Introduction
Liver cancer is the third most fatal cancer in the
world, with mortality rates of ≤0.49‰ and poor prognosis within the
last 5 years (1–3). Although the underlying mechanisms of
liver cancer development remains to be investigated, accumulating
evidence has implicated that tumor cell proliferation contributes
to the process of oncogenesis (4).
Signal transducer and activator of transcription 3 (STAT3) is a
nuclear transcription factor that promotes tumorigenesis, mediates
the occurrence of liver cancer and serves a role in liver cancer
development (5–7). STAT3 is phosphorylated upon
activation, translocates into the nucleus and contributes to the
process of oncogenesis by modulating the expression of various
genes involved in proliferation (4). Inactivation of STAT3 leads to an
inhibition of proliferation in liver cancer cell lines (8–10). The
identification of genes regulating STAT3 activation may provide
novel approaches for the treatment of liver cancer.
17β-Hydroxysteroid dehydrogenase 4 (HSD17B4) is
widely distributed in peroxisomes of mammalian cells, with the
highest levels reported for the liver (11–13).
Previous studies have reported an increase in HSD17B4 expression in
prostate and breast cancer and further tissues and cells (14,15). A
recent study reported that HSD17B4 is upregulated in patients with
liver cancer and HSD17B4 overexpression promotes HepG2
proliferation by enhancing cyclin D1 expression (16). It was demonstrated that STAT3
induces the transcription of cyclin D1 and serves an important role
in hepatocyte proliferation (6,7). It is
suggested that HSD17B4 may enhance STAT3 activation to promote
tumor cell proliferation in liver cancer. However, the mechanism by
which HSD17B4 promotes STAT3 activation in liver cancer requires to
be investigated.
The current study investigated HSD17B4 upregulation
and the correlation of HSD17B4 and the expression of various
proliferation-associated genes in a liver cancer rat model, which
was chemically induced using diethylnitrosamine (DEN). To
understand how HSD17B4 promoted liver cancer cell proliferation,
HSD17B4 and STAT3 levels were evaluated and a positive correlation
between HSD17B4 and phosphorylated (p)-STAT3 in patients with liver
cancer was observed. A connection to the protein kinase B (Akt) and
the mitogen-activated protein kinase
(MEK)/extracellular-signal-regulated kinase (ERK) signaling
pathways was further established. The findings suggested that
HSD17B4 promoted liver cancer cell proliferation via STAT3
activation and HSD17B4 inhibition may inspire liver cancer
treatment in the future.
Materials and methods
Isolation of liver tissue from
patients with liver cancer
Human liver tissues were obtained from 16 male
patients (age, 40–70 years) with liver cancer during surgical
resection in The Second and The Third Affiliated Hospital of Hebei
Medical University (Shijiazhuang, China) between August 2012 and
December 2013. Written informed consent was obtained from each
patient prior to resection and experiments were approved by the
Ethics Committee of Hebei Medical University (Shijiazhuang, China).
Patients with metastatic liver cancer were excluded from the
current study. Tumor and adjacent (1 cm from tumor) liver tissues
were identified according to pathology results. Tissues were fixed
in 10% neutral-buffered formalin overnight at 4°C and embedded in
paraffin fro 1 h at 60–62°C.
Animals
All animal studies were approved by the
Institutional Animal Care and Use Committee of Hebei Medical
University (approval no. HebMU20080026; Shijiazhuang, China) and
efforts were made to minimize suffering. A total of 16 male Wistar
rats (age, 4–5 weeks; weight, 100–120 g) were obtained from the
Central Laboratorial Animal Facility at the Faculty of Medicine of
Hebei University (Shijiazhuang, China). Rats were housed in cages
under controlled environmental conditions at 25°C with 55–65%
humidity, a 12-h light/dark cycle and had free access to food and
water. Following acclimatization for 1 week, rats were divided into
two groups (8/group) as follows: Control group (NC), not receiving
DEN, injected with an equal volume of saline; and DEN group
(Cancer), receiving a weekly intraperitoneal dose of DEN (70 mg/kg)
for 10 weeks. All animals were sacrificed at 20 weeks (17) by exsanguination through cardiac
puncture under urethane anesthesia (20%; 1.2 g/kg;
intraperitoneally). Livers were isolated and fixed for
histopathological analysis.
Histopathological evaluation
Human and rat specimens were processed routinely in
10% formalin buffer overnight at 4°C and embedded in paraffin 1 h
at 60–62°C. Tissue sections (4-µm) were obtained and stained with
hematoxylin and eosin 2–3 min/each at room temperature.
Histopathological examinations were performed under a light
microscope (magnification, ×200).
Western blotting
Precut rats livers were placed in Total Protein
Isolation Buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40,
0.1% SDS, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 mM
Na3VO4 and 1 mM NaF) containing protease
inhibitors (leupeptin and aprotinin, 1 µg/ml each), homogenized
using an ice-chilled glass homogenizer with Teflon pestle and lysed
for 40 min on ice with vortex mixing every 10 min. Supernatants
were collected (15,000 × g; 30 min; 4°C), the protein concentration
was determined using the Lowry method and samples were aliquoted
and stored at −70°C until further analysis.
Cells were lysed in lysis buffer (50 mM PBS pH 7.5,
200 mM NaCl, 0.5 mM EDTA pH 8.0, 2 mM β-mercaptoethanol, 1 mM
phenylmethylsulfonyl fluoride and 0.5% Tween-20) for protein
extraction. Following incubation on ice for 30 min, cell lysates
were centrifuged (12,000 × g; 20 min; 4°C), the protein
concentration was determined as indicated above and the
supernatants were stored at −20°C until use.
A total of 20 or 40 µg protein was used in the
analysis of HSD17B4 overexpressing or knockdown samples,
respectively. Proteins were separated on 10% SDS-PAGE gels and
transferred to polyvinylidene fluoride membranes. Membranes were
blocked with 5% skim milk powder for 1 h at room temperature and
incubated overnight at 4°C with the following primary antibodies:
Mouse anti-HSD17B4 (cat. no. sc-365167; 1:1,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), rabbit anti-cyclin D1 (cat.
no. 2922S; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA,
USA), rabbit antiproliferating cell nuclear antigen (PCNA; cat. no.
13110; 1:1,000; Cell Signaling Technology, Inc.), mouse
anti-β-actin (cat. no. sc-47778; 1:1,000; Santa Cruz Biotechnology,
Inc.), rabbit anti-p-STAT3 (cat. no. sc-135649; 1:500; Santa Cruz
Biotechnology, Inc.), rabbit anti-STAT3 (cat. no. sc-482; 1:500;
Santa Cruz Biotechnology, Inc.), rabbit anti-p-Akt (cat. no.
sc-7985; 1:500; Santa Cruz Biotechnology, Inc.), rabbit anti-Akt
(cat. no. sc-8312; 1:500; Santa Cruz Biotechnology, Inc.), rabbit
anti-p-MEK (cat. no. sc-101733; 1:500; Santa Cruz Biotechnology,
Inc.), rabbit anti-MEK (cat. no. sc-9259; 1:50; Santa Cruz
Biotechnology, Inc.), rabbit anti-p-ERK (cat. no. sc-23759; 1:500;
Santa Cruz Biotechnology, Inc.), rabbit anti-ERK (cat. no.
sc-292838; 1:500; Santa Cruz Biotechnology, Inc.), rabbit
anti-p-c-Jun N-terminal kinase (JNK; cat. no. sc-135642; 1:500;
Santa Cruz Biotechnology, Inc.), rabbit anti-JNK (cat. no. sc-571;
1:500; Santa Cruz Biotechnology, Inc.), rabbit anti-p-p38 (cat. no.
