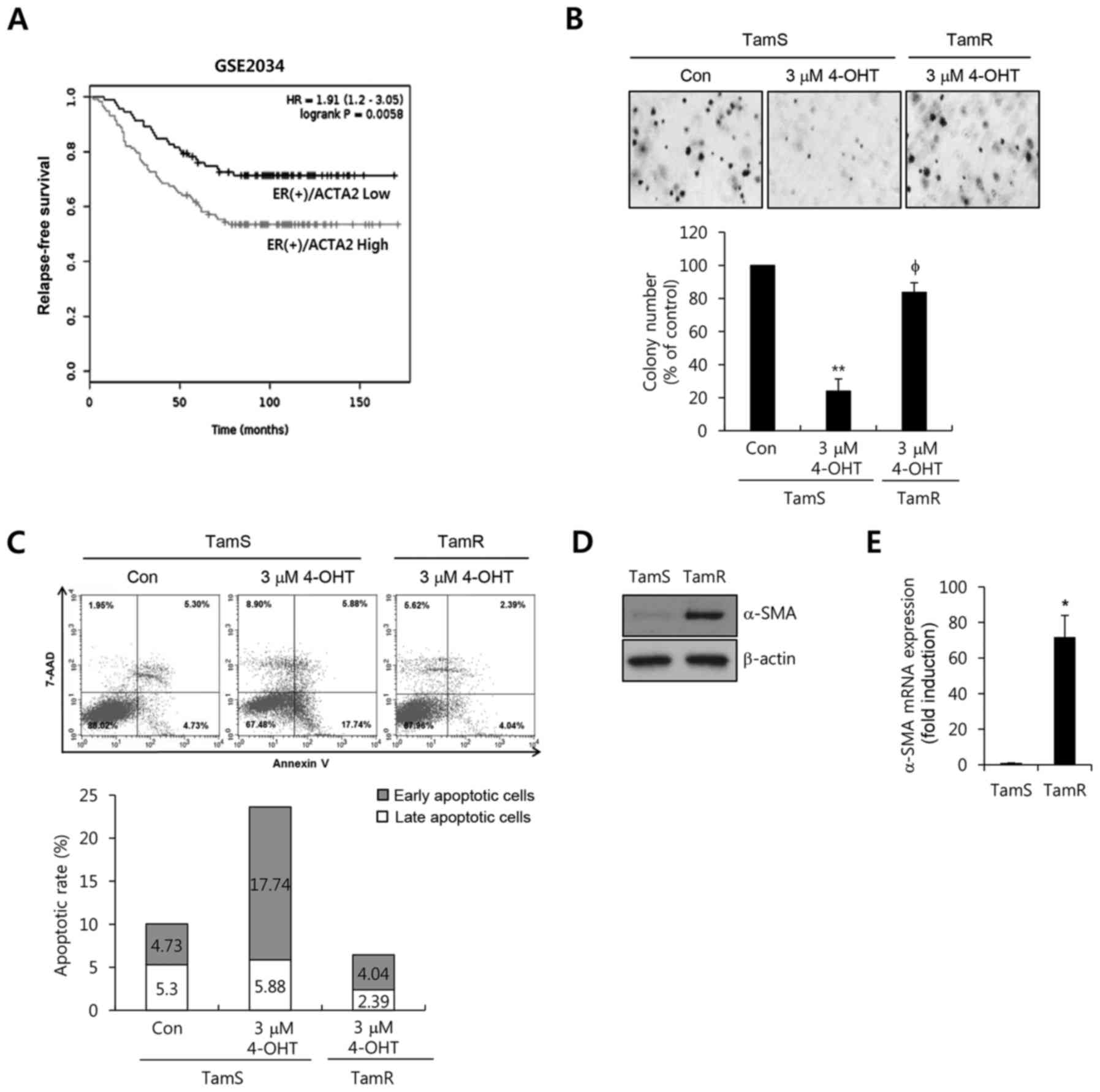
breast cancer patients using the GSE2034 dataset (Fig. 1A). ER(+) breast cancer patients with
a high expression of α-SMA exhibited poorer relapse-free survival
compared to the patients with low expression (P=0.0058, Fig. 1A).
Next, we analyzed the effect of tamoxifen on TamS
and TamR cells. As shown in Fig. 1B and
C, the anchorage-independent growth and apoptotic cell death of
TamS were completely inhibited by 4-OHT treatment. However, cell
growth and the death of TamR cells were not affected by 4-OHT.
Furthermore, we examined the level of α-SMA expression in the TamS
and TamR cells. α-SMA expression was significantly increased in the
TamR cells (Fig. 1D). Under the
same condition, the level of α-SMA mRNA expression was increased by
71.8-fold compared with the TamS cells (Fig. 1E). Therefore, we demonstrated that
α-SMA expression was significantly increased in TamR cells and was
associated with survival in ER(+) breast cancer patients.
