Introduction
The phospholipid bilayer is the basic scaffold of
cell membranes, and inositol phospholipids serve an important role
in cell transmembrane signal transduction in eukaryotes (1). The abnormal expression or dysfunction
of inositol kinases and phosphatases are implicated in a number of
human diseases, including preeclampsia, inflammation and cancer
(1–4). As an inositol phosphatase, Src
homology 2 domain-containing inositol 5′-phosphatase (SHIP) is
encoded by the inositol polyphosphate-5-phosphatase D gene and was
identified in 1996 (5,6). SHIP1 is primarily expressed in the
hematopoietic system (7), where it
functions as a key regulator of immunoreceptor signaling (8), a negative controller of hematopoietic
progenitor cell proliferation (9)
and an inducer of cellular apoptosis (10). In recent years, the abnormal
expression or activity of SHIP1 has been demonstrated to be
involved in a variety of diseases, including Alzheimer's disease
(11), Crohn's disease (12), diabetic kidney disease (13) and hematological tumors (14).
As a hematopoietic-restricted phosphatidylinositol
phosphatase, SHIP1 serves a vital role in driving the occurrence
and development of hematological tumors (10,14).
SHIP1 downregulates PI3K-mediated signaling and suppresses cell
proliferation, differentiation, survival and migration of
hematopoietic cells (15). PI3K is
a central signal component for this major cell signaling pathway
and influences various cellular functions, including cell
proliferation, metastasis and apoptosis (16). Furthermore,
phosphatidylinositol-3,4,5-trisphosphate [PI(3,4,5)P3] is a key
PI3K-generated secondary messenger that is required for PI3K
pathway activity (17). Once
activated by extracellular stimuli, PI3K can quickly synthesize
PI(3,4,5)P3, PI(3,4,5)P3 is located on the plasma membrane and
directly binds and thereby recruits signaling proteins, including
protein kinase B (AKT) and phosphoinositide-dependent kinase 1
(PDK1) (16,18). The activation of these proteins
initiates more signaling cascades to induce various cellular
functions, including cell proliferation, migration and cell cycle
transition (19). SHIP antagonizes
PI3K activity by dephosphorylating the key PI3K-generated secondary
messenger PI(3,4,5)P3, resulting in the suppression of cell
proliferation, migration and cell cycle progression in
hematological tumors (15).
Notably, recent research on SHIP1 is primarily focused on
hematological tumors (7,9,10,14,15),
and there is limited knowledge regarding the role of SHIP1 in
various solid tumors, including lung cancer.
Lung cancer is one of the most common carcinomas
globally and is characterized by hidden onset, high malignancy and
potent invasion (20,21). Lung cancer represents the leading
cause of cancer-associated mortalities worldwide, according to the
epidemiological report published in 2018 (20). As the most common type of lung
cancer, non-small cell lung cancer (NSCLC) accounts for ~85% of all
lung cancer types (21). Emerging
evidence indicates that NSCLC formation and progression is a
multistage process involving the activation of proto-oncogenes and
the inactivation of tumor suppressor genes (21,22).
Thus, gene therapy and small molecule inhibitors targeting
epidermal growth factor receptor (EGFR) have become the most
prevalent approach for the treatment of NSCLC (22). However, although EGFR-targeted
therapy offers new hope to patients with NSCLC, drug resistance has
become increasingly prominent (22). Recent studies have identified a
number of mechanisms of resistance to EGFR-tyrosine kinase
inhibitors (EGFR-TKIs), including secondary EGFR mutations, the
activation of alternative signaling pathways (Met and hepatocyte
growth factor), the aberrant activation of downstream pathways,
including AKT mutations, and the loss of suppressors, such as
phosphatase and tensin homology (PTEN) (21–25).
PTEN and SHIP negatively regulate the EGFR-mediated PI3K pathway
(24,25), but the expression pattern and
functional mechanisms of SHIP1 in NSCLC have not been
investigated.
In the present study, it was demonstrated that SHIP1
expression is reduced in NSCLC and is associated with tumor
progression. However, no notable association with EGFR and Kirsten
rat sarcoma (KRAS) mutations was observed. Furthermore, in the
present study, it was determined that SHIP1 overexpression
suppresses cell proliferation, migration, invasion and
tumorigenicity via the PI3K/AKT pathway. These data provide novel
insights into the molecular function of SHIP1 in NSCLC and solid
tumors.
Materials and methods
Cell culture and sample
collection
The A549 and H1975 cell lines were obtained from the
American Type Culture Collection (Manassas, VA, USA), and the
SPCA-1 and 16HBE cell lines were purchased from the Chinese Academy
of Sciences Cell Bank (Shanghai, China). These cells were confirmed
to be free of mycoplasma with a Polymerase Chain Reaction (PCR) kit
(cat. no. Myco-P-50; Shanghai Inflammation XI Biological Technology
Co., Ltd., Shanghai, China) (http://biothrive2016.cnbio.net/) (26). A549 cells were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Shanghai ExCell Biology, Inc., Shanghai, China).
SPCA-1 and H1975 cells were cultured in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% FBS.
Additionally, 16HBE cells, an immortalized human bronchial
epithelial cell line, were grown in DMEM supplemented with 20% FBS.
All cells were incubated in a humidified chamber with 5%
CO2 at 37°C.
To examine the expression of SHIP1 mRNA in NSCLC
tissues, a total of 26 surgically-resected fresh primary lung
adenocarcinoma tissues and paired normal lung tissues were obtained
from the Third Affiliated Hospital of Kunming Medical University
(Kunming, China). Clinical protocols were approved by the Ethics
Committees of the Third Affiliated Hospital of Kunming Medical
University, and patients provided informed consent. The tissue
array included 146 NSCLC tissues (68 paraffin-embedded primary
adenocarcinoma specimens and 78 squamous carcinoma specimens) and
59 normal specimens, was obtained from Shanghai Outdo Biotech Co.,
Ltd. (Shanghai, China). Patients with a diagnosed relapse, as well
as those who received preoperative radiation, chemotherapy or
biotherapy were excluded. Demographic and clinical data were
obtained from the patients' medical records.
RNA isolation, reverse
transcription-PCR (RT-PCR), RT-quantitative PCR (RT-qPCR), and
primers
Analysis of relative gene expression data was
conducted using RT-qPCR and the 2−∆∆Cq method (27). Total RNA isolation, RT-PCR and
RT-qPCR were performed as described in our previous study (28). SHIP1-specific primers sequences were
as follows: Sense, 5′-TTTACGTGATCGGCACCCAA-3′; and antisense,
5′-GTGGCTGTTGACGAACCCTA-3′. ADP ribosylation factor 5 was used as
an internal control based on the following primers: Sense,
5′-ATCTGTTTCACAGTCTGGGACG-3′; and antisense,
5′-CCTGCTTGTTGGCAAATACC-3′. Experiments were performed according to
the instructions of a RT-qPCR kit (RR064A; Takara Bio, Inc., Otsu,
Japan). The RT-qPCR reactions for each sample were repeated thrice.
Independent experiments were performed in triplicate.
Western blot analysis, reagent and
antibodies
Western blot analysis was performed as previously
described (28), using: Anti-SHIP1
(cat. no. 19694-1-AP), α-tubulin (cat. no. 11224-1-AP; internal
control) and KRAS (cat. no. 12063-1-AP) antibodies (1:1,000;
ProteinTech Group, Inc., Chicago, IL, USA); anti-cyclin D1 (cat.
no. ab134175), cyclin dependent kinase 4 (CDK4; cat. no. ab108357),
CDK6 (cat. no. ab124821), EGFR (cat. no. ab52894), phospho(p)-EGFR
(cat. no. ab40815), AKT (cat. no. ab8805) and p-AKT (Ser473; cat.
no. ab18206) antibodies (1:1,000; Abcam, Cambridge, MA, USA); and
anti-Vimentin (cat. no. 5741), β-catenin (cat. no. 8480),
N-cadherin (cat. no. 13116) and E-cadherin (cat. no. 3195)
antibodies (1:1,000; Cell Signaling Technology, Inc., Danvers, MA,
USA). Horseradish peroxidase-conjugated anti-rabbit/mouse IgG
antibodies were used as the secondary antibodies (1:8,000; Cell
Signaling Technology, Inc.). Signals were detected using enhanced
chemiluminescence reagents (EMD Millipore, Billerica, MA, USA).
Recombinant human EGF (cat. no. PHG0311) was purchased from Gibco
(Thermo Fisher Scientific, Inc.) and dissolved in PBS and stored at
−20°C. The usage concentration of EGF was 100 ng/ml. LY294002, an
inhibitor of the PI3K/AKT pathway, which was used to inactivate the
PI3K/AKT signals in the present study, was purchased from Beyotime
Institute of Biotechnology (Haimen, China).
Inhibition of the PI3K/AKT
pathway
To analysis the effect of PI3K/AKT pathway on cell
proliferation and metastasis, LY294002 was added to DMEM at a final
concentration of 20 µM. Untreated A549 cells were taken as the
control group and the treated cells as the suppression group. After
culturing for 48 h in a humidified chamber with 5% CO2
at 37°C, the cell cycle- and EMT-associated factors were analyzed
by western blot analysis, according to the aforementioned
protocol.
Lentivirus and plasmid production,
infection and transient transfection
SHIP1 plasmid or control vectors containing green
fluorescence protein that could be packaged for recombinant
lentivirus were purchased from TranSheepBio (Shanghai, China).
Lentiviral particles carrying full-length hsa-SHIP1 vector and
their flanking control (NC) were constructed. Subsequently, A549
and SPCA-1 cells were infected with lentiviral vector as described
in our previous study (28). The
transient transfection of plasmid was performed as described
previously (26).
