Introduction
Bladder cancer is a malignancy of the urinary tract
and is the ninth most common malignancy worldwide, with an
estimated 430,000 new cases, and resulting in 165,000 deaths in
2012 (1). At diagnosis, ~75% of
patients have non-invasive bladder cancer and 25% have
muscle-invasive or metastatic disease (2). For many years, cisplatin-based
combination chemotherapy, such as dose-dense methotrexate,
vinblastine, doxorubicin and cisplatin (ddMVAC) or gemcitabine and
cisplatin (GC) regimens, has been the standard treatment for
patients with metastatic urothelial carcinoma. Although the overall
response rate is ~40–60%, the median survival is only one year
(3,4). Due to drug resistance and considerable
side-effects, combination therapies of cisplatin with other cancer
drugs have been applied as novel therapeutic regimens for many
types of cancers (5).
Angiogenesis is required for continued tumor growth,
progression and metastasis of a variety of solid tumors (6,7).
Therefore, one of the approaches in cancer therapy has been the
targeting of angiogenesis. A number of angiogenic factors are
expressed in bladder cancer, including basic and acidic fibroblast
growth factor (FGF), vascular endothelial growth factor (VEGF),
hepatocyte growth factor, interleukin-8, and transforming growth
factor-α (8). VEGF and its
corresponding receptors (VEGFR) are the most prominent regulators
of angiogenesis (6). In particular,
activation of VEGFR-2 promotes the proliferation and survival of
endothelial cells. VEGFR-3 appears to promote lymphangiogenesis and
metastatic spread (9). The
platelet-derived growth factor (PDGF) and FGF signaling pathways
also control angiogenesis, tumor growth, and metastasis, and
compensatory mechanisms may come into play when VEGF signaling is
blocked (6). Research has revealed
that both VEGFRs and the VEGF ligands are expressed in bladder
cancer tissue and cells (10).
Heightened levels of VEGF resulted in significantly decreased
survival compared with normal levels of VEGF expression (11,12).
Motesanib (AMG 706) is an orally administered,
small-molecule angiogenesis inhibitor of multiple targets including
VEGFR-1, −2 and −3, PDGF receptor (PDGFR), and stem cell factor
receptor (13,14). Monotherapy and combination with
chemotherapy resulted in tumor regression and inhibition of
angiogenesis in various xenograft models such as non-small cell
lung, thyroid and colorectal cancer, and breast cancer models
(9,13,15–17).
Motesanib has also exhibited anticancer activity in phase 1 and/or
phase 2 studies in solid tumors including ovarian, fallopian tube
and primary peritoneal carcinoma, metastatic breast, thyroid and
non-squamous non-small-cell lung cancer (18,19).
To the best of our knowledge, the anticancer effect
of motesanib on bladder cancer is still unclear. The present study
was designed to investigate the efficacy of motesanib alone or in
combination with cisplatin in bladder cancer cell lines and to
investigate the mechanisms that mediate these effects.
Materials and methods
Cell lines and reagents
The human bladder cancer cell lines T24, 253J and
HTB9 were obtained from the American Type Culture Collection (ATCC;
Rockville, MD, USA). The cisplatin-resistant cell line T24R2 was
generated by serial desensitization (20). The cells were maintained in
RPMI-1640 medium (Gibco; Invitrogen, Carlsbad, CA, USA)
supplemented with 10% heat-inactivated fetal bovine serum and 1%
penicillin/streptomycin (Gibco; Invitrogen) in a humidified
atmosphere of 95% air and 5% CO2 at 37°C. Motesanib was
purchased from Selleck Chemicals (Houston, TX, USA; Fig. 1). It was dissolved in dimethyl
sulfoxide (DMSO; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and
diluted to obtain the working concentration. The final
concentration of DMSO in the culture media was 0.1% (v/v). Media
containing 0.1% DMSO were used as a control. Cisplatin was obtained
from JW Pharmaceutical (Seoul, Korea).
Cell viability assay
Bladder cancer cells were seeded at 2×103
in 96-well plates. After 24 h, the cells were incubated with
motesanib, cisplatin, and a combination of the two for 48 and 72 h.
