Introduction
Glioblastoma (GBM) is one of the most prevalent
malignant cancers and is the most lethal primary intrinsic brain
tumor (1). Currently, even
following standard treatment, namely, surgical excision followed by
radiotherapy alone or adjuvant chemotherapy, patients exhibit a
poor prognosis, with long-term survival rates of <5% (2,3). The
clinical course of GBM is very rapid and fatal. According to a
previous investigation concerning glioblastoma survival in the
United States, the median survival following standard treatment and
adjuvant chemotherapy with temozolomide (TMZ) was only 15 months
(4). TMZ is an alkylating agent
(current front-line GBM therapeutic drug) administered orally which
causes DNA damage and induces cell death. TMZ exhibits anti-glioma
activity when used alone or in combination with other agents
(5). TMZ has been approved for the
clinical treatment of patients with newly diagnosed malignant
glioma and recurrent malignant glioma (3,6).
Athanassiou et al revealed the therapeutic mechanisms of
TMZ. The authors demonstrated that TMZ can efficiently inhibit the
proliferation and induce the apoptosis of glioma cells (3). However, GBM cells usually achieve
resistance to TMZ which alleviates the cell toxicity of TMZ. The
occurrence of chemoresistance to TMZ is the main cause of the poor
prognosis of GBM patients. Improving the sensitivity of GBM cells
to TMZ has become a promising strategy for GBM treatment.
STAT3 is a transcription factor and one of the major
intracellular signaling proteins. STAT3 has been found to be
upregulated in GBM (7). STAT3 can
be activated by phosphorylation of the STAT3 tyrosine 705 residue,
which is stimulated by JAK2 activation under any of a variety of
cytokines including IL-6 and IL-11 as well as growth factors such
as EGF, TGF-α, PDGF and HGF (8).
Upon activation, Tyr705-phosphorylated STAT3 (p-STAT3) translocates
from the cytoplasm to the nucleus, thus regulating the
transcription of downstream target genes by binding to the promoter
of these genes, including Bcl-2, Bcl-xL and vascular endothelial
growth factor (VEGF) (7).
Constitutive activation of STAT3 has been confirmed to be
positively correlated with the grade of glioma (9). JAK2/STAT3 inhibition leads to
apoptosis (10) and autophagy
(11). JAK2/STAT3 is a well-known
target of GBM treatment (12,13).
Momelotinib (MTB; also named CYT387). is a
small-molecule, adenosine triphosphate (ATP)-competitive inhibitor
of janus kinase (JAK)-1/2 (14,15).
MTB has been evaluated in phase 3 myelofibrosis clinical trials
(NCT01969838 and NCT02101268) (16). MTB was reported to inhibit the
growth of pancreatic ductal adenocarcinoma (PDAC) (17) and ovarian carcinoma cells (18). In addition, a previous study showed
that MTB and dasatinib synergistically inhibited renal cell
carcinoma (19). Similarly, MTB was
reported to enhance the antitumor activity of cetuximab against
non-small cell lung cancer (20).
These results indicated that MTB has anticancer potential. In the
present study, we aimed to investigate whether MTB sensitizes GBM
cells to TMZ and the underlying mechanisms.
Materials and methods
Cell culture and reagents
The human GBM cell lines U373-MG-ATCC, U251, SHG-44
and LN229 (CRL-2611) were purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA). The human GBM cell lines
U251, SHG-44 and LN229 were cultured in Dulbecco's modified
essential medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.), 100 U/ml penicillin + streptomycin
(Gibco; Thermo Fisher Scientific, Inc.). U373-MG-ATCC cells were
maintained in complete EMEM medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 10% FBS and 100 U/ml penicillin +
streptomycin (Gibco; Thermo Fisher Scientific, Inc.). All cell
cultures were maintained at 37°C in a 5% CO2 incubator.
MTB was purchased from Selleckchem (S2219) and TMZ was purchased
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Stock
solutions were made in dimethyl sulfoxide (DMSO; Sigma-Aldrich;
Merck KGaA) and subsequently diluted in culture medium for use.
Working concentrations were freshly prepared daily by diluting the
stock with phosphate-buffered saline (PBS).
Cell Counting Kit-8 cell viability
assay
Cell Counting Kit-8 (CCK-8; Beyotime Institute of
Biotechnology, Shanghai, China) assay was performed to test cell
toxicity of MTB and proliferation of U373-MG-ATCC, U251, SHG-44 and
LN229 cells in this study. For toxicity test, we chose astrocytes
as a control. U-251MG cells treated with MTB (0, 0.1, 0.025, 0.05,
0.1, 0.25, 0.5, 1.0, 2.5, 5, 10, 25, 50, 100 and 200 µM) for 48 h,
were collected to detect the optical density (OD) 490. For
proliferation determination, U-251MG cells were pretreated with NC,
2.5 µM MTB, 100 µM TMZ and MTB + TMZ for 24 h, and then cells were
trypsinized and seeded into 96-well plates at a density of
4×103 cells in 200 µl of complete medium per well. Next,
the 96-well plates were placed at 37°C in a 5% CO2
incubator. CCK-8 assay was performed according to the
manufacturer's instructions provided in the CCK-8 kit for the next
continuous 4 days. The value of 450 nm was measured by ELISA. This
assay was repeated for 3 times.
Treatments of GBM cells
U-251MG cells were divided into 4 groups and treated
with NC (Ctrl), 2.5 µM MTB (MTB), 100 µM TMZ (TMZ) and combination
treatment (2.5 µM MTB + 100 µM TMZ), separately. TMZ at a
concentration of 100 µM was used as previously reported (21).
Hoechst 33342 staining
U-251MG cells were treated as described above for 48
h. Briefly, cells in each group were detached from the culture dish
and resuspended in DMEM containing 2% FBS at a density of
1×106 cells/ml. Then, the cells were incubated with
Hoechst 33342 staining solution (Sigma-Aldrich; Merck KGaA) for 60
min at 37°C according to the manufacturer's instructions. Finally,
the cells were observed under a fluorescence microscope (scale bar,
50 µm). Apoptotic cell death was determined by counting the number
of cells with condensed nuclei in 6 randomly selected areas.
Western blot analysis
U-251MG cells were treated with MTB (2.5 µM) or TMZ
(100 µM) for 48 h. Proteins were extracted with RIPA lysis buffer
adding cocktail tablets (Sigma Aldrich; Merck KGaA) and
phenylmethanesulfonyl fluoride (PMSF; Sigma-Aldrich; Merck KGaA).
An equivalent amount of protein (40 µg) was resolved on SDS-PAGE
(6–12%) gel, transferred onto polyvinylidene fluoride (PVDF)
membranes (EMD Millipore, Billerica, MA, USA) and probed with
specific primary antibodies. The primary antibodies against cleaved
caspase-9, Ki-67, PCNA, cleaved caspase-3, Beclin1, P62, LC3,
phosphorylated (Tyr705) STAT3 (p-STAT3), STAT3, phosphorylated
(Tyr1007/1008) JAK2 (p-JAK2), JAK2, were obtained from the Cell
Signaling Technology, Inc. (Danvers, MA, USA) (Table I). After 1 h of incubation with the
appropriate horseradish peroxidase (HRP)-conjugated secondary
antibody (Santa Cruz Biotechnology, Inc., Dallas, TX, USA).
Finally, the blots were visualized by ECL and detected using a
ChemiDoc XRS imaging system (Bio-Rad Laboratories, Hercules, CA,
USA). Protein bands were normalized with GAPDH as an internal
control.
 | Table I.Primary and secondary antibody
information. |
Table I.
Primary and secondary antibody
information.
| Antibodies | Company | Catalog no. | Dilution |
|---|
| Primary cleaved
caspase-9 | Cell Signaling
Technology (CST), Inc., Danvers, MA, USA | 52,873 | 1:2,000 |
| Cleaved
caspase-3 | CST | 9,662 | 1:2,000 |
|
Ki-67 | Abcam, Cambridge,
UK | ab16667 | 1:2,000 |
|
PCNA | CST | 13,110 | 1:2,000 |
|
Beclin1 | CST | 3,495 | 1:1,000 |
|
p62 | CST | 8,025 | 1:1,000 |
|
LC3 | CST | 4,108 | 1:1,000 |
|
STAT3 | CST | 9,139 | 1:2,000 |
| p-STAT3
(Tyr705) | CST | 9,145 | 1:2,000 |
|
p-JAK2 | CST | 3,771 | 1:1,000 |
| Secondary
(Tyr1007/1008) |
|
JAK2 | CST | 3,230 | 1:1,000 |
| m-IgGκ
BP-HRP | Santa Cruz
Biotechnology, Inc. | sc-516102 | 1:10,000 |
|
| Dallas, TX,
USA |
|
|
Immunofluorescence staining
Immunofluorescence staining was performed to confirm
the localization of p-STAT3 in U-251MG cells. U-251MG cells were
treated and grouped as previously described in the manuscript.
