Introduction
Globally gastric cancer is ranked fifth in cancer
mortality (1). Despite recent
progress, most patients are diagnosed at advanced stages and are
not eligible for curative surgery, and hence chemotherapy is
essential for their treatment (2).
However, conventional chemotherapies display only modest effects on
the survival of patients with advanced gastric cancer. Cisplatin
(CDDP) is one of the most commonly used chemotherapeutic agents in
the clinical management of gastric cancer (2), and included in the first-line regimen
for advanced gastric cancer in Japan (3). Unfortunately, resistance to CDDP
largely limits its beneficial effects, representing a major
obstacle to effective treatments. Therefore, exploring the
mechanisms for this resistance is required for devising therapeutic
strategies to combat gastric cancer.
Phosphoprotein enriched in astrocytes 15 (PEA-15),
also known as phosphoprotein enriched in diabetes (PED), is
ubiquitously expressed in mammals and was originally identified in
primary cultured astrocytes (4).
PEA-15 was initially found to play a prominent role in controlling
cell survival and glucose metabolism (5), and later it was demonstrated to
participate in regulating multiple biological behaviors of cells
from several types of malignancies including breast and colorectal
cancer, and hepatocellular carcinoma (HCC) (6–9). The
well-known mechanism accounting for its function is the regulation
of the activation of extracellular signal-regulated protein kinases
1 and 2 (ERK1/2), which mediate cell proliferation, migration and
apoptosis (6,10). However, its role in gastric cancer
remains unknown. Therefore, the present study was designed to
examine the expression of PEA-15 in human gastric cancer tissues
and investigate its functional role in gastric cancer cells.
In addition, PEA-15 has recently been demonstrated
to contribute to the insensitivity of a panel of chemotherapeutic
agents including CDDP in breast cancer (11), resistance to fluorouracil and CDDP
in colon cancer (8), and sorafenib
resistance in HCC (9). However, it
is unclear whether PEA-15 contributes to the mechanisms of CDDP
resistance in gastric cancer. We have previously reported that
CDDP-resistant gastric cancer cells expressed higher levels of
phosphorylated AKT (p-AKT) (12).
PEA-15 features an AKT phosphorylation motif upstream from Ser116
and the phosphorylation by AKT affects its antiapoptotic function
(11,13,14).
Therefore, we hypothesized that PEA-15 may be involved in AKT
activation-regulated mechanisms of CDDP resistance in gastric
cancer cells.
Materials and methods
Patients
The general information of the patients used in the
present study has been previously described (15). Briefly, a total of 141 consecutive
patients with gastric cancer received surgical treatments at
Qingdao Municipal Hospital in China. The diagnosis of gastric
cancer was pathologically confirmed and histological classification
was performed according to the 2010 World Health Organization (WHO)
histological classification system and cell differentiation
(16). The disease was staged in
accordance with the staging system of the American Joint Committee
on Cancer (16). None of the
patients had received any preoperative anticancer treatments. The
study analyzing human specimens was approved by the Ethics
Committee of Qingdao Municipal Hospital (no. 20150819), and
informed consents were obtained (Qingdao, China). The animal
experiments were approved (permit no. SYXK20020009) by the Animal
Ethics Committee of Harbin Medical University (Harbin, China).
Cell culture, antibodies and
reagents
Human gastric cancer cells MGC-803, SGC7901, BGC823,
AGS, NCI-N87 and HGC-27 were obtained from the Type Culture
Collection Cell Bank (Chinese Academy of Sciences Committee,
Shanghai, China). Cells were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum at 37°C in a humidified
atmosphere of 5% CO2. All cell lines were confirmed
negative for mycoplasma infection using a PCR-based Universal
Mycoplasma Detection kit (American Type Culture Collection,
Manassas, VA, USA). An antibody (Ab) against PEA-15 (SAB4503451),
FAST DAB (3,3′-diaminobenzidine tetrahydrochloride) and
CoCl2 enhancer tablets, and a TUNEL (Terminal
deoxynucleotidyl transferase-mediated dUTP nick end labeling agent)
kit were purchased from Sigma-Aldrich (Shanghai, China). An
anti-PEA-15 (Ser116) Ab (cat. no. PA5-38314) was purchased from
Thermo Fisher Scientific Inc. (Shanghai, China). Abs against
p-PEA-15 (Ser104) (cat. no. 2776), ERK1/2 (cat. no. 4695), p-ERK1/2
(Thr202/Thyr204) (cat. no. 4370), AKT (cat. no. 4691), p-AKT
(Ser473) (cat. no. 4060), caspase-3 (cat. no. 9662), p27 (cat. no.
3688), cyclin D1 (cat. no. 2922), GSK-3 (cat. no. 9315), p-GSK-3
(cat. no. 5558) and caspase-9 (cat. no. 9508) were provided by Cell
Signaling Technology (Boston, MA, USA). Abs against β-actin (cat.
no. sc-130065), caspase-9 (cat. no. sc-56073), caspase-8 (cat. no.
sc-56070), caspase-8 (cat. no. sc-73526) were obtained from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). An anti-Ki-67 Ab (cat.
no. ab15580) and a caspase-3 activity kit (cat. no. ab39401) were
purchased from Abcam (Shanghai, China).
Immunohistochemical analysis of human
specimens
The methods have been previously described (15). Briefly, tissues collected from
patients during surgery were formalin-fixed, paraffin-embedded,
sectioned and mounted on 3-aminopropyltriethoxysilane-coated
slides. After antigen retrieval, the sections were blocked and
incubated with a rabbit anti-human PEA-15 Ab (diluted at 1:250) at
4°C overnight. A standard horseradish peroxidase staining procedure
was performed using a biotinylated secondary Ab (diluted at 1:250),
and immunoreactivity developed with Sigma FAST DAB and
CoCl2 enhancer tablets. Normal rabbit sera (diluted at
1:10) were used for blocking and dilution of Abs. Negative controls
were achieved using irrelevant goat IgG (diluted at 1:50).
Immunostaining was assessed in 20 randomly selected fields per
specimen using a semi-quantitative grading system. The staining
intensity (Value A) was graded in a four-tier grading system: No
staining (0), faint yellow (1),
yellow (2) and brown (3); while the extent of positive staining
(Value B) was graded in a four-tier grading system based on the
percentage of positive cells: ≤10% (1), 11–40% (2), 41–70% (3) and ≥70% (4). The immunohistological score was
calculated by A × B, and each specimen was graded lower (≤5) or
higher level (>5).
Quantitative reverse-transcription
polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the cells, and cDNA was
synthesized. The reaction mixtures for qRT-PCR were prepared with
the primers for PEA-15 (forward, 5′-AGGAAGACATCCCCAGCGAA-3′ and
reverse, 5′-CCATAGTGAGTAGGTCAGGACG-3′); and β-actin (forward,
5′-TTAGCACCCCTGGCCAAGG-3′ and reverse, 5′-CTTACTCCTTGGAGGCCATG-3′)
as previously described (17).
Reaction solutions were analyzed by MX3000P Real-Time PCR Systems
(Stratagene; Agilent Technologies, Inc., North Billerica, MA, USA).
Experiments were performed in triplicate, and the data were
calculated using the ∆∆Cq method (18).
shRNA expression vectors
The previously reported sequences were selected to
specifically target PEA-15 (5′-GCGAAAAGAGUGAGGAGAU-3′ and
5′-AUCUCCUCACUCUUUUCGC-3′) corresponding to nucleotides 611–629 of
human PEA-15 (GenBank NM_001297576.1) as previously reported
(19). The oligonucleotides were
introduced into the pSuppressorNeo vector to generate PEA-shRNA. A
scrambled shRNA vector (Sc-shRNA) targeting non-specific sequences
(5′-GAAGACGAAGAGUGAGGAU-3′ and 5′-AUCUCCACUCUCUGUUCUC-3′) served as
a control.
Animal experiments
A total of 46 male nude BALB/c mice (H-2b) (aged 6–8
weeks; body weight, 20.3±2.5 g) were obtained from the Animal
Research Center, The First Affiliated Hospital of Harbin Medical
University (Harbin, China). Animals were housed in cages under
pathogen-free conditions at a temperature of 20–25°C and 12-h
light/dark cycle, and fed commercial pellets and water ad
libitum. The experimental protocol has been previously
described (12,15,20).
