Introduction
Endometriosis is defined as the presence of the
ectopic implantation of endometrial glands and stroma at
extra-uterine sites. Endometriosis is a common, chronic
inflammatory disease that affects ~10% of women of reproductive age
(1,2). The common symptoms of this disease are
dysmenorrhea, dyspareunia, chronic pelvic pain and infertility
(1,2). Epidemiologically, endometriosis has
been reported to increase the risk of certain types of
malignancies, particularly for ovarian endometrioid carcinoma (EC),
clear cell carcinoma (CCC), low-grade serous carcinoma and
seromucinous neoplasms (3–6). CCC and EC of the ovary are the two
most common types of ovarian cancer, which arise from endometriosis
(4–6). Endometriosis is found in approximately
20% of EC and CCC cases, presents adjacent to the tumor, and has
direct topological continuity with the carcinoma (3). Patients with endometriosis-associated
ovarian cancer (EAOC) belong to the relatively younger-aged
population, and have early-stage and low histological grade tumors
compared with non-EAOC patients (7). EAOC tumors frequently occur in
perimenopausal and early postmenopausal women. Ovarian cancer is
known to develop in approximately 1% of women with endometriosis
(4). Endometriosis may be related
to an increased risk of EAOC; however, the underlying mechanism
remains largely unknown. Over the past decade, a dramatic shift has
occurred in our understanding of the pathophysiology of EAOC.
The aim of the present study was to provide an
overview of the current pathophysiological concepts of the
malignant transformation of endometriosis. We summarize recent
knowledge about the role of the shared and independent (epi)genetic
background between EC and CCC, and the current hypotheses regarding
the pathophysiology of the malignant processes.
Data collection methods
A computerized literature search was conducted to
identify relevant studies reported in the English language. We
collected a comprehensive literature search from the PubMed and
Embase databases up to April, 2018, combining the keywords
‘endometriosis’, ‘endometriosis-associated ovarian cancer’,
‘endometrioid carcinoma’, ‘clear cell carcinoma’, ‘pathogenesis’,
‘carcinogenesis’, ‘oxidative stress’, ‘hemoglobin’, ‘iron’,
‘inflammation’, ‘endothelial cells’, ‘extracellular matrix’ and
‘microenvironment’. A variety of combinations of these terms were
used, depending on which database was searched. Furthermore, the
references of each article were searched to identify potentially
relevant studies. Publications of original studies and review
articles were included, while those documenting opinions, points of
view or anecdotes were discarded.
Results
The mechanisms underlying the
malignant transformation of endometriosis
The gene expression profile-based clustering divided
ovarian cancer into two groups, type I and type II, which is
generally based on the potential clinical and translational value
of the dualistic model of ovarian carcinogenesis (8,9). Type
I ovarian cancer consists of patients with low-grade serous,
mucinous, EC, CCC and slow-growing tumors, while type II is
composed of patients with rapidly-growing high-grade serous
carcinoma (HGSC) and highly aggressive malignancies (8,9). EAOC
belongs to the type I category and consists of two major subtypes
originating from EC and CCC, which exhibits different pathological
and clinical features, characterized by unique morphologies and
responses to treatment (6).
EC may occur during an estrogenic mode of action due
to the observed induction of estrogen receptor (ESR) isoforms
(10–14). Estrogen is considered to be involved
in ovarian cancer progression (15). The Wnt/β-catenin signaling pathway
regulated by estrogen is highly activated in EC and inhibits
oxidative stress-induced cell apoptosis (16–18).
By contrast, estrogens are known to produce reactive oxygen species
(ROS) and are implicated in cellular carcinogenesis, as chronic
oxidative stress promotes cell growth, survival and the tumorigenic
potential of breast cancer cells (19). In the present study, we provide an
update on the recent advances in the understanding of the
reduction-oxidation (redox)-related molecular signaling and
imbalance in the cellular redox state in malignant transformation
of endometriosis (8,20–27).
The pathogenesis of the malignant transformation of endometriosis
remains obscure; however, the results of several studies support
the hypothesis that the redox imbalance, inflammatory/immune
response, cell cycle regulation and hormone activity are the
deregulated functions and act in a dynamic epigenetic network
(20,22,23,24).
Redox imbalance: Possible unexpected
results
Repeated episodes of hemorrhage occur in
endometriosis throughout menstruation (23). Red blood cells accumulate in ovarian
endometrioma (OE) and in the pelvic cavity through retrograde
menstruation. The destruction of red blood cells leads to the
release of Hb, heme and free iron (22,28).
While Hb provides life-sustaining oxygen delivery, extracellular
free Hb produces toxic heme degradation products and is a source of
ROS due to inherent peroxidase activity (22,28,29).
These findings are consistent with and are supported by in
vitro experimental data (30)
and in vivo clinical data (31). Yamaguchi et al presented, for
the first time, that the iron-induced persistent oxidative stress
within the endometriotic cyst leads to dynamic changes in the
oxidative environment, which may play a crucial role in the process
of endometriosis carcinogenesis (30). The authors reported that patients
with endometriotic cysts had significantly higher cyst fluid
concentration of free iron (100.9 mmol/l = 5,635 mg/l), compared to
those with non-endometriotic cysts (0.075 mmol/l = 4.19 mg/l)
(30). Free iron concentrations in
CCC (4.27 mmol/l = 238 mg/l) were 20-fold lower than those in
endometriotic cysts (30). Since
free iron has a propensity to induce oxidative stress, DNA damage,
protein modification and lipid peroxidation, we hypothesized that
patients with EAOC would have much higher levels of iron-related
compounds compared with those with benign OE. Therefore, Yoshimoto
et al extensively investigated cyst fluid levels of
iron-related compounds in benign OE and EAOC (31). The median ± SD concentrations of
total iron, heme iron and free iron for OE and EAOC cysts were
244.4±204.9 mg/l vs. 14.2±36.6 mg/l (total iron), 303.9±324.4 mg/l
vs. 27.6±53.4 mg/l (heme iron), and 13.5±16.2 mg/l vs. 3.9±2.7 mg/l
(free iron), respectively (31).
The concentrations of total iron, heme and free iron in EAOC were
17-, 11- and 3-fold lower than those in OE, respectively (31). There are no significant differences
in cyst fluid concentrations of iron-related compounds between
patients with CCC and those with EC. Several assays for the
measurement of iron-related compounds are available: In a previous
study, there was a significant difference in the cyst fluid iron
levels between the two methods (30,31),
which may be due to the different chelate colorimetric assay
methods and differences in their analytical performances.
Notwithstanding these limitations, patients with EAOC had much
lower levels of iron-related compounds compared with those with
benign OE.
Hemoglobin, heme and free iron in endometriotic
cysts can lead to distortion in the homeostatic redox balance, the
so-called redox imbalance (22).
Total iron is composed of heme iron and nonheme iron (free
catalytic form of iron, Fe2+). Free iron is labile and
catalyzes the Fenton chemical reaction, resulting in the generation
of hydroxyl radical (•OH) as follows: Fe2+ +
H2O2 Ú Fe3+ + OH− +
•OH. The iron-dependent Fenton reaction has been shown
to lead to genomic alterations, including a Cdkn2a/2b
deletion and a Met amplification, during carcinogenesis in
an animal model (32). Yoshimoto
et al found, for the first time, that the great majority of
iron in the cyst fluid is considered to be heme iron, but not free
iron (31). Hemoglobin and heme
iron are oxidized from the oxyhemoglobin (oxyHb-Fe2+) to
the methemoglobin (metHb-Fe3+) with generation of the
superoxide anion (O2−) as an autoxidation as
follows: Hb-Fe2+ (oxyHb) + O2 Ú
Hb-Fe3+ (metHb) + O2−. Since heme
iron is abundant in the cyst fluid of benign OE, autoxidation,
rather than the Fenton reaction, may be the main process
accomplishing the oxidative reaction.
It would be of interest to determine the origin and
biological function of metHb that is abundantly expressed in benign
OE. Peritoneal concentrations of nitric oxide (NO) metabolites
(nitrite and nitrate) in patients with endometriosis have been
shown to be significantly higher than those in patients with
non-endometriosis (33). Inducible
NO synthase (iNOS) is an enzyme that catalyzes the production of NO
from L-arginine. The mRNA level of iNOS has also been shown
to be higher in the endometriosis group than in the
non-endometriosis group (34).