MTT assay
Cell proliferation was assessed using MTT as
previously described (29). The
cells were seeded at a density of 3×103 cells/well for 4
days. For each experimental condition, five parallel wells were
assigned to each group. Experiments were performed in
triplicate.
Cell cycle analysis
Cell cycle examination was performed as previously
described (30). NSCLC cells were
cultured for 24 h with serum-free DMEM. Subsequently, the SHIP1 and
control plasmids were separately transfected into NSCLC cells for
48 h, and the DNA content of labeled cells was measured using a BD
FACSCanto™ II Flow Cytometer (BD Biosciences; Beckon, Dickinson and
Company, Franklin Lakes, NJ, USA) and analyzed with ModFit LT
software version 3.2 (Verity Software House, Inc., Topsham, ME,
USA).
Cell migration and invasion
assays
In vitro cell migration and invasion assays
were examined according to our previous study (28). For Transwell assays,
1×105 cells in a 100 µl DMEM without serum were seeded
on a fibronectin-coated polycarbonate membrane insert in a
Transwell apparatus (Corning, Inc., Corning, NY, USA). In the lower
surface, 500 µl DMEM with 10% FBS was added as chemoattractant.
After the cells were incubated for 10 h at 37°C in a 5%
CO2 atmosphere, Giemsa stained cells adhering to the
lower surface were counted under an optical microscope (DMI 4000B;
Leica GmbH, Wetzlar, Germany) in five predetermined fields (×100
magnification). All assays were independently repeated at least
thrice. For Boyden assays, the procedure was similar to the cell
migration assay, except that the Transwell membranes were
pre-coated with 24 µg/ml Matrigel (R&D Systems, Inc.,
Minneapolis, MN, USA).
In vivo tumorigenesis and metastasis
assays
In vivo tumorigenesis in nude mice was
performed based on our previous study (28), and a total of 24 mice were purchased
from Experimental Animal Center of Kunming Medical University. Mice
were maintained in a barrier facility on high efficiency
particulate air-filtered racks, which is a specific pathogen-free
animal facility with a light/dark cycle of 12/12 h, temperature of
22±2°C, humidity of 50±10%, and had unlimited access to irradiated
food and sterilized water. D-Hank's solution was prepared with
sodium chloride (8 g), potassium chloride (0.4 g), potassium
dihydrogen phosphate (0.06 g), sodium bicarbonate (0.35 g) and
disodium hydrogen phosphate dodecahydrate (0.15 g) in 1,000 ml
distilled water. Subsequently, a total of 5×106
logarithmically-growing stable SHIP1-overexpressing or control A549
cells in 0.1 ml D-Hank's solution were subcutaneously inoculated
into the left-right symmetric flank of 4- to 6-week-old female
BALB/c-nu/nu mice (n=6/group). Tumor volumes were measured every 3
days. When the largest tumor diameter reached 1.5 cm or progressive
tumor growth was evident, the mice were sacrificed by cervical
dislocation and tumors were excised and weighed. All animal studies
were approved by the Animal Research and Care Committee of Kunming
Medical University (Kunming, China).
In vivo metastasis assays were performed
according to our previous study (31). A total of 5×106 stable
SHIP1-overexpressing or control A549 cells were injected under the
liver capsule of 4- to 6-week-old female BALB/c-nu/nu mice (n=6 for
SHIP1-overexpressing group or control group). The optical
fluorescence images (×1) were visualized to monitor primary tumor
growth and formation of metastatic lesions by Living Image Software
(version 2.50; PerkinElmer, Inc., Waltham, MA, USA). After 40 days,
all mice were sacrificed by cervical dislocation. Livers were
removed, and metastatic tissues were analyzed by hematoxylin and
eosin staining according to our previous study (28).
Immunohistochemistry
Immunohistochemistry was performed based on our
previous study (28). The tissues
were fixed in 10% formalin within 48 h at room temperature and
embedded in paraffin. Paraffin sections (4 µm) from samples were
deparaffinized in 100% xylene and rehydrated in descending ethanol
series (100, 100, 95, 85, 80 and 75%) and water. Heat-induced
antigen retrieval was performed in 10 mM citrate buffer (cat. no.
MVS-0100; Fuzhou Maixin Biotech Co., Ltd., Fuzhou, China) for 2 min
at 100°C. Endogenous peroxidase activity and non-specific antigens
were blocked with peroxidase blocking reagent containing 3%
hydrogen peroxide and goat serum (cat. no. SP KIT-B3; Fuzhou Maixin
Biotech Co., Ltd.), followed by incubation with a SHIP1 antibody
(1:500; ProteinTech Group, Inc.) overnight at 4°C. After washing 3
times, sections were incubated with undiluted rabbit secondary
antibodies from a Dako REAL EnVision detection system/Horseradish
Peroxidase for rabbit/mouse secondary antibodies kit (cat. no.
k5007; DAKO; Agilent Technologies, Inc., Santa Clara, CA, USA) for
30 min followed by streptavidin-conjugated horseradish peroxidase
for 30 min at 37°C. The peroxidase reaction was developed using
3,3-diaminobenzidine (DAB) chromogen solution with DAKO. Sections
were visualized with DAB, counterstained with hematoxylin for 2 min
at room temperature, mounted in neutral gum and analyzed using an
optical microscope (DMI 4000B; Leica GmbH).
Evaluation of staining
Stained tissue sections were reviewed and scored
independently by two investigators blinded to the clinical data.
The score was based on the sum of staining intensity and the
percentage of stained cells. The staining intensity was scored as
previously described (0–3) (28),
and the percentage of positive staining areas of cells was defined
as a scale of 0–3 (0, <10%; 1, 10–25%; 2, 26–75%; and 3,
>76%). The sum of the staining intensity and staining extent
scores (0–6) was used as the final staining score. For statistical
analysis, a final staining score of 0–2 and 3–6 were considered to
be negative and positive expression levels, respectively.
Use of database
To assess the expression of SHIP1 in NSCLC,
Affymetrix HG-U133_Plus_2 array data were extracted from Gene
Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/gds/?term). The GSE19188
dataset (32) was included in the
present study and the genome-wide gene expression analysis from 91
NSCLC and 65 normal lung tissue samples were extracted. To analyze
the association between SHIP1 expression and EGFR/KRAS mutations in
NSCLC, the GSE75037 dataset (33)
which used Illumina WG6-V3 expression arrays to profile the gene
expression signature in 83 lung adenocarcinomas and 83 matched
adjacent non-malignant lung tissues, was included in the present
study. To investigate the prognostic effect of SHIP1 on patients
with NSCLC, the Kaplan-Meier Plotter database (http://kmplot.com/analysis/index.php?cancer=lung&p=service)
was use to predict the overall survival rates. To analyze whether
low SHIP1 expression results from SHIP1 promoter methylation, the
EMBOSS Cpgplot (http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/)
was use to predict the CpG islands in the SHIP1 promoter.
Statistical analysis
All data were independently repeated in triplicate.
SPSS 13.0 (SPSS, Inc., Chicago, IL, USA) and Graph Pad Prism 5.0
software (GraphPad Software, Inc., La Jolla, CA, USA) were used for
statistical analysis. Data are expressed as the mean ± standard
deviation from at least 3 independent experiments. Student's t-test
was employed for analysis between two groups, one-way analysis of
variance with post hoc contrasts by Dunnett's test was employed for
analysis between 3 groups, and a parametric generalized linear
model with random effects was employed for analysis of tumor growth
and MTT assay results. Analysis of SHIP1 expression in 26 fresh
primary NSCLC tissues and 83 lung adenocarcinoma tissues (GSE75037)
were performed using a paired-samples Student's t-test. The
χ2 test was used to determine the differences in SHIP1
protein expression between NSCLC tissues and non-cancerous lung
tissues. Survival analysis was performed using the Kaplan-Meier
method with the log-rank test. A multivariate Cox proportional
hazards method was used to analyze the association between the
variables and patient's survival time. P<0.05 was considered to
indicate a statistically significant difference.