Two sets of experiments were designed for evaluating the functional
role of PEA-15 in gastric cancer cells in vivo.
Tumorigenicity
Cells (2×106) were subcutaneously
injected into the flanks of mice, which were monitored to check the
appearance of tumors, and tumor size was measured every 4 days.
Tumor volumes were calculated according to the following formula:
π/6 × a2 × b, where a is the short axis and b the long
axis. Mice were sacrificed 28 days later, and tumors were harvested
and weighed.
Therapeutic study
Cells (2×106) were subcutaneously
injected into the flanks of mice. When tumors reached ~100
mm3, the mice were assigned to 4 groups: Control (n=8),
CDDP (n=6), PEA-shRNA (n=8) and CDDP + PEA-shRNA (n=6). Each mouse
in each group received both intratumoral and intraperitoneal
injections. Normal saline (200 µl) and CDDP (0.25 mg/mouse,
dissolved in 0.9% sodium chloride 0.9%) were intraperitoneally
injected every 3 days. The shRNA transfection solution was prepared
by mixing the shRNA vector, Lipofectamine 2000 and serum-free
medium, and was intratumorally injected at a dose of 50 ml
containing 200 µg shRNA. The doses have been previously reported
(12,15). Sc-shRNA was administered in the
control and CDDP groups; saline, in the control and PEA-shRNA
groups; CDDP, in the CDDP and CDDP + PEA-shRNA groups; and
PEA-shRNA, in the PEA-shRNA and CDDP + PEA-shRNA groups. Four days
after commencement of treatments, 2 mice from the control and
PEA-shRNA groups each were sacrificed to examine tumoral expression
of PEA-15. The other mice were monitored and euthanized by carbon
dioxide exposure on day 20.
Establishment of stable transfectants, cell
viability analysis, bromodeoxyuridine incorporation proliferation
assay, cell cycle assessment, transfection of siRNA, CDDP-resistant
cells, immunoblotting, immunohistochemistry, in situ Ki-67
proliferation and in situ cell death detection. The detailed
methods have been previously described (21–23),
and are available upon request.
Statistical analysis
The association between the expression of PEA-15 and
clinicopathology of gastric cancer was analyzed using a
Mann-Whitney U test. The Kaplan-Meier method was used to estimate
the association between the expression levels of PEA-15 and the
survival of patients, and a log-rank test was used to compare the
survival curves of 2 groups of patients with low (grade ≤5) and
high (grade >5) expression levels of PEA-15. Other data were
expressed as the mean values ± standard deviation, and statistical
analysis was performed using a one-way analysis of variance (ANOVA)
followed by Dunnett's test. P<0.05 was considered to indicate a
statistically significant result.
Results
PEA-15 expression is associated with
the clinicopathology and prognosis of gastric cancer
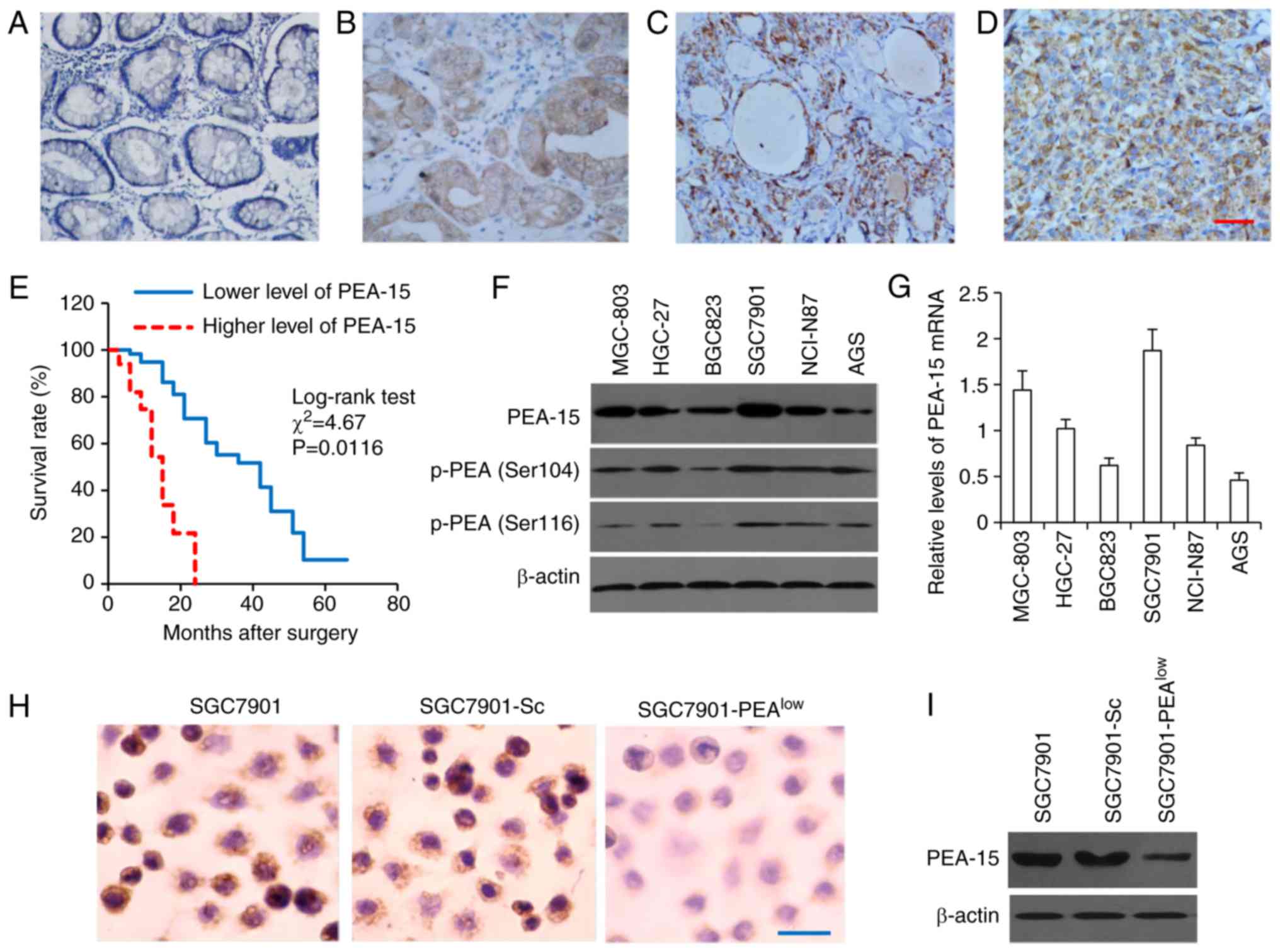
The expression of PEA-15 in tissues was examined
using immunohistochemistry, which revealed that normal gastric
mucosa had weaker PEA-15 expression, while gastric cancer tissues
expressed higher levels of PEA-15 (Fig.
1 A-D), although the expression levels varied (Table I). The results of Mann-Whitney
analysis revealed that the expression levels of PEA-15 were not
significantly associated with the sex or age of the patients, but
significantly associated with TNM staging, tumor differentiation
and pathological types according to the criteria of WHO
histological classification (Table
I). These results indicated that high PEA-15 expression may be
associated with advanced stages and poor differentiation of gastric
tumors. In addition, through a follow-up of patients after surgery,
Kaplan-Meier survival curves were used to analyze the association
between the expression levels of PEA-15 and the prognosis of
patients. We found that patients with lower PEA-15 expression had a
significantly longer overall survival time (median 42.5 months)
than those with higher PEA-15 expression (median 13.7 months)
(Fig. 1E), indicating that higher
PEA-15 expression may be associated with a poor prognosis in
gastric cancer.
 | Table I.Association between PEA-15 expression
and clinicopathology of gastric cancer. |
Table I.