Then, the cells were collected and washed with PBS twice and fixed
with 4% paraformaldehyde for 20 min at room temperature. Next, the
samples were permeabilized with 0.4% Triton X-100 (Sigma-Aldrich;
Merck KGaA) for 10 min, washed with PBS and then blocked with 2%
bovine serum albumin (BSA; Beijing Solarbio Science and Technology
Co., Ltd., Beijing, China) in PBS for 1 h at 37°C. After this, the
samples were incubated with anti-p-STAT3 in 1% BSA (Beijing
Solarbio Science and Technology Co., Ltd.) in PBS overnight at 4°C.
The samples were subsequently washed with PBS and incubated with
fluorescein-conjugated secondary antibody in 1% BSA (Beijing
Solarbio Science and Technology Co., Ltd.) in PBS for 1 h. Finally,
the samples were stained with DAPI (1 µg/ml) for 10 min to stain
the cell nuclei. The samples were then visualized a confocal laser
scanning microscope at ×400 magnification (Leica Microsystems,
GmbH, Wetzlar, Germany).
Cell cycle assay
U-251MG cells were seeded in 6-well cell culture
plates. After 48 h of treatment with TMZ with or without MTB, the
cells were analyzed by flow cytometry (BD FACSCanto II; BD
Biosciences, San Diego, CA, USA) using CycleTEST™ Plus DNA Reagent
kit (BD Biosciences) according to the manufacturer's recommended
protocol. The obtained results were analyzed using ModFit LT
(version 4.0; Verity Software House, Topsham, ME, USA).
Xenograft tumor model
For testing of the efficacy of MTB combined with TMZ
in vivo, orthotopic xenograft models were established.
Twenty 6-week-old male BALB/c-nu/nu mice weighing 16–18 g were bred
in aseptic cages, which were kept in an isolator unit with filtered
air. The mice had access to water and food ad libitum. U251
cell suspension (0.1 ml) [2-5×107/ml Hank's balanced
salt solution (HBSS), Gibco; Thermo Fisher Scientific, Inc.] was
stereotypically injected into the left striata of mice. After the
tumor was established, the mice were randomized into the following
groups: Ctrl; MTB (50 mg/kg/day body weight) (19); TMZ (20 mg/kg/day body weight); MTB +
TMZ (50 mg/kg/day MTB + 20 mg/kg/day TMZ). All mice were
administered the drug or saline daily by oral gavage 1 day after
tumor cell injection (18). All
animals were sacrificed at 30 days and tumors were collected. Tumor
weight was measured. All animal protocols were approved by the
Beijing Tiantan Hospital and Capital Medical university laws
governing animal care.
TUNEL
Terminal deoxynucleotidyl transferase-mediated
dUTP-biotin nick-end labeling (TUNEL) staining was performed to
detect apoptosis according to the manufacturer's instructions
(TUNEL Apoptosis Detection Kit; Upstate Biotechnology, Inc., Lake
Placid, NY, USA). TUNEL-positive cells were quantified by counting
brown staining within the nucleus of apoptotic cells. The extent of
apoptosis was calculated with the Motic Image software (version
1.2; Micro Optical Group Co., Haimen, China) and expressed as a
number of TUNEL-positive cells from the same field under an Olympus
BX60 microscope and Spot camera (Olympus Optical Co., Tokyo, Japan;
magnification, ×400).
Immunohistochemistry
Tumors were cut into 2-µm sections and
paraffin-embedded tumor sections were constructed. Sections were
deparaffinized and blocked with 3% H2O2 for
10 min at room temperature followed by incubation with anti-Ki-67
and anti-cleaved caspase-3 at 4°C overnight. Slides were rinsed
with PBS and incubated at room temperature for 30 min with
biotinylated goat anti-mouse immunglobulin G secondary antibody
(Dako, Glostrup, Denmark). After washing in Tris-hydrochloric acid
buffer (TBS), the slides were incubated with
streptavidin-peroxidase reagent (Dako) and treated with
3,3′-diaminobenzidine (DAB; Sigma Aldrich; Merck KGaA) for 5 min.
The slides were counterstained and dehydrated and the final
percentage of positive cells was calculated with the Motic Image
software (version 1.2; Micro Optical Group Co.)
Autophagy detection
Autophagy was assessed by western blot analysis and
immunofluorescence staining for LC3. Detailed methods were shown in
paragraph Western blot analysis and Immunofluorescence
staining. All stained specimens were observed under a confocal
laser scanning microscope (Leica Microsystems, GmbH). For the
quantitative analysis, LC3 positive puncta were analyzed by ImageJ
(version 1.50b; National Institutes of Health, Bethesda, ME, USA;
http://imagej.nih.gov/ij/).
Statistical analysis
Statistically significant differences between the
control and other groups were evaluated by means of analysis of
variance (ANOVA). For comparisons among >2 groups, the one-way
ANOVA followed by post hoc Tukey's test was applied. For the CCK-8
assay, the one-way ANOVA with Dunnett's post hoc test was used.
Data are illustrated in bar graphs, values are presented as mean ±
SEM from at least 3 independent experiments and post hoc test.
Asterisks/pound symbols denote statistical significance in the
figures. Statistically significant differences in Kaplan-Meier
survival were determined by a log-rank test using GraphPad Prism.
Statistical significance was considered if P<0.05 in any
analysis. All the data were analyzed using GraphPad Prism 6
(GraphPad Software, Inc., San Diego, CA, USA).
Results
MTB inhibits the cell viability of 4
human GBM cell lines
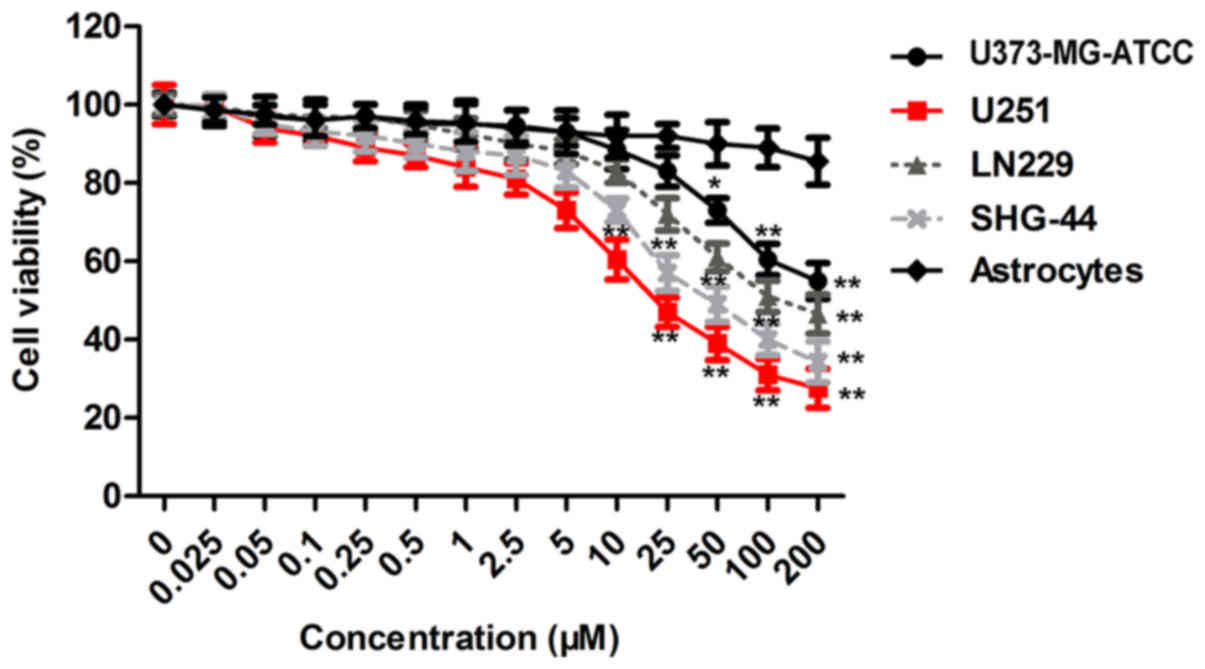
To clarify the influence of MTB on GBM cells, we
conducted a CCK-8 assay using U373-MG-ATCC, U251, SHG-44 and LN229
cells and astrocytes treated with MTB at concentrations of 0 to 200
µM for 24 h, respectively. The result (Fig. 1) showed that MTB exerted an
inhibitory effect on the viability of GBM cells in a dose-dependent
manner (P<0.01; compared to the astrocyte group). Particularly,
U251 cells showed more sensitivity to MTB than the other cell
lines. The concentration of MTB that inhibited cell viability was
2.5 µM in U251 cells. In addition, treatment with MTB at 2.5 µM did
not induce significant cytotoxicity of U251 cells. These data
suggest that MTB is a potential anticancer agent against GBM cells,
particularly U251 cells. Thus, 2.5 µM of MTB and U251 cells were
used for subsequent experiments.