At the end of the drug exposure, 10 µl of the Cell Counting Kit-8
(CCK-8) solution (Dojindo Molecular Technologies, Inc.,
Gaithersburg, MD, USA) was added to each well. After 4 h of
incubation, the optical density of each well was measured at 450 nm
using a microplate reader (Molecular Devices, LLC, Sunnyvale, CA,
USA). Cell viability was calculated as the percentage of viable
cells in the total population.
Synergism determination
The synergistic effect between motesanib and
cisplatin was determined based on a combination index (CI) using
CalcuSyn software (version 2.1; Biosoft, Cambridge, UK). The CI
indicates synergism at <1.0, antagonism at >1.0, and additive
effects at 1.0.
Clonogenic assays
T24 and T24R2 cells were plated at 4×102
in 6-well plates, incubated with either motesanib or cisplatin, and
combined for 48 h. The cells were cultured for another 10–14 days
in motesanib- and cisplatin-free medium. The colonies were fixed
with methanol and stained with 0.1% crystal violet solution. The
plates were photographed, and colonies >0.2 mm in diameter were
counted.
Cell cycle analysis
T24R2 cells were cultured at 3×105/60 mm
dish, grown for 24 h, and then incubated with motesanib or
cisplatin alone and combined for 48 h. The cells were trypsinized,
fixed in 70% ethanol, and stained with propidium iodide (PI;
Sigma-Aldrich; Merck KGaA) solution for 30 min at 37°C. The cell
cycle distribution was determined on a FACSCalibur instrument (BD
Biosciences, San Jose, CA, USA). The resultant data were analyzed
with the BD CellQuest Pro software (BD Biosciences).
RNA extraction and real-time
polymerase chain reaction (PCR)
Total RNA from T24R2 cells was extracted using the
RNeasy Mini Kit (Qiagen, Inc., Valencia, CA, USA). cDNA was
produced from 1 µg RNA using the Omniscript RT kit (Qiagen, Inc.)
according to the manufacturer's instructions. Real-time PCR was
performed for target genes using the Power SYBR-Green PCR Master
Mix (Applied Biosystems, Warrington, UK) with a 7500 Real-Time PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The specific primer sequences are presented in
Table I.
 | Table I.Sequences of primers. |
Table I.
Sequences of primers.
| Gene | Sequences |
|---|
| VEGFR-1 | F:
TCATGAATGTTTCCCTGCAA |
|
| R:
TTTGTTGCAGTGCTCACCTC |
| VEGFR-2 | F:
TGATCGGAAATGACACTGGA |
|
| R:
CACGACTCCATGTTGGTCAC |
| VEGFR-3 | F:
GAGACAAGGACAGCGAGGAC |
|
| R:
CTGTGTCGTTGGCATGTACC |
| PDGFR-α | F:
AGCTGATCCGTGCTAAGGAA |
|
| R:
ATCGACCAAGTCCAGAATGG |
| GAPDH | F:
TGCACCACCAACTGCTTAG |
|
| R:
AGAGGCAGGGATGATGTTC |
Western blotting
Cells were lysed with radio immunoprecipitation
assay buffer, consisting of 50 mM Tris-HCl (pH 8.0), 150 mM sodium
chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl
sulfate, and 1 mM phenylmethylsulfonyl fluoride. Protein
concentrations from cell extracts were assessed using a
bicinchoninic acid protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Equal amounts of total protein (30 µg) were
separated using sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (8–12% SDS-PAGE) and transferred onto
polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA,
USA). The membranes were blocked in 5% (w/v) non-fat dry milk for 1
h at room temperature, and then incubated with primary antibodies
(dilution 1:1,000) against poly(ADP-ribose) polymerase (PARP, cat.