Association between PEA-15 expression
and clinicopathology of gastric cancer.
|
| n | Lower level of
PEA-15 | Higher level of
PEA-15 | P-value |
|---|
| Sex |
|
|
| 0.355 |
|
Male | 87 | 38 | 49 |
|
|
Female | 54 | 20 | 34 |
|
| Age median
(range): |
|
|
| 0.387 |
| 53.4 (27–82) |
|
<60 | 93 | 32 | 61 |
|
|
≤60 | 48 | 21 | 27 |
|
| TNM stage |
|
|
| 0.011 |
| 0 | 5 | 5 | 0 |
|
| I | 26 | 18 | 8 |
|
| II | 38 | 25 | 13 |
|
|
III | 49 | 22 | 37 |
|
| IV | 23 | 5 | 18 |
|
| WHO histological
classification |
|
|
| 0.039 |
|
Tubular | 67 | 41 | 26 |
|
|
Papillary | 42 | 19 | 23 |
|
|
Mucinous | 13 | 3 | 10 |
|
| Poorly
cohesive | 11 | 2 | 9 |
|
|
Uncommon histologic
variants | 8 | 4 | 4 |
|
|
Differentiation |
|
|
| 0.008 |
|
Well | 35 | 28 | 7 |
|
|
Moderate | 74 | 40 | 34 |
|
|
Poor | 32 | 6 | 26 |
|
PEA-15 expression in gastric cancer
cell lines
To gain insight into the functional role of PEA-15
in gastric cancer, we examined PEA-15 expression in a panel of
available human gastric cancer cell lines. PEA-15 protein
expression was variable among these cell lines, thus SGC7901 and
MGC-803 cells expressed higher levels of PEA-15, while BGC823 and
AGS, lower levels of PEA-15 (Fig.
1F). Notably, the bi-phosphorylation forms of PEA-15, p-PEA-15
(Ser104) and p-PEA-15 (Ser116) were also observed (Fig. 1F). In addition, we assessed PEA-15
mRNA expression by qRT-PCR, which revealed a similar variability to
that of protein expression (Fig.
1G). Since SGC7901 cells were revealed to express the highest
level of PEA-15 among the 6 types of gastric cells, they were
selected for generating stable transfectants. SGC7901 cells stably
transfected with PEA-15 shRNA or Sc-shRNA were termed
SGC7901-PEAlow and SGC7901-Sc, respectively. Compared
with the parental SGC7901 cells, SGC7901-PEAlow
expressed significantly lower, while SGC7901-Sc cells expressed
similar, levels of PEA-15, when examined by immunocytochemistry
(Fig. 1H) and immunoblotting
(Fig. 1I).
PEA-15 depletion increases the
sensitivity of gastric cancer cells to CDDP by inducing cell cycle
arrest at the G1 phase
We first demonstrated that PEA-15 depletion
inhibited the growth of gastric cancer cells in culture. As
revealed in Fig. 2A,
SGC7901-PEAlow cells had a significantly lower
viability, while SGC7901-Sc cells had a similar viability, compared
with parental cells. CDDP treatment reduced the viability of the 3
cell lines in a concentration-dependent manner (Fig. 2B). SGC7901-PEAlow cells
were more sensitive to CDDP than parental or SGC7901-Sc cells
(Fig. 2B). CDDP treatment
significantly reduced the viability of both SGC7901 and
SGC7901-PEAlow cells, but the viability of
SGC7901-PEAlow cells was significantly lower than that
of SGC7901 cells after incubation for 48 h with CDDP (15 µM)
(Fig. 2C). SGC7901 and SGC7901-Sc
cells had revealed similar cell viabilities when they were
incubated in the absence or presence of CDDP (data not shown and
available upon request).
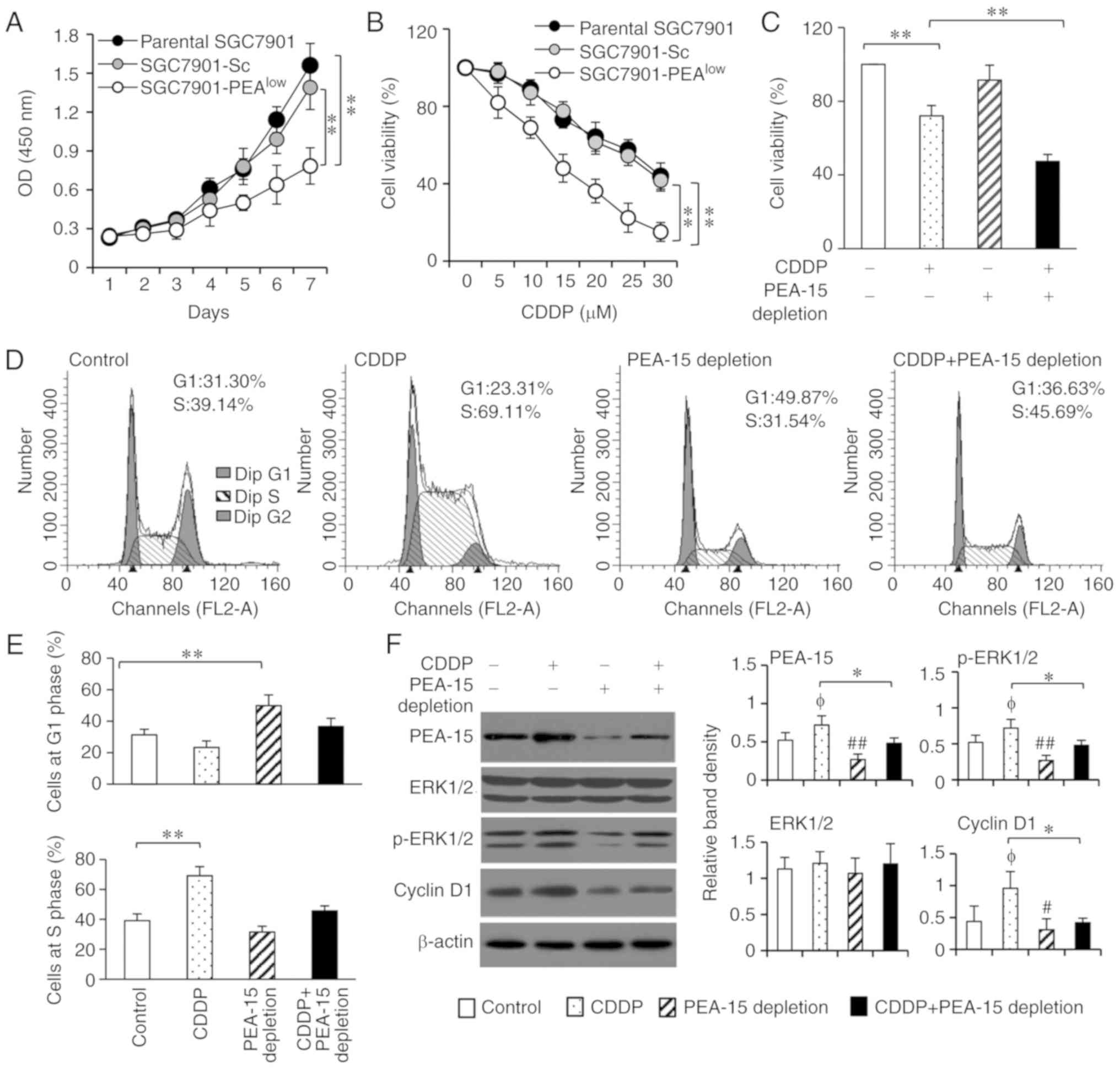 | Figure 2.PEA-15 depletion enhances the
sensitivity of gastric cancer cells to CDDP by inducing cell cycle
arrest at phase G1 through the ERK pathway. (A) Parental SGC7901,
SGC7901-Sc and SGC7901-PEAlow cells were cultured for 7
days, and their viability was assessed by a CCK-8 assay at the
indicated time-points. Cell viability was represented by optical
density (OD) at 450 nm. (B) Cells were incubated with increasing
concentrations of CDDP for 48 h, and their percentage viability was
compared to that of untreated respective cells. (C) Cells were
cultured in the presence or absence of CDDP (15 µM) for 48 h, and
their viability was assessed. (D and E) The aforementioned cells
were subjected to flow cytometry to assess cell cycle distribution.