MTB enhances the
proliferation-inhibiting and apoptosis-promoting effect of TMZ
A concentration of 100 µM TMZ was used as previously
described (21). As shown in
Fig. 2A, both MTB and TMZ promoted
the apoptosis of U251 cells compared with the control (P<0.05).
As shown in Fig. 2B, both MTB and
TMZ suppressed the growth of U-251MG cells compared with the
control (P<0.05). However, the combination of MTB and TMZ
enhanced the proliferation-inhibiting effect compared with either
the MTB or TMZ group (P<0.05). In addition, the combination of
MTB and TMZ enhanced the apoptosis-promoting effect compared to TMZ
or MTB used alone at the same dose (P<0.05). The present results
were verified by western blot analysis of Ki-67, PCNA, cleaved
caspase-3 and cleaved caspase-9 (Fig.
2C). The relative expression of Ki-67 and PCNA were
significantly reduced by MTB and TMZ, and the combination therapy
(P<0.05), while the expression of cleaved caspase-3 and cleaved
caspase-9 exhibited inverse results. We also assessed the effect of
MTB on cell cycle distribution (Fig.
2D). MTB and TMZ combination significantly arrested the cell
cycle at the G2 phase.
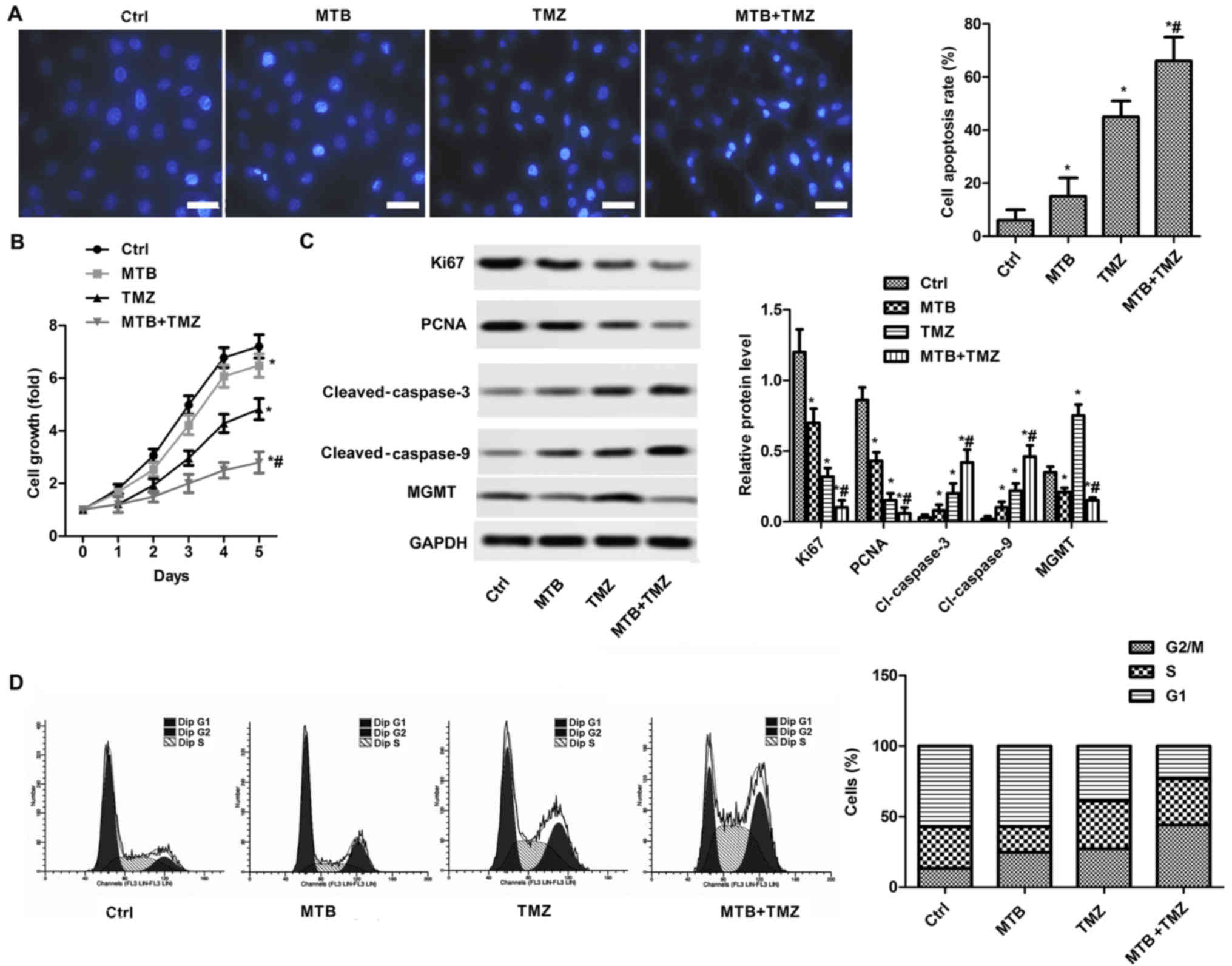 | Figure 2.MTB enhances the
proliferation-inhibiting and apoptosis-promoting effect of TMZ in
GBM cells. The U251 cell line was selected for subsequent
experiments. U251 cells were treated with DMSO (control), 2.5 µM
MTB, 100 µM TMZ, or 2.5 µM MTB combined with 100 µM TMZ (MTB +
TMZ). (A) Apoptosis of cells under different treatments was
detected by Hoechst 33258 staining (left panels) Scale bar, 50 µm.
The representative histogram showing the results of the apoptosis
rate of U251 cells (right panel). (B) Cells were cultured in
96-well plates and then treated with solvent only (control; Ctrl)
or 100 µM TMZ with 2.5 µM MTB for 48 h. OD490 of U251
cells treated with MTB and TMZ were determined by CCK-8 assay. (C)
The protein expression of cells under different treatments was
detected by western blot analysis. GAPDH was used as the loading
control (left image). The representative column diagrams showing
results of relative protein expression (right histogram). (D) Cell
cycle distribution was detected by FACS cell cycle analysis (left
panels) and the representative histogram showing cell cycle
distribution (right histogram). *P<0.05, compared with the Ctrl;
#P<0.05, compared with TMZ. Data are represented as
mean ± SEM for 3 replicates. MTB, momelotinib; TMZ, temozolomide;
GBM, glioblastoma; PCNA, proliferating cell nuclear antigen; MGMT,
O-methylguanine-DNA methyltransferase; Ctrl, control. |
MTB enhances the autophagy-promoting
effect of TMZ
To elucidate the role of MTB in autophagy,
expression levels of Beclin1, LC3-II, LC3-I and p62 were detected
by western blot assay. As shown in Fig.
3A, both MTB and TMZ increased the level of Beclin1 and LC3-II
and decreased the level of p62 compared to the control (P<0.05).
Expectedly, the combination of MTB and TMZ enhanced the
autophagy-promoting effect, evidenced by higher levels of Beclin1
and LC3-II/I ratio, as well as a lower level of P62 compared with
the MTB and TMZ group (P<0.05). In addition, the combination of
MTB and TMZ obviously increased the number of LC3-positive puncta
per cell when compared to that when TMZ or MTB was used alone at
the same dose (Fig. 3B, P<0.05).