no. 9542); cleaved caspases-3 (cat. no. 9664), −8 (cat. no. 9496),
and −9 (cat. no. 9505); cytochrome c (cat. no. 4272); Bcl-2
(cat. no. 15071); Bad (cat. no. 9268); cyclin E1 (cat. no. 4129);
phosphoinositide 3-kinase (PI3K; cat. no. 4257); phospho-PI3 kinase
p85 (Tyr458)/p55 (Tyr199; cat. no. 4228); protein kinase B (Akt,
cat. no. 4685); phospho-Akt (Ser473; cat. no. 4060); extracellular
signal-regulated kinases (Erk; cat. no. 4695);
phosphorylated-p44/42 MAPK (Erk1/2) (Thr202/Tyr204; cat. no. 4376);
VEGF (cat. no. 2445); VEGFR-1 (cat. no. 2893); VEGFR-2 (cat. no.
2479) (Cell Signaling Technology, Inc., Beverly, MA, USA); and
VEGFR-3 (cat. no. sc-28297) (Santa Cruz Biotechnology Inc., Santa
Cruz, CA, USA) at 4°C overnight. After incubation with horseradish
peroxidase (HRP)-conjugated anti-mouse IgG (dilution 1:10,000; cat.
no. sc-516102) or anti-rabbit IgG (dilution 1:5,000; cat no.
sc-2004) for 1 h, protein expression was detected with the Enhanced
Chemiluminescence Western Blot substrate kit (Pierce; Thermo Fisher
Scientific, Inc.). The blots were analyzed using ImageJ 1.48v
software (NIH; National Institutes of Health, Bethesda, MD,
USA).
Statistical analysis
The data are presented as the mean ± standard
deviation of three independent experiments. Statistical analysis
was performed using SPSS statistical software package (IBM SPSS
statistics 20; IBM Corp., Armonk, NY, USA). P<0.05 was