sc-101759; 1:500; Santa Cruz Biotechnology, Inc.), and rabbit
anti-p38 (cat. no. sc-156091; 1:500; Santa Cruz Biotechnology,
Inc.). Following incubation with goat anti-mouse or anti-rabbit IgG
horseradish peroxidase (HRP)-conjugated secondary antibody (cat.
nos. 610-703-002 and 611-142-002, respectively; 1:5,000; Rockland
Immunochemicals Inc., Limerick, Pennsylvania, USA) for 1 h at room
temperature, bands were visualized using an enhanced
chemiluminescence kit (Fuazon Fx; Vilber Lourmat, Marne-la-Vallée,
France). Images were captured and processed by FusionCapt Advance
Fx5 (Vilber Lourmat). The relative gray scale indicated the
expression of various genes, with β-actin as the protein loading
control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from rat livers using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) according to the manufacturer's instructions. cDNA was
synthesized using a SuperScript Reverse Transcription kit
(Invitrogen; Thermo Fisher Scientific, Inc.) with the following
protocol: 5 min at 65°C, 2 min at 37°C, 10 min at 25°C, 50 min at
37°C and 15 min at 70°C. cDNA was used as a template for qPCR with
a SYBR Green PCR Master Mix kit (Takara Biotechnology Co., Ltd.,
Dalian, China). qPCR was performed using a Rotor-Gene 3000
Detection System (GeneBio Systems, Inc., Oakville, ON, Canada)
using the following protocol: Initial denaturation step at 95°C for
30 sec, followed by 40 cycles of denaturation for 5 sec at 95°C,
annealing for 30 sec at 55°C and elongation for 20 sec at 72°C. 18S
ribosomal RNA was used as internal control and for normalization.
All reactions were performed in triplicate. Relative mRNA
expression was determined using the 2−ΔΔCq method
(18). Primer details are presented
in Table I.
 | Table I.Polymerase chain reaction
primers. |
Table I.
Polymerase chain reaction
primers.
|
| Primer sequence
(5′-3′) |
|---|
|
|
|
|---|
| Gene | Forward | Reverse |
|---|
| 18S rRNA
(M11188) |
CGCCGCTAGAGGTGAAATTC |
CCAGTCGGCATCGTTTATGG |
| HSD17B4
(NM_024392) |
GGTGGTAAAGAAAGTAAATG |
AATTGTGATGGTCGTGTC |
| cyclin D1
(NM_171992.4) |
GGAGCAGAAGTGCGAAGA |
GGGTGGGTTGGAAATGAA |
| PCNA
(NM_022381.3) |
ACAGAGCATGGATTCGTCTCAC |
AGAAAACTTCACCCCGTCCTTT |
Immunohistochemistry (IHC) assays
Immunohistochemistry was performed with
paraffin-embedded human or rat tissue sections (4-µm) as previously
described (19). Briefly, liver
sections were blocked with goat serum (1:1; OriGene Technologies,
Inc., Rockville, MD, USA) at 37°C in a wet box for 30 min and
incubated with a mouse anti-HSD17B4 antibody (cat. no. PA1727;
1:1,000; Santa Cruz Biotechnology, Inc.) and rabbit anti-STAT3
antibody (cat. no. sc-135649; 1:500; Santa Cruz Biotechnology,
Inc.) overnight at 4°C in moist chambers. Following washing with
0.01 mol/l PBS (pH 7.2) three times, slides were incubated with a
biotinylated secondary anti-mouse (cat. no. PV-6000; 1:1; OriGene
Technologies, Inc.) or anti-rabbit antibodies (cat. no. PV-9000;
1:1; OriGene Technologies, Inc.) for 30 min at 37°C. Samples were
then incubated with HRP-avidin D (cat. no. A-2004; Vector
Laboratories, Inc.; Maravai LifeSciences, San Diego, CA, USA) at
37°C for 30 min and developed using a diaminobenzidine kit (Vector
Laboratories, Inc.; Maravai LifeSciences). Sections were
counterstained with hematoxylin for 30 sec at room temperature.
Staining intensities were determined by measuring the integrated
optical density (IOD) with light microscopy (magnification, ×200)
with associated software (SPOT Basic™ image capture software
version 3.2.4; cat. no. SPOT53BE; SPOT Imaging; Diagnostic
Instruments, Inc., Sterling Heights, MI, USA).
Cell culture and treatment
The human liver cancer cell line HepG2 was obtained
from the American Type Culture Collection (cat. no. HB-8065;
Manassas, VA, USA). HepG2 cells were maintained in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal bovine
serum (FBS; Abgent Biotech Co., Ltd., Suzhou, China), penicillin
and streptomycin (100 U/ml, each) in a humidified environment at
37°C with 5% CO2. For inhibitor studies, all inhibitors
were dissolved in dimethyl sulfoxide (DMSO) and control experiments
were performed with equal volumes of DMSO. Cells were treated for 2
h with MEK inhibitor PD98059 (5 µmol/; Calbiochem; Merck KGaA,
Darmstadt, Germany) or phosphoinositide 3-kinase (PI3K) inhibitor
LY294002 (5 µmol/l; Calbiochem; Merck KGaA) prior to transformation
with the HSD17B4 overexpression plasmid.
Cell proliferation assay
HepG2 cell proliferation assays were performed using
a bromodeoxyuridine (BrdU) Cell Proliferation Assay kit (Millipore;
Merck KGaA) according to the manufacturer's instructions. Cells
were labeled with BrdU for 6 h at 24 h following HSD17B4 plasmid
transfection. Optical density readings were performed at 450 nm to
measure the incorporation of BrdU. All groups were evaluated with
≥3 separate wells/experiment.
Plasmid constructs and
transfection
HSD17B4 cDNA was cloned into the pLL3.7 vector
(Addgene, Inc., Cambridge, MA, USA). HepG2 cells were plated at
5×106/well in 6-well culture dishes for 18 h to reach
~70% confluence in Dulbecco's modified Eagle's medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc.) containing 10% FBS without
antibiotics. Cells were washed twice with serum-free RPMI-1640 and
1 ml serum-free Opti-MEM I (Gibco; Thermo Fisher Scientific, Inc.)
was added to each well. A DNA-Lipofectamine 2000 complex was
prepared according to the manufacturer's instructions (Invitrogen;
Thermo Fisher Scientific, Inc.). A total of 6 µg transfected
plasmid and 30 µl Lipofectamine reagent were added to each
reaction. The empty vector control is indicated using ‘−’ and the
HSD17B4 plasmid is highlighted using ‘+’ in the following. Cells
were incubated at 37°C for 6 h. Following transfection, cells were
maintained in DMEM containing 1% FBS for 24 h and the transfection
efficiency was determined using western blotting. Cells were
collected for further analysis.
Small interfering (si) RNA
transfection
siRNA targeting HSD17B4 (siHSD17B4; forward,
5′-GUACCUUUGUAUUUGAGGAdTdT-3′ and reverse,
5′-UCCUCAAAUACAAAGGUACdTdT-3′) and non-specific siRNA (siNC;
forwards, 5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′), were purchased from Sigma-Aldrich
(Merck KGaA). Transfections with 10 µmol/l siRNA (siNC as ‘−’ and
siHSD17B4 as ‘+’) were performed using the Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) following the
manufacturer's instructions, when cells reached 50–60% confluence.
Following 24 h of transfection, the transfection efficiency was
determined using western blotting. Cells were harvested and lysed
as described for RT-qPCR and western blotting.
Statistical analysis
Data are presented as the mean ± standard deviation
of ≥3 independent experiments. All statistical analyses were
performed using SPSS 13.0 (SPSS, Inc., Chicago, IL, USA).