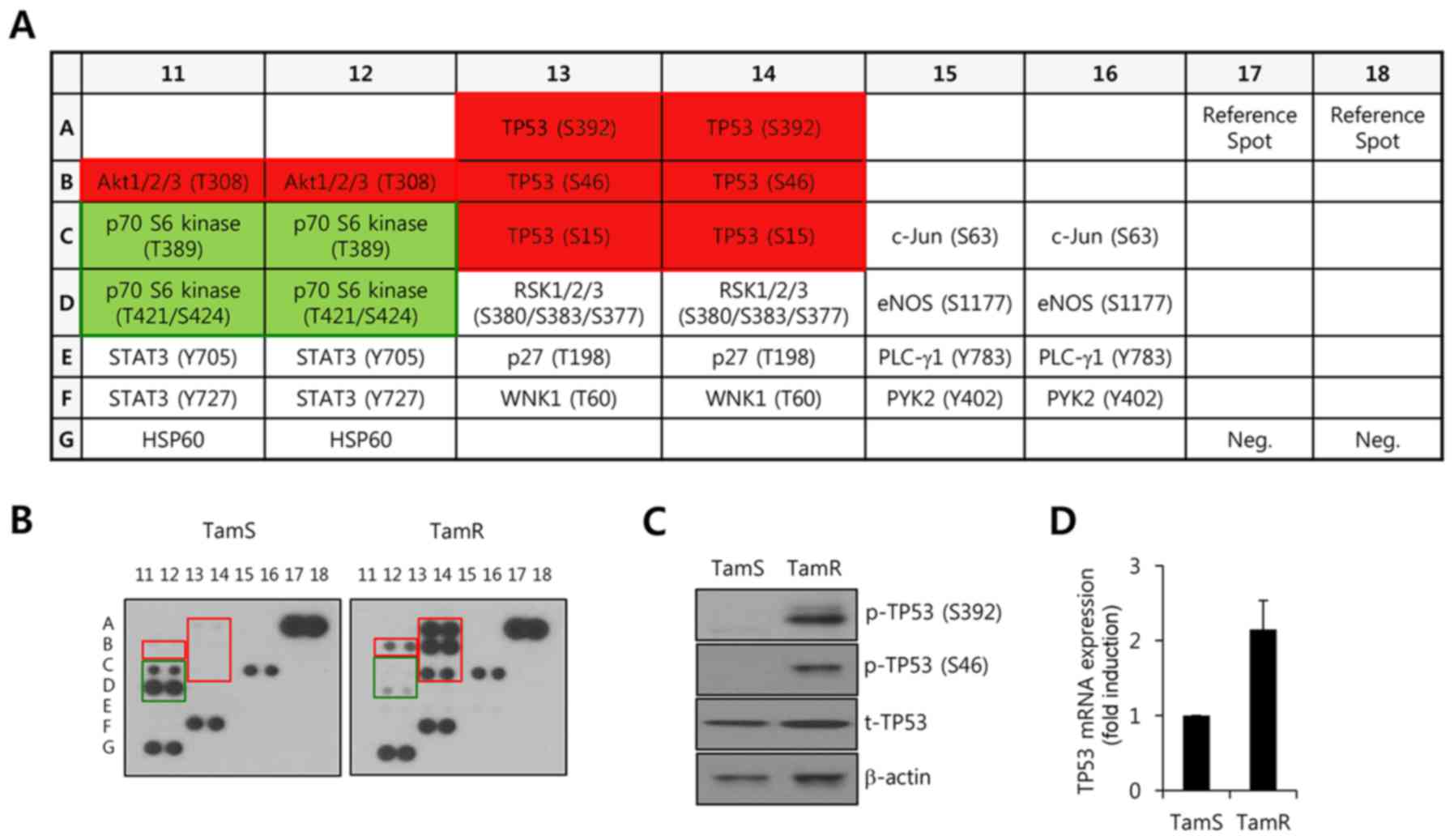
Analysis of kinase activities between
TamS and TamR cells
To characterize α-SMA expression-related protein
kinases, we analyzed the phosphorylated levels of various protein
kinases in TamS and TamR cells using the Proteome Profiler Human
Phospho-Kinase Array. Protein lysates were loaded to the Proteome
Profiler Human Phospho-Kinase Array Kit membranes. A schematic
model of the membrane is indicated in Fig. 2A. In the present study, we found
that the phosphorylation levels of Akt and TP53 were significantly
increased in TamR cells when compared with TamS cells (Fig. 2B, red square). To confirm the
results obtained by the Phospho-Kinase array, we examined the
levels of pS392-p53 and pS46-p53 by western blot analysis. As
expected, our results showed that the levels of pS392-p53 and
pS46-p53 were markedly increased in the TamR cells (Fig. 2C). In addition, we observed that the
level of TP53 protein (Fig. 2C) and
mRNA (Fig. 2D) expression were
slightly increased in the TamR cells. However, the phosphorylation
level of p70 S6 kinase (T421/S424) was observably decreased in the
TamR cells (Fig. 2B, green square).