3-Methyladenine (3-MA) was used to verifiy the autophagy-promoting
effect of MTB. As shown in Fig. 3C,
TMZ alone significantly increased the number of LC3-positive
puncta, and 3-MA totally inhibited the autophagy induced by TMZ,
while the suppression was reversed by MTB.
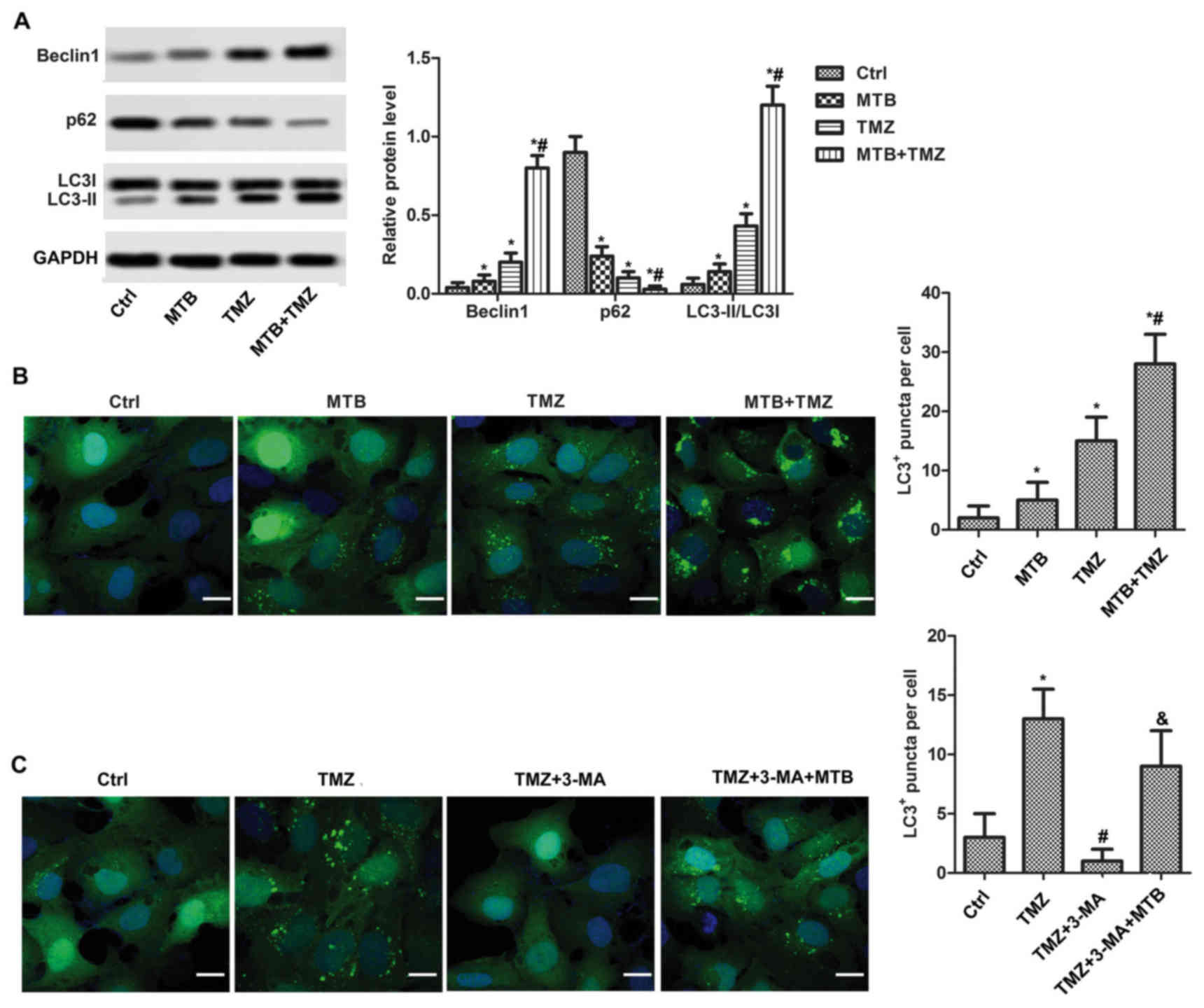 | Figure 3.MTB enhances the autophagy-promoting
effect of TMZ in GBM cells. 3-MA was used to verify the
autophagy-promoting effect of TMZ. (A) U251 cells were treated with
DMSO (Ctrl), 2.5 µM MTB, 100 µM TMZ, or 2.5 µM MTB combined with
100 µM TMZ (MTB + TMZ). Relative protein levels of Beclin1, LC3
I/II and p62 were detected by western blot analysis. GAPDH was used
as loading control. The quantification of relative protein
expression was performed by ImageJ software. (B) Expression of LC3
was visualized with fluorescence microscopy after
immunofluorescence staining with the LC3 antibody (green); scale
bar, 20 µm. The number of LC3+ puncta/cell was compared
between the different groups. *P<0.05, compared with Ctrl;
#P<0.05, compared with TMZ. (C) U251 cells were
treated with DMSO (Ctrl), 100 µM TMZ, 100 µM TMZ + 10 mM 3-MA (TMZ
+ 3-MA) and 2.5 µM MTB + 100 µM TMZ + 10 mM 3-MA (TMZ + 3-MA +
MTB). Expression of LC3 was visualized with fluorescence microscopy
after immunofluorescence staining with LC3 antibody (green); scale
bar, 20 µm. The number of LC3+ puncta/cell was compared
between the different groups. *P<0.05, compared with Ctrl;
#P<0.05, compared with TMZ;
&P<0.05, compared with TMZ + 3-MA. The results
are presented as the mean of 3 independent experiments. MTB,
momelotinib; TMZ, temozolomide; GBM, glioblastoma; 3-MA,
3-methyladenine; LC3, microtubule-associated protein l light chain
3; Ctrl, control. |
MTB inactivates the JAK2/STAT3
pathway
To elucidate the mechanism of MTB in autophagy
induction, expression levels of JAK2 and STAT3 were determined by
western blot analysis. As shown in Fig.
4A, western blot analysis showed that the combination of MTB
and TMZ decreased the phosphorylation levels of JAK2 and STAT3
compared to that following treatment with TMZ or MTB used alone at
the same dose (P<0.05). In particular, MTB was observed to
suppress STAT3 nuclear translocation. As shown in Fig. 4B, the nuclear level of p-STAT3 was
high in the untreated U251 cells; whereas, the nuclear level of
p-STAT3 was significantly reduced by MTB and TMZ. In addition, the
nuclear level of p-STAT3 in the MTB + TMZ group was significantly
decreased compared to that following TMZ or MTB used alone at the
same dose (P<0.05).