Thus, the serum NO level is elevated in the endometriosis group as
compared to the control group due to the induction of iNOS. NO can
oxidize oxyHb to metHb as follows (35): HNO + 2[HbO2]2+
Ú 2[Hb]3+ + NO3− +
HO2−. Therefore, metHb is known as an
oxidative stress marker and causes production of free radicals to
induce oxidative stress by reacting with peroxides (hydrogen
peroxide, lipid hydroperoxides) (29). MetHb is downregulated in EAOC when
compared to benign OE, which supports the hypothesis that
paradoxically, a shift in the balance between oxidants and
antioxidants in EAOC is in favor of antioxidants (24). Glutathione, one of the most abundant
endogenous antioxidant, is responsible for the conversion of metHb
to oxyHb (36). It plays a role in
repairing damage induced by oxidative stress in cancer cells and
stops the process of cancer cachexia (36). Glutathione constitutes the survival
advantage for cancer cells and is required for cancer initiation
and progression (37,38). To date, a number of studies have
demonstrated that the oxidant-antioxidant imbalance plays a
critical role in the initiation and progression of multistage
carcinogenesis of endometriosis (23,30,39–41).
These studies support the redox imbalance hypothesis that there are
at least two stages of iron carcinogenesis and tumor progression:
The initial wave of iron-induced oxidative stress would be followed
by the second big wave of subsequent synthesis of the antioxidants,
which diminishes cellular oxidative stress capacity, increases
apoptosis resistance and promotes tumor initiation and
progression.
Similar epigenetic modifications:
Gene-environment interactions
The practical and theoretical implications was
discussed with regard to the current knowledge of epigenetic
modifications in benign OE and EAOC. An excess of heme iron and
non-heme iron is toxic to cell and tissue components. Hackett et
al reported the quantification of total iron from the hematoma
folllowing an intracerebral hemorrhage in an animal model (42). Neuronal cell death was observed at a
concentration of ~1.0 µg/cm2 (42). The iron concentration of the brain
tissue at the periphery of the hematoma has also been shown to be
~400 mg/l in two human subjects (43). The iron concentration in OE (244.4
mg/l) is almost similar to intracerebral hemorrhage (400 mg/l),
suggesting that endometriotic cells may face the crisis of death.
Under prolonged stressful conditions, endometriotic cells must cope
with various internal and external ROS for survival. The choice
between cell survival and death depends on the net result of ROS
production and their elimination by antioxidative enzymes (44). High levels of ROS promote DNA damage
and cell death, although perhaps surprisingly, low levels of ROS
are known to be associated with the development of tumors and then
the process of carcinogenesis (22–24,31).
Thus, an imbalance in the cellular redox state may play an
important role in the mechanism of its long-term carcinogenic
effect; gene-environment interactions may modify an individuals
susceptibility to this type of cancer.
High ROS levels damage the mitochondrial DNA and
promote its mutation, which affects the epigenetic control
mechanisms of nuclear DNA, by decreasing the activity of some
methyltransferases, thus causing DNA hypomethylation (45). ROS-induced oxidative stress is
associated not only with global/genome-wide DNA hypomethylation,
but also with tumor suppressor gene promoter-specific aberrant
hypermethylation via the upregulation of the expression of DNA
methyltransferases (46). CpG
clusters are susceptible to oxidative DNA damage to cytosine in the
Fenton reaction, which is the main cause of cytosine-to-thymine
transition mutations (47,48). Therefore, the dynamics of DNA
methylation at CpG clusters can drive an increased likelihood of
genetic mutations. Epigenetic mechanisms and then genetic mutations
are considered to contribute to the necessary plasticity of
endometriotic cells. Recent studies have attempted to link genetic
modifications with epigenetic or environmental risk factors for
EAOC (23,49). These results shed light onto the
mechanisms underlying the associations of environmental stimuli and
redox imbalance with risk of developing EAOC.
Defective CpG methylation affects several genes
involved in endometriosis malignant transformation, such as
Runt-related transcription factor 3 (RUNX3), human mutL
homolog 1 (hMLH1), E-cadherin (CDH1), Ras-association
domain family of gene 2 (RASSF2), p16, AT-rich
interactive domain-containing protein 1A (ARID1A) and
phosphatase and tensin homolog deleted on chromosome 10
(PTEN) by promoter hypermethylation (50). By contrast, steroidogenic factor-1
(SF-1), a transcriptional factor essential for estrogen
biosynthesis, has been shown to be hypomethylated and aberrantly
expressed (51). Furthermore,
oxidative stress (exogenous H2O2)
downregulates ARID1A mRNA and protein expression (39,52,53).
Oxidative stress recruits DNA methyltransferase to chromatin
(54), and also modifies the
expression of CpG demethylases, such as ten-eleven translocation
(TET) and jumonji (JMJ) genes (49). These genes may be involved in the
development of endometriosis and its malignant transformation. The
epigenetic switch occurs even in benign endometriosis (49).
Similar genetic abnormalities
Furthermore, endometriosis and EAOC harbor not only
multiple somatic gene mutations, but also epigenetic modifications.
Herein, we provide overview of the possible pathogenesis of
malignant transformation of endometriosis that have exhibited
distinct tumor morphological and phenotypical features, but have
suggested similar (epi)genetic abnormalities. EC is distinguished
from CCC due to different morphologies, but both represent common
environmental profiles (53) and
maintain the similar genomic abnormalities with multiple overlaps
and share similar molecular signatures (54). Recent microarray, targeted
sequencing and whole genome studies have identified that somatic
mutations of AT-rich interaction domain 1A (ARID1A),
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha (PIK3CA), PTEN, KRAS proto-oncogene, GTPase
(KRAS), catenin beta 1 (CTNNB1) and mutL homolog 1
(MLH1) were commonly found across EAOC (55–61).
EAOC and adjacent endometriotic lesions exhibited common multiple
cancer driver gene mutations, suggesting that they can share
extensive genetic similarity, a common genomic origin and a common
lineage (62–65).
We hypothesized that endometriotic cells would
acquire (epi)genetic modifications required for survival among the
harshest and poorest environments. The cells selected for oxidative
resistance enable clonal expansion/differentiation and could
survive (66). Indeed,
endometriosis is considered to be monoclonal in origin and
neoplastic in nature (67).
Surprisingly, mutations of classical cancer driver genes have been
observed in 4% of a histopathologically benign OE and 26% of deep
infiltrating endometriosis in cancer driver genes, including
ARID1A, KRAS, PIK3CA and PPP2R1A (64,65).
The existence of somatic driver mutations occurring in the
epithelial glandular cells but not the stromal cells of the same
endometriosis lesion (64) implies
that endometriotic epithelial cells might incur an advantage
through selective survival or proliferation and that the stromal
cells are resistant to environmental hazards.
CCC-specific (epi)genetic profile
Despite a similar genetic profile, EAOC tumors
present a different biological profile (68). Several studies have provided new
insight into the signaling pathway of genes differentially
expressed between EC and CCC. The interpretation of differentially
expressed genes has verified the dysregulated biological functions
related to glucose utilization, cell cycle regulation and hormone
metabolism, that plays important role in the development of EAOC
(60,68–70).
Transcription factor hepatocyte nuclear factor-1β (HNF1B)
was identified as a biomarker of ovarian CCC histology, but not EC,
with the hypomethylation of the HNF1B promoter influencing
the characteristic biology (60,68,70–75).
Endometriosis is composed of two subgroups: HNF-1β-positive and
-negative cells (71). The
expression patterns have been shown to be similar in a contiguous
transition from endometriotic cells to atypical cells to CCC
(60). HNF-1β-positive
endometriotic cells may represent a prototypical lineage of CCC
cells. Much interest has been focused on HNF-1β, which is commonly
upregulated in endometriosis and CCC. HNF-1β is associated with the
normal development of the liver, pancreas, gut, lungs and kidneys,
and its mutations represents the frequent occurrence of familial
forms of type 2 diabetes (68). The
exact biological function of the HNF-1β gene in CCC has been widely
reported: This gene plays key roles in glycogen synthesis,
anti-oxidative defense, anti-apoptosis, resistance to anticancer
agents and cell cycle regulators at G2/M transition (73,76,77).
First, in the elegant review, Mandai et al
provided new insight into the biological impact of CCC in a tumor
microenvironment via the upregulation of HNF-1β expression
(68). HNF-1β upregulates glucose
uptake and glycolysis to give rise to an increased yield of lactate
in CCC, which is known as the Warburg metabolic phenotype (68,78).