Based on these results, it was theorized that TP53 activity may be
involved with α-SMA expression in breast cancer cells.
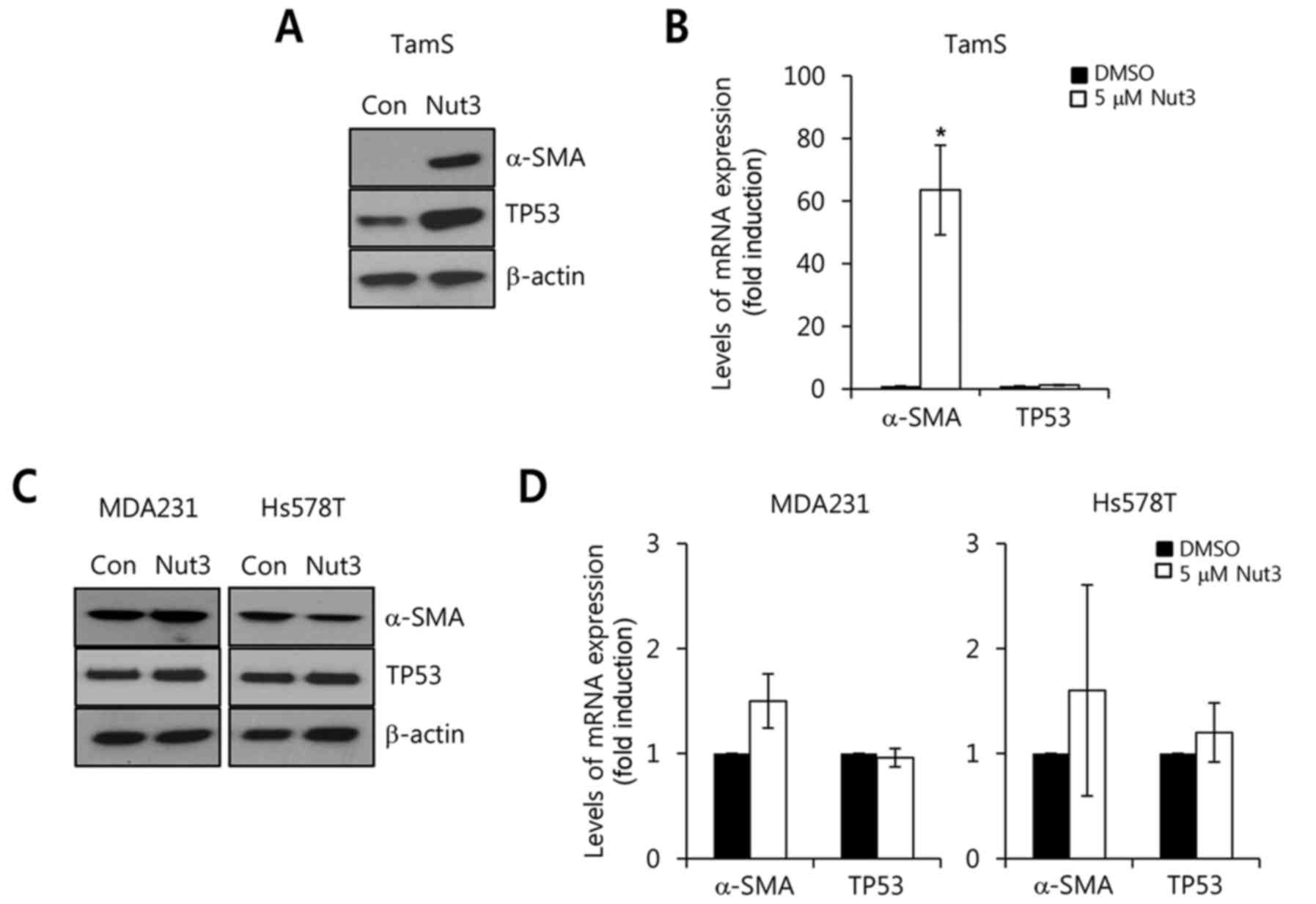
TP53 activator, nutlin3, upregulates
α-SMA expression in breast cancer cells with wild-type TP53
To verify the role of TP53 on α-SMA expression,
activator of TP53, Nut3, was administered to block the interaction
of MDM2 and TP53 in TamS cells with wild-type TP53 for 24 h. After
24 h, we harvested whole cell lysates for the detection of α-SMA
and TP53 protein and mRNA expression. As shown in Fig. 3A, the level of α-SMA expression was
observably increased by Nut3 treatment. Under the same conditions,
we analyzed the levels of α-SMA and TP53 mRNA expression. As
expected, the level of α-SMA mRNA expression increased by
63.5±14.3-fold with Nut3 relative to the control (Fig. 3B). However, TP53 mRNA levels were
not affected by Nut3 treatment, although the basal level of TP53
expression was slightly increased (Fig.