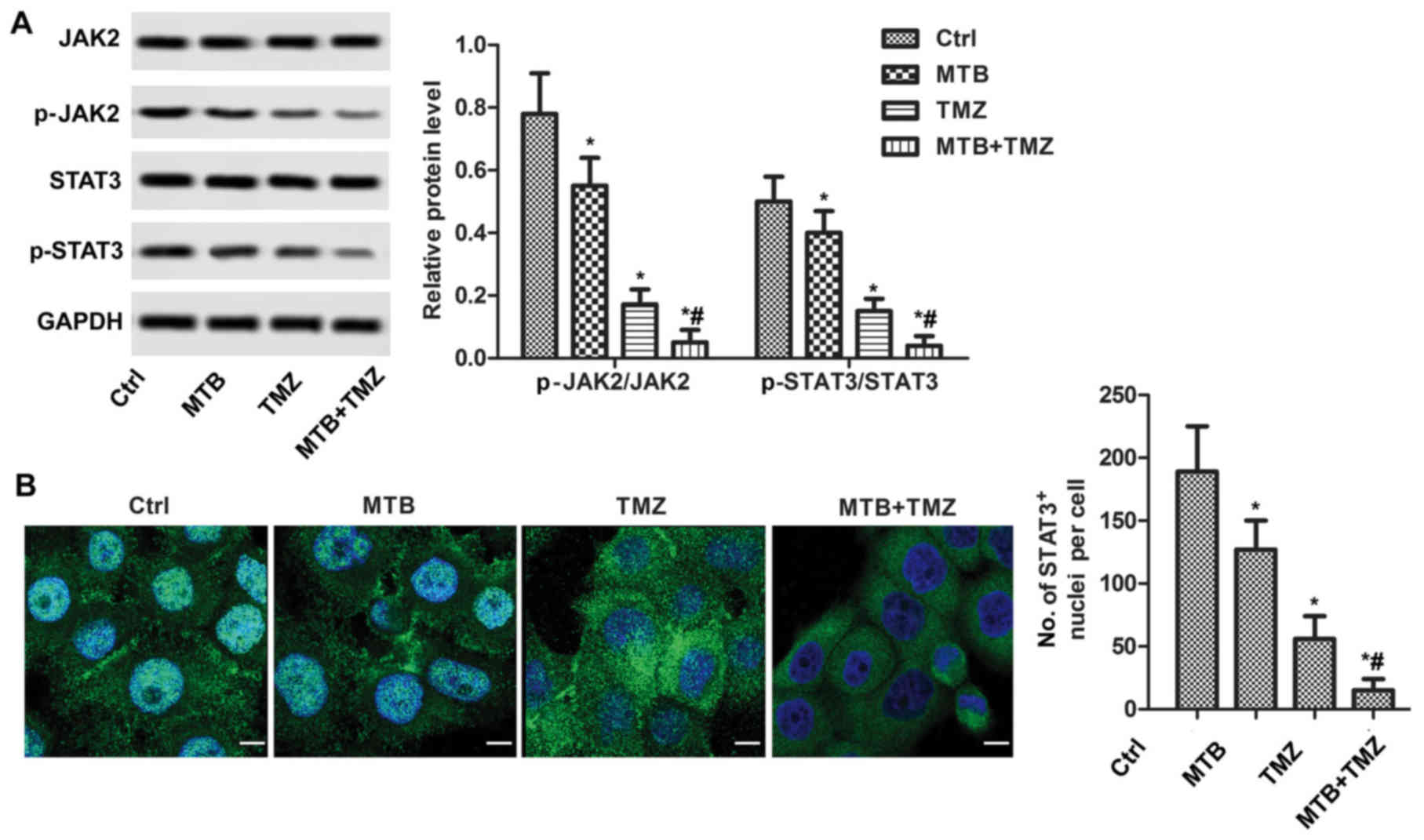 | Figure 4.Treatment with the combination of MTB
and TMZ inhibits the JAK2/STAT3 pathway in GBM cells in
vitro. (A) Relative protein levels of JAK2, STAT3, p-JAK2 and
p-STAT3 were detected by western blot analysis. GAPDH was used as
loading control. The quantification of relative protein expression
was performed by ImageJ. (B) Localization of STAT3 was visualized
with fluorescence microscopy after immunofluorescence staining with
STAT3 antibody (green). Cells were stained with DAPI for
visualization of the cell nuclei (blue). The number of
STAT3-positive staining nuclei were counted. *P<0.05, compared
with Ctrl; #P<0.05, compared with TMZ. Data are
presented as the mean ± SEM for 3 replicates; scale bar, 20 µm.
MTB, momelotinib; TMZ, temozolomide; GBM, glioblastoma; STAT3,
signal transducers and activators of transcription; p-STAT3,
Tyr705-phosphorylated STAT3; JAK2, Janus kinase 2; p-JAK2,
phosphorylated Janus kinase 2; Ctrl, control. |
MTB enhances the growth-inhibiting
effect and apoptosis-promoting of TMZ in U251 orthotopic xenograft
models
We employed U-251MG orthotopic xenograft models to
confirm the results of the combination treatment of MTB + TMZ in
vivo. MTB at 50 mg/kg/day (body weight) (19) and TMZ at 20 mg/kg/day (MTB + TMZ)
were administered by oral gavage. At day 30 after tumor cell
implantation, mice were sacrificed for tumor weight analysis,
immunohistochemistry, TUNEL and western blot assay. As shown in
Fig. 5A, the combination treatment
with MTB + TMZ significantly decreased xenograft tumor weight
compared to that following TMZ or MTB used alone at the same dose
(P<0.05). As shown in Fig. 5B,
the combination treatment with MTB and TMZ significantly increased
the survival rate of the mice (P<0.05). In addition, the results
of the TUNEL assay combined with immunohistochemistry demonstrated
that the combination treatment with MTB and TMZ significantly
promoted the apoptosis of the xenograft tumor cells (Fig. 5C and D; P<0.05);
immunohistochemistry of cleaved caspase-3 confirmed an elevated
apoptosis rate (Fig. 5D).
Meanwhile, the low expression of Ki-67 indicated suppression of
proliferation following treatment with the combination of MTB and
TMZ (Fig. 5D). Similarly, the
combination of MTB and TMZ enhanced the autophagy-promoting effect
in the xenograft tumors (Fig. 6A).
Both TMZ and MTB decreased the phosphorylation of JAK2 and STAT3,
while the combination of MTB and TMZ almost totally inhibited
p-JAK2/p-STAT3 (Fig. 6B). The in
vivo results were consistent with the in vitro
findings.
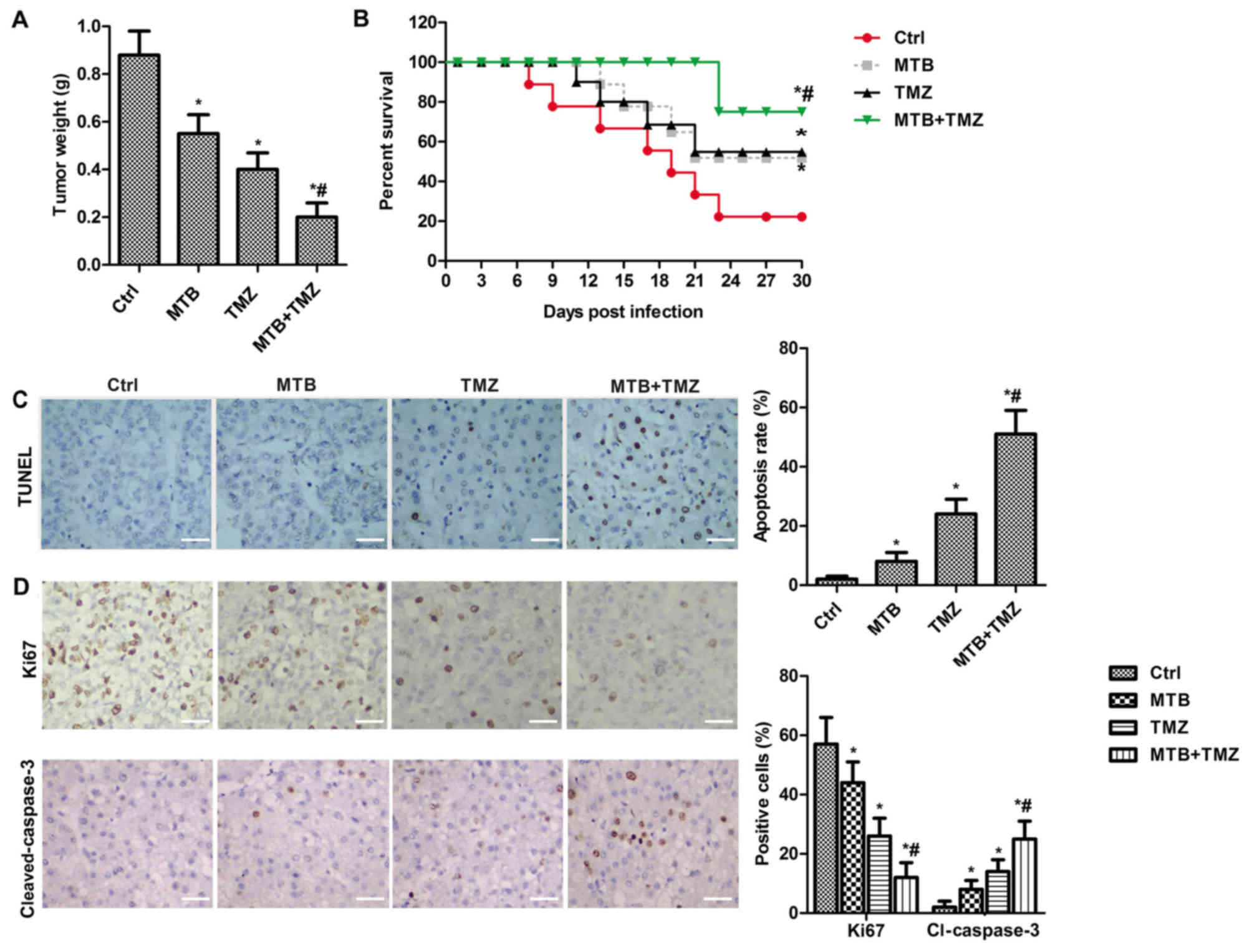 | Figure 5.MTB enhances the growth-inhibiting
effect of TMZ in vivo. (A) Nude mice bearing subcutaneous
xenograft GBM tumors were treated with Ctrl, MTB (50 mg/kg of body
weight), TMZ (20 mg/kg/day body weight) or MTB + TMZ. The weight of
the tumors (g) was determined at day 30 after mice were sacrificed.