The Warburg effect benefits cancer cells to avoid excess ROS
generation and thus gains increased survival advantage in iron-rich
stressful environments such as endometriosis. The genes involved in
glucose homeostasis, including dipeptidyl peptidase 4 (DPP4)
(79) aldolase B (ALDOB)
(80), glucose transporter-1
(GLUT-1) gene and several key enzymes in the glycolytic
process (78), are downstream
targets of HNF1B. HNF-1β thus may be a key regulator of
glycolysis, gluconeogenesis and glucose homeostasis.
Second, HNF-1β actually reduces and protects cancer
cells from oxidative stress by markedly changing antioxidant
activity (78,81,82).
HNF-1β may repair damage caused by oxidative stress and can promote
survival by upregulating antioxidant proteins via binding with
antioxidant response element (37,68,81,83,84).
This gene upregulates the synthesis of glutathione (GSH), a
powerful antioxidant (85). HNF-1β
also triggers ROS resistance in CCC cells via rBAT, a cystine
transporter (68). Thus, HNF-1β
reduces oxidative stress and confers ROS resistance and a survival
advantage in CCC cells (37,68).
Finally, it would also be of interest to determine
the mechanisms underlying the protective effects of HNF-1β on cells
against any cytotoxicity and genotoxicity caused by ROS when CCC
cells were exposed to a stressful environment. DNA damage occurs
continually through various intrinsic and extrinsic stressors such
as ROS, ultraviolet radiation, smoking and errors during
replication (86). In
endometriosis, environmental hazards, including hemoglobin, heme
and iron, induce lesions in genomic DNA. The cellular DNA damage
response (DDR) comprises the coordinated actions of DNA repair and
checkpoint systems that regulate a spectrum of processes before
replication, where cell cycle arrest enables DNA repair to occur
(86). The DDR also promotes cell
death when the damage is beyond repair. If excessive damage exists,
the DDR activates cell death and eliminates the damaged cells by
apoptosis. Two key regulators of the DDR cell cycle checkpoints
include ataxia telangiectasia mutated (ATM) and ataxia
telangiectasia mutated and rad3-related (ATR) (86). ATR responds to a broad spectrum of
DNA damage, including replication-associated DNA damage, while ATM
is activated by DNA double-strand breaks (86). A number of studies have vigorously
investigated the association between redox imbalance and cell cycle
signaling pathways in CCC (73,77,83,87–89),
while no studies have focused on the influence of ROS on the
pathogenesis of EC, at least to the best of our knowledge.
Shigetomi et al investigated the role of HNF-1β in
regulation of the cell cycle arrest in response to DNA damage in
the CCC cell line, TOV21G (87).
Flow cytometric analysis of cell cycle profiles indicated that
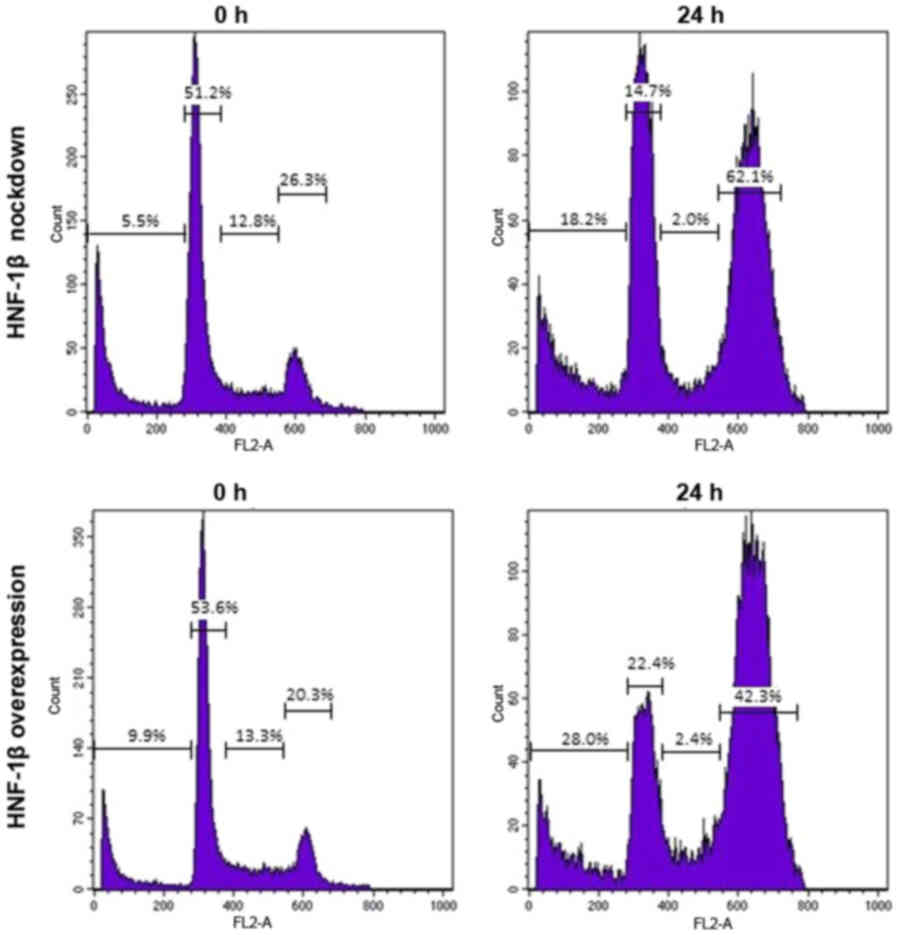
HNF-1β inhibited cell cycle progression (87). Fig.
1 illustrates the typical flow cytometry histograms of the
results. HNF-1β-expressing TOV21G cells exhibited a marked increase
in the proportion of cells in the G2/M phase following exposure to
a genotoxic agent, bleomycin, for 24 h (62.1 vs. 42.3%) (87). The knockdown of endogenous HNF-1β
attenuated G2/M phase cell cycle arrest and stimulated cell death
(28.0 vs. 18.2%) (87). It would
also be of interest to determine which genes and their signaling
pathways enhance and accelerate cell cycle arrest at G2/M phase.
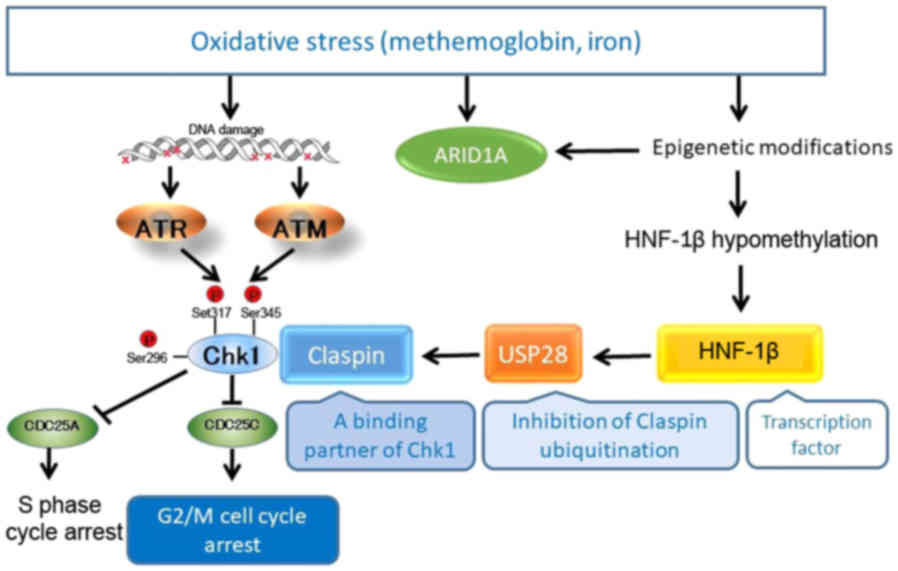
Shigetomi et al (87) and
Ito et al (89) explored the
activated and interconnected signaling network of HNF-1β to
identify novel downstream targets (Fig.
2). HNF-1β promotes TOV21G cell survival through Chk1
phosphorylation (87,89). As previously demonstrated, the
inactivation of HNF1B with siRNA suppressed Claspin protein
expression, but failed to inhibit Claspin mRNA expression
(89). Claspin transmits a
replication stress signal from ATR to Chk1 and functions as an
adaptor protein required for Chk1 activation. In CCC cells, HNF-1β,
but not ATR, are essential for the upregulation of Claspin protein
expression, suggesting that this gene functions as a Claspin
protein post-translational modification. Ito et al
vigorously identified potential modifiers of Claspin protein
relevant to HNF-1β biology (89).