3A and B).
Next, we examined the effect of Nut3 on α-SMA
expression in TP53-mutant breast cancer cells. The TP53 status of
breast cancer cell lines was determined from the database
http://p53.free.fr/Database/Cancer_cell_lines/Breast_cancer.html,
which revealed the TP53-mutant breast cancer cells (MDA-MB-231 and
Hs578T). As shown in Fig. 3C, the
levels of α-SMA and TP53 protein expression were not observably
altered by Nut3 treatment in MDA-MB-231 and Hs579T cells. In
addition, the levels of α-SMA and TP53 mRNA expression were also
markedly changed by Nut3 (Fig. 3D).
Therefore, we demonstrated that the TP53 status plays an important
role on α-SMA expression in breast cancer cells.
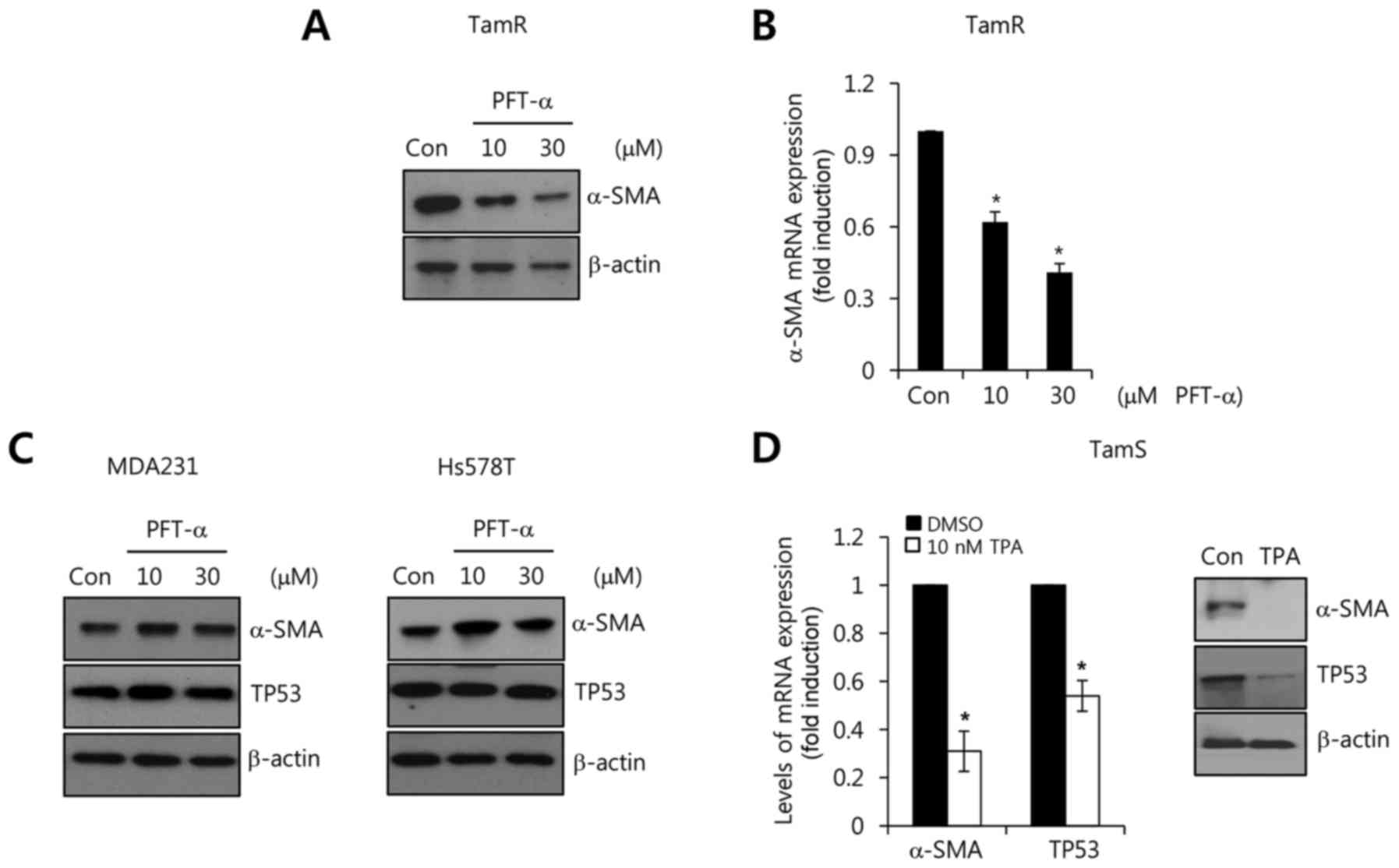
Suppression of TP53 downregulates
α-SMA expression in tamoxifen-resistant cells
Conversely, we investigated the effect of a TP53
inhibitor, pifithrin-α (PFT-α), on α-SMA expression in TamR cells.