Results
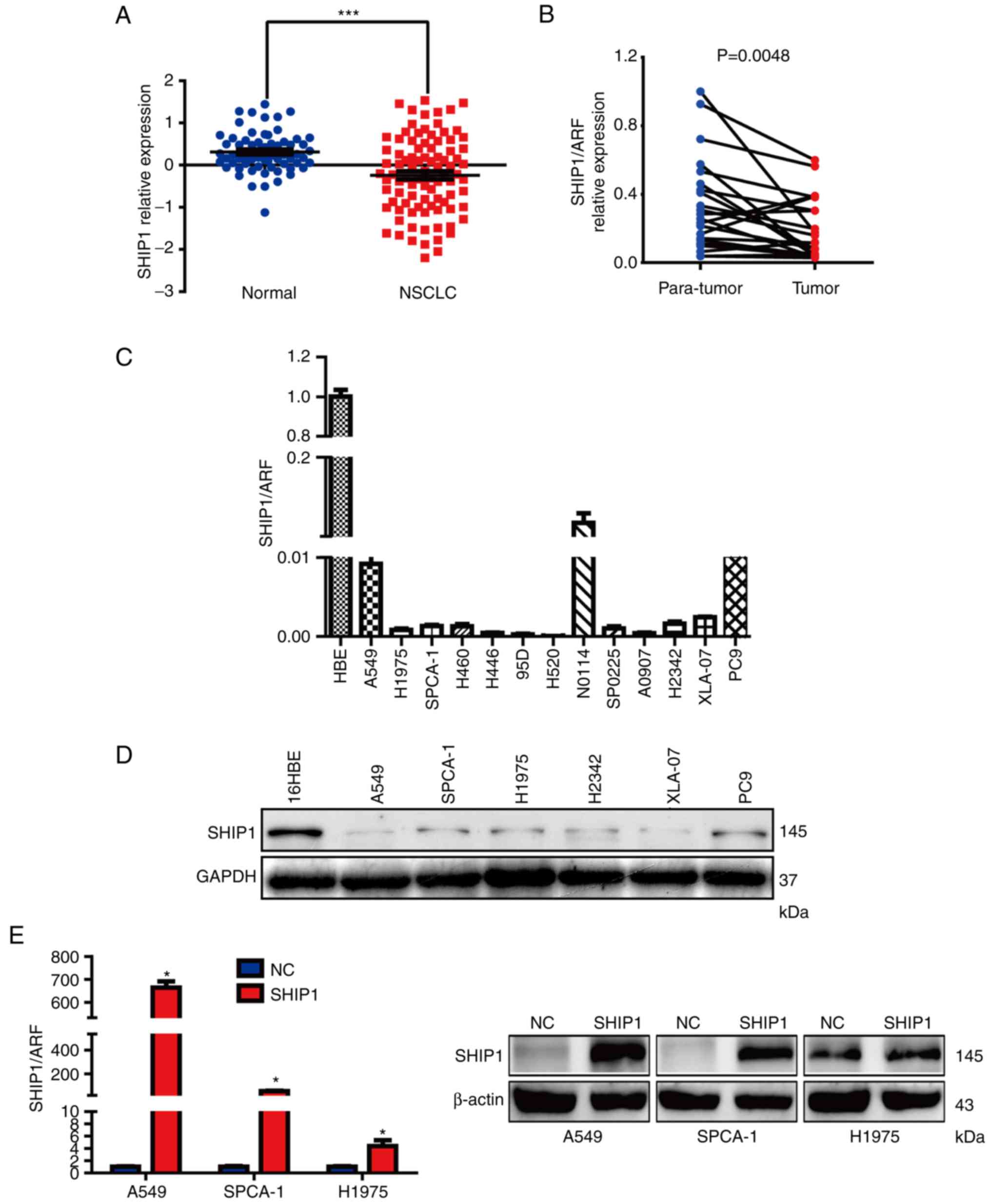
SHIP1 is downregulated in NSCLC
To assess the expression of SHIP1 in NSCLC,
Affymetrix HG-U133_Plus_2 array data were extracted from the GEO
GSE19188 dataset. The genome-wide gene expression analysis from 91
NSCLC and 65 that normal lung tissue samples was included in the
GSE19188 dataset. SHIP1 expression was analyzed in these samples
and it was determined that SHIP1 expression was significantly
reduced in NSCLC samples, compared with normal lung tissues
(Fig. 1A). Furthermore, RT-qPCR was
used to detect SHIP1 mRNA levels in 26 fresh primary NSCLC and
paired adjacent normal lung tissues. Consistently, mean SHIP1 mRNA
levels were significantly reduced in NSCLC tissues, compared with
the paired adjacent normal lung tissues (Fig. 1B). A panel of human NSCLC cell lines
was also analyzed for SHIP1 expression. Compared with the
immortalized human bronchial epithelial cell line 16HBE, SHIP1 mRNA
and protein expression were notably reduced in NSCLC cell lines
(Fig. 1C and D).
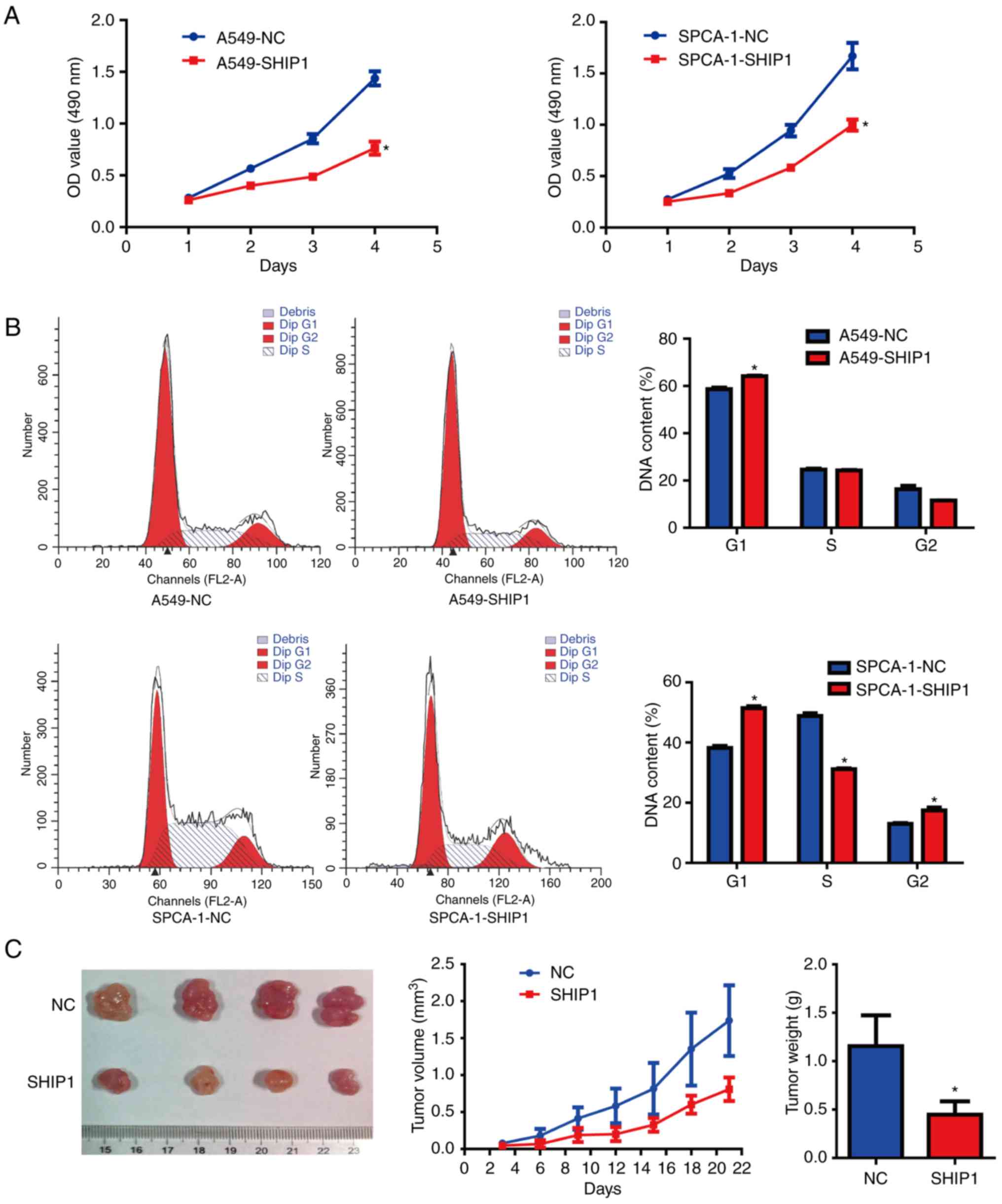
SHIP1 inhibits NSCLC cell
proliferation in vitro and in vivo
To assess SHIP1's biological functions in NSCLC, a
SHIP1 inducible plasmid was used to upregulate SHIP1 expression in
3 NSCLC cell lines, and the results demonstrated that SHIP1 mRNA
and protein were notably overexpressed in A549 and SPCA-1 cells,
but in H1975 cells, only SHIP1 mRNA was notably overexpressed
(Fig. 1E); thus,
SHIP1-overexpressing A549 and SPCA-1 cells were the appropriate
cell models for the following experiments. Cell growth was assessed
using MTT assays. These assays demonstrated that the induction of
SHIP1 expression from the plasmid significantly inhibited cell
viability, compared with cells transfected with control plasmid
(Fig. 2A). To clarify the influence
of SHIP1 on cell cycle progression, NSCLC cells were grown for 24 h
in serum-free DMEM medium. Subsequently, SHIP1 and control plasmids
were separately transfected into NSCLC cells for 48 h. Flow
cytometry was used to detect DNA content and demonstrated that the
G1/S cell cycle phase transition was significantly inhibited by
SHIP1 overexpression in A549 and SPCA-1 cells (Fig. 2B). To determine the effect of SHIP1
on the growth of NSCLC cells in vivo, an in vivo
tumorigenesis study was established by inoculating stable
SHIP1-overexpressing or control A549 cells into nude mice. Tumor
volumes were measured every 3 days, and the growth curves were
generated to assess tumor growth rates. A total of 3 weeks after
injection, the mice were sacrificed and the tumors were weighed.
The tumor growth curves revealed that the tumor growth rate of the
SHIP1-overexpression group was notably reduced, compared with the
control group. Furthermore, the mean tumor weights of the
SHIP1-overexpression group were significantly reduced, compared
with the control group (1.156±0.112 vs. 0.448±0.122 g; Fig. 2C). These results supported the
hypothesis that SHIP1 significantly inhibits cell growth in
vitro and in vivo.
SHIP1 inhibits NSCLC cell metastasis
in vitro and in vivo
To assess the metastasis effect of SHIP1 in NSCLC
cells, the Transwell and Boyden chamber assays were used.
SHIP1-overexpressing cells and control cells were cultured in a
Transwell apparatus or Boyden chambers coated with Matrigel for 10
h. Subsequently, migrated cells were counted, and the number of
migrating cells in the SHIP1-overexpressing group was significantly
reduced, compared with the control group (Fig. 3A and B). To further assess the
effect of SHIP1 on NSCLC metastasis in vivo, intrahepatic
metastasis assays were performed by inoculating stable
SHIP1-overexpression or control A549 cells under the liver capsule
of mice, and fluorescent image detection was used to confirm
intra-hepatic tumor dissemination. Additionally, intra-hepatic
dissemination in the SHIP1-overexpressing group was notably
reduced, compared with the control group (Fig. 3C). The aforementioned results
indicate that SHIP1 inhibits NSCLC cell metastasis in vitro
and in vivo.