To date, >450 unique protein modifications have been identified,
including phosphorylation, acetylation, ubiquitination and
SUMOylation through post-translational modification (90). Phosphorylation is one of the most
common and reversible intracellular post-translational
modifications of serine and threonine residues (91). Acetylation is a modification of the
lysine residues (92).
Ubiquitination is a widely studied method of post-translational
protein modification (4). Claspin
is reportedly regulated by ubiquitin-dependent proteasomal
degradation, whereas the ubiquitin-specific processing protease
(USP) 28- and USP29-mediated deubiquitination inhibits its
degradation (93). Martín et
al reported that USP29 controls the stability of Claspin by
deubiquitination (94). However,
HNF-1β did not stimulate the upregulation of USP29 protein in CCC
cells. Ito et al identified, for the first time, a novel
regulator of Claspin, USP28, as a direct downstream target of HNF-1
(89). USP28 interacts with Claspin
and is able to deubiquitinate it. With these results, USP28 is
identified as a novel player in the HNF-1β-Chk1 pathway and the
control of DNA replication, via the
HNF-1β-USP28-Claspin-Chk1-CDC25C pathway (89). This pathway contributes to a loss of
G2/M checkpoint control, which accumulates genomic and chromosomal
instability and then paves the way for further major genetic
changes.
Although the impact of HNF-1β on the cell cycle
arrest at G2/M phase under oxidative stress conditions is
recognized in CCC, the role of HNF-1β in oxidative stress-induced
endometriosis carcinogenesis remains poorly defined. When
endometriotic cells are exposed to genotoxic oxidative stressors
such as hemoglobin, heme or free iron, the HNF-1β gene may also be
epigenetically hypomethylated and is demonstrated as a positive
modulator of cell survival, through the HNF-1β signaling pathway.
This hypothesis needs to be verified in future studies.
EC-specific (epi)genetic profile
Endometriosis is an estrogen-dependent disease. The
enzyme aromatase P450 is expressed aberrantly in endometriosis and
catalyzes the final step of estrogen production and upregulates the
expression of prostaglandin E2 (PGE2) and macrophage migration
inhibitory factor (MIF), which, in turn, induces the expression of
aromatase within endometriotic lesions (55). The effects of estrogen on stromal
cell PGE2 production may be mediated in a feed-forward manner. MIF
is a cytokine marker of M2 polarization of macrophage which
facilitates the onset and progression of endometriosis. Such an
interplay with a positive feedback cycle is involved in cell
proliferation and apoptotic resistance of endometriosis, and then
its malignant transformation.
There are two types of estrogen receptors (ESRs),
ESR1 (also known as ERα) and ESR2 (ERβ). The ESR expression level
has been shown to be higher in EC and high-grade serous than in CCC
and mucinous carcinoma (95). Among
EAOC, ESR positivity has been shown to be significantly higher in
EC (91%), but lower in CCC (8%) (60). ESR gene expression is modulated by a
number of factors, such as DNA methylation of the promoter region,
histone deacetylation, chromatin remodeling, or heme and iron
binding (58). The interrelation
between the ESR expression and these factors is complex, as genetic
characteristics and environmental factors can mutually impact upon
each other. The significant up- and downregulation of ESR has been
shown to be associated with marked epigenomic alterations: ESR2 is
the predominant ESR in endometriosis due to the hypomethylation of
promoter CpG islands, whereas ESR1 levels are lower in
endometriosis (69). EC shares
estrogen-dependent oncogenic pathways and signaling network. A
hyperestrogenic state or the upregulation of ESR expression may be
shared in common with benign and malignant endometriosis, which may
denote that endometriosis has carcinogenic potential. Furthermore,
G-protein-coupled estrogen receptor-30 (GPR30) is the novel
estrogen-responsive receptor G protein-coupled estrogen receptor 1,
GPER (96). GPR30 expression is
higher in EAOC than in benign OE (96). The upregulation of ESR expression is
associated with a better clinical outcome in ovarian cancer
(95) and CCC (14), suggesting the role of ESR in tumor
initiation or the early development of primary EC, but not in EC
progression.
Taken together, EAOC is a heterogeneous disease,
with at least two intrinsic subtypes, EC and CCC. Although the role
of DNA methylation in EAOC development is not yet fully understood,
its profiling defines cancer subclasses differing in
clinicopathologic characteristics, molecular profiles and survival.
Genes with promoter hypermethylation and hypomethylation are
consistent in cancer function and characteristics of concordant
methylation. The promoter hypomethylated ESR gene is reversely
correlated with the promoter hypermethylated HNF1B gene in
EC (58,60,97). A
low expression of ESR (95) and
high expression of HNF-1β (71) are
identified as potential biomarkers for CCC.
Other crucial aspects
We discuss the potential involvement of
microenvironment in endometriosis and its malignant transformation.
The dysfunctional regulation of immune and inflammatory
microenvironment, extracellular matrix remodeling, or new blood
vessel formation is a crucial aspect of pathogenesis of
endometriosis and its malignant transformation. Ovarian cancer is
initially associated with pelvic inflammatory disease, such as
endometriosis, demonstrating a similarity between the processes of
inflammation and carcinogenesis. Endometriotic cells adapt to
survive in the unique microenvironment conditions with high levels
of iron, inflammatory cytokines and chemokines (98). Microenvironment-cell interplay may
modulate the major signaling pathways associated with cell cycle
regulation, growth factor signaling, immune and inflammatory
pathways, and the extracellular matrix remodeling, which results in
phenotype transformation (99).
Researchers have focused on the function of matrix
metalloproteinase (MMPs) (100),
lysyl oxidases (LOXs) and nuclear factor κ-light-chain-enhancer of
activated B (NF-κB) in the pathophysiology of inflammation and EAOC
(101). Several studies have
identified the NF-κB-dependent multiple oncogenic pathways in
endometriosis (102) and highlight
its malignant transformation (103). MMP-2 promotes angiogenesis during
endometriosis progression via the cyclooxygenase (COX)-2/PGE2/pAKT
axis (104). The upregulation of
lysyl oxidase (LOX) expression is involved in extracellular
membrane degradation, invasive and metastatic potential of
endometriosis (105). The
disruption of epithelial-stromal communication networks elicits a
feed-forward loop involving endometriosis to drive inflammation,
which may be relevant in diseases such as EAOC.
In addition, chemokines are key players in the
activation and are recruitment of immune cells at sites of
inflammation. The CXCR4/CXCL12 axis is functional in endometriosis
and plays a role in a number of diverse cellular functions,
including immune surveillance, inflammation response, tissue
homeostasis, and tumor growth and metastasis (106). CXCR4 expression is upregulated by
vascular endothelial growth factor (VEGF), and plays an important
role in the malignant transformation of endometriosis (107). Furthermore, the microvascular
endothelium of ectopic endometrial tissue originates from
circulating endothelial progenitor cells mobilized from the bone
marrow, which is also controlled by the CXCL12/CXCR4 axis (108). The neovascularization of
endometriotic lesions is not only driven by angiogenesis, but also
vasculogenesis from circulating endothelial progenitor cells
(108). Thus, angiogenesis and
vasculogenesis play an integral part in the establishment and
growth of endometriotic lesions and malignant transformation
(108,109). Therefore, these changes in the
microenvironment are necessary to accumulate enough epigenetic,
genetic and pathological alterations for malignant transformation
of endometriosis (110).
Discussion
In the present study, we provide a literature
review of various lines of evidence supporting the concept of an
altered redox environmental model for malignant transformation of
endometriosis. Fig. 3 summarizes
the current knowledge about the role of the shared and independent
(epi)genetic background between EAOC tumors, and their interaction
with environmental stimuli. We initially updated the epigenetic,
genetic and environmental backgrounds of EAOC and surveyed the
examples of environmentally induced epigenetic changes. Despite the
differences in morphology between EC and CCC, they share remarkable
(epi)genetic similarities and enrichment for driver somatic
mutations affecting ARID1A, PTEN and KRAS genes
(55–61). The hemoglobin, heme and free iron
accumulated during endometriosis development are a prerequisite to
modification of genomic DNA for prompt cellular responses to
oxidative stress (58). An excess
of heme iron and nonheme iron participates in the Fenton reaction
generating the toxic hydroxyl radical. Autoxidation of oxyHb to
metHb always occurs due to abundant heme iron in the contents of
benign OE. Autoxidation, rather than the Fenton reaction, might be
the main process accomplishing the oxidative reaction in
endometriosis (31). Redox biology
is considered to alter (epi)genetic events. Environmentally-induced
epigenetic alterations may result in a change of the adaptive gene
function, leading to phenotypic plasticity. Endometriosis is
predisposed to develop into EAOC through the progressive
accumulation of epigenetic alterations (3,4,6,9)
during obvious redox imbalance (20,23).