(B) The survival of mice was determined every 3 days for 30 days.
(C) TUNEL assay was used to determine the apoptosis in the MTB, TMZ
and combination treatment tumors compared with the NC
(Ctrl)-treated tumors (magnification, ×400); scale bar, 50 µm. (D)
Immunohistochemical analysis of expression of Ki-67,
cleaved-caspase-3 in the MTB, TMZ and combination treatment tumors
compared with the NC (Ctrl)-treated tumors (magnification, ×400);
scale bar, 50 µm. Positive cells (brown) were counted. *P<0.05,
compared with Ctrl; #P<0.05, compared with TMZ. The
results are presented as the mean of 3 independent experiments.
MTB, momelotinib; TMZ, temozolomide; GBM, glioblastoma; TUNEL,
terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end
labeling; Ctrl, control; caspase-3, cleaved-caspase-3. |
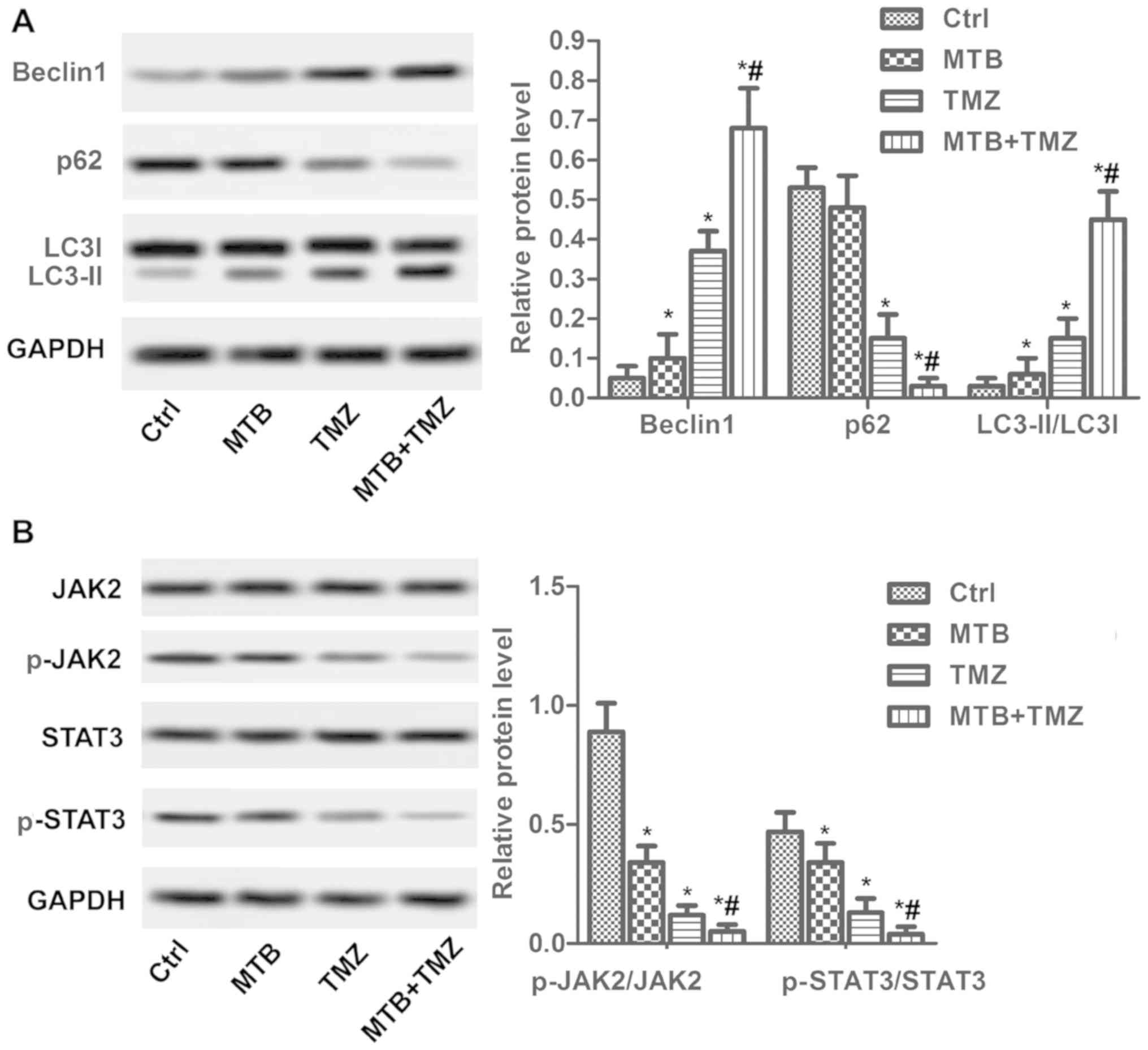 | Figure 6.Treatment with the combination of MTB
and TMZ inhibits the JAK2/STAT3 pathway in vivo. Nude mice
(n=60) were treated with NC (Ctrl), MTB (50 mg/kg body weight), TMZ
(2 mg/kg body weight) or MTB + TMZ combination. At day 30, mice
were sacrificed for immunohistochemistry, TUNEL and western blot
assay. (A) Relative protein levels of Beclin1, LC3 I/II and p62
were detected by western blot analysis. GAPDH was used as loading
control. The quantification of relative protein expression was
performed by ImageJ. (B) Relative protein levels of JAK2, STAT3,
p-JAK2 and p-STAT3 were detected by western blot analysis. GAPDH
was used as a loading control. The quantification of relative
protein expression was performed by ImageJ. *P<0.05, compared
with Ctrl; #P<0.05, compared with TMZ. The results
were presented as the mean of 3 independent experiments. MTB,
momelotinib; TMZ, temozolomide; GBM, glioblastoma; STAT3, signal
transducers and activators of transcription; p-STAT3,
Tyr705-phosphorylated STAT3; JAK2, Janus kinase 2; p-JAK2,
phosphorylated janus kinase 2; LC3, microtubule-associated protein
l light chain 3; Ctrl, control. |
Discussion
Temozolomide (TMZ) has been applied for the clinic
treatment of gliomas as a first-line drug. However, TMZ resistance
has become the most threatening challenge to glioblastoma
multiforme (GBM) treatment. Therefore, it is vital to enhance the
chemosensitivity of GBM to TMZ, which may contribute to prolong the
survival time of these patients. In the present study, we
demonstrated that 2.5 µM/50 mg/kg momelotinib (MTB) enhanced the
chemosensitivity of GBM to TMZ both in vitro and in
vivo.
The role of autophagy in GBM cells in response to
metabolic and therapeutic stress seems to be different, including
prosurvival and prodeath (22).
Moreover, accumulating evidence has revealed a positive correlation
between chemoresistance and autophagy reduction in GBM cells
(23,24). Therefore, autophagy induction may be
a promising strategy to enhance the chemosensitivity of GBM to TMZ
and improve the effectiveness of chemotherapy. For example, Bak
et al reported that vitamin D enhanced autophagy in
TMZ-based GBM chemotherapy (25).