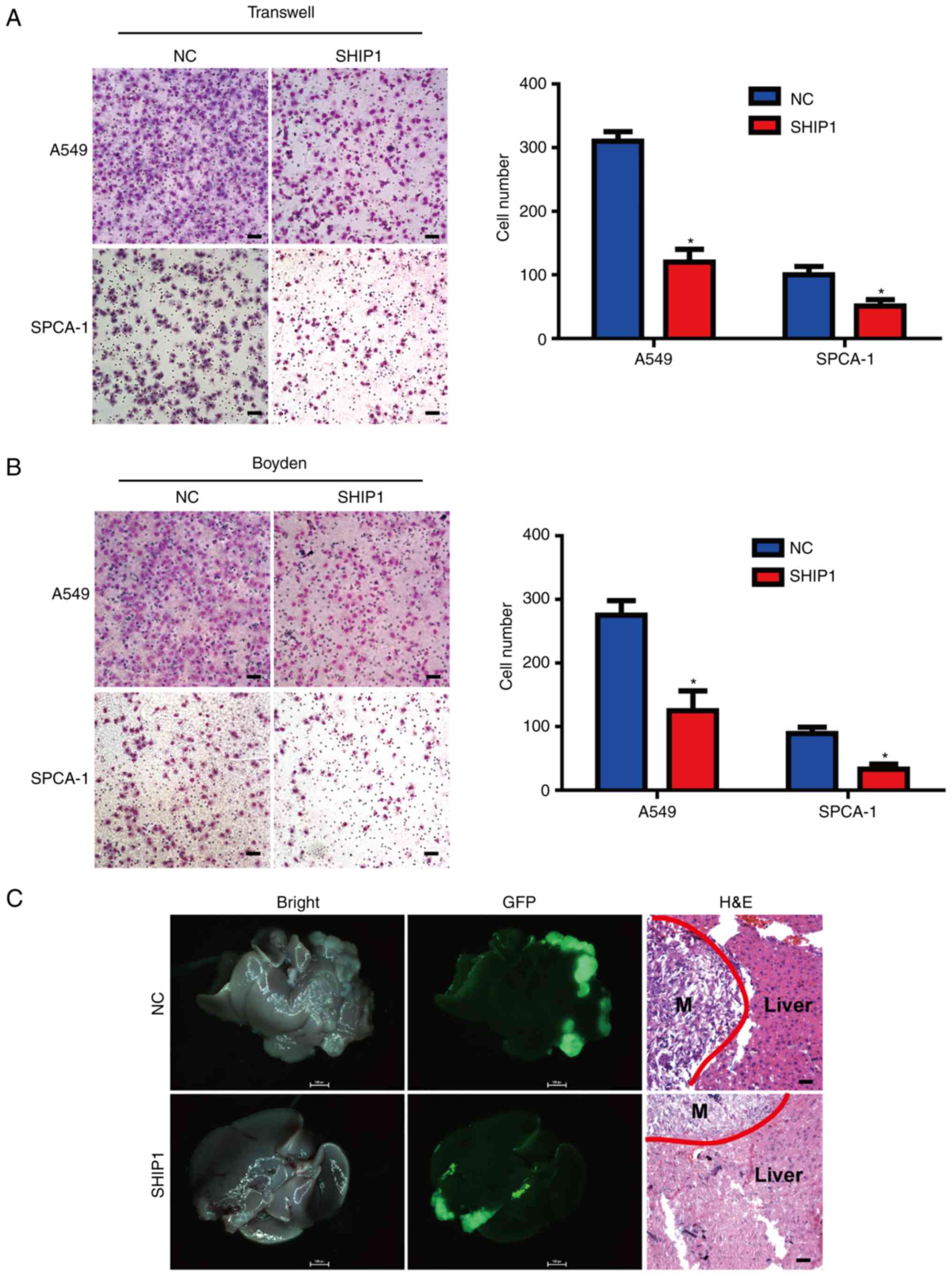 | Figure 3.SHIP1 inhibits NSCLC cell metastasis
in vitro and in vivo. (A) SHIP1 upregulation reduced
NSCLC cell migration in vitro by Transwell assay. Student's
t-test, *P<0.05. (B) SHIP1 upregulation reduced NSCLC cell
invasion in vitro by Boyden assay. Student's t-test,
*P<0.05. (C) Intrahepatic metastasis assays were performed by
inoculating stable SHIP1-overexpressing or control A549 cells under
the liver capsule of mice, and external optical fluorescence images
of liver were obtained 40 days after injection. The livers were
observed under a microscope (×1 magnification). Representative
images of H&E staining of metastatic cancer tissues are
presented. Scale bars, 25 µm. Data are presented as the mean ±
standard deviation for 3 independent experiments. M, metastatic
cancer tissue; H&E, hematoxylin and eosin; SHIP1, Src homology
2-containing inositol-5′-phosphatase 1; NSCLC, non-small cell lung
cancer; NC, control; GFP, green fluorescence protein. |
SHIP1 suppresses PI3K/AKT-mediated
cell cycle and epithelial-mesenchymal transition (EMT) signals
To further investigate the mechanism by which SHIP1
functions as a tumor suppressor in NSCLC, the protein levels of
cell cycle- and EMT-associated genes were examined in
SHIP1-overexpression and control cells. Cyclin D1 and CDK4/6 levels
were reduced in SHIP1-overexpression cells, indicating that SHIP1
suppresses the association between cyclin D1 and CDK4/6 (Fig. 4A). The EMT-associated protein
E-cadherin was increased in SHIP1-overexpression cells, compared
with control cells, whereas N-cadherin and Vimentin levels were
downregulated by SHIP1. These results indicate that SHIP1 regulates
NSCLC metastasis via EMT-associated proteins (Fig. 4B). Further pathway analysis revealed
that SHIP1 overexpression notably reduced β-catenin and p-AKT
levels but not AKT, KRAS, EGFR and p-EGFR levels (Fig. 4C), indicating that as an antagonizer
of PI3K activity, SHIP1 suppresses PI3K-mediated downstream
pathways. Furthermore, β-catenin, cyclin D1 and CDK6 levels were
gradually restored when using EGF to activate the PI3K/AKT pathway
in SHIP1-overexpressing cells (Fig.
4D). Subsequently, LY294002, an inhibitor of the PI3K/AKT
pathway, was used to inactivate the PI3K/AKT pathway in A549 cells
and detect the levels of its downstream cell cycle- and
EMT-associated factors. The results demonstrated that β-catenin,
cyclin D1, and CDK6 levels were decreased, but the E-cadherin level
was increased (Fig. 4E).
Collectively, these results indicate that SHIP1 suppresses
PI3K/AKT-mediated cell cycle and EMT signals to inhibit cell
proliferation and metastasis in NSCLC.
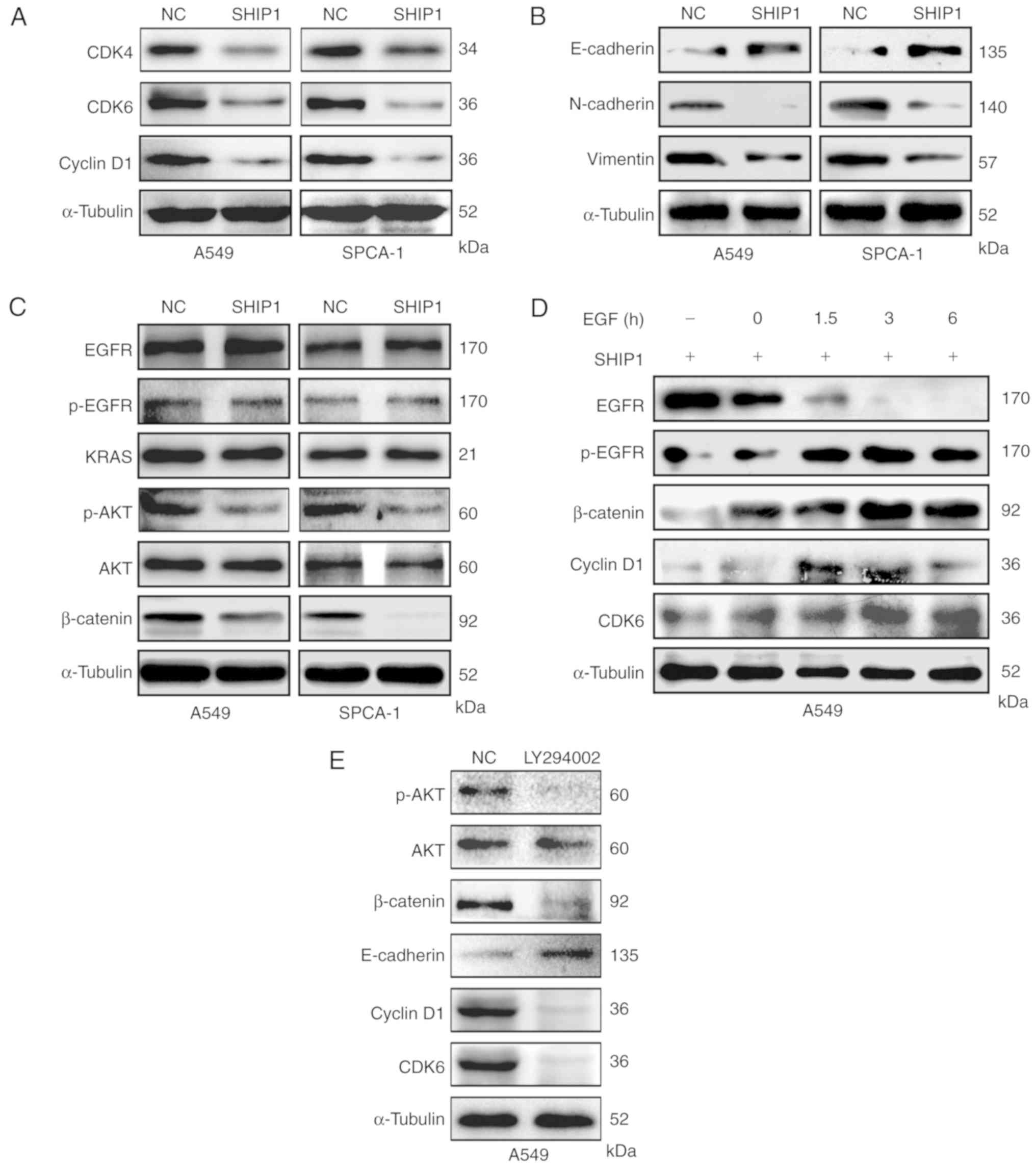 | Figure 4.SHIP1 suppresses PI3K/AKT-mediated
cell cycle and EMT signals. (A) Expression of the cell
cycle-associated proteins cyclin D1, CDK4 and CDK6 in
SHIP1-overexpressing NSCLC and control cells. (B) SHIP1
upregulation reduced the expression of EMT-associated proteins,
including Vimentin and N-cadherin, and increased the expression of
E-cadherin. (C) In NSCLC cells, upregulated SHIP1 inhibited
β-catenin and p-AKT levels, but not EGFR, p-EGFR and KRAS levels.