We treated the TamR cells with the indicated concentrations (10 and
30 µM) of PFT-α for 72 h. As shown in Fig. 4A, the basal levels of α-SMA protein
expression were dose-dependently decreased by PFT-α treatment. In
addition, the levels of α-SMA mRNA expression were also decreased
by PFT-α (Fig. 4B). The reduction
of α-SMA mRNA expression by PFT-α was decreased to 0.62 and
0.41-fold relative to the control (Fig.
4B). However, the MDA-MB-231 and Hs578T cells with mutant TP53
were not altered by PFT-α treatment (Fig. 4C). In a previous study, TPA
stimulated the ubiquitination and degradation of TP53 through the
downregulation of PKC-d and the suppression of TP53 transcriptional
activity (21,22). Therefore, we treated the TamS cells
with 10 nM TPA for 24 h with wild-type TP53. As expected, the level
of α-SMA mRNA expression was decreased to 0.31-fold relative to the
control (Fig. 4D). In addition, the
level of TP53 mRNA expression decreased 0.54-fold relative to the
control (Fig. 4D). Based on these
results, we demonstrated that the level of wild-type TP53 plays an
important role in the α-SMA expression in breast cancer cells.
Wild-type TP53 upregulates α-SMA
expression in breast cancer cells
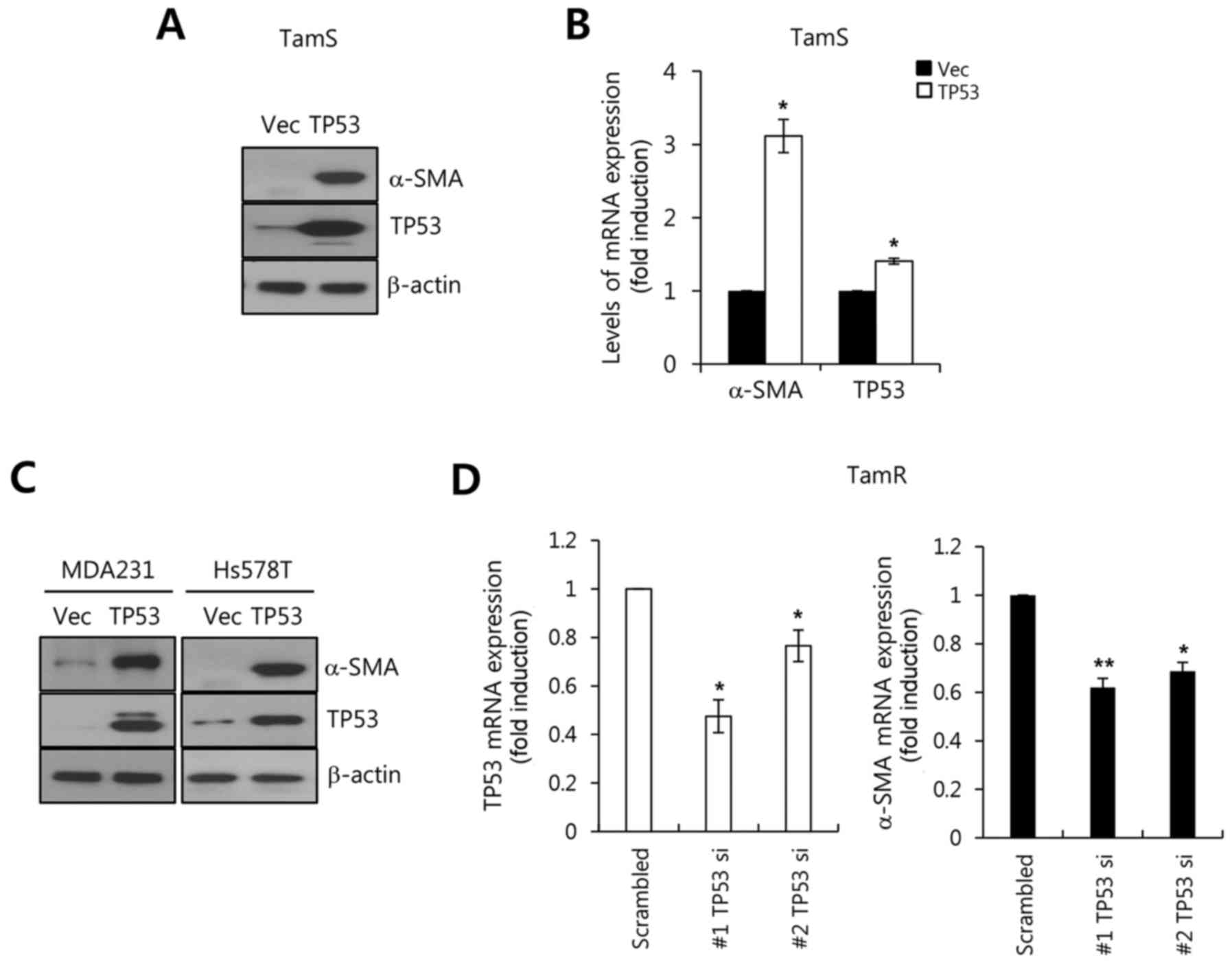
We investigated whether TP53 upregulates α-SMA
expression in breast cancer cells. In the present study, breast
cancer cells were transfected with adenoviral LacZ and
TP53 genes for 48 h and further incubated for 24 h in fresh
serum-free media. As expected, our results showed that the level of
α-SMA protein expression was significantly increased by TP53
overexpression (Fig. 5A). Under the
same conditions, the level of α-SMA mRNA expression was increased
by TP53 overexpression (Fig. 5B).