(D) Representative histograms were shown and (E) the percentages of
cells at the G1 and S phases were plotted. (F) Lysates from the
aforementioned cells were immunoblotted. The density of each band
was measured and normalized to β-actin. *P<0.05 and **P<0.001
indicate a significant difference. φP<0.05 indicates
a significant increase, while #P<0.05 and
##P<0.01, indicate a significant reduction, from the
control. PEA-15, phosphoprotein enriched in astrocytes 15; CDDP,
cisplatin; ERK, extracellular signal-regulated kinase; CCK-8, Cell
Counting Kit-8. |
Analyses of cell cycle distribution revealed that
CDDP treatment induced more cells arrested at the S phase, while
PEA-15 depletion induced more cells arrested at the G1 phase,
compared with the controls (Fig. 2D and
E). The anticancer mechanism of CDDP mainly relies on its
activity to inhibit DNA replication, and cells in phase G1 appear
to be maximally sensitive to CDDP (24). Thus, unsurprisingly PEA-15 depletion
enhanced the sensitivity of SGC7901 cells to CDDP and worked
together with CDDP to further reduce cell viability (Fig. 2C).
PEA-15 depletion and CDDP inhibits
cell proliferation by regulating ERK/cyclin D1
The results of viability and cell cycle distribution
were consistent with cell proliferation as examined by BrdU
incorporation assays. CDDP treatment and PE-15 depletion resulted
in significant decreases in the percentage of BrdU-positive cells,
and CDDP treatment further reduced the percentage of BrdU-positive
SGC7901-PEAlow cells, compared with the controls (data
not shown and available upon request). Previous studies have
revealed that PEA-15 promotes cell proliferation by regulating ERK
phosphorylation (9,25). As revealed in Fig. 2F, exposure of CDDP significantly
increased the expression of p-ERK1/2 though it had no effect on
total ERK1/2, resulting in a sequential upregulation of cyclin D1,
which is a well-known downstream factor of ERK1/2 and plays key
roles in cell cycle transition from the G1 to S phase (26). PEA-15 depletion downregulated the
expression of p-ERK1/2 and cyclin D1 expression and could reverse
the increased expression of p-ERK1/2 and cyclin D1 induced by CDDP,
in SGC7901-PEAlow cells (Fig. 2F). Notably, SGC7901 and SGC7901-Sc
cells revealed similar alterations of gene expression upon CDDP
incubation (data not shown and available upon request). The
aforementioned results indicated that PEA-15 depletion enhanced the
sensitivity of gastric cancer cells to CDDP and were confirmed
using another cell line MGC-803 (data not shown and available upon
request).
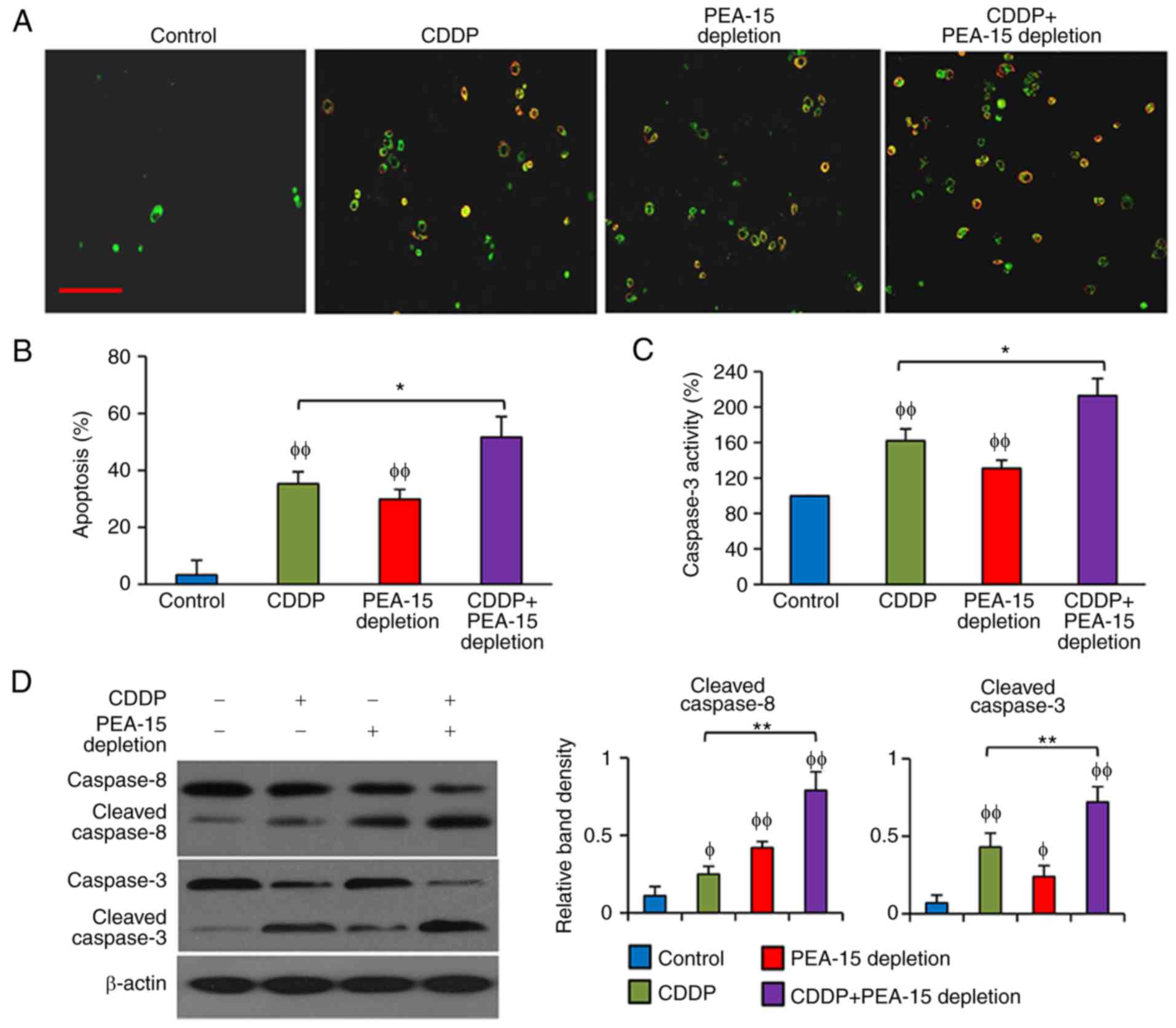
PEA-15 depletion enhances the
pro-apoptotic activity of CDDP in gastric cancer cells
CDDP and PEA-15 depletion significantly increased
apoptosis of SGC7901 cells, and CDDP treatment further increased
the apoptosis rate of SGC7901-PEAlow cells (Fig. 3A and B). In accordance, both CDDP
and PEA-15 increased caspase-3 activity in SGC7901 cells, and CDDP
further increased caspase-3 activity in SGC7901-PEAlow
cells (Fig. 3C). CDDP had little
effect on caspase-8, but significantly increased the cleavage of
caspase-3, while PEA-15 depletion increased the cleavage of
caspase-8 and caspase-3, and CDDP treatments further elevated the
cleavage of caspase-3 in SGC7901-PEAlow cells (Fig. 3D).