considered to indicate a statistically significant difference as
determined by ANOVA followed by Tukey's multiple-range test.
Results
Combination treatment of motesanib and
cisplatin suppresses proliferation of human bladder cancer
cells
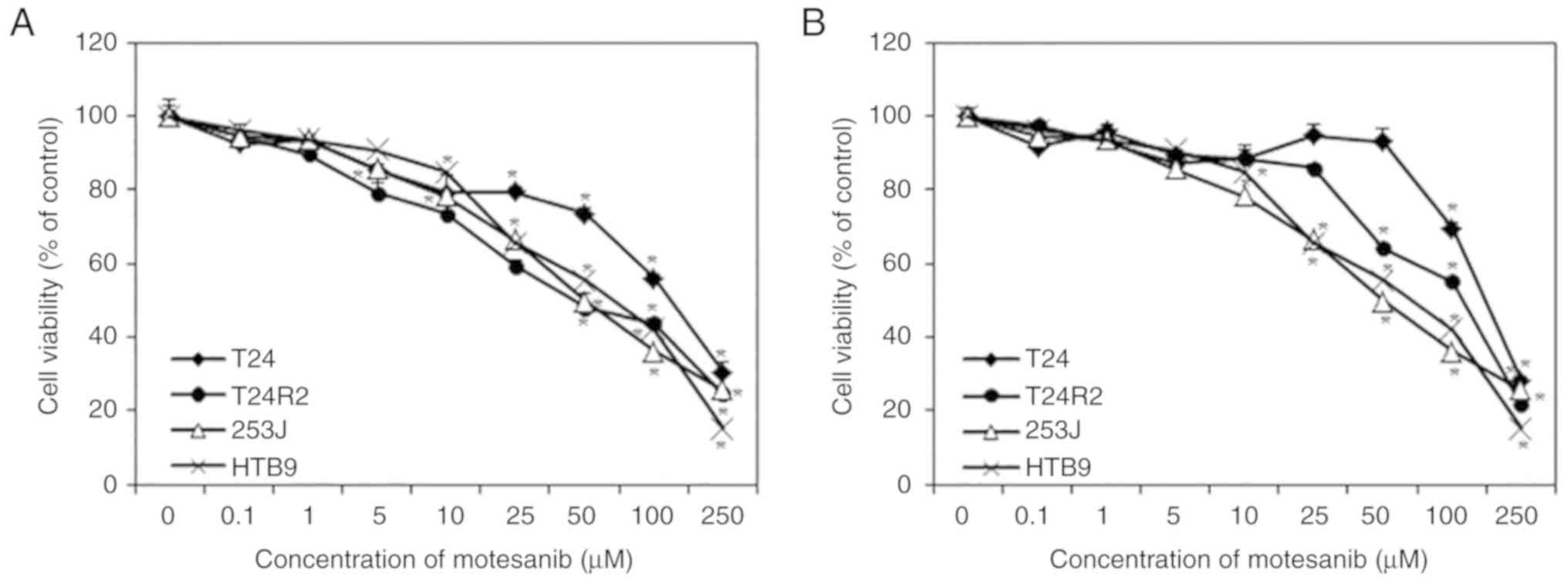
Bladder cancer cell lines were exposed to motesanib
(0, 0.1, 1, 5, 10, 25, 50, 100 and 250 µM), with cell viability
assessed by CCK-8 assay. As revealed in Fig. 2, treatment with motesanib for 48 and
72 h inhibited the proliferation of bladder cancer cells in a
dose-dependent manner when compared with that of non-treated cells
(control). Highly sensitive cell line T24R2 was selected at 48 h
for further investigation. Compared with the individual drug, the
drug combination of 50 µM motesanib with 2.5 µg/ml cisplatin
induced significant inhibition of cell proliferation (Fig. 3).
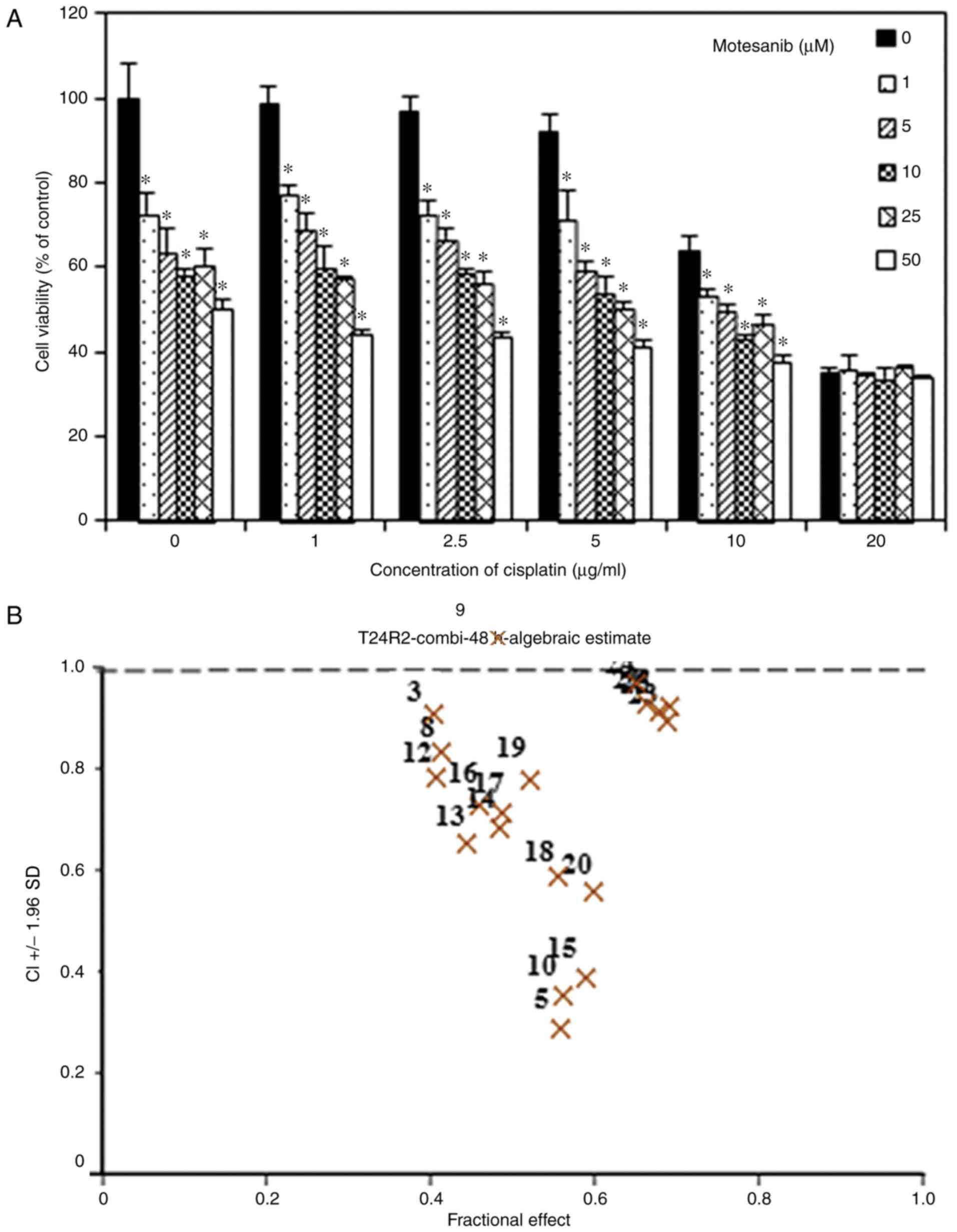 | Figure 3.Effect of combination treatment of
motesanib and cisplatin on T24R2 cell viability. (A) The cells were
co-treated with motesanib (0, 1, 5, 10, 25 and 50 µM) and cisplatin
(0, 1, 2.5, 5, 10 and 20 µg/ml) for 48 h, and viability was
evaluated by CCK-8 assay. (B) Combination index of motesanib and
cisplatin was <1.0, revealing synergism. The data are
represented as the mean ± standard deviation (SD) of three
independent experiments. *P<0.05, statistically significant
difference compared with the non-treated control. CCK-8, Cell
Counting Kit-8. |