Statistical differences between groups were assessed using a
one-way analysis of variance, followed by Dunnett's or Bonferroni's
multiple comparison tests. Correlation analysis was performed using
the Pearson's correlation. P<0.05 was considered to indicate a
statistically significant difference.
Results
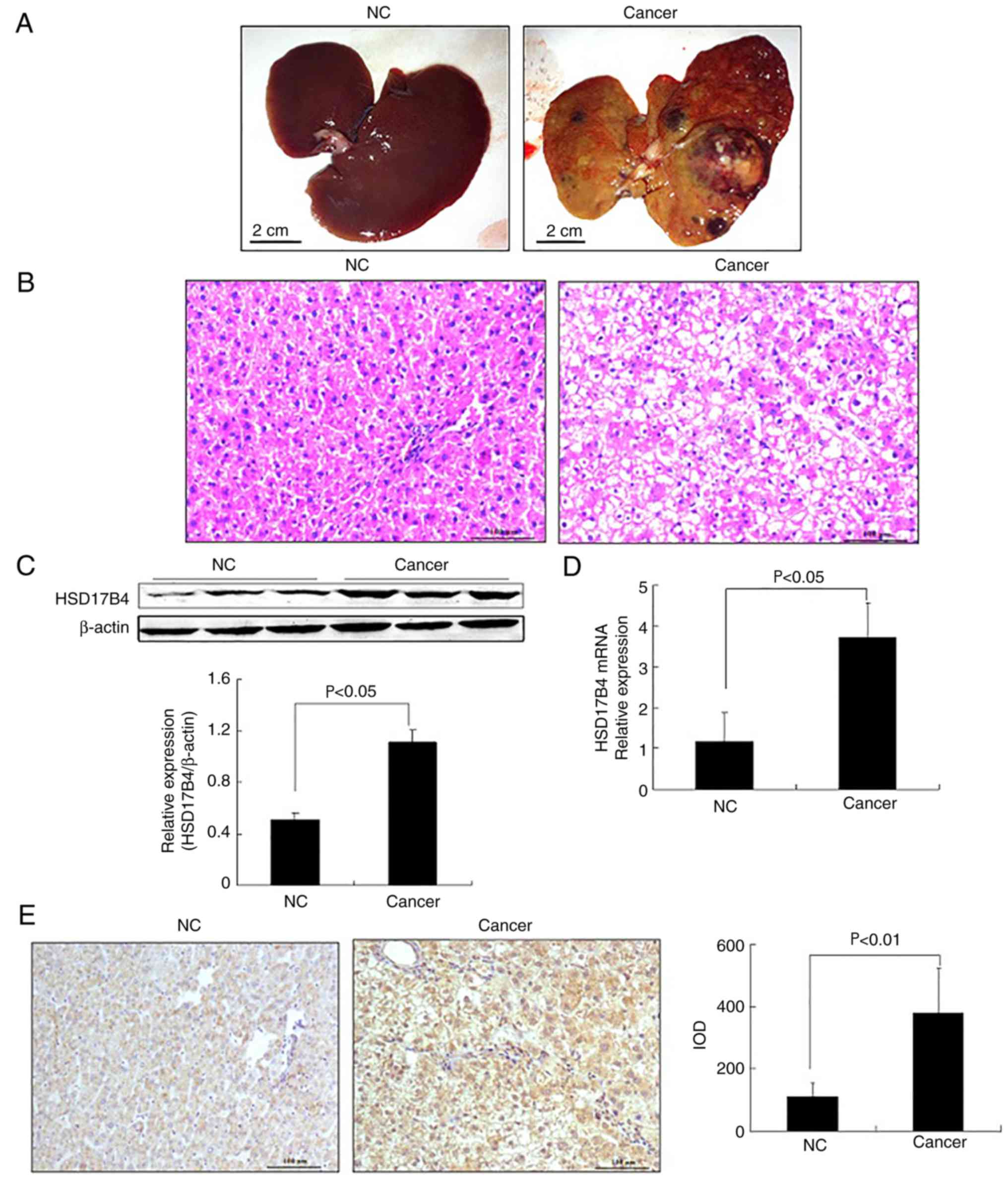
HSD17B4 expression is increased in
tissues from rats with liver cancer
To evaluate HSD17B4 expression during liver cancer
development, rats with DEN-induced liver cancer were investigated.
All rats in the Cancer group developed liver cancer while the
livers of rats in the NC group exhibited a normal lobular
architecture with central vein and radiating hepatic cords
(Fig. 1A and B). Hepatocytes were
polyhedral in shape and the cytoplasm was granulated with small
uniform nuclei. The Cancer group exhibited extensive cell swelling
and single cell necrosis; necrotic cells were small with basophilic
nuclei and dark cytoplasm (Fig.
1B). These results suggested that the rat liver cancer model
using DEN was successfully established.
HSD17B4 expression in the liver tissues of the rats
was determined. HSD17B4 protein and mRNA levels were significantly
increased in the Cancer compared with the NC group (P<0.05;
Fig. 1C and D). DEN administration
resulted in HSD17B4 accumulation in the cytoplasm of liver tissues
from rats with liver cancer (Fig.
1E). Compared with the NC, the IOD was significantly increased
in the Cancer group (P<0.01; Fig.
1E). The results indicated that HSD17B4 expression was
increased during liver cancer development.
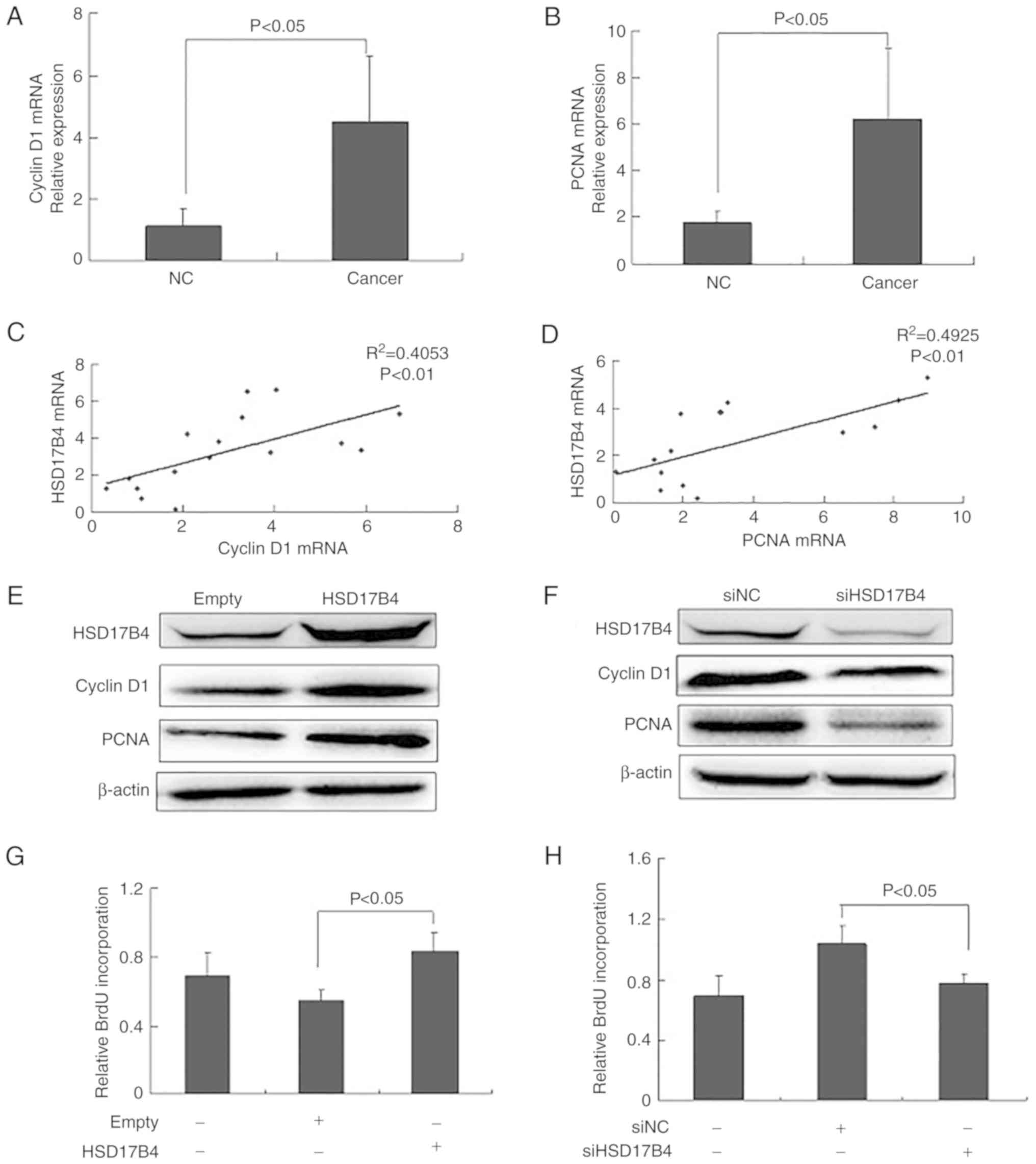
HSD17B4 enhances proliferation and
expression of proliferation-associated genes
To evaluate an association between HSD17B4
expression and proliferation-associated genes, mRNA levels of
cyclin D1 and PCNA were evaluated. It was observed that expression
was significantly increased in rats with liver cancer compared with
the NC group (P<0.05; Fig. 2A and
B). The correlation analysis between HSD17B4 and cyclin D1 or
PCNA mRNA expression revealed a positive correlation (P<0.01;
Fig. 2C and D).