The level of α-SMA mRNA expression increased 3.11±0.23-fold
relative to the Vec alone (Fig.
5B). As shown in Fig. 3C and D,
triple-negative breast cancer (TNBC) cells with mutant TP53 did not
exhibit changes in the levels of α-SMA expression by Nut3
treatment. However, these cells also exhibited significantly
increased levels of α-SMA expression following wild-type TP53
overexpression (Fig. 5C). In
contrast, we examined the effects of TP53 knockdown by two
different TP53 siRNAs on α-SMA expression in TamR cells. As
expected, our results showed that the downregulation of TP53 by
TP53 siRNA decreased the levels of α-SMA mRNA expression in TamR
cells (Fig. 5D). Therefore, we
demonstrated that wild-type TP53 expression upregulates the level
of α-SMA expression in breast cancer cells.
Discussion
Breast cancer is the most common cancer in women
worldwide accounting for approximately 25% of all cancer cases and
15% of all cancer-related deaths (23). Although therapeutic strategies for
breast cancer patients involve systemic therapy including
chemotherapy, hormonal therapy and targeted therapy, their
therapeutic efficacy is still limited by either intrinsic or
acquired resistances (24,25). To date, the level of α-SMA
expression has been known as a prognostic marker for a variety of
cancers, including oral tongue squamous cell carcinoma and lung
adenocarcinomas (4,26). Aberrant α-SMA expression is
significantly associated with worse overall survival and
metastasis-free survival rates in lung adenocarcinomas (4). Consistent with these reports, we also
observed that α-SMA expression is associated with survival in
breast cancer. ER-α(+) breast cancer patients with high expression
of α-SMA exhibited decreased relapse-free survival compared to
α-SMA-low expressing patients. In addition, we found that the
levels of α-SMA mRNA and protein expression were increased in TamR
cells when compared to TamS cells. Therefore, we focused on the
underlying mechanism of α-SMA expression which is highly expressed
in tamoxifen-resistant breast cancer cells.
The level of TP53 expression is also regulated by a
wide variety of cellular stress, such as UV, hypoxia and phorbol
ester (11,22,27).
Our results showed that the basal level of TP53 expression
decreased while α-SMA mRNA and protein expression were
significantly increased through TPA treatment. In previous studies,
the basal level of α-SMA expression was upregulated through focal
adhesion kinase (FAK) as well as the JAK2/STAT1-dependent pathway
in breast cancer cells and fibroblasts (2,28,29).
In the present study, we analyzed kinase activities to identify the
regulatory kinases on α-SMA expression in TamS and TamR cells. Our
results showed that the activities of TP53 and Akt were
significantly increased in TamR cells, but did not affect the
activity of p70 S6 kinase. Therefore, we investigated the role of
TP53 in the regulation of α-SMA expression in breast cancer
cells.
Nutlin3 (Nut3), a cis-imidazoline analog,
interferes with the binding between MDM2 and p53 as an antagonist
of MDM2, an E3 ubiquitin ligase of protein of the TP53 family
(30). Nut3 induces prominent
p21WAF1 expression by upregulating the phosphorylation
of Ser 46 on p53 (31). Although we
did not assess the phosphorylation of TP53 by Nut3, our results
showed that the basal level of TP53 expression was slightly
increased through Nut3 treatment in breast cancer cells with
wild-type TP53, but not in breast cancer cells with mutant TP53. In
addition, α-SMA expression was significantly increased following
Nut3 treatment in breast cancer cells with wild-type TP53. In
contrast, the basal level of α-SMA mRNA and protein expression
dose-dependently decreased with PFT-α in TamR cells that blocked
the transcriptional activity of TP53. Therefore, we demonstrated
that the activity and expression of wild-type TP53 plays an
important role in the regulation of α-SMA expression in breast
cancer cells.