To evaluate the antiproliferative effect of
motesanib combined with cisplatin on T24R2 cells, a clonogenic
assay was performed. The colony-forming ability of T24R2 cells was
significantly inhibited by 56.9 and 28.3% when treated with 50 µM
motesanib or 2.5 µg/ml cisplatin only, respectively, in comparison
with that of the non-treated control (Fig. 4). Particularly, the combination of
50 µM motesanib and 2.5 µg/ml cisplatin demonstrated improved
suppression of clonogenic formation, compared to motesanib or
cisplatin alone. These data indicated that motesanib and cisplatin
treatment could synergistically suppress the proliferation of
bladder cancer cells.
Cell cycle alteration in bladder
cancer cells caused by treatment with motesanib and cisplatin
Flow cytomety was performed to assess changes in the
cell cycle and apoptosis in bladder cancer cells. As revealed in
Fig. 5, the combined treatment of
50 µM motesanib and 2.5 µg/ml cisplatin significantly increased the
sub-G1 cell percentage. These results revealed that the combination
treatment increased the sub-G1 population, corresponding to
apoptotic cells, in T24R2 bladder cancer cells. In addition,
cisplatin alone and combination treatment primarily increased the
S-phase cell percentage on compared with the non-treated control.
Thus it was demonstrated that the combined treatment of motesanib
and cisplatin strongly altered the cell cycle progression of the
bladder cancer cell line.