However, there is only limited knowledge of the mechanisms through
which environmental factors affect gene function.
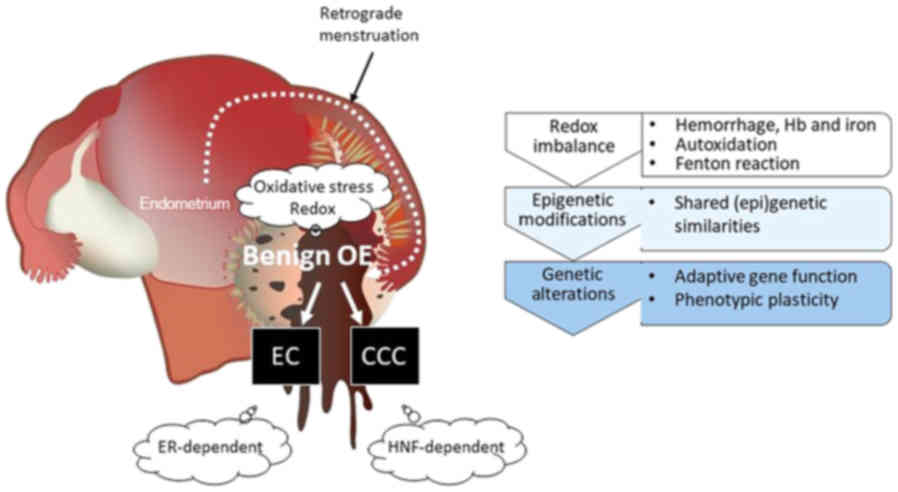 | Figure 3.The concept of an altered redox
environmental model for malignant transformation of endometriosis.
EAOC consists of different histological subtypes mainly originating
from EC and CCC. First, repeated episodes of hemorrhage occur in
endometriosis throughout menstruation. Extracellular free
hemoglobin produces toxic heme degradation products and is a source
of ROS. Hemoglobin, heme and free iron in endometriotic cysts cause
redox imbalance. Second, there is a link between environmental
stimuli and (epi)genetic modifications. EC is distinguished from
CCC due to different morphologies, but both represent common
environmental profiles and maintain the similar genomic
abnormalities with multiple overlaps and share similar molecular
signatures, including ARID1A, PIK3CA, PTEN, or KRAS. Finally, ESR
and HNF-1β proteins are mutually exclusive in EAOC. HNF-1β-positive
and ESR-negative endometriotic cells may represent a prototypical
lineage of CCC cells. The positive ESR expression and negative
HNF-1β expression is a frequent finding in EC. EAOC tumors had
enrichment of cancer-specific gene signatures corresponding with
each histological subtype: ESR induces EC cell proliferation (‘go’)
and HNF-1β induces CCC cell cycle arrest (‘stop’) for a survival
mechanism in response to several stresses. EAOC,
endometriosis-associated ovarian cancer; EC, endometrioid
carcinoma; CCC, clear cell carcinoma; ARID1A, AT-rich interactive
domain-containing protein 1A; PIK3CA,
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha; PTEN, phosphatase and tensin homolog deleted on chromosome
10; HNF-1β, hepatocyte nuclear factor-1β. |
In addition, a previous approach identified
different genetic backgrounds between EAOC tumors (60). By comparing the gene expression
profile, at least two differentially expressed genes were
identified in EC and CCC. A positive ESR expression and negative
HNF-1β expression is a frequent finding in EC, but not in CCC
(58,60). EC may develop in the setting of
estrogen-driven pathway (111). On
the other hand, HNF-1β-dependent ovarian cancer arising from
endometriosis is substantially more associated with CCC than with
EC (11,60,77,83,87,89).
Immunohistochemical data have indicated that atypical endometriosis
is a precursor lesion molecularly similar to adjacent invasive
cancer (60). Pre-malignant
endometriotic cells exposed to mixture of genotoxic oxidative
stressors inhibit cell proliferation and promote cell cycle arrest
at G2/M phase for DNA damage repair through the
HNF-1β/USP28/Clasipin/Chk1 pathway (89). Therefore, HNF-1β induces cell cycle
arrest, DNA damage and genomic instability, thereby promoting
erroneous DNA repair and can predispose to CCC. EAOC tumors had
enrichment of cancer-specific gene signatures corresponding with
each histological subtype: ESR induces EC cell proliferation (‘go’)
and HNF-1β induces CCC cell cycle arrest (‘stop’). This model
underscores a subtype-dependent dichotomy between ‘go’ and ‘stop’
in EAOC, through potentially better ability to adapt in a changing
environment (Fig. 3).
In conclusion, a special emphasis is given to
current pathophysiological concepts of malignant transformation of
endometriosis, including redox imbalance, environmental
stimuli-induced (epi)genetic modifications and mutually exclusive
expression of ESR and HNF-1β genes for a survival mechanism in
response to several stresses.
Acknowledgements
Not applicable.
Funding
The present study was supported by JSPS KAKENHI
(grant no. JP16K11150) and Tohoku Bureau of Economy, Trade and
Industry (Tohoku 1607028).
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors contributions
YY, NK and KO collected the data regarding the
epigenetic and genetic abnormalities and the underlying mechanism
of endometriosis transformation using the PubMed database. NK, KO
and CY performed the literature search and supervised the study. HK
and CY made substantial contributions to the conception of the
study. HK contributed to the study design and interpretation of the
included research studies. The final version of the manuscript has
been read and approved by all authors.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no potential competing
interests with respect to the research, authorship and publication
of this article.
References
|
1
|
Bulletti C, Coccia ME, Battistoni S and
Borini A: Endometriosis and infertility. J Assist Reprod Genet.
27:441–447. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Giudice LC and Kao LC: Endometriosis.
Lancet. 364:1789–1799. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brinton LA, Sakoda LC, Sherman ME,
Frederiksen K, Kjaer SK, Graubard BI, Olsen JH and Mellemkjaer L:
Relationship of benign gynecologic diseases to subsequent risk of
ovarian and uterine tumors. Cancer Epidemiol Biomarkers Prev.
14:2929–2935. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kobayashi H, Sumimoto K, Kitanaka T,
Yamada Y, Sado T, Sakata M, Yoshida S, Kawaguchi R, Kanayama S,
Shigetomi H, et al: Ovarian endometrioma--risks factors of ovarian
cancer development. Eur J Obstet Gynecol Reprod Biol. 138:187–193.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim HS, Kim TH, Chung HH and Song YS: Risk
and prognosis of ovarian cancer in women with endometriosis: A
meta-analysis. Br J Cancer. 110:1878–1890. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilbur MA, Shih IM, Segars JH and Fader
AN: Cancer implications for patients with endometriosis. Semin
Reprod Med. 35:110–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Munksgaard PS and Blaakaer J: The
association between endometriosis and gynecological cancers and
breast cancer: A review of epidemiological data. Gynecol Oncol.
123:157–163. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kurman RJ and Shih IeM: Pathogenesis of
ovarian cancer: Lessons from morphology and molecular biology and
their clinical implications. Int J Gynecol Pathol. 27:151–160.
2008.PubMed/NCBI
|
|
9
|
Kurman RJ and Shih IeM: The origin and
pathogenesis of epithelial ovarian cancer: A proposed unifying
theory. Am J Surg Pathol. 34:433–443. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu RC, Veras E, Lin J, Gerry E,
Bahadirli-Talbott A, Baras A, Ayhan A, Shih IM and Wang TL:
Elucidating the pathogenesis of synchronous and metachronous tumors
in a woman with endometrioid carcinomas using a whole-exome
sequencing approach. Cold Spring Harb Mol Case Stud. 3:a0016932017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kajihara H, Yamada Y, Shigetomi H,
Higashiura Y and Kobayashi H: The dichotomy in the histogenesis of
endometriosis-associated ovarian cancer: Clear cell-type versus
endometrioid-type adenocarcinoma. Int J Gynecol Pathol. 31:304–312.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van der Horst PH, van der Zee M,
Heijmans-Antonissen C, Jia Y, DeMayo FJ, Lydon JP, van Deurzen CH,
Ewing PC, Burger CW and Blok LJ: A mouse model for endometrioid
ovarian cancer arising from the distal oviduct. Int J Cancer.