TMZ has been confirmed to induce autophagy in both glioma cells and
surgical specimens (26,27). Recently, another study demonstrated
that 100 µM TMZ induced autophagy AMPK-ULK1 pathways (28). MTB at 2.0 µM was reported to induce
autophagy in a human renal cell carcinoma (RCC) cell line by
inhibiting mTOR complex 1 (29). In
agreement with the previous results, our data found induction of
autophagy with TMZ at a concentration of 100 µM in vitro. In
particular, 2.5 µM MTB enhanced the autophagy of U251 cells induced
by TMZ in vitro. Preclinical analysis has exhibited that MTB
was well tolerated when administered to mice orally at doses up to
50 mg/kg of body weight, without obvious toxicity (30). In our in vivo study, 50 mg/kg
MTB enhanced the autophagy induced by TMZ in nude mouse tumors.
Thus, MTB enhanced the chemosensitivity of GBM to TMZ by
potentiating TMZ-induced autophagy.
It has been well established that apoptosis is one
of the promising therapeutic strategies for cancer treatment
(11,31). Recently, research has demonstrated
that apoptosis and the potentiating effects of chemotherapy
response in GBM are positively related with STAT3 pathway
inactivation (13,32,33).
Previous studies have revealed that Stat3 activation was fully
suppressed following the addition of MTB (19,34,35).
Importantly, MTB was reported to synergize with dasatinib to
inhibit proliferation and induce apoptosis in RCC cell lines and
suppress tumor growth in mouse xenograft models (19). In addition, MTB was reported to
inhibit proliferation and induce apoptosis in myeloma cells via
IL-6/JAK/STAT pathway inactivation (35). Research has indicated that apoptosis
induction may lead to cancer cell impairment along with
chemosensitivity enhancement (36–38).
In the present study, it was found that MTB reduced the
chemoresistance of GBM cells to TMZ by potentiating TMZ-induced
apoptosis. The results were further supported by the finding that
MTB + TMZ-treated xenografts had a significant decrease in tumor
weight compared with that following TMZ alone treatment. Therefore,
MTB may not only decease TMZ resistance in vitro, but also
confine cancer growth in vivo.
JAK2/STAT3 pathway has been reported to play an
important role in regulating cell growth, autophagy and apoptosis
in various types of cancers, including breast cancer, esophageal
carcinoma, colon cancer, osteosarcoma and ovarian cancer (39–43).
In particular, STAT3 can be transiently activated by specific
growth factors and cytokines in normal cells, while STAT3 remains
constitutively active in certain populations of tumor cells,
including breast, ovarian and prostate (44,45).
Notably, there exists a positive association between STAT3
constitutive activation and tumor grade in gliomas (9). Moreover, activation of STAT3 in cancer
cells contributed to upregulation of multidrug resistance gene thus
resulting in concomitant chemoresistance (34). It is well studied that the
JAK2/STAT3 signaling pathway may be activated thus inducing tumor
cell apoptosis (43) and autophagy
(11). Our present study showed
that MTB inhibited the phosphorylation of JAK2 and STAT3 in U251
cells, suggesting that MTB suppresses cell growth and
chemoresistance through inactivation of the JAK2/STAT3 pathway.
Collectively, it was demonstrated that MTB reduced the resistance
of GBM to TMZ via induction of apoptosis induction and an increase
in autophagy. Mechanistically, our findings indicate that MTB
inactivates JAK2/STAT3 signaling thus triggering apoptosis and
autophagy. As Bcl-2 and Bcl-xL are the downstream targets of STAT3,
it is reasonable that MTB enhances the chemosensitivity of GBM via
the MTB/JAK2/STAT3/Bcl2 axis.
In conclusion, MTB has anti-GBM potential both in
GBM cells and U251 ×enograft mouse models, and MTB enhances the
antitumor effect of TMZ and chemosensitivity of GBM to TMZ via
apoptosis and autophagy induction. The underlying mechanism may be
attributed to the MTB/JAK2/STAT3/Bcl2 axis. Our data indicated that
combination treatment with MTB and TMZ could provide a more
effective therapeutic approach for GBM. In particularly, we found
that MTB reduced the MGMT protein level. Due to the limited
funding, we did not perform methylation detection. MTB may affect
MGMT promoter methylation and this will be clarified in future
research.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81672481).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
YXu and YXi conceived and designed the study. TL, AL
and YXu performed the experiments. TL and YXi wrote the paper. YXu
and YXi reviewed and edited the manuscript. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All animal protocols were approved by the Beijing
Tiantan Hospital and Capital Medical University laws governing
animal care.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MTB
|
momelotinib
|
|
TMZ
|
temozolomide
|
|
GBM
|
glioblastoma multiforme
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP-biotin nick end labeling
|
|
p-STAT3
|
Tyr705-phosphorylated STAT3
|
|
DMSO
|
dimethyl sulfoxide
|
|
JAK2
|
janus kinase 2
|
References
|
1
|
Lathia JD, Mack SC, Mulkearns-Hubert EE,
Valentim CL and Rich JN: Cancer stem cells in glioblastoma. Genes
Dev. 29:1203–1217. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mahaley MS Jr, Mettlin C, Natarajan N,
Laws ER Jr and Peace BB: National survey of patterns of care for
brain-tumor patients. J Neurosurg. 71:826–836. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Athanassiou H, Synodinou M, Maragoudakis
E, Paraskevaidis M, Verigos C, Misailidou D, Antonadou D, Saris G,
Beroukas K and Karageorgis P: Randomized phase II study of
temozolomide and radiotherapy compared with radiotherapy alone in
newly diagnosed glioblastoma multiforme. J Clin Oncol.
23:2372–2377. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Johnson DR and O'Neill BP: Glioblastoma
survival in the United States before and during the temozolomide
era. J Neurooncol. 107:359–364. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ostermann S, Csajka C, Buclin T, Leyvraz
S, Lejeune F, Decosterd LA and Stupp R: Plasma and cerebrospinal
fluid population pharmacokinetics of temozolomide in malignant
glioma patients. Clin Cancer Res. 10:3728–3736. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stupp R, Dietrich PY, Ostermann Kraljevic
S, Pica A, Maillard I, Maeder P, Meuli R, Janzer R, Pizzolato G,
Miralbell R, et al: Promising survival for patients with newly
diagnosed glioblastoma multiforme treated with concomitant
radiation plus temozolomide followed by adjuvant temozolomide. J
Clin Oncol. 20:1375–1382. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mukthavaram R, Ouyang X, Saklecha R, Jiang
P, Nomura N, Pingle SC, Guo F, Makale M and Kesari S: Effect of the
JAK2/STAT3 inhibitor SAR317461 on human glioblastoma tumorspheres.
J Transl Med. 13:2692015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Darnell JE Jr, Kerr IM and Stark GR:
Jak-STAT pathways and transcriptional activation in response to
IFNs and other extracellular signaling proteins. Science.
264:1415–1421. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Atkinson GP, Nozell SE and Benveniste ET:
NF-kappaB and STAT3 signaling in glioma: Targets for future
therapies. Expert Rev Neurother. 10:575–586. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Harada D, Takigawa N and Kiura K: The role
of STAT3 in non-small cell lung cancer. Cancers. 6:708–722. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song Y, Kong L, Sun B, Gao L, Chu P, Ahsan
A, Qaed E, Lin Y, Peng J, Ma X, et al: Induction of autophagy by an
oleanolic acid derivative, SZC017, promotes ROS-dependent apoptosis
through Akt and JAK2/STAT3 signaling pathway in human lung cancer
cells. Cell Biol Int. 41:1367–1378. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng Q, Han L, Dong Y, Tian J, Huang W,
Liu Z, Jia X, Jiang T, Zhang J, Li X, et al: JAK2/STAT3 targeted
therapy suppresses tumor invasion via disruption of the
EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in
EGFRvIII-expressing glioblastoma. Neurooncol. 16:1229–1243.
2014.
|
|
13
|
Stechishin OD, Luchman HA, Ruan Y, Blough
MD, Nguyen SA, Kelly JJ, Cairncross JG and Weiss S: On-target
JAK2/STAT3 inhibition slows disease progression in orthotopic
xenografts of human glioblastoma brain tumor stem cells. Neuro
Oncol. 15:198–207. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Höfener M, Pachl F, Kuster B and Sewald N:
Inhibitor-based affinity probes for the investigation of JAK
signaling pathways. Proteomics. 15:3066–3074. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iurlo A and Cattaneo D: Treatment of
myelofibrosis: Old and new strategies. Clin Med Insights Blood
Disord. 10:1179545×176952332017. View Article : Google Scholar
|
|
16
|
Pardanani A, Abdelrahman RA, Finke C,
Lasho TT, Begna KH, Al-Kali A, Hogan WJ, Litzow MR, Hanson CA,
Ketterling RP, et al: Genetic determinants of response and survival
in momelotinib-treated patients with myelofibrosis. Leukemia.