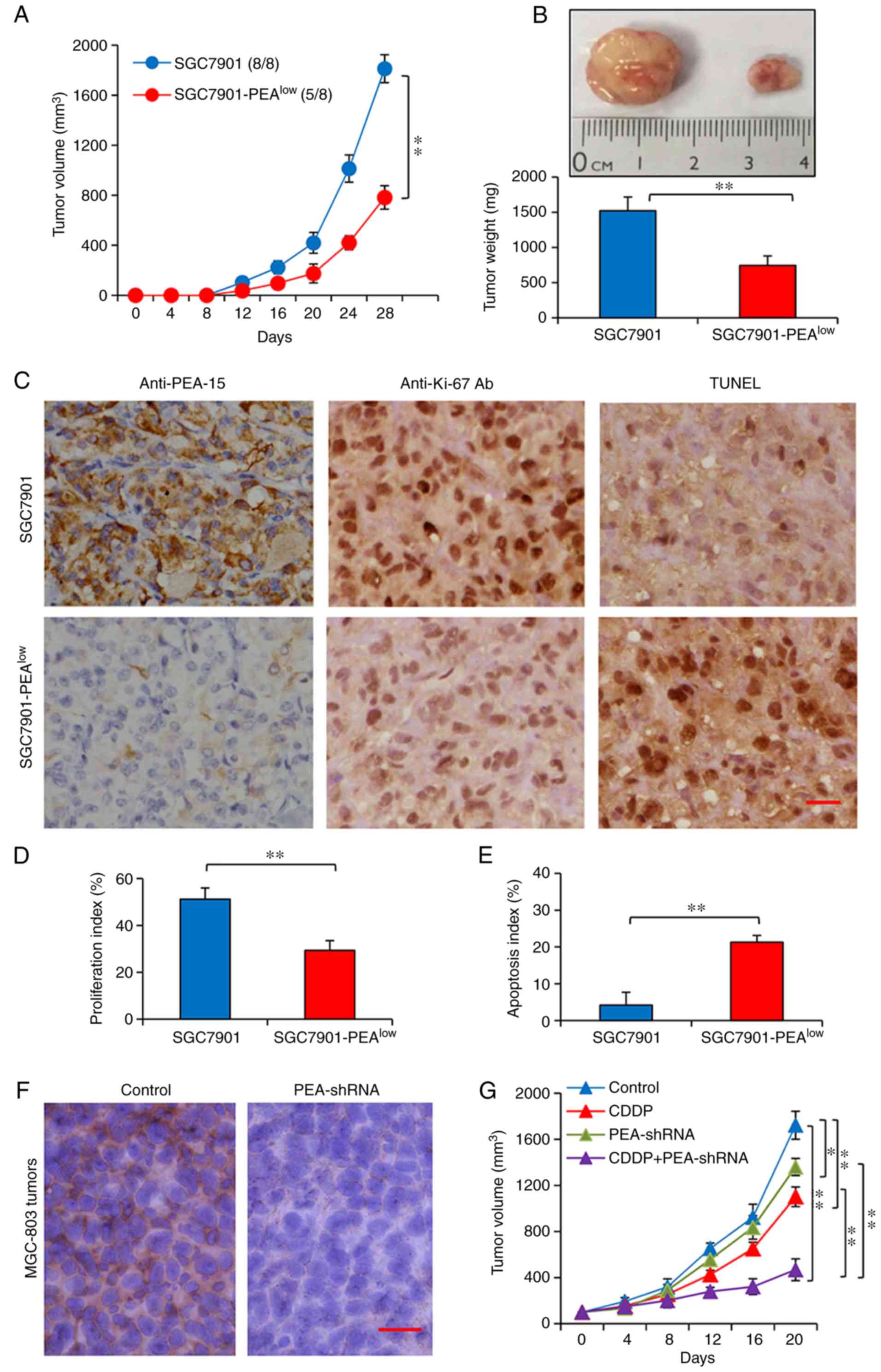
PEA-15 depletion enhances the
therapeutic effects of CDDP in gastric cancer animal models
The aforementioned results led us to investigate the
function of PEA-1 in vivo. The study of tumorigenesis
revealed that tumors were observed in all the 8 mice receiving
subcutaneous injection of SGC7901 cells, but in only 5 out of 8
mice receiving SGC7901-PEAlow cells (Fig. 4A). Furthermore,
SGC7901-PEAlow tumors grew to 782.3±94.5 mm3
(736.4±138.6 mg in weight), significantly smaller than SGC7901
tumors whose size was 1,813.6±114.7 mm3 (1,524±187.8 mg
in weight), 4 weeks after cell inoculation (Fig. 4A and B). The tumors harvested in
Fig. 4B were subjected to
immunohistochemistry to examine PEA-15 expression, cell
proliferation and apoptosis in situ. Consistent with the
in vitro results (Fig. 1F and
G), SGC7901-PEAlow tumors expressed markedly lower
levels of PEA-1 protein than SGC7901 tumors (Fig. 4C). PEA-1 depletion significantly
inhibited cell proliferation and promoted apoptosis (Fig. 4C-E). The expression of key molecules
in vivo was consistent with that obtained in vitro;
and notably p-PEA-15 at both Ser104 and Ser116 residues was also
downregulated in SGC7901-PEAlow tumors (data not shown
and available upon request).
In the present study, to assess the therapeutic
effects, we established subcutaneous tumors using another gastric
cancer cell line, MGC-803, which was also revealed to express
higher levels of PEA-15 (Fig. 1F and
G). When tumors reached ~100 mm3, they were assigned
to different treatments as indicated in Materials and methods.
Intratumoral delivery of PEA-shRNA resulted in downregulation of
PEA-1 in situ, compared with control tumors injected with
Sc-shRNA (Fig. 4F). MGC-803 tumors
injected with PEA-shRNA were significantly smaller (1,360.4±75.3
mm3) than control tumors (1719.5±123.1 mm3),
20 days after the commencement of treatments (Fig. 4G). CDDP treatment also led to a
significant reduction in the size of tumors injected with Sc-shRNA
(1,098.6±123.1 mm3) and further reduced the size of
tumors injected with PEA-shRNA (467.9±88.7 mm3)
(Fig. 4G).
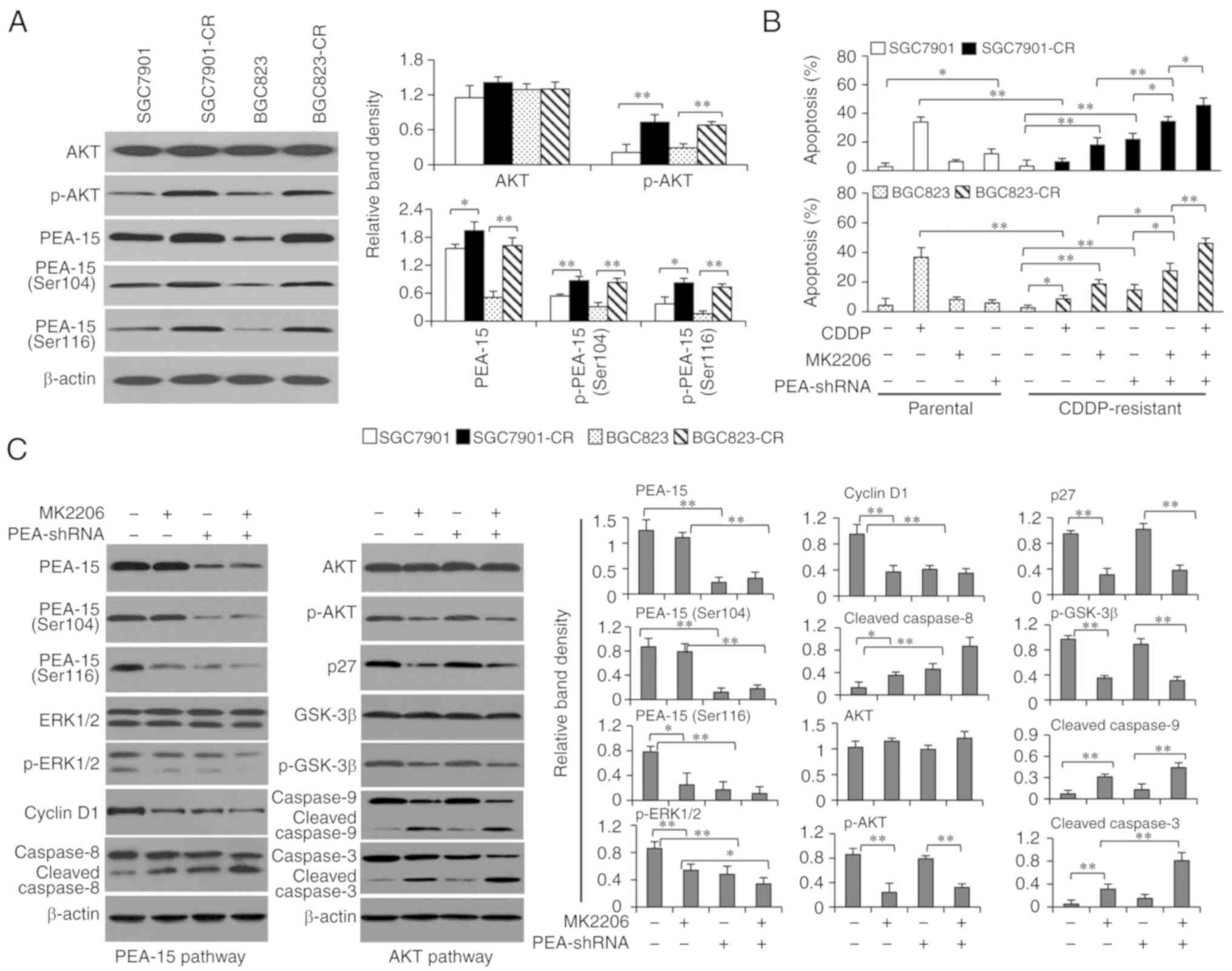
PEA-15 regulated by AKT participates
in the mechanisms for CDDP resistance of gastric cancer cells
We previously reported that activation of AKT
contributes to the resistance of gastric cancer cells to CDDP
(12), and AKT is an upstream
factor of PEA-15 by regulating its phosphorylation (11,13,14).