Protein expression of HSD17B4, cyclin D1 and PCNA
was determined in HepG2 HSD17B4 overexpression and knockdown cells.
Compared with the empty vector, cyclin D1 and PCNA levels were
increased in the HSD17B4 overexpressing cells (Fig. 2E). In the siHSD17B4-treated cells
cyclin D1 and PCNA expression was downregulated compared with the
siNC (Fig. 2F). The results
indicated that HSD17B4 expression affected proliferation-associated
genes in HepG2 cells.
To further investigate the role of HSD17B4 in
proliferation, BrdU incorporation assays were performed with
HSD17B4 overexpression and knockdown HepG2 cells. Proliferation of
the HSD17B4 overexpressing cells was significantly increased
compared with the empty vector control (P<0.05; Fig. 2G) and BrdU incorporation was
significantly decreased in the siHSD17B4-treated cells compared
with the siNC (P<0.05; Fig. 2H).
The results indicated that HSD17B4 promoted HepG2 proliferation by
affecting proliferation-associated genes.
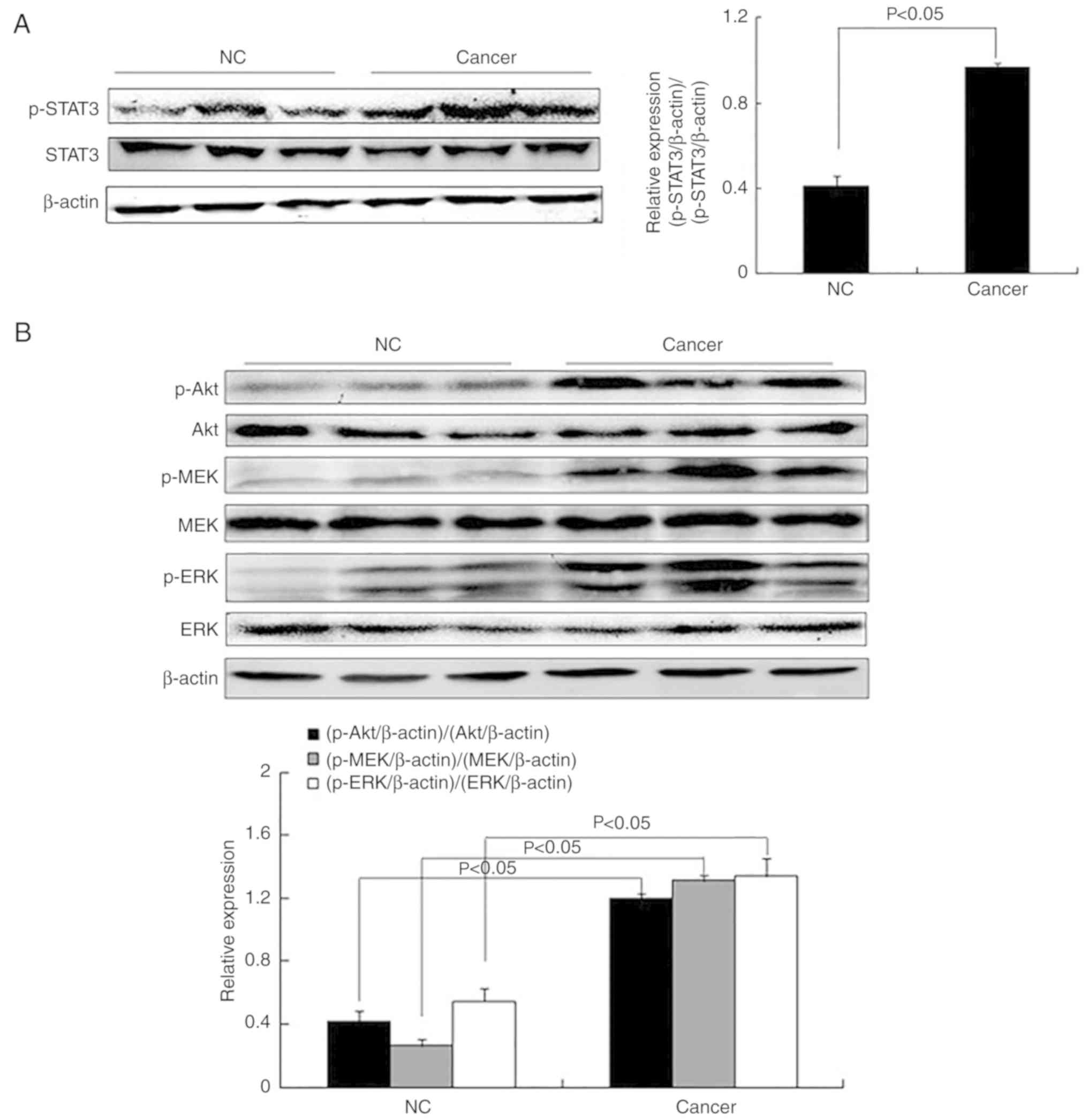
STAT3 activation is increased in liver
tissues from rats with liver cancer
STAT3 phosphorylation contributes to liver cancer
progression (5–7). STAT3 phosphorylation was significantly
increased in the Cancer compared with the NC group (P<0.05;
Fig. 3A). As STAT3 is involved in
various signaling pathways (20,21),
proteins associated with the Akt and the MEK/ERK signaling pathways
were investigated. Western blot analysis revealed that levels of
p-Akt, p-ERK and p-MEK were significantly increased in tissues from
the Cancer compared with the NC group (P<0.05; Fig. 3B).