(D) SHIP1-overexpressing A549 cells were treated with EGF (100
ng/ml) for different times to activate EGFR phosphorylation, and
cellular EGFR, p-EGFR, β-catenin, cyclin D1 and CDK6 levels were
assessed by western blot analysis. (E) A549 cells were treated with
LY294002 to inactivate the PI3K/AKT pathway, and cellular
β-catenin, cyclin D1, CDK6 and E-cadherin levels were assessed by
western blot analysis. α-Tubulin served as an internal control. All
of the experiments were repeated at least thrice. CDK, cyclin
dependent kinase; p-, phospho-; EMT, epithelial-mesenchymal
transition; PI3K, phosphoinositide 3-kinase; SHIP1, Src homology
2-containing inositol-5′-phosphatase 1; NSCLC, non-small cell lung
cancer; NC, control; EGFR, epidermal growth factor receptor. |
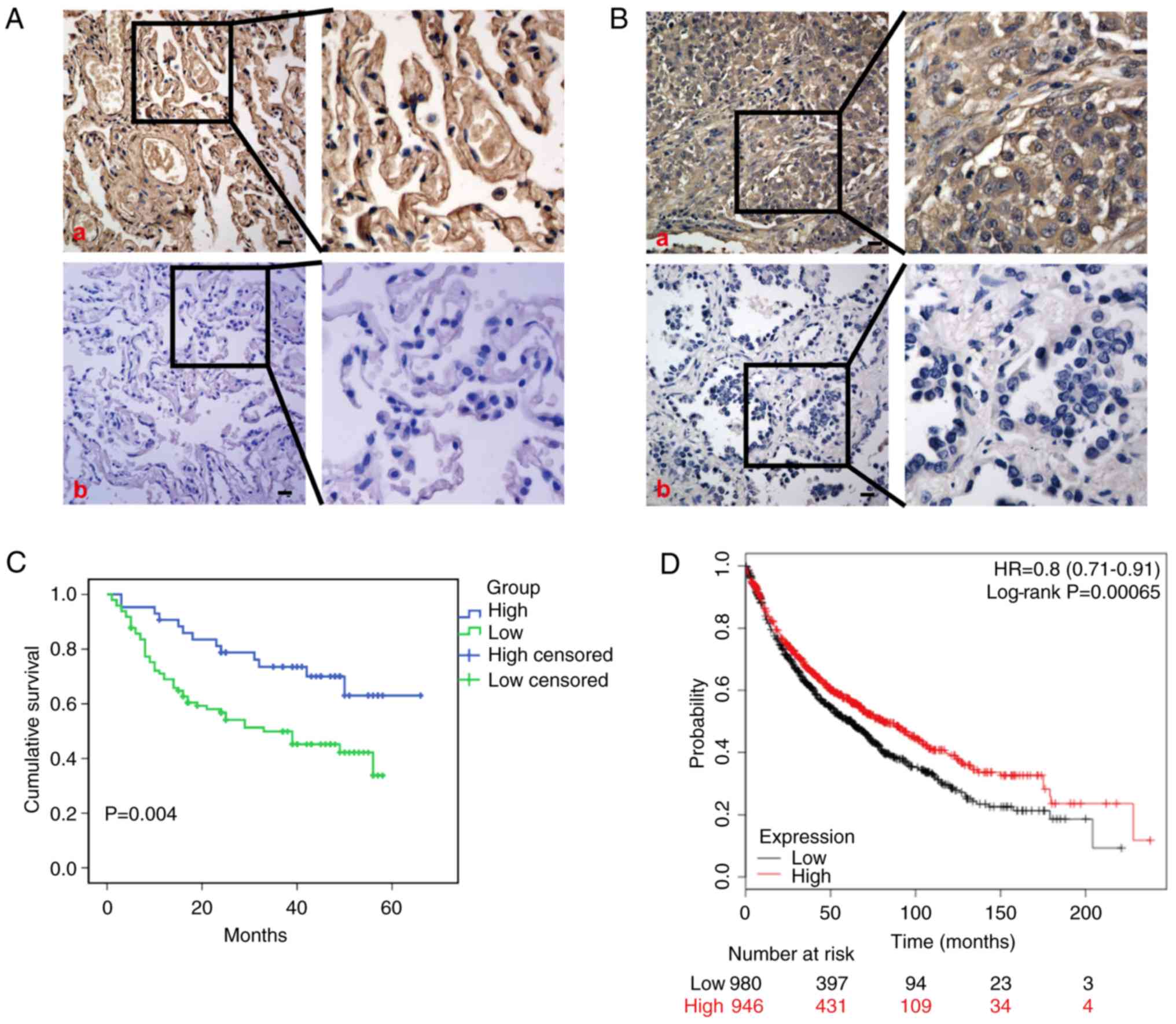
Pathoclinical characteristics of SHIP1
expression in NSCLC
Immunohistochemistry was performed to examine SHIP1
protein expression in 146 NSCLC tissues and 59 non-cancerous lung
tissues, and significant downregulation of SHIP1 was observed in
NSCLC tissues, compared with non-cancerous lung tissues (Fig. 5A and B; Table I). Clinical characteristics
associated with SHIP1 were analyzed, revealing that low SHIP1
expression was significantly negatively associated with American
Joint Committee on Cancer (AJCC) clinical stage (AJCC 8th stage
system) (34) (P=0.009), T
classification (P=0.028), and N classification (P=0.043), but no
other clinical features (Table
II). Kaplan-Meier survival analysis revealed that patients with
NSCLC with low SHIP1 expression exhibited significantly reduced
overall survival rates (Fig. 5C).
Consistently, the prediction analysis of microarrays and
constructed Kaplan-Meier plots from Kaplan-Meier Plotter database
(http://kmplot.com/analysis/index.php?cancer=lung&p=service)
also revealed similar overall survival rates (Fig. 5D).
 | Table I.The expression of SHIP1 in non-small
cell lung cancer, compared with non-cancerous lung tissues. |
Table I.
The expression of SHIP1 in non-small
cell lung cancer, compared with non-cancerous lung tissues.
|
|
| SHIP1
expression |
|
|---|
|
|
|
|
|
|---|
| Group | Cases (n) | Negative (%) | Positive (%) | P-value |
|---|
| Cancer | 146 | 101 (69.2) | 45 (30.8) | <0.001 |
| Non-cancerous | 59 | 11 (18.6) | 48 (81.4) |
|
| Table II.Association between the
clinicopathologic factors and expression of SHIP1 in non-small cell
lung cancer. |
Table II.
Association between the
clinicopathologic factors and expression of SHIP1 in non-small cell
lung cancer.
|
|
| SHIP1
expression |
|
|---|
|
|
|
|
|
|---|
| Factors | n | Negative (n) | Positive (n) | P-value |
|---|
| Sex |
|
|
| 0.661 |
|
Male | 101 | 71 (70.3%) | 30 (29.7%) |
|
|
Female | 45 | 30 (66.7%) | 15 (33.3%) |
|
| Age (years) |
|
|
| 0.512 |
|
≤60 | 71 | 48 (67.6%) | 23 (32.4%) |
|
|
>60 | 73 | 53 (72.6%) | 20 (27.4%) |
|
|
Unknown |
2 | 0 | 2 |
|
| Pathology |
|
|
| 0.708 |
|
Squamous carcinoma | 78 | 55 (70.5%) | 23 (29.5%) |
|
|
Adenocarcinoma | 68 | 46 (67.6%) | 22 (32.4%) |
|
| T
classificationb |
|
|
| 0.028a |
|
T1 | 23 | 11 (47.8%) | 12 (52.2%) |
|
|
T2 | 103 | 72 (69.9%) | 31 (30.1%) |
|
|
T3 | 18 | 16 (88.9%) | 2 (11.1%) |
|
|
T4 |
2 | 2 (100%) | 0 |
|
| N
classificationb |
|
|
| 0.043a |
|
N0 | 63 | 38 (60.3%) | 25 (39.7%) |
|
|
N1+N2+N3 | 83 | 63 (75.9%) | 20 (24.1%) |
|
| Clinical
stageb |
|
|
| 0.009a |
| I | 33 | 18 (54.5%) | 15 (45.5%) |
|
| II | 88 | 60 (68.2%) | 28 (31.8%) |
|
|
III | 25 | 23 (92%) | 2 (8%) |
|
Further univariate and multivariate Cox regression
analyses were performed to assess independent prognostic factors in
patients with NSCLC. It was observed that SHIP1 expression levels
and AJCC stage were significantly associated with overall survival
rate. Patients with low SHIP1 levels exhibited significantly
reduced survival, compared with patients with high levels, and
patients with advanced AJCC stage exhibited significantly reduced
survival, compared with those with early stage disease (Table III). Collectively, these
observations indicate that low SHIP1 expression is a negative
prognostic factor in NSCLC.
| Table III.The univariate and multivariate Cox
regression analysis of overall survival time. |
Table III.