135:1028–1037. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu R, Zhai Y, Kuick R, Karnezis AN, Garcia
P, Naseem A, Hu TC, Fearon ER and Cho KR: Impact of oviductal
versus ovarian epithelial cell of origin on ovarian endometrioid
carcinoma phenotype in the mouse. J Pathol. 240:341–351. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rambau P, Kelemen LE, Steed H, Quan ML,
Ghatage P and Köbel M: Association of hormone receptor expression
with survival in ovarian endometrioid carcinoma: Biological
validation and clinical implications. Int J Mol Sci. 18:E5152017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lukanova A and Kaaks R: Endogenous
hormones and ovarian cancer: Epidemiology and current hypotheses.
Cancer Epidemiol Biomarkers Prev. 14:98–107. 2005.PubMed/NCBI
|
|
16
|
Wu R, Hendrix-Lucas N, Kuick R, Zhai Y,
Schwartz DR, Akyol A, Hanash S, Misek DE, Katabuchi H, Williams BO,
et al: Mouse model of human ovarian endometrioid adenocarcinoma
based on somatic defects in the Wnt/beta-catenin and PI3K/Pten
signaling pathways. Cancer Cell. 11:321–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tanwar PS, Zhang L, Kaneko-Tarui T, Curley
MD, Taketo MM, Rani P, Roberts DJ and Teixeira JM: Mammalian target
of rapamycin is a therapeutic target for murine ovarian
endometrioid adenocarcinomas with dysregulated Wnt/β-catenin and
PTEN. PLoS One. 6:e207152011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tao GZ, Lehwald N, Jang KY, Baek J, Xu B,
Omary MB and Sylvester KG: Wnt/β-catenin signaling protects mouse
liver against oxidative stress-induced apoptosis through the
inhibition of forkhead transcription factor FoxO3. J Biol Chem.
288:17214–17224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mahalingaiah PK and Singh KP: Chronic
oxidative stress increases growth and tumorigenic potential of
MCF-7 breast cancer cells. PLoS One. 9:e873712014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mandai M, Matsumura N, Baba T, Yamaguchi
K, Hamanishi J and Konishi I: Ovarian clear cell carcinoma as a
stress-responsive cancer: Influence of the microenvironment on the
carcinogenesis and cancer phenotype. Cancer Lett. 310:129–133.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Suryawanshi S, Huang X, Elishaev E, Budiu
RA, Zhang L, Kim S, Donnellan N, Mantia-Smaldone G, Ma T, Tseng G,
et al: Complement pathway is frequently altered in endometriosis
and endometriosis-associated ovarian cancer. Clin Cancer Res.
20:6163–6174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Iwabuchi T, Yoshimoto C, Shigetomi H and
Kobayashi H: Oxidative stress and antioxidant defense in
endometriosis and its malignant transformation. Oxid Med Cell
Longev 2015. 8485952015.
|
|
23
|
Kobayashi H: Potential scenarios leading
to ovarian cancer arising from endometriosis. Redox Rep.
21:119–126. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Iwabuchi T, Yoshimoto C, Shigetomi H and
Kobayashi H: Cyst fluid hemoglobin species in endometriosis and its
malignant transformation: The role of metallobiology. Oncol Lett.
11:3384–3388. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bandini S, Macagno M, Hysi A, Lanzardo S,
Conti L, Bello A, Riccardo F, Ruiu R, Merighi IF, Forni G, et al:
The non-inflammatory role of C1q during Her2/neu-driven mammary
carcinogenesis. Oncoimmunology. 5:e12536532016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McKinnon BD, Kocbek V, Nirgianakis K,
Bersinger NA and Mueller MD: Kinase signalling pathways in
endometriosis: Potential targets for non-hormonal therapeutics. Hum
Reprod Update. 22:382–403. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jung M, Weigert A, Mertens C, Rehwald C
and Brüne B: Iron handling in tumor-associated macrophages: Is
there a new role for lipocalin-2? Front Immunol. 8:11712017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ngô C, Chéreau C, Nicco C, Weill B,
Chapron C and Batteux F: Reactive oxygen species controls
endometriosis progression. Am J Pathol. 175:225–234. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alayash AI: Oxygen therapeutics: Can we
tame haemoglobin? Nat Rev Drug Discov. 3:152–159. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamaguchi K, Mandai M, Toyokuni S,
Hamanishi J, Higuchi T, Takakura K and Fujii S: Contents of
endometriotic cysts, especially the high concentration of free
iron, are a possible cause of carcinogenesis in the cysts through
the iron-induced persistent oxidative stress. Clin Cancer Res.
14:32–40. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yoshimoto C, Iwabuchi T, Shigetomi H and
Kobayashi H: Cyst fluid iron-related compounds as useful markers to
distinguish malignant transformation from benign endometriotic
cysts. Cancer Biomark. 15:493–499. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Akatsuka S, Yamashita Y, Ohara H, Liu YT,
Izumiya M, Abe K, Ochiai M, Jiang L, Nagai H, Okazaki Y, et al:
Fenton reaction induced cancer in wild type rats recapitulates
genomic alterations observed in human cancer. PLoS One.
7:e434032012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kianpour M, Nematbakhsh M and Ahmadi SM:
Asymmetric dimethylarginine (ADMA), nitric oxide metabolite, and
estradiol levels in serum and peritoneal fluid in women with
endometriosis. Iran J Nurs Midwifery Res. 20:484–489. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yeo SG, Won YS, Lee HY, Kim YI, Lee JW and
Park DC: Increased expression of pattern recognition receptors and
nitric oxide synthase in patients with endometriosis. Int J Med
Sci. 10:1199–1208. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bellavia L, DuMond JF, Perlegas A, Bruce
King S and Kim-Shapiro DB: Nitroxyl accelerates the oxidation of
oxyhemoglobin by nitrite. Nitric Oxide. 31:38–47. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
den Boer PJ, Bleeker WK, Rigter G,
Agterberg J, Stekkinger P, Kannegieter LM, de Nijs IM and Bakker
JC: Intravascular reduction of methemoglobin in plasma of the rat
in vivo. Biomater Artif Cells Immobilization Biotechnol.
20:647–650. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Amano Y, Mandai M, Yamaguchi K, Matsumura
N, Kharma B, Baba T, Abiko K, Hamanishi J, Yoshioka Y and Konishi
I: Metabolic alterations caused by HNF1β expression in ovarian
clear cell carcinoma contribute to cell survival. Oncotarget.
6:26002–26017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Harris IS, Treloar AE, Inoue S, Sasaki M,
Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA,
et al: Glutathione and thioredoxin antioxidant pathways synergize
to drive cancer initiation and progression. Cancer Cell.
27:211–222. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Winarto H, Tan MI, Sadikin M and Wanandi
SI: ARID1A Expression is down-regulated by oxidative stress
in endometriosis and endometriosis-associated ovarian cancer.
Transl Oncogenomics. February 24–2017.doi. 1177272716689818.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yan S, Sorrell M and Berman Z: Functional
interplay between ATM/ATR-mediated DNA damage response and DNA
repair pathways in oxidative stress. Cell Mol Life Sci.
71:3951–3967. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Toyokuni S: Iron and thiols as two major
players in carcinogenesis: Friends or foes? Front Pharmacol.
5:2002014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hackett MJ, DeSouza M, Caine S, Bewer B,
Nichol H, Paterson PG and Colbourne F: A new method to image
heme-Fe, total Fe, and aggregated protein levels after
intracerebral hemorrhage. ACS Chem Neurosci. 6:761–770. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chaudhary N, Pandey AS, Merchak K, Gemmete
JJ, Chenevert T and Xi G: Perihematomal cerebral tissue iron
quantification on MRI following intracerebral hemorrhage in two
human subjects: Proof of principle. Acta Neurochir Suppl (Wien).
121:179–183. 2016. View Article : Google Scholar
|
|
44
|
Nagano O, Okazaki S and Saya H: Redox
regulation in stem-like cancer cells by CD44 variant isoforms.