HSD17B4 overexpression increases STAT3
activation
A recent study revealed that HSD17B4 is upregulated
in patients with liver cancer and HSD17B4 over-expression promotes
HepG2 proliferation (16). To
determine whether HSD17B4 serves a role in liver cancer
progression, HSD17B4 expression in adjacent and tumor tissues from
patients with liver cancer were evaluated. IHC images revealed that
HSD17B4 expression was increased in tumor tissues compared with
adjacent normal tissues obtained from patients with liver cancer
and the determined IOD was significantly increased in the cancerous
tissues (P<0.01; Fig. 4A).
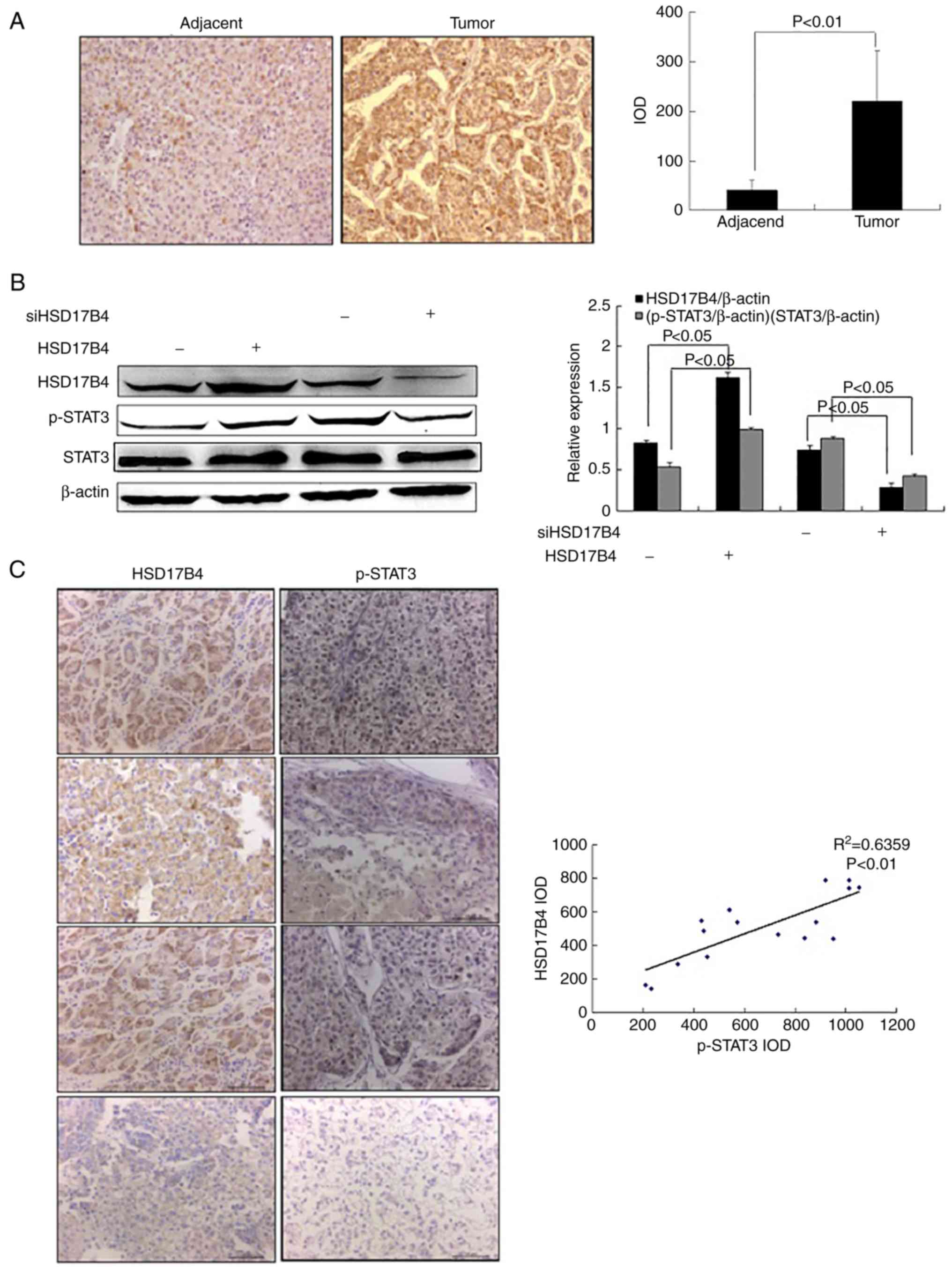 | Figure 4.HSD17B4 expression induces STAT3
activation. (A) IHC staining targeting HSD17B4 in liver tumor and
adjacent normal tissues from patients with liver cancer (n=16;
magnification, ×200). (B) STAT3 activation in HepG2 cells treated
with HSD17B4 overexpression vector or treated with siHSD17B4
determined by western blotting (n=3/group). (C) IHC analysis of
HSD17B4 and p-STAT3 levels in liver tumor and adjacent normal
tissues from patients with liver cancer (n=16; magnification, ×200)
and Pearson's correlation analysis of IODs. HSD17B4,
17β-hydroxysteroid dehydrogenase 4; IOD, integrated optical
density; si, small interfering RNA; p, phosphorylated; STAT3,
signal transducer and activator of transcription 3; NC, negative
control; -, transfected empty vector or siNC; +, transfected
HSD17B4 vector or siHSD17B4. |
An association between HSD17B4 expression and
activation of STAT3 was evaluated in HSD17B4 overexpression and
knockdown HepG2 cells. As presented in Fig. 4B, HSD17B4 overexpression
significantly increased STAT3 phosphorylation in HepG2 cells
compared with the empty vector group (P<0.05). In contrast,
p-STAT3 levels were significantly decreased in the
siHSD17B4-treated cells compared to the siNC group (P<0.05;
Fig. 4B).
To verify these finding, HSD17B4 and p-STAT3
expression in tumor and adjacent normal tissues of patients liver
cancer were evaluated using IHC. IODs determined for HSD17B4 and
p-STAT3 suggested a positive correlation between the HSD17B4
expression and STAT3 activation (P<0.01; Fig. 4C). The results suggested that
HSD17B4 expression induced STAT3 activation.
HSD17B4 expression induces Akt and
MEK/ERK activation
To determine the mechanism by which HSD17B4 promotes
STAT3 activation in liver cancer, activation of various signaling
pathways involved in liver cancer progression were assessed.
HSD17B4 overexpression increased the phosphorylation of Akt, MEK
and ERK in HepG2 cells compared with the empty vector group
(P<0.05; Fig. 5A). In
siHSD17B4-treated cells, Akt, MEK and ERK phosphorylation was
significantly decreased compared with the siNC group (P<0.05;
Fig. 5A). Activation of JNK and p38
were not affected by the induction or knockout of HSD17B4
expression (P>0.05; Fig. 5A).
The results suggested that activation of the Akt and the MEK/ERK
signaling pathways was associated with HSD17B expression.