The univariate and multivariate Cox
regression analysis of overall survival time.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Factors | P-value | HR | 95% CI | P-value | HR | 95% CI |
|---|
| Sex |
| Male
vs. female | 0.407 | 1.322 | 0.683–2.557 | – | – | – |
| Age (years) |
| ≤60 vs.
>60 | 0.546 | 1.172 | 0.701–1.960 | – | – | – |
| T
classificationb |
|
T1+T2
vs. T3+T4 | 0.789 | 0.923 | 0.511–1.665 | – | – | – |
| N
classificationb |
|
N0 vs.
N1+N2+N3 | 0.923 | 1.022 | 0.655–1.595 | – | – | – |
|
Pathologyb |
|
Squamous carcinoma vs.
adenocarcinoma |
<0.001a | 0.313 | 0.167–0.586 |
<0.001a | 0.345 | 0.196–0.606 |
| Stageb |
| I+II
vs. III |
<0.001a | 3.692 | 1.783–7.644 |
<0.001a | 3.515 | 2.176–5.678 |
| SHIP1
expression |
|
Positive vs. negative | 0.008a | 2.361 | 1.255–4.444 | 0.007a | 2.352 | 1.262–4.384 |
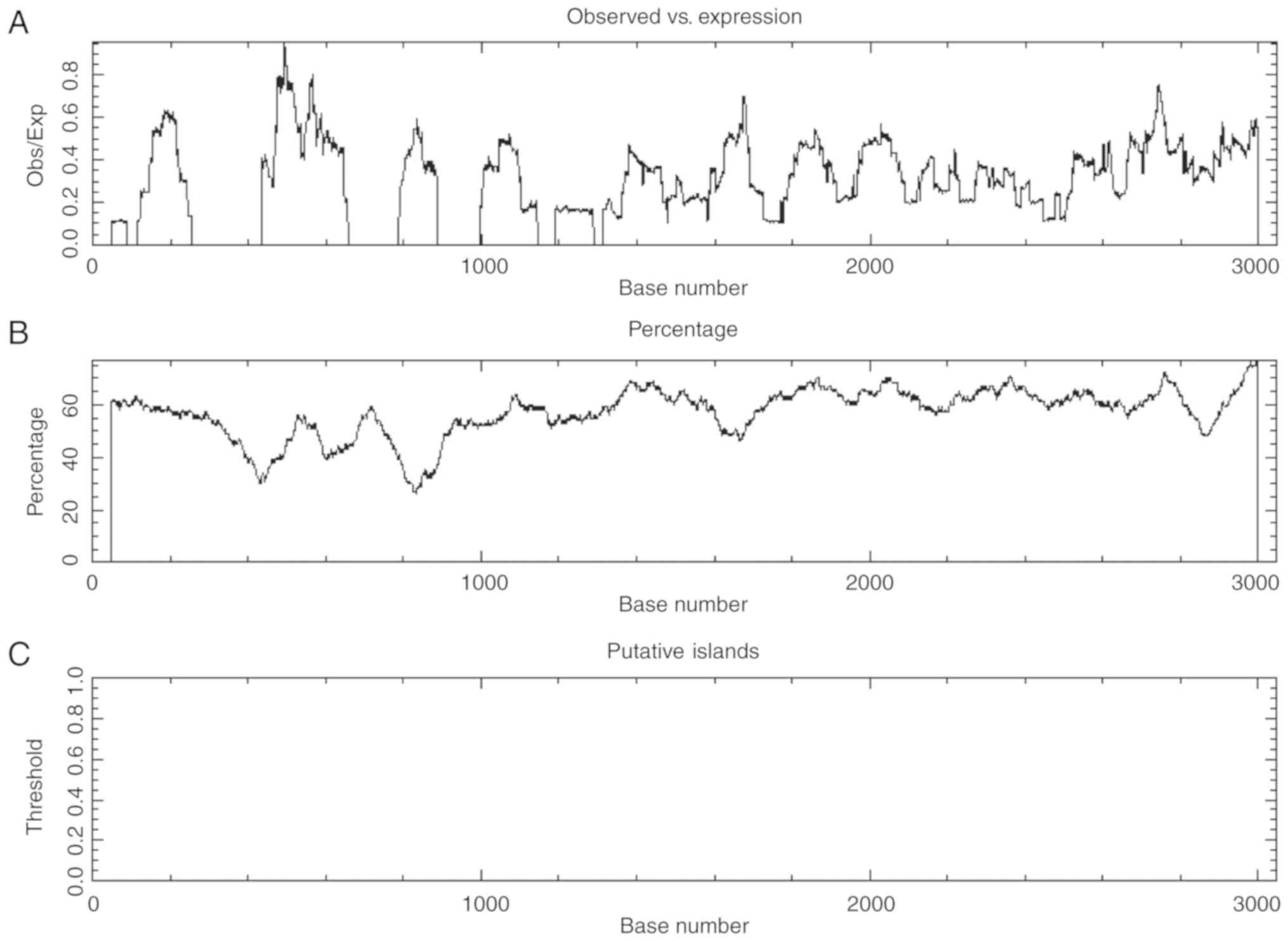
The SHIP1 promoter lacks methylation
sites
Emerging research indicates that hypermethylation of
CpG islands in gene promoters frequently results in transcriptional
silencing of genes (35). To
analyze whether low SHIP1 expression results from SHIP1 promoter
methylation, bioinformatics was used to predict CpG islands in the
SHIP1 promoter. CpG islands are defined as sequences >200 bp in
length with a (G+C) (%GC) content >50% and a ratio of CpG
dinucleotide frequencies (CpGobs/CpGexp)
>0.6 (36). EMBOSS Cpgplot
(http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/)
results predicted that no CpG islands were observed in the SHIP1
promoter (Fig. 6A-C), indicating
that reduced SHIP1 expression in NSCLC is not associated with its
promoter methylation.
Reduced SHIP1 expression is not
associated with EGFR or KRAS mutations
To analyze the association between SHIP1 expression
and EGFR/KRAS mutations in NSCLC, the gene expression signature of
83 matched pairs of lung adenocarcinomas and non-malignant adjacent
tissue from GSE75037 were analyzed using Illumina WG6-V3 expression
arrays. SHIP1 gene expression information was extracted and it was
determined that the SHIP1 gene has significantly reduced expression
in lung adenocarcinomas tissues, compared with non-malignant
adjacent tissues (Fig. 7A). SHIP1
expression is progressively lost during tumor progression (Fig. 7B). Additionally, the association
between SHIP1 expression and EGFR/KRAS mutations was further
analyzed and it was determined that no significant differences were
observed between the EGFR/KRAS-mutated tissues and wild-type
tissues (Fig. 7C-E). Collectively,
these results indicated that reduced SHIP1 expression is not
associated with EGFR or KRAS mutations in NSCLC.