The tumor-suppressor protein TP53 is a transcription
factor that regulates anti-carcinogenesis programs associated with
apoptosis, cell cycle arrest, and DNA repair in genotoxic and
not-genotoxic cellular injuries (32,33).
In recent studies, we reported that the level of fibronectin
expression is associated with the status and expression of TP53 in
breast cancer cells (34,35). Wu et al reported that Slug is
transcriptionally induced by TP53 and then escapes from apoptosis
by repressing the TP53-mediated transcription of Puma (36). We found for the first time that the
levels of α-SMA expression are increased by wild-type TP53
overexpression in all breast cancer cells with wild-type or mutant
TP53. Therefore, we demonstrated that wild-type TP53 is involved
with α-SMA expression in breast cancer cells.
In conclusion, aberrant α-SMA expression is
associated with the survival of ER(+) breast cancer patients. Nut3
significantly increased the levels of α-SMA mRNA and protein
expression in TamS cells with wild-type TP53, but not in MDA-MB-231
and Hs578T cells with mutant TP53. In addition, the overexpression
of wild-type TP53 augmented the level of α-SMA expression in all
breast cancer cells with wild-type or mutant TP53. In contrast, we
observed that PFT-α dose-dependently suppressed α-SMA expression in
TamR cells. Based on these results, we demonstrated that wild-type
TP53 expression augments the level of α-SMA expression in breast
cancer cells. We also will investigate whether TP53 directly
regulates α-SMA expression in breast cancer cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (2016R1D1A1B01010508) and
by a National Research Foundation of Korea (NRF) grant funded by
the Korean Government (MSIP) (2016R1A5A2945889).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SK and JEL designed the study, interpreted the data
and wrote the manuscript. DY and YJ were responsible for the
laboratory experiments such as real-time PCR and western blotting;
JY, SWK and SJN analyzed the data and developed the prognostic
signature. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Lambrechts A, Van Troys M and Ampe C: The
actin cytoskeleton in normal and pathological cell motility. Int J
Biochem Cell Biol. 36:1890–1909. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jeon M, You D, Bae SY, Kim SW, Nam SJ, Kim
HH, Kim S and Lee JE: Dimerization of EGFR and HER2 induces breast
cancer cell motility through STAT1-dependent ACTA2 induction.
Oncotarget. 8:50570–50581. 2016.PubMed/NCBI
|
|
3
|
Kim S, Lee J, Oh SJ, Nam SJ and Lee JE:
Differential effect of EGFR inhibitors on tamoxifen-resistant
breast cancer cells. Oncol Rep. 34:1613–1619. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee HW, Park YM, Lee SJ, Cho HJ, Kim DH,
Lee JI, Kang MS, Seol HJ, Shim YM, Nam DH, et al: Alpha-smooth
muscle actin (ACTA2) is required for metastatic potential of human
lung adenocarcinoma. Clin Cancer Res. 19:5879–5889. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sinn M, Denkert C, Striefler JK, Pelzer U,
Stieler JM, Bahra M, Lohneis P, Dorken B, Oettle H, Riess H and
Sinn BV: α-Smooth muscle actin expression and desmoplastic stromal
reaction in pancreatic cancer: results from the CONKO-001 study. Br
J Cancer. 111:1917–1923. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsujino T, Seshimo I, Yamamoto H, Ngan CY,
Ezumi K, Takemasa I, Ikeda M, Sekimoto M, Matsuura N and Monden M:
Stromal myofibroblasts predict disease recurrence for colorectal
cancer. Clin Cancer Res. 13:2082–2090. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aoki H, Ohnishi H, Hama K, Shinozaki S,
Kita H, Osawa H, Yamamoto H, Sato K, Tamada K and Sugano K:
Cyclooxygenase-2 is required for activated pancreatic stellate
cells to respond to proinflammatory cytokines. Am J Physiol Cell
Physiol. 292:C259–C268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lim MJ, Ahn J, Yi JY, Kim MH, Son AR, Lee
SL, Lim DS, Kim SS, Kang MA, Han Y, et al: Induction of galectin-1
by TGF-β1 accelerates fibrosis through enhancing nuclear retention
of Smad2. Exp Cell Res. 326:125–135. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: Lessons from recent developments in the IARC TP53
database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davidoff AM, Humphrey PA, Iglehart JD and
Marks JR: Genetic basis for p53 overexpression in human breast
cancer. Proc Natl Acad Sci USA. 88:5006–5010. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sugrue MM, Shin DY, Lee SW and Aaronson
SA: Wild-type p53 triggers a rapid senescence program in human
tumor cells lacking functional p53. Proc Natl Acad Sci USA.
94:9648–9653. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Royds JA and Iacopetta B: p53 and disease:
When the guardian angel fails. Cell Death Differ. 13:1017–1026.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sur S, Pagliarini R, Bunz F, Rago C, Diaz
LA Jr, Kinzler KW, Vogelstein B and Papadopoulos N: A panel of
isogenic human cancer cells suggests a therapeutic approach for
cancers with inactivated p53. Proc Natl Acad Sci USA.