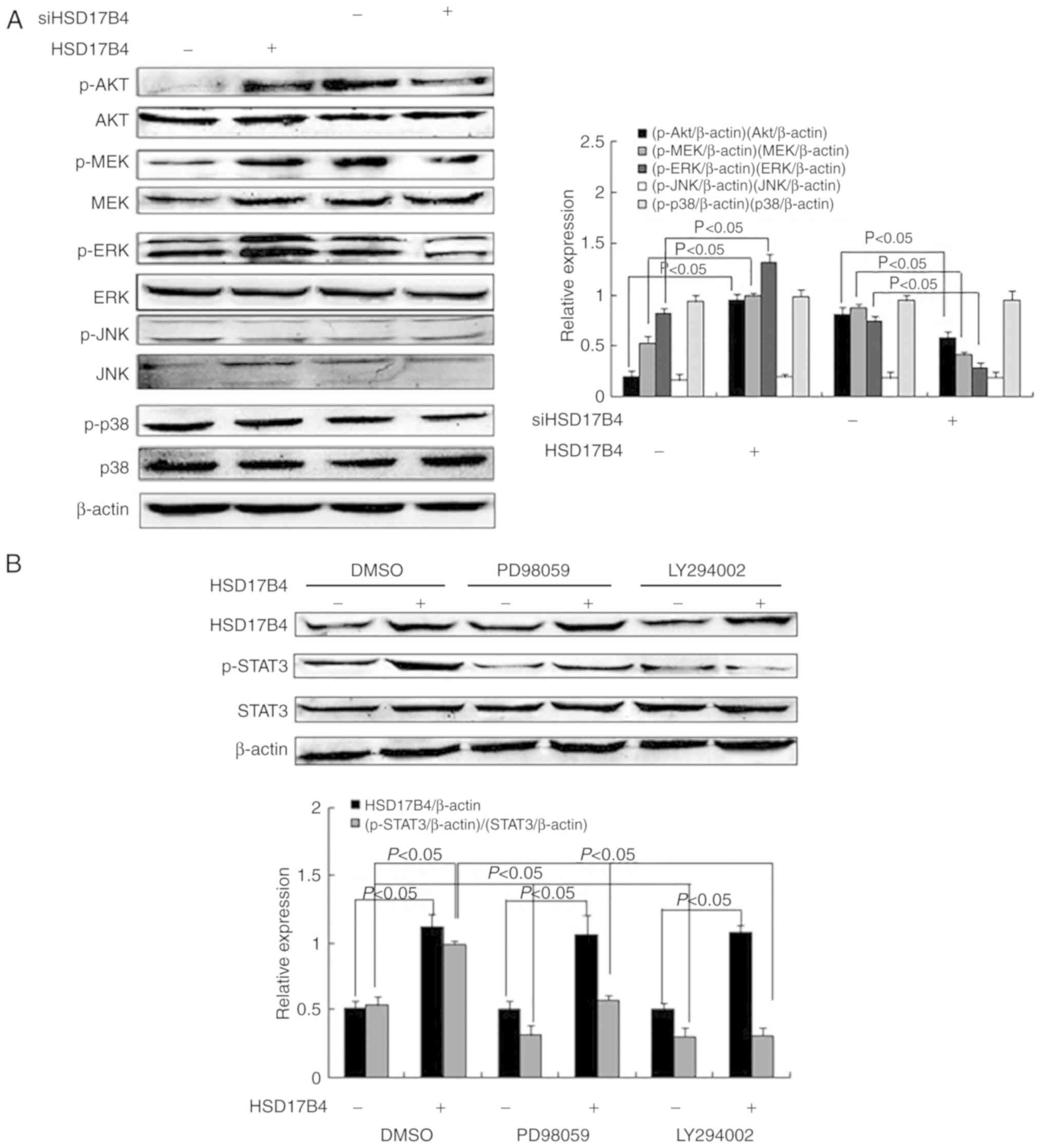 | Figure 5.HSD17B4 expression is associated the
Akt and the MEK/ERK signaling pathways. (A) Expression and
activation of the Akt, MEK, ERK, JNK and p38 in HepG2 cells treated
with HSD17B4 overexpression vector or treated with siHSD17B4
determined by western blotting (n=3/group). (B) HSD17B4
overexpressing HepG2 cells were pretreated with LY294002 and
PD98059 inhibitors and HSD17B4 expression and STAT3 activation were
assessed by western blotting (n=3/group). HSD17B4,
17β-hydroxysteroid dehydrogenase 4; si, small interfering RNA; p,
phosphorylated; STAT3, signal transducer and activator of
transcription 3; Akt, protein kinase B; MEK, mitogen-activated
protein kinase; ERK, extracellular-signal-regulated kinase; JNK,
c-Jun N-terminal kinase; LY294002, phosphoinositide 3-kinase
inhibitor; PD98059, MEK inhibitor; NC, negative control; -,
transfected empty vector or siNC; +, transfected HSD17B4 vector or
siHSD17B4. |
To investigate which signaling pathways are involved
in STAT3 activation, HepG2 cells were incubated with PD98059 and
LY294002, inhibitors of the MEK/ERK and the PI3K/Akt signaling
pathway, respectively, prior to transfection with the HSD17B4
overexpression plasmid (22). STAT3
phosphorylation was determined and it was observed that levels
significantly decreased in HSD17B4 overexpressing cells in the
presence of MEK/ERK and PI3K/Akt signaling pathway inhibitors
compared with the DMSO treated cells and a significant decrease was
further observed for empty vector-transfected cells in the presence
of inhibitors compared with the DMSO-treated cells (P<0.05;
Fig. 5B). The results suggested
HSD17B4 expression promoted STAT3 activation via the PI3K/Akt and
the MEK/ERK signaling pathways.
Discussion
As an important oxidoreductase, HSD17B4 is widely
distributed in peroxisomes of mammalian cells (11–13).
HSD17B4 is ubiquitous and increased levels were detected in
mammalian liver, heart, brain and prostate tissues under normal
physiological conditions (12,13).
Initial research focused on HSD17B4 function associated with the
inactivation of the estrogen metabolism (23,24).
It was further discovered that HSD17B4 is an important enzyme in
the fatty acid β-oxidation pathway in peroxisomes; it is involved
in the oxidative decomposition of very long-chain fatty acids and
branched-chain fatty acids and in the biosynthesis of
docosahexaenoic acid (25). An
increase of HSD17B4 was observed in a variety of tumor cells and
tissue suggesting that HSD17B4 may serve a role in tumor
development (26–28).
In a previous study, it was demonstrated that
HSD17B4 expressed is increased in patients with liver cancer
(16). In the current study, it was
observed that HSD17B4 expression was upregulated rats with liver
cancer compared with healthy control animals. It was revealed that
HSD17B4 overexpression and knockdown in HepG2 increased and
decreased expression of proliferation-associated genes,
respectively. The current study further revealed a positive
correlation between HSD17B4 and cyclin D1 and PCNA mRNA expression
in rats with liver cancer. The results suggested that HSD17B4 may
promote liver cancer proliferation and may serve a crucial role in
liver cancer development.
STAT3 is a nuclear transcription factor that binds
to specific sequences of target gene promoters (29,30)
and induces cancer cell proliferation by upregulating the
expression of various genes (4,31).
STAT3 promotes liver tumor formation by mediating multiple cellular
processes and enhancing the development of liver cancer (32,33).
With accumulating research, STAT3 has become an attractive target
for the treatment and prevention of human liver cancer (34,35).
The current study suggested that HSD17B4 upregulation mediated
STAT3 activation and a correlation was determined between these
factors in tumor and adjacent normal tissues from patients with
liver cancer.
STAT3 is active in liver tumor cells and is involved
in various signaling pathways (20,21).
STAT3 phosphorylation is involved the MEK and mitogen-activated
protein kinase kinase 4 signaling cascade and is induced
independently of ERK-1 or JNK-1 activity by interleukin-6 (36). Sorafenib inhibits liver cancer
growth by blocking the MEK/ERK/STAT3 and the PI3K/Akt/STAT3
signaling pathways, independent of Janus kinase 2 and
tyrosine-protein phosphatase non-receptor type 11 activation
(37). Various studies have
confirmed that the MEK/ERK/STAT3 and the PI3K/Akt/STAT3 signaling
pathways serve important roles in promoting liver cancer cell
proliferation (37,38). Experiments using HepG2 revealed that
HSD17B4 induced STAT3 phosphorylation through the MEK/ERK and the
PI3K/Akt signaling pathways without affecting the JNK and the p38
signaling pathways. The specific mechanism by which HSD17B4 affects
the signal pathways requires further investigation.