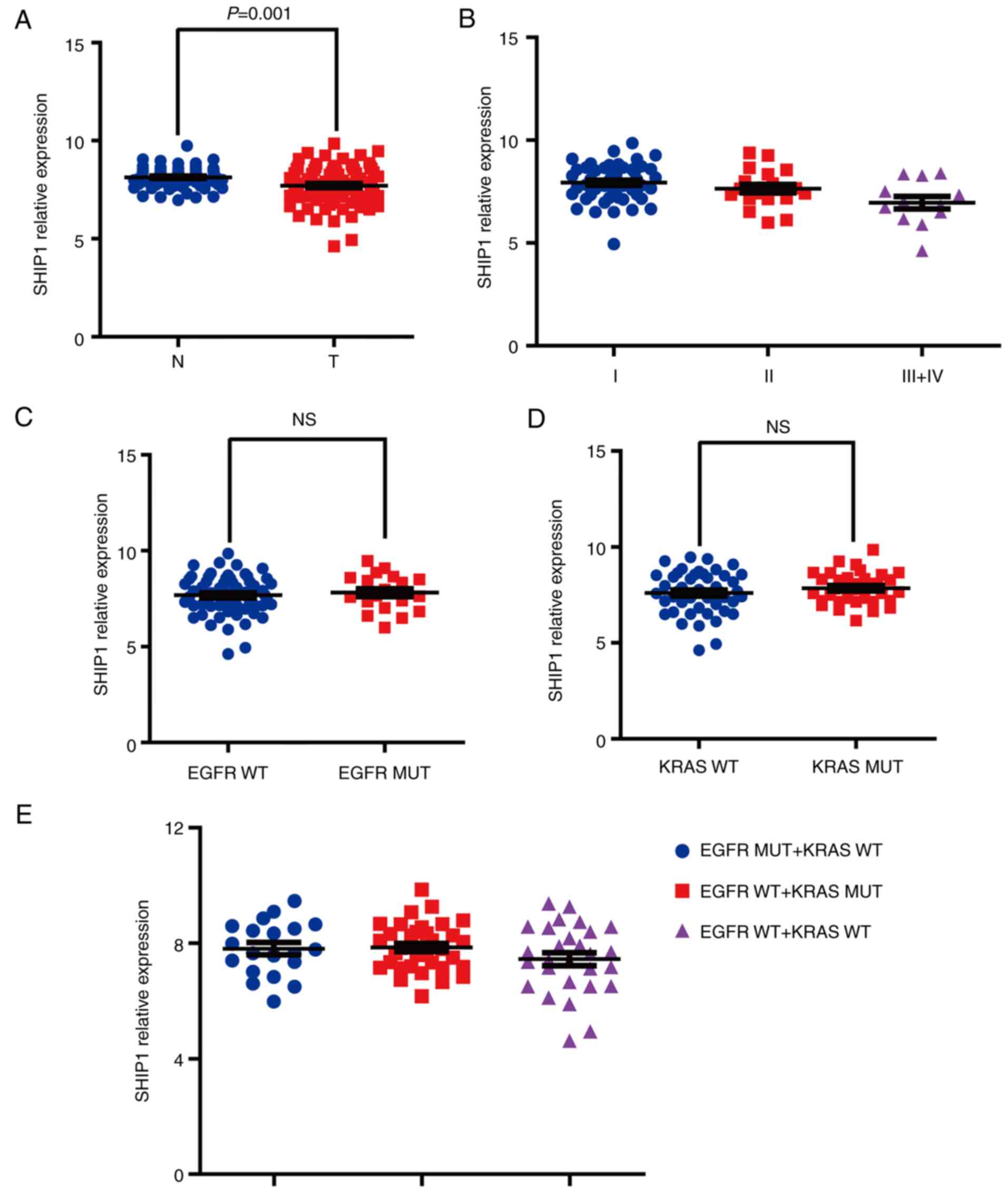 | Figure 7.SHIP1 downregulation is not
associated with EGFR or KRAS mutations. (A) SHIP1 gene expression
was analyzed in 83 matched pairs of lung adenocarcinoma and
non-malignant adjacent tissues from Gene Expression Omnibus
GSE75037. Paired-sample Student's t-test, P=0.001. (B) SHIP1
expression is progressively lost during tumor progression with
American Joint Committee on Cancer 8th stage system (34). One-way ANOVA, P=0.0052; Dunnett-t
test (P=0.0491 for I vs. II; P=0.0335 for II vs. III+IV). (C) The
association between SHIP1 expression and EGFR mutations. Student's
t-test, P>0.05. (D) The association between SHIP1 expression and
KRAS mutations. Student's t-test, P>0.05. (E) The association
between SHIP1 expression and EGFR or KRAS mutations. One-way ANOVA,
P>0.05. ANOVA, analysis of variance; KRAS, Kirsten rat sarcoma;
EGFR, epidermal growth factor receptor; NS, not significant; WT,
wild-type; MT, mutated; SHIP1, Src homology 2-containing
inositol-5′-phosphatase 1; N, normal; T, tumor. |
Discussion
The aberrant expression of SHIP1 has been observed
in various diseases, including malignant tumors (11–14).
The investigation of SHIP1 in tumors is limited to hematological
tumors, where SHIP1 was identified as a suppressor of the
occurrence and development of hematological tumors by inhibiting
hematopoietic cell proliferation and metastasis (15). SHIP1 downregulation was observed in
acute myeloid leukemia and associated with poor survival rates
(9). However, the role of SHIP1 in
solid tumors remains poorly understood.
As the leading cause of cancer-associated
mortalities globally, according to the epidemiological report
published in 2018 (20), lung
cancer has been a hotly pursued area of cancer research. However,
the expression and roles of SHIP1 in NSCLC remain unclear. In the
present study, it was determined that SHIP1 gene expression was
reduced in NSCLC tissues, compared with normal lung tissues, based
on GEO database analysis. Furthermore, SHIP1 levels are
progressively reduced during tumor progression. Additionally,
RT-qPCR and western blot analysis also revealed that SHIP1 mRNA and
protein levels were reduced in NSCLC tissues and cell lines. These
results strongly support the hypothesis that SHIP1 serves a vital
role in NSCLC.
Functional analysis revealed that SHIP1 inhibits
cell proliferation, invasion and migration of NSCLC in vitro
and in vivo. Furthermore, flow cytometry detection revealed
G1-S phase arrest upon SHIP1 overexpression. Mechanistically,
reductions in G1-S phase-associated proteins cyclin D1, CDK4 and
CDK6, and EMT-associated proteins N-cadherin and Vimentin were
observed in SHIP1-overexpression NSCLC cells, whereas the EMT
antagonistic factor E-cadherin was increased. The cell cycle and
EMT are the key pathways involved in tumor cell growth and
metastasis (37,38). These results demonstrated that SHIP1
inhibits cell proliferation and metastasis via cell cycle- and
EMT-associated proteins.
The PI3K/AKT pathway is a key signal mediator during
cell cycle transitions (39) and
promotes the progression of the EMT (26). As an antagonist of PI3K activity
(15), SHIP1 suppresses
PI3K-mediated downstream pathways by dephosphorylating the key
PI3K-generated secondary messenger in hematological tumors and a
number of non-malignant diseases, including acute myelocytic
leukemia and diabetic kidney disease (9,13). In
the present study, it was determined that β-catenin and p-AKT
levels, but not AKT, KRAS, EGFR and p-EGFR levels, were reduced in
SHIP1-overexpressing NSCLC cells, indicating that PI3K/AKT are
downstream factors of SHIP1. Furthermore, EGF was used to activate
the PI3K/AKT pathway in SHIP1-overexpressing NSCLC cells, and it
was determined that β-catenin, cyclin D1 and CDK6 levels were
restored. These data support the hypothesis that SHIP1
dephosphorylates the key PI3K-generated secondary messenger to
inactivate the PI3K/AKT pathway, and the downstream cell cycle and
EMT pathway, ultimately inducing the inhibition of cell growth and
metastasis in NSCLC.
Previous studies reported that SHIP1 levels were
significantly reduced in acute myeloid leukemia tissues and has
been implicated as a suppressor of hematopoietic transformation
(40), but the association between
SHIP1 expression and pathoclinical characteristics of hematopoietic
tumors remains unclear. In the present study, it was observed that
SHIP1 was negatively associated with AJCC clinical stage, T
classification and N classification. Notably, low SHIP1 expression
acts as a negative prognostic factor in patients with NSCLC.
As an important modification of proteins and nucleic
acids, methylation of CpG islands in gene promoters frequently
results in silencing of suppressor genes (41). The methylation rates of the CpG
sites in the upstream region of SHIP1 exon 1 did not significantly
differ between Alzheimer's disease and control subjects (42). However, SHIP1 promoter methylation
levels were not reported. In the present study, bioinformatics was
used to predict CpG islands in the SHIP1 promoter and it was
determined that the SHIP1 promoter lacks CpG islands, indicating
that the reduced expression of SHIP1 in NSCLC was not associated
with the methylation of its promoter.
EGFR and KRAS mutations are frequently exhibited in
patients with NSCLC, particularly in patients with lung
adenocarcinoma (43). EGFR-TKIs
have become a key treatment for patients with NSCLC with different
EGFR mutations (44); however, drug
resistance is becoming increasingly prominent (22). Recent research has identified the
dysregulation of downstream pathways, including loss of
suppressors, as a mechanism of resistance (23). In the present study, the association
between SHIP1 expression and EGFR/KRAS mutations was analyzed in
NSCLC and it was determined that reduced SHIP1 expression was not
associated with EGFR or KRAS mutations in NSCLC.
The present data demonstrated that SHIP1 is
suppressed in NSCLC and inhibits cell proliferation, migration,
invasion and tumorigenicity via the PI3K/AKT pathway. Furthermore,
the downregulation of SHIP1 exhibits no notable association with
EGFR and KRAS mutations. Thus, SHIP1 may be considered as a tumor
suppressor, and low SHIP1 expression is a poor prognostic factor in
NSCLC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the China
Postdoctoral Science Foundation (grant no. 2017M613008), the
National Natural Science Foundation of China and Yunnan Joint
Foundation (grant nos. U1502222, 81702295, 81602029 and 81660389),
the Applied Basic Science Research Foundation of Yunnan Province
(grant nos. 2017FB12 and 2017FB127) and the Application of genomic
technology based early diagnosis and treatment of Qujing Xuanwei
Lung Cancer (grant no. 2016RA037).
Availability of data and material
Not applicable.
Authors' contributions
QF, YH, CG, ZL, HT, QL, YW and GY performed the
research. XS and WF designed the research study. HL, RL, XT and YX
performed the statistical analysis. QF, YH, WF and XS wrote the
paper. CG, ZL, HT and QL collected the clinical specimens. YW, GY,
HL, RL, XT and YX performed the database analysis. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
The tissues were approved by the Ethics Committees
of the Third Affiliated Hospital of Kunming Medical University.
Informed consent was obtained from patients.
Patient consent for publication
Patients provided informed consent for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Conduit SE, Ramaswamy V, Remke M, Watkins
DN, Wainwright BJ, Taylor MD, Mitchell CA and Dyson JM: A
compartmentalized phosphoinositide signaling axis at cilia is
regulated by INPP5E to maintain cilia and promote Sonic Hedgehog
medulloblastoma. Oncogene. 36:5969–5984. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
D'Oria R, Laviola L, Giorgino F, Unfer V,
Bettocchi S and Scioscia M: PKB/Akt and MAPK/ERK phosphorylation is
highly induced by inositols: Novel potential insights in
endothelial dysfunction in preeclampsia. Pregnancy Hypertension.
10:107–112. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang F, Wang Y, Hemmings BA, Ruegg C and
Xue G: PKB/Akt-dependent regulation of inflammation in cancer.
Semin Cancer Biol. 48:62–69. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang H and Shears SB: Structural features
of human inositol phosphate multikinase rationalize its inositol
phosphate kinase and phosphoinositide 3-kinase activities. J Biol
Chem. 292:18192–18202. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chacko GW, Tridandapani S, Damen JE, Liu
L, Krystal G and Coggeshall KM: Negative signaling in B lymphocytes
induces tyrosine phosphorylation of the 145-kDa inositol
polyphosphate 5-phosphatase, SHIP. J Immunol. 157:2234–2238.