Therefore, the functional role of PEA-15 in the mechanisms of CDDP
resistance was investigated using SGC7901 cells expressing higher
levels of PEA-15, BGC823 cells expressing lower levels of PEA-15,
and 2 CDDP-resistant cell lines (SGC7901-CR and BGC823-CR) that
were previously established (12).
As anticipated, CDDP-resistant cells expressed a similar level of
AKT but an increased level of p-AKT, compared with respective
parental cells (Fig. 5A), in
accordance with our previous study (12). CDDP-resistant cells had higher
expression of PEA-15 than their respective parental cells, but the
difference was less pronounced for SGC7901 cells which had a high
baseline of PEA-15 expression (Fig.
5A). Notably, the expression of p-PEA-15 at both Ser116 and
Ser104 residues was also increased in CDDP-resistant cells
(Fig. 5A). In addition, SGC7901-CR
and BGC823-CR cells had significantly lower rates of apoptosis,
compared with respective parental cells, when they were incubated
for 48 h with CDDP (Fig. 5B).
Specific inhibition of AKT by a specific AKT inhibitor (MK2206), a
novel selective inhibitor of pan-Akt (27), resulted in significantly higher
apoptosis rates of SGC7901-CR and BGC823-CR cells (Fig. 5B). Transfection of PEA-shRNA also
significantly increased apoptosis rates of SGC7901-CR and BGC823-CR
cells. In addition, combination of MK2206 and PEA-shRNA
transfection resulted in higher apoptosis rates of CDDP-resistant
cells than either MK220 or PEA-shRNA alone (Fig. 5B).
In exploring the regulatory mechanisms between
PEA-15 and AKT pathways, SGC7901-Sc and SGC7901-PEAlow
cells were incubated with MK2206 and the expression of key
molecules involved in the 2 pathways was detected. As revealed in
Fig. 5C, MK2206 significantly
inhibited the AKT pathway, as evidenced by the reduced expression
of p-AKT and its downstream factors including p27, phosphorylated
glycogen synthase kinase 3β (p-GSK-3β), and the increased cleavage
of caspase-9 and −3. MK2206 had little effect on the expression of
PEA-15, p-PEA-15 at Ser104 residue or total ERK1/2 however it could
significantly reduce the expression of p-PEA-15 at Ser116 residue,
resulting in sequential downregulation of p-ERK1/2, cyclin D1 and
increased cleavage of caspase-8 (Fig.
5C). The results indicated that inhibition of AKT could
regulate the PEA-15 pathway by reducing the phosphorylation of
PEA-15 at Ser116 residue. However, depletion of PEA-15 by PEA-shRNA
reduced the expression of PEA-15 and the phosphorylation of PEA-15
at both Ser104 and Ser116 residues but had little effect on either
the expression of AKT, or p-AKT, or AKT downstream factors
including p27, GSK-3β and the cleavage of caspase-9 (Fig. 5C). The results indicated that
depletion of PEA-15 had little effect on the AKT pathway,
indicating the regulatory effects between PEA-15 and AKT may be
unidirectional in gastric cancer cells.
Discussion
As a multifunctional phosphoprotein, PEA-15 has
displayed an important role in several cancer entities (6–9), but
its expression and function have not yet been investigated in
gastric cancer. The present study demonstrated that PEA-15 was
overexpressed in gastric cancer tissues and cells. In addition,
clinical gastric tumors with higher expression of PEA-15 had more
advanced TNM stages and poorer cell differentiation, and the
patients had a shorter survival time. PEA-15 was also associated
with histopathological types of gastric cancer. Similarly, it has
been reported that PEA-15 overexpression is observed in breast
cancer (11,28), HCC (9), lung cancer (29), esophageal carcinoma (30) and colorectal cancer (8). In these tumors, PEA-15 acted as a
tumor promoter and was associated with poor prognosis. Notably,
PEA-15 overexpression has been revealed to be associated with a
good prognosis in ovarian cancer, where PEA-15 exists in
unphosphorylated status (31,32).
The difference of its role in tumor promotion or suppression may
depend on its phosphorylation status (9). The present results revealed that
gastric cancer cells expressed p-PEA-15 at both Ser104 and Ser116
residues, which had also been revealed in HCC samples (9). Therefore, the results may indicate
that PEA-15 acts as a tumor promoter in gastric cancer similar to
other types of malignancies excluding ovarian cancer.
It has been demonstrated that p-PEA-15 enhanced cell
proliferation by activating the ERK pathway (9,14,25),
which regulates cyclin D1, a key molecule controlling cell cycle
transition from the G1 to S phase (26). Similarly, it was revealed herein
that PEA-15 depletion resulted in reduced expression of p-ERK1/2,
leading to downregulation of cyclin D1 and cell cycle arrest at the
G1 phase. CDDP belongs to the alkylating agent family and its major
mechanism involves inhibiting DNA replication, thus CDDP exposure
induces cell cycle arrest at the S/G2 phases. Consistently our
results revealed that CDDP incubation induced more cells arrested
at the S phase. It has been reported that cells in phase G1 appear
to be the most sensitive to CDDP (24). This may partially explain the
mechanism involved in the enhanced activity of CDDP by PEA-15
depletion.
CDDP displays its anticancer activity by inducing
apoptosis and caspase-3 activation (33). However, the resistance to CDDP
largely limits its therapeutic benefits. PEA-15 has been revealed
to contribute to the insensitivity or resistance to various
chemotherapeutic agents including CDDP in various types of cancer
(8,9,11). In
the present study, it was demonstrated that PEA-15 depletion
re-sensitized CDDP-resistant gastric cancer cells to CDDP. The
analysis exploring the mechanisms revealed that PEA-15 depletion
inhibited the cleavage of caspase-8, leading to the inhibition of
caspase-3 activation. Supportively it has been revealed that PEA-15
displays its anti-apoptotic activity mainly through its binding to
Fas-associated protein with death domain (FADD), which regulates
the activation of caspase-8 (9).
The AKT pathway has emerged as a potent molecular
target for overcoming acquired resistance to cancer chemotherapy,
since AKT is a cancer multidrug resistance locus, by
phosphorylating many proteins involved in cancer hallmarks
including apoptosis resistance (34,35).
We have previously reported that CDDP-resistant gastric cancer
cells expressed higher levels of p-AKT, whose activation regulates
the resistance to CDDP (12).
Conversely, several studies have indicated that AKT is an upstream
factor of PEA-15 by phosphorylating it at the site of Ser116
(11,13,14).
In the present study, we further confirmed the overexpression of
p-AKT and p-PEA-15 at Ser104 and Ser116 residues in CDDP-resistant
gastric cancer cells. Specific inhibition of AKT by MK2206
(27) not only inhibited the AKT
pathway, but also downregulated p-PEA-15 at Ser116 and the related
downstream factors. However, depletion of PEA-15 had little effect
on the expression of either total AKT or p-AKT, or the related
downstream factors. Combination of AKT inhibition and PEA-15
depletion further increased apoptosis rates of CDDP-resistant
gastric cancer cells by activating caspase cascades via both
caspase-8 and −9. The results may indicate that both PEA-15 and AKT
contribute to the mechanisms of CDDP resistance but their
regulatory effect is unidirectional.