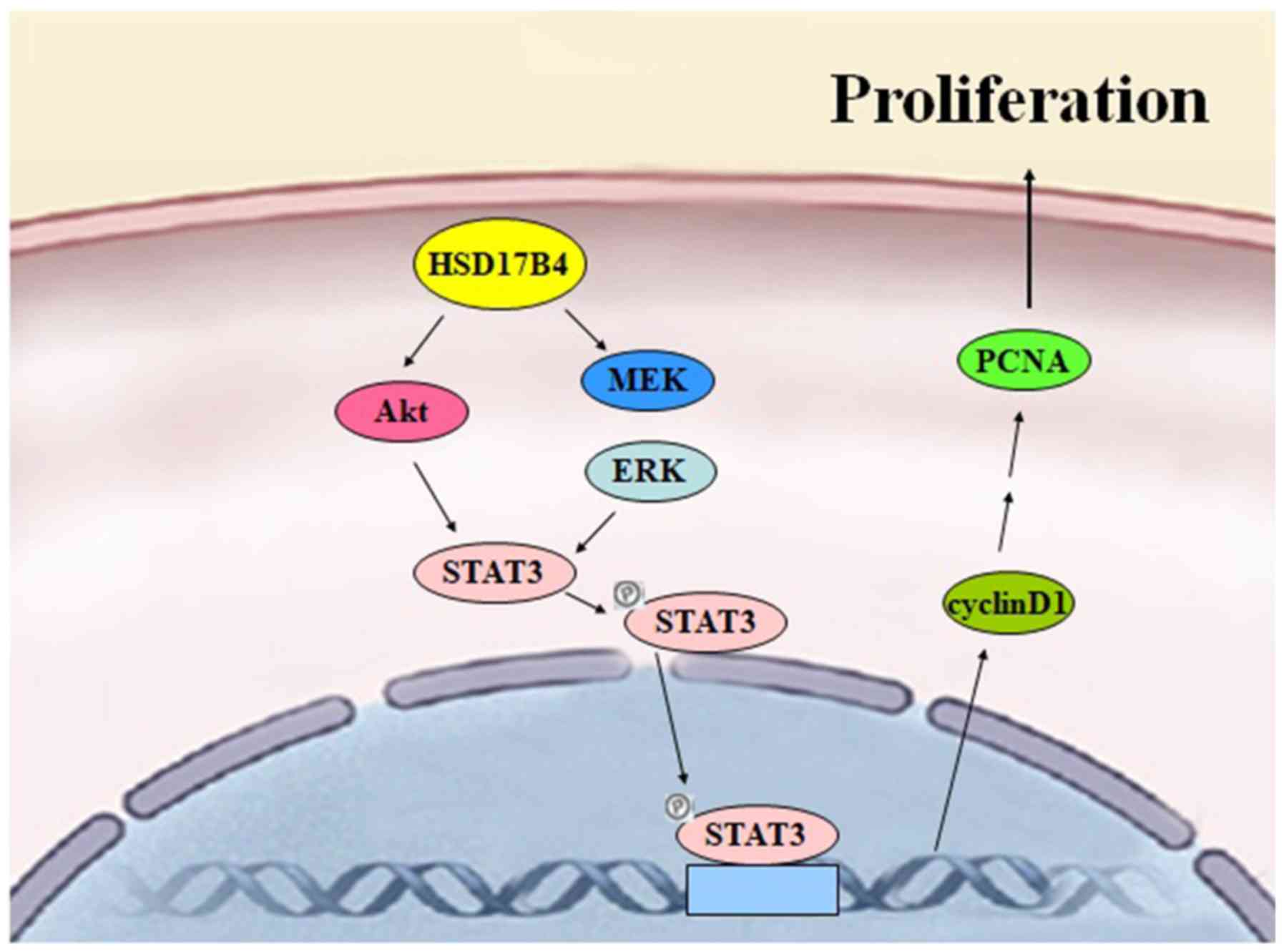
In conclusion, the data collected in the current
study indicated that HSD17B4 may be a novel proliferation-promoting
protein and the following mechanism is proposed: HSD17B4
overexpression promotes activation of STAT3 via the PI3K/Akt and
the MEK/ERK signaling pathways, which stimulate STAT3 binding to
the response element. In turn, cell proliferation is promoted via
cyclin D1 and PCNA upregulation (Fig.
6). The presented results may describe an experimental basis
for novel approaches in the prevention and treatment of liver
cancer using HSD17B4.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
XL and PM performed the majority of the experiments,
collected the clinical samples and acquired patients' information.
LK, XW and WL made substantial contributions to acquisition,
analysis and interpretation of data. LJ conceived, designed the
study and revised the manuscript. All authors read and approved the
final version of the manuscript.
Ethical approval and consent to
participate
Experiments involving human samples and animal
experiments were approved by the Ethics Committee of Hebei Medical
University (Shijiazhuang, China). All experiments were conducted
according to relevant national and international guidelines.
Written informed consent was obtained from all participants
included in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shukla SK, Singh G, Shahi KS, Bhuvan and
Pant P: Staging, treatment, and future approaches of gallbladder
carcinoma. J Gastrointest Cancer. 49:9–15. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cervello M, Augello G, Cusimano A, Emma
MR, Balasus D, Azzolina A, McCubrey JA and Montalto G: Pivotal
roles of glycogen synthase-3 in hepatocellular carcinoma. Adv Biol
Regul. 65:59–76. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kang KJ and Ahn KS: Anatomical resection
of hepatocellular carcinoma: A critical review of the procedure and
its benefits on survival. World J Gastroenterol. 23:1139–1146.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, Gupta SR, Tharakan ST, Koca C, Dey S and Sung B: Signal
transducer and activator of transcription-3, inflammation, and
cancer: how intimate is the relationship? Ann NY Acad Sci 1171.
59–76. 2009. View Article : Google Scholar
|
|
5
|
Calvisi DF, Ladu S, Gorden A, Farina M,
Conner EA, Lee JS, Factor VM and Thorgeirsson SS: Ubiquitous
activation of Ras and Jak/Stat pathways in human HCC.
Gastroenterology. 130:1117–1128. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barre B, Avril S and Coqueret O: Opposite
regulation of myc and p21waf1 transcription by STAT3 proteins. J
Biol Chem. 278:2990–2996. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang HY, Cheng Z and Malbon CC:
Overexpression of mitogen-activated protein kinase phosphatases
MKP1, MKP2 in human breast cancer. Cancer Lett. 191:229–237. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Johnson FM, Saigal B, Tran H and Donato
NJ: Abrogation of signal transducer and activator of transcription
3 reactivation after Src kinase inhibition results in synergistic
antitumor effects. Clin Cancer Res. 13:4233–4244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oh SB, Hwang CJ, Song SY, Jung YY, Yun HM,
Sok CH, Sung HC, Yi JM, Park DH, Ham YW, et al: Anti-cancer effect
of tectochrysin in NSCLC cells through overexpression of death
receptor and inactivation of STAT3. Cancer Lett. 353:95–103. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carie AE and Sebti SM: A chemical biology
approach identifies a beta-2 adrenergic receptor agonist that
causes human tumor regression by blocking the Raf-1/Mek-1/Erk1/2
pathway. Oncogene. 26:3777–3788. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moeller G and Adamski J:
Multifunctionality of human 17beta-hydroxysteroid dehydrogenases.