Oncogene. 32:5191–5198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Szumiel I: Ionizing radiation-induced
oxidative stress, epigenetic changes and genomic instability: The
pivotal role of mitochondria. Int J Radiat Biol. 91:1–12. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu Q and Ni X: ROS-mediated DNA
methylation pattern alterations in carcinogenesis. Curr Drug
Targets. 16:13–19. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bandaru B, Gopal J and Bhagwat AS:
Overproduction of DNA cytosine methyltransferases causes
methylation and C --> T mutations at non-canonical sites. J Biol
Chem. 271:7851–7859. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Samson-Thibault F, Madugundu GS, Gao S,
Cadet J and Wagner JR: Profiling cytosine oxidation in DNA by
LC-MS/MS. Chem Res Toxicol. 25:1902–1911. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ito F, Yamada Y, Shigemitsu A, Akinishi M,
Kaniwa H, Miyake R, Yamanaka S and Kobayashi H: Role of oxidative
stress in epigenetic modification in endometriosis. Reprod Sci.
24:1493–1502. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
He J, Chang W, Feng C, Cui M and Xu T:
Endometriosis malignant transformation: Epigenetics as a probable
mechanism in ovarian tumorigenesis. Int J Genomics 2018.
14653482018.
|
|
51
|
Bulun SE, Monsivais D, Kakinuma T,
Furukawa Y, Bernardi L, Pavone ME and Dyson M: Molecular biology of
endometriosis: From aromatase to genomic abnormalities. Semin
Reprod Med. 33:220–224. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xie H, Chen P, Huang HW, Liu LP and Zhao
F: Reactive oxygen species downregulate ARID1A expression via its
promoter methylation during the pathogenesis of endometriosis. Eur
Rev Med Pharmacol Sci. 21:4509–4515. 2017.PubMed/NCBI
|
|
53
|
Kolin DL, Dinulescu DM and Crum CP: Origin
of clear cell carcinoma: nature or nurture? J Pathol. 244:131–134.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chang CM, Yang YP, Chuang JH, Chuang CM,
Lin TW, Wang PH, Yu MH and Chang CC: Discovering the deregulated
molecular functions involved in malignant transformation of
endometriosis to endometriosis-associated ovarian carcinoma using a
data-driven, function-based analysis. Int J Mol Sci.
18:23452017.doi: 10.3390/ijms18112345. View Article : Google Scholar :
|
|
55
|
Bulun SE: Endometriosis. N Engl J Med.
360:268–279. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ayhan A, Mao TL, Seckin T, Wu CH, Guan B,
Ogawa H, Futagami M, Mizukami H, Yokoyama Y, Kurman RJ, et al: Loss
of ARID1A expression is an early molecular event in tumor
progression from ovarian endometriotic cyst to clear cell and
endometrioid carcinoma. Int J Gynecol Cancer. 22:1310–1315. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Veillat V, Sengers V, Metz CN, Roger T,
Leboeuf M, Mailloux J and Akoum A: Macrophage migration inhibitory
factor is involved in a positive feedback loop increasing aromatase
expression in endometriosis. Am J Pathol. 181:917–927. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tanase Y, Yamada Y, Shigetomi H, Kajihara
H, Oonogi A, Yoshizawa Y, Furukawa N, Haruta S, Yoshida S, Sado T,
et al: Modulation of estrogenic action in clear cell carcinoma of
the ovary (Review). Exp Ther Med. 3:18–24. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Worley MJ Jr, Welch WR, Berkowitz RS and
Ng SW: Endometriosis-associated ovarian cancer: A review of
pathogenesis. Int J Mol Sci. 14:5367–5379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lai CR, Hsu CY, Chen YJ, Yen MS, Chao KC
and Li AF: Ovarian cancers arising from endometriosis: A
microenvironmental biomarker study including ER, HNF1β, p53, PTEN,
BAF250a, and COX-2. J Chin Med Assoc. 76:629–634. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Takeda T, Banno K, Okawa R, Yanokura M,
Iijima M, Irie-Kunitomi H, Nakamura K, Iida M, Adachi M, Umene K,
et al: ARID1A gene mutation in ovarian and endometrial cancers
(Review). Oncol Rep. 35:607–613. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Bissell MJ: Modelling molecular mechanisms
of breast cancer and invasion: Lessons from the normal gland.
Biochem Soc Trans. 35:18–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kobayashi H, Kajiwara H, Kanayama S,
Yamada Y, Furukawa N, Noguchi T, Haruta S, Yoshida S, Sakata M,
Sado T, et al: Molecular pathogenesis of endometriosis-associated
clear cell carcinoma of the ovary (Review). Oncol Rep. 22:233–240.
2009.PubMed/NCBI
|
|
64
|
Anglesio MS, Papadopoulos N, Ayhan A,
Nazeran TM, Noë M, Horlings HM, Lum A, Jones S, Senz J, Seckin T,
et al: Cancer-associated mutations in endometriosis without cancer.
N Engl J Med. 376:1835–1848. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zou Y, Zhou JY, Guo JB, Wang LQ, Luo Y,
Zhang ZY, Liu FY, Tan J, Wang F and Huang OP: The presence of KRAS,
PPP2R1A and ARID1A mutations in 101 Chinese samples with ovarian
endometriosis. Mutat Res. 809:1–5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Jamnongkan W, Thanee M, Yongvanit P,
Loilome W, Thanan R, Kimawaha P, Boonmars T, Silakit R, Namwat N
and Techasen A: Antifibrotic effect of xanthohumol in combination
with praziquantel is associated with altered redox status and
reduced iron accumulation during liver fluke-associated
cholangiocarcinogenesis. PeerJ. 6:e42812018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jimbo H, Hitomi Y, Yoshikawa H, Yano T,
Momoeda M, Sakamoto A, Tsutsumi O, Taketani Y and Esumi H: Evidence
for monoclonal expansion of epithelial cells in ovarian endometrial
cysts. Am J Pathol. 150:1173–1178. 1997.PubMed/NCBI
|
|
68
|
Mandai M, Amano Y, Yamaguchi K, Matsumura
N, Baba T and Konishi I: Ovarian clear cell carcinoma meets
metabolism; HNF-1β confers survival benefits through the Warburg
effect and ROS reduction. Oncotarget. 6:30704–30714. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bukulmez O, Hardy DB, Carr BR, Word RA and
Mendelson CR: Inflammatory status influences aromatase and steroid
receptor expression in endometriosis. Endocrinology. 149:1190–1204.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kato N, Tamura G and Motoyama T:
Hypomethylation of hepatocyte nuclear factor-1beta (HNF-1beta) CpG
island in clear cell carcinoma of the ovary. Virchows Arch.
452:175–180. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kato N, Sasou S and Motoyama T: Expression
of hepatocyte nuclear factor-1beta (HNF-1beta) in clear cell tumors
and endometriosis of the ovary. Mod Pathol. 19:83–89. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Takano M, Kikuchi Y, Kudoh K, Goto T,
Furuya K, Kikuchi R, Kita T, Fujiwara K, Shiozawa T and Aoki D:
Weekly administration of temsirolimus for heavily pretreated
patients with clear cell carcinoma of the ovary: A report of six
cases. Int J Clin Oncol. 16:605–609. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Shigetomi H, Tsunemi T, Haruta S, Kajihara
H, Yoshizawa Y, Tanase Y, Furukawa N, Yoshida S, Sado T and
Kobayash H: Molecular mechanisms linking endometriosis under
oxidative stress with ovarian tumorigenesis and therapeutic
modalities. Cancer Invest. 30:473–480. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kao YC, Lin MC, Lin WC, Jeng YM and Mao
TL: Utility of hepatocyte nuclear factor-1β as a diagnostic marker
in ovarian carcinomas with clear cells. Histopathology. 61:760–768.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ye S, Yang J, You Y, Cao D, Huang H, Wu M,
Chen J, Lang J and Shen K: Clinicopathologic significance of
HNF-1β, AIRD1A, and PIK3CA expression in ovarian clear cell
carcinoma: A tissue microarray study of 130 cases. Medicine
(Baltimore). 95:e30032016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Barbacci E, Chalkiadaki A, Masdeu C,
Haumaitre C, Lokmane L, Loirat C, Cloarec S, Talianidis I,
Bellanne-Chantelot C and Cereghini S: HNF1beta/TCF2 mutations
impair transactivation potential through altered co-regulator
recruitment. Hum Mol Genet. 13:3139–3149. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kobayashi H, Yamada Y, Kanayama S,
Furukawa N, Noguchi T, Haruta S, Yoshida S, Sakata M, Sado T and Oi
H: The role of hepatocyte nuclear factor-1beta in the pathogenesis
of clear cell carcinoma of the ovary. Int J Gynecol Cancer.