29:741–744. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang S, Imamura Y, Jenkins RW, Cañadas I,
Kitajima S, Aref A, Brannon A, Oki E, Castoreno A, Zhu Z, et al:
Autophagy inhibition dysregulates TBK1 signaling and promotes
pancreatic inflammation. Cancer Immunol Res. 4:520–530. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chan E, Luwor R, Burns C, Kannourakis G,
Findlay JK and Ahmed N: Momelotinib decreased cancer stem cell
associated tumor burden and prolonged disease-free remission period
in a mouse model of human ovarian cancer. Oncotarget.
9:16599–16618. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lue HW, Cole B, Rao SA, Podolak J, Van
Gaest A, King C, Eide CA, Wilmot B, Xue C, Spellman PT, et al: Src
and STAT3 inhibitors synergize to promote tumor inhibition in renal
cell carcinoma. Oncotarget. 6:44675–44687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu Y, Dong XZ, Liu X, Liu P and Chen YB:
Enhanced antitumor activity of cetuximab in combination with the
Jak inhibitor CYT387 against non-small-cell lung cancer with
various genotypes. Mol Pharm. 13:689–697. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Cai J, Zhao S, Yao K, Sun Y, Li Y,
Chen L, Li R, Zhai X, Zhang J, et al: GANT61, a GLI inhibitor,
sensitizes glioma cells to the temozolomide treatment. J Exp Clin
Cancer Res. 35:1842016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li H, Chen L, Li JJ, Zhou Q, Huang A, Liu
WW, Wang K, Gao L, Qi ST and Lu YT: miR-519a enhances
chemosensitivity and promotes autophagy in glioblastoma by
targeting STAT3/Bcl2 signaling pathway. J Hematol Oncol. 11:702018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi F, Guo H, Zhang R, Liu H, Wu L, Wu Q,
Liu J, Liu T and Zhang Q: The PI3K inhibitor GDC-0941 enhances
radiosensitization and reduces chemoresistance to temozolomide in
GBM cell lines. Neuroscience. 346:298–308. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fu J, Liu ZG, Liu XM, Chen FR, Shi HL,
Pangjesse CS, Ng HK and Chen ZP: Glioblastoma stem cells resistant
to temozolomide-induced autophagy. Chin Med J. 122:1255–1259.
2009.PubMed/NCBI
|
|
25
|
Bak DH, Kang SH, Choi DR, Gil MN, Yu KS,
Jeong JH, Lee NS, Lee JH, Jeong YG, Kim DK, et al: Autophagy
enhancement contributes to the synergistic effect of vitamin D in
temozolomide-based glioblastoma chemotherapy. Exp Ther Med.
11:2153–2162. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Natsumeda M, Aoki H, Miyahara H, Yajima N,
Uzuka T, Toyoshima Y, Kakita A, Takahashi H and Fujii Y: Induction
of autophagy in temozolomide treated malignant gliomas.
Neuropathology. 31:486–493. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin CJ, Lee CC, Shih YL, Lin CH, Wang SH,
Chen TH and Shih CM: Inhibition of mitochondria- and endoplasmic
reticulum stress-mediated autophagy augments temozolomide-induced
apoptosis in glioma cells. PLoS One. 7:e387062012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zou Y, Wang Q, Li B, Xie B and Wang W:
Temozolomide induces autophagy via ATMAMPKULK1 pathways in glioma.
Mol Med Rep. 10:411–416. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lue HW, Podolak J, Kolahi K, Cheng L, Rao
S, Garg D, Xue CH, Rantala JK, Tyner JW, Thornburg KL, et al:
Metabolic reprogramming ensures cancer cell survival despite
oncogenic signaling blockade. Genes Dev. 31:2067–2084. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tyner JW, Bumm TG, Deininger J, Wood L,
Aichberger KJ, Loriaux MM, Druker BJ, Burns CJ, Fantino E and
Deininger MW: CYT387, a novel JAK2 inhibitor, induces hematologic
responses and normalizes inflammatory cytokines in murine
myeloproliferative neoplasms. Blood. 115:5232–5240. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cotter TG: Apoptosis and cancer: The
genesis of a research field. Nat Rev Cancer. 9:501–507. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu J, Feng X, Zhang B, Li J, Xu X, Liu J,
Wang X, Wang J and Tong X: Blocking the bFGF/STAT3 interaction
through specific signaling pathways induces apoptosis in
glioblastoma cells. J Neurooncol. 120:33–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sai K, Wang S, Balasubramaniyan V, Conrad
C, Lang FF, Aldape K, Szymanski S, Fokt I, Dasgupta A, Madden T, et
al: Induction of cell-cycle arrest and apoptosis in glioblastoma
stem-like cells by WP1193, a novel small molecule inhibitor of the
JAK2/STAT3 pathway. J Neurooncol. 107:487–501. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abubaker K, Luwor RB, Zhu H, McNally O,
Quinn MA, Burns CJ, Thompson EW, Findlay JK and Ahmed N: Inhibition
of the JAK2/STAT3 pathway in ovarian cancer results in the loss of
cancer stem cell-like characteristics and a reduced tumor burden.
BMC Cancer. 14:3172014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Monaghan KA, Khong T, Burns CJ and Spencer
A: The novel JAK inhibitor CYT387 suppresses multiple signalling
pathways, prevents proliferation and induces apoptosis in
phenotypically diverse myeloma cells. Leukemia. 25:1891–1899. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Beaujouin M, Baghdiguian S, Glondu-Lassis
M, Berchem G and Liaudet-Coopman E: Overexpression of both
catalytically active and -inactive cathepsin D by cancer cells
enhances apoptosis-dependent chemo-sensitivity. Oncogene.
25:1967–1973. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sun T, Jia Y and Xiao D: Interference of
STAT 5b expression enhances the chemo-sensitivity of gastric cancer
cells to gefitinib by promoting mitochondrial pathway-mediated cell
apoptosis. Oncol Rep. 34:227–234. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen Y, Li R, Pan M, Shi Z, Yan W, Liu N,
You Y, Zhang J and Wang X: MiR-181b modulates chemosensitivity of
glioblastoma multiforme cells to temozolomide by targeting the
epidermal growth factor receptor. J Neurooncol. 133:477–485. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim MS, Lee WS, Jeong J, Kim SJ and Jin W:
Induction of metastatic potential by TrkB via activation of
IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget.
6:40158–40171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sun KX, Xia HW and Xia RL: Anticancer
effect of salidroside on colon cancer through inhibiting JAK2/STAT3
signaling pathway. Int J Clin Exp Pathol. 8:615–621.
2015.PubMed/NCBI
|
|
41
|
Abubaker K, Luwor RB, Escalona R, McNally
O, Quinn MA, Thompson EW, Findlay JK and Ahmed N: Targeted
disruption of the JAK2/STAT3 pathway in combination with systemic
administration of paclitaxel inhibits the priming of ovarian cancer
stem cells leading to a reduced tumor burden. Front Oncol.
4:752014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu JR, Wu WJ, Liu SX, Zuo LF, Wang Y,
Yang JZ and Nan YM: Nimesulide inhibits the growth of human
esophageal carcinoma cells by inactivating the JAK2/STAT3 pathway.
Pathol Res Pract. 211:426–434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Park KR, Yun HM, Quang TH, Oh H, Lee DS,
Auh QS and Kim EC: 4-Methoxydalbergione suppresses growth and
induces apoptosis in human osteosarcoma cells in vitro and in vivo
xenograft model through down-regulation of the JAK2/STAT3 pathway.
Oncotarget. 7:6960–6971. 2016.PubMed/NCBI
|
|
44
|
Walker SR, Xiang M and Frank DA: Distinct
roles of STAT3 and STAT5 in the pathogenesis and targeted therapy
of breast cancer. Mol Cell Endocrinol. 382:616–621. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu C, Zhu Y, Lou W, Cui Y, Evans CP and
Gao AC: Inhibition of constitutively active Stat3 reverses
enzalutamide resistance in LNCaP derivative prostate cancer cells.
Prostate. 74:201–209. 2014. View Article : Google Scholar : PubMed/NCBI
|