Mol Cell Endocrinol. 248:47–55. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peltoketo H, Luu-The V, Simard J and
Adamski J: 17beta-hydroxysteroid dehydrogenase (HSD)/17-ketosteroid
reductase (KSR) family; nomenclature and main characteristics of
the 17HSD/KSR enzymes. J Mol Endocrinol. 23:1–11. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Markus M, Husen B, Leenders F, Jungblut
PW, Hall PF and Adamski J: The organelles containing porcine 17
beta-estradiol dehydrogenase are peroxisomes. Eur J Cell Biol.
68:263–267. 1995.PubMed/NCBI
|
|
14
|
Rasiah KK, Gardiner-Garden M, Padilla EJ,
Möller G, Kench JG, Alles MC, Eggleton SA, Stricker PD, Adamski J
and Sutherland RL: HSD17B4 overexpression, an independent biomarker
of poor patient outcome in prostate cancer. Mol Cell Endocrinol.
301:89–96. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maleki J, Nourbakhsh M, Shabani M, Korani
M, Nourazarian SM, Ostadali DM and Moghadasi MH: 17β-estradiol
stimulates generation of reactive species oxygen and nitric oxide
in ovarian adenocarcinoma cells (OVCAR 3). Iran J Cancer Prev.
8:e23322015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu X, Ma P, Shi Y, Yao M, Hou L, Zhang P
and Jiang L: NF-κB increased expression of 17beta-hydroxysteroid
dehydrogenase 4 promotes HepG2 proliferation via inactivating
estradiol. Mol Cell Endocrinol. 401:1–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim NH, Heo JD, Kim TB, Rho JR, Yang MH
and Jeong EJ: Protective effects of ethyl acetate soluble fraction
of Limonium tetragonum on diethylnitrosamine-induced liver
fibrosis in rats. Biol Pharm Bull. 39:1022–1028. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bookout AL and Mangelsdorf DJ:
Quantitative real-time PCR protocol for analysis of nuclear
receptor signaling pathways. Nucl Recept Signal. 1:e0122003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fishbein MC, Wang T, Matijasevic M, Hong L
and Apple FS: Myocardial tissue troponins T and I. An
immunohistochemical study in experimental models of myocardial
ischemia. Cardiovasc Pathol. 12:65–71. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song H, Wang R, Wang S and Lin J: A
low-molecular-weight compound discovered through virtual database
screening inhibits Stat3 function in breast cancer cells. Proc Natl
Acad Sci USA. 102:4700–4705. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jia H, Li Y, Zhao T, Li X, Hu J, Yin D,
Guo B, Kopecko DJ, Zhao X, Zhang L, et al: Antitumor effects of
Stat3-siRNA and endostatin combined therapies, delivered by
attenuated Salmonella, on orthotopically implanted hepatocarcinoma.
Cancer Immunol Immunother. 61:1977–1987. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fang L, Li G, Liu G, Lee SW and Aaronson
SA: p53 induction of heparin-binding EGF-like growth factor
counteracts p53 growth suppression through activation of MAPK and
PI3K/Akt signaling cascades. EMBO J. 20:1931–1939. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Breitling R, Marijanovic Z, Perovic D and
Adamski J: Evolution of 17beta-HSD type 4, a multifunctional
protein of beta-oxidation. Mol Cell Endocrinol. 171:205–210. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
de Launoit Y and Adamski J: Unique
multifunctional HSD17B4 gene product: 17beta-hydroxysteroid
dehydrogenase 4 and D-3-hydroxyacyl-coenzyme A
dehydrogenase/hydratase involved in Zellweger syndrome. J Mol
Endocrinol. 22:227–240. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Veldhoven PP, Casteels M, Mannaerts GP and
Baes M: Further insights into peroxisomal lipid breakdown via
alpha- and beta-oxidation. Biochem Soc Trans. 29:292–298. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Romanuik TL, Wang G, Morozova O, Delaney
A, Marra MA and Sadar MD: LNCaP Atlas: Gene expression associated
with in vivo progression to castrationrecurrent prostate cancer.
BMC Med. Genomics. 3:432010.
|
|
27
|
True L, Coleman I, Hawley S, Huang CY,
Gifford D, Coleman R, Beer TM, Gelmann E, Datta M, Mostaghel E, et
al: A molecular correlate to the Gleason grading system for
prostate adenocarcinoma. Proc Natl Acad Sci USA. 103:10991–10996.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zha S, Ferdinandusse S, Hicks JL, Denis S,
Dunn TA, Wanders RJ, Luo J, De Marzo AM and Isaacs WB: 2005.
Peroxisomal branched chain fatty acid beta-oxidation pathway is
upregulated in prostate cancer. Prostate. 63:316–323. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ihle JN: STATs: Signal transducers and
activators of transcription. Cell. 84:331–334. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Costantino L and Barlocco D: STAT 3 as a
target for cancer drug discovery. Curr Med Chem. 15:834–843. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aggarwal BB, Sethi G, Ahn KS, Sandur SK,
Pandey MK, Kunnumakkara AB, Sung B and Ichikawa H: Targeting
signal- transducer-and-activator-of-transcription-3 for prevention
and therapy of cancer: Modern target but ancient solution. Ann NY
Acad Sci 1091. 151–169. 2006. View Article : Google Scholar
|
|
32
|
Subramaniam A, Shanmugam MK, Perumal E, Li
F, Nachiyappan A, Dai X, Swamy SN, Ahn KS, Kumar AP, Tan BK,
Perumal E, Chen L, Vali S, Abbasi T, Kapoor S, Ahn KS, Kumar AP, et
al: Potential role of signal transducer and activator of
transcription (STAT)3 signaling pathway in inflammation, survival,
proliferation and invasion of hepatocellular carcinoma. Biochim
Biophys Acta 1835. 46–60. 2013.
|
|
33
|
Subramaniam A, Shanmugam MK, Ong TH, Li F,
Perumal E, Chen L, Vali S, Abbasi T, Kapoor S, Ahn KS, et al:
Emodin inhibits growth and induces apoptosis in an orthotopic
hepatocellular carcinoma model by blocking activation of STAT3. Br
J Pharmacol. 170:807–821. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schindler C and Darnell JE Jr:
Transcriptional responses to polypeptide ligands: the JAK-STAT
pathway. Annu Rev Biochem. 64:621–651. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chiarle R, Simmons WJ, Cai H, Dhall G,
Zamo A, Raz R, Karras JG, Levy DE and Inghirami G: Stat3 is
required for ALK-mediated lymphomagenesis and provides a possible
therapeutic target. Nat Med. 11:623–629. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schuringa JJ, Jonk LJ, Dokter WH, Vellenga
E and Kruijer W: Interleukin-6-induced STAT3 transactivation and
Ser727 phosphorylation involves Vav, Rac-1 and the kinase
SEK-1/MKK-4 as signal transduction components. Biochem J 347 Pt.
1:89–96. 2000. View Article : Google Scholar
|
|
37
|
Gu FM, Li QL, Gao Q, Jiang JH, Huang XY,
Pan JF, Fan J and Zhou J: Sorafenib inhibits growth and metastasis
of hepatocellular carcinoma by blocking STAT3. World J
Gastroenterol. 17:3922–3932. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chiablaem K, Lirdprapamongkol K,
Keeratichamroen S, Surarit R and Svasti J: Curcumin suppresses
vasculogenic mimicry capacity of hepatocellular carcinoma cells
through STAT3 and PI3K/AKT inhibition. Anticancer Res.
34:1857–1864. 2014.PubMed/NCBI
|