19:471–479. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Okamoto T, Mandai M, Matsumura N,
Yamaguchi K, Kondoh H, Amano Y, Baba T, Hamanishi J, Abiko K,
Kosaka K, et al: Hepatocyte nuclear factor-1β (HNF-1β) promotes
glucose uptake and glycolytic activity in ovarian clear cell
carcinoma. Mol Carcinog. 54:35–49. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Senkel S, Lucas B, Klein-Hitpass L and
Ryffel GU: Identification of target genes of the transcription
factor HNF1beta and HNF1alpha in a human embryonic kidney cell
line. Biochim Biophys Acta 1731. 179–190. 2005.
|
|
80
|
Gregori C, Porteu A, Mitchell C, Kahn A
and Pichard AL: In vivo functional characterization of the aldolase
B gene enhancer. J Biol Chem. 277:28618–28623. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yamaguchi K, Mandai M, Oura T, Matsumura
N, Hamanishi J, Baba T, Matsui S, Murphy SK and Konishi I:
Identification of an ovarian clear cell carcinoma gene signature
that reflects inherent disease biology and the carcinogenic
processes. Oncogene. 29:1741–1752. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Shigetomi H, Higashiura Y, Kajihara H and
Kobayashi H: A potential link of oxidative stress and cell cycle
regulation for development of endometriosis. Gynecol Endocrinol.
28:897–902. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Akasaka J, Uekuri C, Shigetomi H, Koike M
and Kobayashi H: Hepatocyte nuclear factor (HNF)-1β and its
physiological importance in endometriosis. Biomed Rep. 1:13–17.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wang X, Wu H, Yu W, Liu J, Peng J, Liao N,
Zhang J, Zhang X and Hai C: Hepatocyte nuclear factor 1b is a novel
negative regulator of white adipocyte differentiation. Cell Death
Differ. 24:1588–1597. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lopes-Coelho F, Gouveia-Fernandes S,
Gonçalves LG, Nunes C, Faustino I, Silva F, Félix A, Pereira SA and
Serpa J: HNF1β drives glutathione (GSH) synthesis underlying
intrinsic carboplatin resistance of ovarian clear cell carcinoma
(OCCC). Tumour Biol. 37:4813–4829. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Sundar R, Brown J, Ingles Russo A and Yap
TA: Targeting ATR in cancer medicine. Curr Probl Cancer.
41:302–315. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Shigetomi H, Sudo T, Shimada K, Uekuri C,
Tsuji Y, Kanayama S, Naruse K, Yamada Y, Konishi N and Kobayashi H:
Inhibition of cell death and induction of G2 arrest accumulation in
human ovarian clear cells by HNF-1β transcription factor:
Chemosensitivity is regulated by checkpoint kinase CHK1. Int J
Gynecol Cancer. 24:838–843. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Koshiyama M, Matsumura N and Konishi I:
Recent concepts of ovarian carcinogenesis: Type I and type II.
BioMed Res Int 2014. 9342612014.
|
|
89
|
Ito F, Yoshimoto C, Yamada Y, Sudo T and
Kobayashi H: The HNF-1β-USP28-Claspin pathway upregulates DNA
damage-induced Chk1 activation in ovarian clear cell carcinoma.
Oncotarget. 9:17512–17522. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Venne AS, Kollipara L and Zahedi RP: The
next level of complexity: Crosstalk of posttranslational
modifications. Proteomics. 14:513–524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Terman JR and Kashina A:
Post-translational modification and regulation of actin. Curr Opin
Cell Biol. 25:30–38. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhang D, Zaugg K, Mak TW and Elledge SJ: A
role for the deubiquitinating enzyme USP28 in control of the
DNA-damage response. Cell. 126:529–542. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Martín Y, Cabrera E, Amoedo H,
Hernández-Pérez S, Domínguez-Kelly R and Freire R: USP29 controls
the stability of checkpoint adaptor Claspin by deubiquitination.
Oncogene. 34:1058–1063. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen S, Dai X, Gao Y, Shen F, Ding J and
Chen Q: The positivity of estrogen receptor and progesterone
receptor may not be associated with metastasis and recurrence in
epithelial ovarian cancer. Sci Rep. 7:169222017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Long L, Cao Y and Tang LD: Transmembrane
estrogen receptor GPR30 is more frequently expressed in malignant
than benign ovarian endometriotic cysts and correlates with MMP-9
expression. Int J Gynecol Cancer. 22:539–545. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yamaguchi K, Huang Z, Matsumura N, Mandai
M, Okamoto T, Baba T, Konishi I, Berchuck A and Murphy SK:
Epigenetic determinants of ovarian clear cell carcinoma biology.
Int J Cancer. 135:585–597. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wendel JRH, Wang X and Hawkins SM: The
endometriotic tumor microenvironment in ovarian cancer. Cancers
(Basel). 10:E2612018. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Kobayashi H, Sugimoto H, Onishi S and
Nakano K: Novel biomarker candidates for the diagnosis of ovarian
clear cell carcinoma. Oncol Lett. 10:612–618. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Liu H, Wang J, Wang H, Tang N, Li Y, Zhang
Y and Hao T: Correlation between matrix metalloproteinase-9 and
endometriosis. Int J Clin Exp Pathol. 8:13399–13404.
2015.PubMed/NCBI
|
|
101
|
Kisielewski R, Tołwińska A, Mazurek A and
Laudański P: Inflammation and ovarian cancer--current views.
Ginekol Pol. 84:293–297. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Kaponis A, Iwabe T, Taniguchi F, Ito M,
Deura I, Decavalas G, Terakawa N and Harada T: The role of
NF-kappaB in endometriosis. Front Biosci (Schol Ed). 4:1213–1234.
2012.PubMed/NCBI
|
|
103
|
Suzuki E, Kajita S, Takahashi H, Matsumoto
T, Tsuruta T and Saegusa M: Transcriptional upregulation of HNF-1β
by NF-κB in ovarian clear cell carcinoma modulates susceptibility
to apoptosis through alteration in bcl-2 expression. Lab Invest.
95:962–972. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Jana S, Chatterjee K, Ray AK, DasMahapatra
P and Swarnakar S: Regulation of matrix metalloproteinase-2
activity by COX-2-PGE2-pAKT axis promotes angiogenesis in
endometriosis. PLoS One. 11:e01635402016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ruiz LA, Báez-Vega PM, Ruiz A, Peterse DP,
Monteiro JB, Bracero N, Beauchamp P, Fazleabas AT and Flores I:
Dysregulation of lysyl oxidase expression in lesions and
endometrium of women with endometriosis. Reprod Sci. 22:1496–1508.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Ruiz A, Ruiz L, Colón-Caraballo M,
Torres-Collazo BJ, Monteiro JB, Bayona M, Fazleabas AT and Flores
I: Pharmacological blockage of the CXCR4-CXCL12 axis in
endometriosis leads to contrasting effects in proliferation,
migration, and invasion. Biol Reprod. 98:4–14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Del Carmen MG, Smith Sehdev AE, Fader AN,
Zahurak ML, Richardson M, Fruehauf JP, Montz FJ and Bristow RE:
Endometriosis-associated ovarian carcinoma: Differential expression
of vascular endothelial growth factor and estrogen/progesterone
receptors. Cancer. 98:1658–1663. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Laschke MW and Menger MD: Basic mechanisms
of vascularization in endometriosis and their clinical
implications. Hum Reprod Update. 24:207–224. 2018. View Article : Google Scholar
|
|
109
|
Becker CM, Beaudry P, Funakoshi T, Benny
O, Zaslavsky A, Zurakowski D, Folkman J, D'Amato RJ and Ryeom S:
Circulating endothelial progenitor cells are up-regulated in a
mouse model of endometriosis. Am J Pathol. 178:1782–1791. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wei JJ, William J and Bulun S:
Endometriosis and ovarian cancer: A review of clinical, pathologic,
and molecular aspects. Int J Gynecol Pathol. 30:553–568. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Mandai M, Yamaguchi K, Matsumura N, Baba T
and Konishi I: Ovarian cancer in endometriosis: Molecular biology,
pathology, and clinical management. Int J Clin Oncol. 14:383–391.
2009. View Article : Google Scholar : PubMed/NCBI
|