The proposed mechanisms for its functions in
influencing the proliferation and apoptosis of gastric cancer
cells, involvement in the mechanisms of CDDP resistance, and
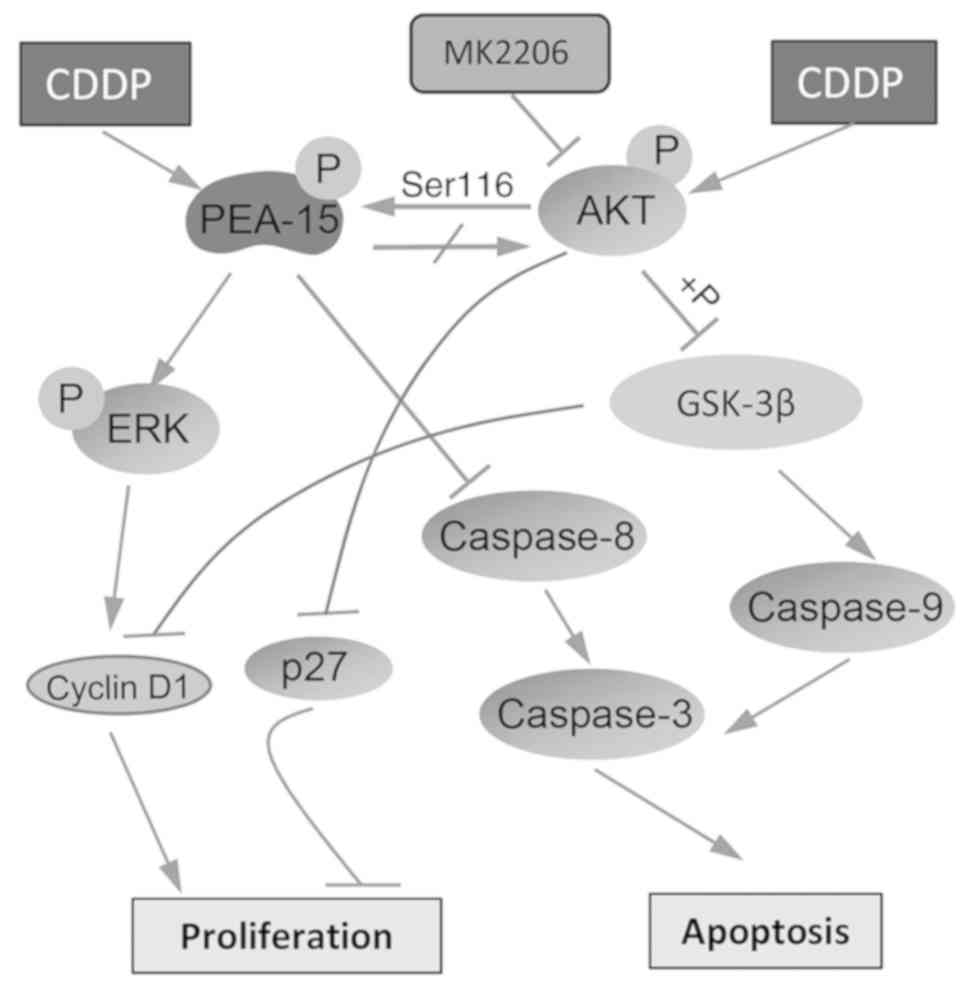
regulation by AKT are summarized in Fig. 6. PEA-15 participates in controlling
cell proliferation by activating ERK, which regulates the
expression of cyclin D1 (9,14,25,26).
PEA-15 displays its anti-apoptotic activity by binding to FADD,
resulting in the inhibition of caspase-8 and −3 (9). Exposure to CDDP leads to the higher
expression of p-PEA-15 and p-AKT, contributing to the mechanisms of
CDDP resistance. Activated AKT regulates GSK-3β, which controls
cell apoptosis and proliferation by regulating p27, cyclin D1 and
caspase-9 (23,36). Activated AKT is also able to mediate
cell proliferation and apoptosis by regulating the PEA-15
pathway.
In summary, PEA-15 is overexpressed in gastric
cancer tissues and associated with the clinicopathology and
prognosis, and contributes to AKT-regulated CDDP resistance.
Although not investigated in the present study, PEA-15 may also
participate in the mechanisms for drug resistance of gastric cancer
by regulating other pathways. For example, PEA-15 was revealed to
regulate autophagy by activating the c-Jun N-terminal kinase (JNK)
pathway in glioma cells and autophagy has been revealed to be
involved in drug resistance in many types of cancer (19). It is well known that PEA-15
activates ERK1/2, which participates in many biological behaviors
and drug resistance of cancer cells (6,10). The
initial function of PEA-15 is to mediate energy metabolism
(5), which is critical for the
therapy resistance of cancer cells (37). The present results indicated that
PEA-15 may be a valuable biomarker and potential therapeutic target
for gastric cancer, particularly for patients expressing higher
levels of PEA-15 and becoming resistant to CDDP.
Acknowledgements
We thank Dr Shiva Reddy from the University of
Auckland (New Zealand) for revising and editing the manuscript.
Funding
The present study was supported by grants from the
National Key Research and Development Program of China (no.
2017YFC1308602), the National Natural Scientific Foundation of
China (nos. 81472321 and 81703055), and the Fundamental Research
Funds for the Provincial Universities in Heilongjiang Province,
China (no. 2017LCZX06).
Availability of data and materials
The majority of the data generated or analyzed
during the present study are included in this published article.
Some data are not presented and available upon request.
Authors' contributions
XJ, XX and XS participated in the conception and
study design. XJ and WL performed bench and animal experiments. CZ,
DJ, ZW and ML participated in examining the clinical specimens and
assisted in bench and animal experiments. XJ and CZ were involved
in the data collection and analyses. XJ and XS drafted the
manuscript. XX revised the manuscript. XX and XS supervised the
study. All authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The study analyzing human specimens was approved by
the Ethics Committee of Qingdao Municipal Hospital (no. 20150819),
and informed consents were obtained (Qingdao, China). The animal
experiments were approved (permit no. SYXK20020009) by the Animal
Ethics Committee of Harbin Medical University (Harbin, China).
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cats A, Jansen EP, van Grieken NC,
Sikorska K, Lind P, Nordsmark M, Meershoek-Klein Kranenbarg E, Boot
H, Trip AK, Swellengrebel HA, et al: Chemotherapy versus
chemoradiotherapy after surgery and preoperative chemotherapy for
resectable gastric cancer (CRITICS): An international, open-label,
randomised phase 3 trial. Lancet Oncol. 19:616–628. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Koizumi W, Tanabe S, Azuma M, Ishido K,
Nishimura K, Sasaki T, Nakatani K, Higuchi K, Nakayama N and Katada
C: Impacts of fluorouracil-metabolizing enzymes on the outcomes of
patients treated with S-1 alone or S-1 plus cisplatin for
first-line treatment of advanced gastric cancer. Int J Cancer.
126:162–170. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Araujo H, Danziger N, Cordier J, Glowinski
J and Chneiweiss H: Characterization of PEA-15, a major substrate
for protein kinase C in astrocytes. J Biol Chem. 268:5911–5920.
1993.PubMed/NCBI
|
|
5
|
Fiory F, Formisano P, Perruolo G and
Beguinot F: Frontiers: PED/PEA-15, a multifunctional protein
controlling cell survival and glucose metabolism. Am J Physiol
Endocrinol Metab. 297:E592–E601. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Greig FH and Nixon GF: Phosphoprotein
enriched in astrocytes (PEA)-15: A potential therapeutic target in
multiple disease states. Pharmacol Ther. 143:265–274. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mohammed HN, Pickard MR and
Mourtada-Maarabouni M: The protein phosphatase 4-PEA15 axis
regulates the survival of breast cancer cells. Cell Signal.