1996.PubMed/NCBI
|
|
6
|
Damen JE, Liu L, Rosten P, Humphries RK,
Jefferson AB, Majerus PW and Krystal G: The 145-kDa protein induced
to associate with Shc by multiple cytokines is an inositol
tetraphosphate and phosphatidylinositol 3,4,5-triphosphate
5-phosphatase. Proc Natl Acad Sci USA. 93:1689–1693. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Geier SJ, Algate PA, Carlberg K, Flowers
D, Friedman C, Trask B and Rohrschneider LR: The human SHIP gene is
differentially expressed in cell lineages of the bone marrow and
blood. Blood. 89:1876–1885. 1997.PubMed/NCBI
|
|
8
|
Veillette A, Latour S and Davidson D:
Negative regulation of immunoreceptor signaling. Ann Rev Immunol.
20:669–707. 2002. View Article : Google Scholar
|
|
9
|
Täger M, Horn S, Latuske E, Ehm P, Schaks
M, Nalaskowski M, Fehse B, Fiedler W, Stocking C, Wellbrock J, et
al: SHIP1, but not an AML-derived SHIP1 mutant, suppresses myeloid
leukemia growth in a xenotransplantation mouse model. Gene Therapy.
24:749–753. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brooks R, Fuhler GM, Iyer S, Smith MJ,
Park MY, Paraiso KH, Engelman RW and Kerr WG: SHIP1 inhibition
increases immunoregulatory capacity and triggers apoptosis of
hematopoietic cancer cells. J Immunol. 184:3582–3589. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McKeever PM, Kim T, Hesketh AR, MacNair L,
Miletic D, Favrin G, Oliver SG, Zhang Z, St George-Hyslop P and
Robertson J: Cholinergic neuron gene expression differences
captured by translational profiling in a mouse model of Alzheimer's
disease. Neurobiol Aging. 57:104–119. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ngoh EN, Weisser SB, Lo Y, Kozicky LK, Jen
R, Brugger HK, Menzies SC, McLarren KW, Nackiewicz D, van Rooijen
N, et al: Activity of SHIP, which prevents expression of
interleukin 1β, is reduced in patients with Crohn's disease.
Gastroenterology. 150:465–476. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li F, Li L, Cheng M, Wang X, Hao J, Liu S
and Duan H: SHIP, a novel factor to ameliorate extracellular matrix
accumulation via suppressing PI3K/Akt/CTGF signaling in diabetic
kidney disease. Biochem Biophys Res Commun. 482:1477–1483. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen Z, Shojaee S, Buchner M, Geng H, Lee
JW, Klemm L, Titz B, Graeber TG, Park E, Tan YX, et al: Signalling
thresholds and negative B-cell selection in acute lymphoblastic
leukaemia. Nature. 521:357–361. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kerr WG: Inhibitor and activator: Dual
functions for SHIP in immunity and cancer. Ann NY Acad Sci.
1217:1–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Janku F: Phosphoinositide 3-kinase (PI3K)
pathway inhibitors in solid tumors: From laboratory to patients.
Cancer Treatment Rev. 59:93–101. 2017. View Article : Google Scholar
|
|
17
|
Viernes DR, Choi LB, Kerr WG and Chisholm
JD: Discovery and development of small molecule SHIP phosphatase
modulators. Med Res Rev. 34:795–824. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marion F, Williams DE, Patrick BO,
Hollander I, Mallon R, Kim SC, Roll DM, Feldberg L, Van Soest R and
Andersen RJ: Liphagal, a Selective inhibitor of PI3 kinase alpha
isolated from the sponge Aka coralliphaga: Structure
elucidation and biomimetic synthesis. Org Lett. 8:321–324. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Catimel B, Yin MX, Schieber C, Condron M,
Patsiouras H, Catimel J, Robinson DE, Wong LS, Nice EC, Holmes AB,
et al: PI(3,4,5)P3 interactome. J Proteome Res. 8:3712–3726. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Walter AO, Sjin RT, Haringsma HJ, Ohashi
K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, et al:
Discovery of a mutant-selective covalent inhibitor of EGFR that
overcomes T790M-mediated resistance in NSCLC. Cancer Discov.
3:1404–1415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Passiglia F, Listi A, Castiglia M, Perez
A, Rizzo S, Bazan V and Russo A: EGFR inhibition in NSCLC: New
findings… and opened questions? Crit Rev Oncol Hematol.
112:126–135. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morgillo F, Della Corte CM, Fasano M and
Ciardiello F: Mechanisms of resistance to EGFR-targeted drugs: Lung
cancer. ESMO Open. 1:e0000602016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Krystal G: Lipid phosphatases in the
immune system. Semin Immunol. 12:397–403. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leslie NR, Biondi RM and Alessi DR:
Phosphoinositide-regulated kinases and phosphoinositide
phosphatases. Chem Rev. 101:2365–2380. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fu Q, Song X, Liu Z, Deng X, Luo R, Ge C,
Li R, Li Z, Zhao M, Chen Y, et al: miRomics and proteomics reveal a
miR-296-3p/PRKCA/FAK/Ras/c-Myc feedback loop modulated by
HDGF/DDX5/β-catenin complex in lung adenocarcinoma. Clin Cancer
Res. 23:6336–6350. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fu QF, Liu Y, Fan Y, Hua SN, Qu HY, Dong
SW, Li RL, Zhao MY, Zhen Y, Yu XL, et al: Alpha-enolase promotes
cell glycolysis, growth, migration, and invasion in non-small cell
lung cancer through FAK-mediated PI3K/AKT pathway. J Hematol Oncol.
8:222015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang H, Wu Q, Liu Z, Luo X, Fan Y, Liu Y,
Zhang Y, Hua S, Fu Q, Zhao M, et al: Downregulation of FAP
suppresses cell proliferation and metastasis through PTEN/PI3K/AKT
and Ras-ERK signaling in oral squamous cell carcinoma. Cell Death
Dis. 5:e11552014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu Z, Luo W, Zhou Y, Zhen Y, Yang H, Yu
X, Ye Y, Li X, Wang H, Jiang Q, et al: Potential tumor suppressor
NESG1 as an unfavorable prognosis factor in nasopharyngeal
carcinoma. PLoS One. 6:e278872011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhen Y, Fang W, Zhao M, Luo R, Liu Y, Fu
Q, Chen Y, Cheng C, Zhang Y and Liu Z:
miR-374a-CCND1-pPI3K/AKT-c-JUN feedback loop modulated by PDCD4
suppresses cell growth, metastasis, and sensitizes nasopharyngeal
carcinoma to cisplatin. Oncogene. 36:275–285. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Philipsen S: Expression data for early
stage NSCLC. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE191882010
April 10–2018
|
|
33
|
Gazdar A, Girard L, Stephen L, Wan L and
Zhang W: Expression profiling of 83 matched pairs of lung
adenocarcinomas and non-malignant adjacent tissue. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE750372016
March 17–2018
|
|
34
|
Amin MB, Edge S, Greene F, Byrd DR,
Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR,
Sullivan DC, et al: AJCC Cancer Staging manual 8th. https://cancerstaging.org/references-tools/deskreferences/Pages/default.aspx)2016
January 26–2018
|
|
35
|
Michaelsen SR, Aslan D, Urup T, Poulsen
HS, Grønbæk K, Broholm H and Kristensen LS: DNA methylation levels
of the ELMO gene promoter CpG islands in human
glioblastomas. Int J Mol Sci. 19:E6792018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gardiner-Garden M and Frommer M: CpG
islands in vertebrate genomes. J Mol Biol. 196:261–282. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Castaneda M, Chen L, Pradhan L, Li S, Zein
R, Lee Y, Lim HS, Nam HJ and Lee J: A forkhead box protein C2
inhibitor: Targeting epithelial-mesenchymal transition and cancer
metastasis. Chembiochem. 19:1359–1364. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou X, Liu J, Zhang J, Wei Y and Li H:
Flubendazole inhibits glioma proliferation by G2/M cell cycle
arrest and pro-apoptosis. Cell Death Disco. 4:182018. View Article : Google Scholar
|
|
39
|
Zhao M, Luo R, Liu Y, Gao L, Fu Z, Fu Q,
Luo X, Chen Y, Deng X, Liang Z, et al: miR-3188 regulates
nasopharyngeal carcinoma proliferation and chemosensitivity through
a FOXO1-modulated positive feedback loop with
mTOR-p-PI3K/AKT-c-JUN. Nat Commun. 7:113092016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xue H, Hua LM, Guo M and Luo JM: SHIP1 is
targeted by miR-155 in acute myeloid leukemia. Oncol Rep.
32:2253–2259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li J, Gong P, Lyu X, Yao K, Li X and Peng
H: Aberrant CpG island methylation of PTEN is an early event in
nasopharyngeal carcinoma and a potential diagnostic biomarker.
Oncol Rep. 31:2206–2212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yoshino Y, Yamazaki K, Ozaki Y, Sao T,
Yoshida T, Mori T, Mori Y, Ochi S, Iga JI and Ueno SI: INPP5D mRNA
expression and cognitive decline in Japanese Alzheimer's disease
subjects. J Alzheimers Dis. 58:687–694. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
McIntyre A and Ganti AK: Lung cancer-a
global perspective. J Surg Oncol. 115:550–554. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Masuzawa K, Yasuda H, Hamamoto J, Nukaga
S, Hirano T, Kawada I, Naoki K, Soejima K and Betsuyaku T:
Characterization of the efficacies of osimertinib and nazartinib
against cells expressing clinically relevant epidermal growth
factor receptor mutations. Oncotarget. 8:105479–105491. 2017.
View Article : Google Scholar : PubMed/NCBI
|