106:3964–3969. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Woods DB and Vousden KH: Regulation of p53
function. Exp Cell Res. 264:56–66. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gasco M, Shami S and Crook T: The p53
pathway in breast cancer. Breast Cancer Res. 4:70–76. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choi HK, Yang JW, Roh SH, Han CY and Kang
KW: Induction of multidrug resistance associated protein 2 in
tamoxifen-resistant breast cancer cells. Endocr Relat Cancer.
14:293–303. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
You D, Jung SP, Jeong Y, Bae SY, Lee JE
and Kim S: Fibronectin expression is upregulated by PI-3K/Akt
activation in tamoxifen-resistant breast cancer cells. BMB Rep.
50:615–620. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gyorffy B, Lanczky A, Eklund AC, Denkert
C, Budczies J, Li Q and Szallasi Z: An online survival analysis
tool to rapidly assess the effect of 22,277 genes on breast cancer
prognosis using microarray data of 1,809 patients. Breast Cancer
Res Treat. 123:725–731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Klijn JG, Zhang Y, Sieuwerts AM,
Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, et
al: Gene-expression profiles to predict distant metastasis of
lymph-node-negative primary breast cancer. Lancet. 365:671–679.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2ΔΔCT method. Methods.
25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abbas T, White D, Hui L, Yoshida K, Foster
DA and Bargonetti J: Inhibition of human p53 basal transcription by
down-regulation of protein kinase Cdelta. J Biol Chem.
279:9970–9977. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim S, Han J, Kim NY, Lee SK, Cho DH, Choi
MY, Kim JS, Kim JH, Choe JH, Nam SJ, et al: Effect of berberine on
p53 expression by TPA in breast cancer cells. Oncol Rep.
27:210–215. 2012.PubMed/NCBI
|
|
23
|
DeSantis C, Ma J, Bryan L and Jemal A:
Breast cancer statistics, 2013. CA Cancer J Clin. 64:52–62. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Musgrove EA and Sutherland RL: Biological
determinants of endocrine resistance in breast cancer. Nat Rev
Cancer. 9:631–643. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pohlmann PR, Mayer IA and Mernaugh R:
Resistance to trastuzumab in breast cancer. Clin Cancer Res.
15:7479–7491. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ding L, Zhang Z, Shang D, Cheng J, Yuan H,
Wu Y, Song X and Jiang H: α-Smooth muscle actin-positive
myofibroblasts, in association with epithelial-mesenchymal
transition and lymphogenesis, is a critical prognostic parameter in
patients with oral tongue squamous cell carcinoma. J Oral Pathol
Med. 43:335–343. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han J, Kim S, Yang JH, Nam SJ and Lee JE:
TPA-induced p21 expression augments G2/M arrest through a
p53-independent mechanism in human breast cancer cells. Oncol Rep.
27:517–522. 2012.PubMed/NCBI
|
|
28
|
Chen R, Zhang Z, Xue Z, Wang L, Fu M, Lu
Y, Bai L, Zhang D and Fan Z: Focal adhesion kinase (FAK) siRNA
inhibits human hypertrophic scar by suppressing integrin alpha,
TGF-β and α-SMA. Cell Biol Int. 38:803–808. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li S, Butler P, Wang Y, Hu Y, Han DC,
Usami S, Guan JL and Chien S: The role of the dynamics of focal
adhesion kinase in the mechanotaxis of endothelial cells. Proc Natl
Acad Sci U S A. 99:3546–3551. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wade M, Li YC and Wahl GM: MDM2, MDMX and
p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 13:83–96.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Enge M, Bao W, Hedstrom E, Jackson SP,
Moumen A and Selivanova G: MDM2-dependent downregulation of p21 and
hnRNP K provides a switch between apoptosis and growth arrest
induced by pharmacologically activated p53. Cancer Cell.
15:171–183. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Menendez D, Inga A and Resnick MA: The
expanding universe of p53 targets. Nat Rev Cancer. 9:724–737. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
You D, Jung SP, Jeong Y, Bae SY and Kim S:
Wild-type p53 controls the level of fibronectin expression in
breast cancer cells. Oncol Rep. 38:2551–2557. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jeong Y, You D, Kang HG, Yu J, Kim SW, Nam
SJ, Lee JE and Kim S: Berberine suppresses fibronectin expression
through inhibition of c-jun phosphorylation in breast cancer cells.
J Breast Cancer. 21:21–27. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu WS, Heinrichs S, Xu D, Garrison SP,
Zambetti GP, Adams JM and Look AT: Slug antagonizes p53-mediated
apoptosis of hematopoietic progenitors by repressing puma.
Cell. 123:641–653. 2005. View Article : Google Scholar : PubMed/NCBI
|