28:1389–1400. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Funke V, Lehmann-Koch J, Bickeboller M,
Benner A, Tagscherer KE, Grund K, Pfeifer M, Herpel E, Schirmacher
P, Chang-Claude J, et al: The PEA-15/PED protein regulates cellular
survival and invasiveness in colorectal carcinomas. Cancer Lett.
335:431–440. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Quintavalle C, Hindupur SK, Quagliata L,
Pallante P, Nigro C, Condorelli G, Andersen JB, Tagscherer KE, Roth
W, Beguinot F, et al: Phosphoprotein enriched in diabetes
(PED/PEA15) promotes migration in hepatocellular carcinoma and
confers resistance to sorafenib. Cell Death Dis. 8:e31382017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buonomo R, Giacco F, Vasaturo A, Caserta
S, Guido S, Pagliara V, Garbi C, Mansueto G, Cassese A, Perruolo G,
et al: PED/PEA-15 controls fibroblast motility and wound closure by
ERK1/2-dependent mechanisms. J Cell Physiol. 227:2106–2116. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stassi G, Garofalo M, Zerilli M,
Ricci-Vitiani L, Zanca C, Todaro M, Aragona F, Limite G, Petrella G
and Condorelli G: PED mediates AKT-dependent chemoresistance in
human breast cancer cells. Cancer Res. 65:6668–6675. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun XP, Dong X, Lin L, Jiang X, Wei Z,
Zhai B, Sun B, Zhang Q, Wang X, Jiang H, et al: Up-regulation of
survivin by AKT and hypoxia-inducible factor 1α contributes to
cisplatin resistance in gastric cancer. FEBS J. 281:115–128. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Trencia A, Perfetti A, Cassese A,
Vigliotta G, Miele C, Oriente F, Santopietro S, Giacco F,
Condorelli G, Formisano P, et al: Protein kinase B/Akt binds and
phosphorylates PED/PEA-15, stabilizing its antiapoptotic action.
Mol Cell Biol. 23:4511–4521. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hayashi N, Peacock JW, Beraldi E, Zoubeidi
A, Gleave ME and Ong CJ: Hsp27 silencing coordinately inhibits
proliferation and promotes Fas-induced apoptosis by regulating the
PEA-15 molecular switch. Cell Death Differ. 19:990–1002. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li L, Jiang X, Zhang Q, Dong X, Gao Y, He
Y, Qiao H, Xie F, Xie X and Sun X: Neuropilin-1 is associated with
clinicopathology of gastric cancer and contributes to cell
proliferation and migration as multifunctional co-receptors. J Exp
Clin Cancer Res. 35:162016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu B, El Hajj N, Sittler S, Lammert N,
Barnes R and Meloni-Ehrig A: Gastric cancer: Classification,
histology and application of molecular pathology. J Gastrointest
Oncol. 3:251–261. 2012.PubMed/NCBI
|
|
17
|
Shin M, Lee KE, Yang EG, Jeon H and Song
HK: PEA-15 facilitates EGFR dephosphorylation via ERK sequestration
at increased ER-PM contacts in TNBC cells. FEBS Lett.
589:1033–1039. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Böck BC, Tagscherer KE, Fassl A, Krämer A,
Oehme I, Zentgraf HW, Keith M and Roth W: The PEA-15 protein
regulates autophagy via activation of JNK. J Biol Chem.
285:21644–21654. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei Z, Jiang X, Qiao H, Zhai B, Zhang L,
Zhang Q, Wu Y, Jiang H and Sun X: STAT3 interacts with Skp2/p27/p21
pathway to regulate the motility and invasion of gastric cancer
cells. Cell Signal. 25:931–938. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma L, Li G, Zhu H, Dong X, Zhao D, Jiang
X, Li J, Qiao H, Ni S and Sun X: 2-Methoxyestradiol synergizes with
sorafenib to suppress hepatocellular carcinoma by simultaneously
dysregulating hypoxia-inducible factor-1 and −2. Cancer Lett.
355:96–105. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu
B, Pan S, Dong X, Tan G, Wei Z, et al: Inhibition of Akt reverses
the acquired resistance to sorafenib by switching protective
autophagy to autophagic cell death in hepatocellular carcinoma. Mol
Cancer Ther. 13:1589–1598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han P, Li H, Jiang X, Zhai B, Tan G, Zhao
D, Qiao H, Liu B, Jiang H and Sun X: Dual inhibition of Akt and
c-Met as a second-line therapy following acquired resistance to
sorafenib in hepatocellular carcinoma cells. Mol Oncol. 11:320–334.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shah MA and Schwartz GK: Cell
cycle-mediated drug resistance: An emerging concept in cancer
therapy. Clin Cancer Res. 7:2168–2181. 2001.PubMed/NCBI
|
|
25
|
Eckert A, Böck BC, Tagscherer KE, Haas TL,
Grund K, Sykora J, Herold-Mende C, Ehemann V, Hollstein M,
Chneiweiss H, et al: The PEA-15/PED protein protects glioblastoma
cells from glucose deprivation-induced apoptosis via the ERK/MAP
kinase pathway. Oncogene. 27:1155–1166. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stottrup C, Tsang T and Chin YR:
Upregulation of AKT3 confers resistance to the AKT inhibitor MK2206
in breast cancer. Mol Cancer Ther. 15:1964–1974. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hindupur SK, Balaji SA, Saxena M, Pandey
S, Sravan GS, Heda N, Kumar MV, Mukherjee G, Dey D and Rangarajan
A: Identification of a novel AMPK-PEA15 axis in the
anoikis-resistant growth of mammary cells. Breast cancer Res.
16:4202014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zanca C, Garofalo M, Quintavalle C, Romano
G, Acunzo M, Ragno P, Montuori N, Incoronato M, Tornillo L,
Baumhoer D, et al: PED is overexpressed and mediates TRAIL
resistance in human non-small cell lung cancer. J Cell Mol Med.
12:2416–2426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang M, Zhu XY, Wang L and Lin Y:
Expression and significance of CDC25B, PED/PEA-15 in esophageal
carcinoma. Cancer Biother Radiopharm. 30:139–145. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bartholomeusz C, Rosen D, Wei C, Kazansky
A, Yamasaki F, Takahashi T, Itamochi H, Kondo S, Liu J and Ueno NT:
PEA-15 induces autophagy in human ovarian cancer cells and is
associated with prolonged overall survival. Cancer Res.
68:9302–9310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee J, Bartholomeusz C, Krishnamurthy S,
Liu P, Saso H, Lafortune TA, Hortobagyi GN and Ueno NT: PEA-15
unphosphorylated at both serine 104 and serine 116 inhibits ovarian
cancer cell tumorigenicity and progression through blocking
β-catenin. Oncogenesis. 1:e222012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Singh M, Chaudhry P, Fabi F and Asselin E:
Cisplatin-induced caspase activation mediates PTEN cleavage in
ovarian cancer cells: A potential mechanism of chemoresistance. BMC
Cancer. 13:2332013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Radisavljevic Z: AKT as locus of cancer
multidrug resistance and fragility. J Cell Physiol. 228:671–674.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li CW, Xia W, Lim SO, Hsu JL, Huo L, Wu Y,
Li LY, Lai CC, Chang SS, Hsu YH, et al: AKT1 inhibits
Epithelial-to-mesenchymal transition in breast cancer through
phosphorylation-dependent Twist1 degradation. Cancer Res.
76:1451–1462. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Serova M, de Gramont A, Tijeras-Raballand
A, Dos Santos C, Riveiro ME, Slimane K, Faivre S and Raymond E:
Benchmarking effects of mTOR, PI3K, and dual PI3K/mTOR inhibitors
in hepatocellular and renal cell carcinoma models developing
resistance to sunitinib and sorafenib. Cancer Chemother Pharmacol.
71:1297–1307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maiso P, Huynh D, Moschetta M, Sacco A,
Aljawai Y, Mishima Y, Asara JM, Roccaro AM, Kimmelman AC and
Ghobrial IM: Metabolic signature identifies novel targets for drug
resistance in multiple myeloma. Cancer Res. 75:2071–2082. 2015.
View Article : Google Scholar : PubMed/NCBI
|