Effect of combined treatment of
motesanib and cisplatin on VEGFR
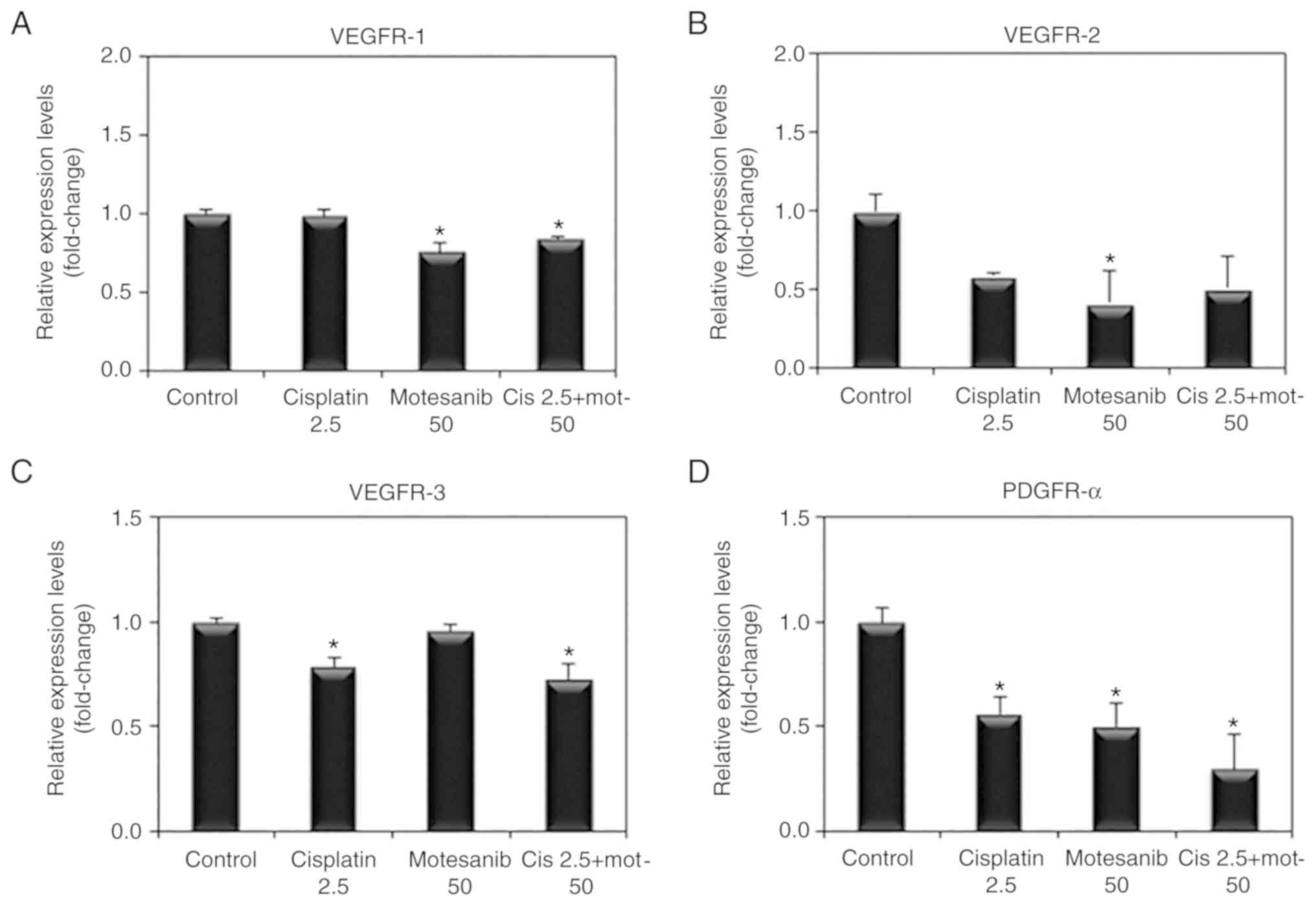
To examine whether VEGFR and PDGFR are affected by
the combined treatment of motesanib and cisplatin, VEGFR and PDGFR
mRNA expression was determined by real-time PCR. The combined
treatment of motesanib and cisplatin significantly reduced the mRNA
expression levels of VEGFR-1, −3 and PDGFR-α compared to those of
the non-treated control (Fig. 6).
VEGFR-2 mRNA expression was also decreased by motesanib alone and
combination treatment of motesanib and cisplatin. However, the
change was not significant. Moreover, VEGFR-1, −2, and −3 protein
levels were also significantly reduced by combination treatment of
motesanib and cisplatin (Fig.
7).
Change in the expression of proteins
regulating apoptosis and the cell cycle in bladder cancer cells
caused by treatment with motesanib and cisplatin
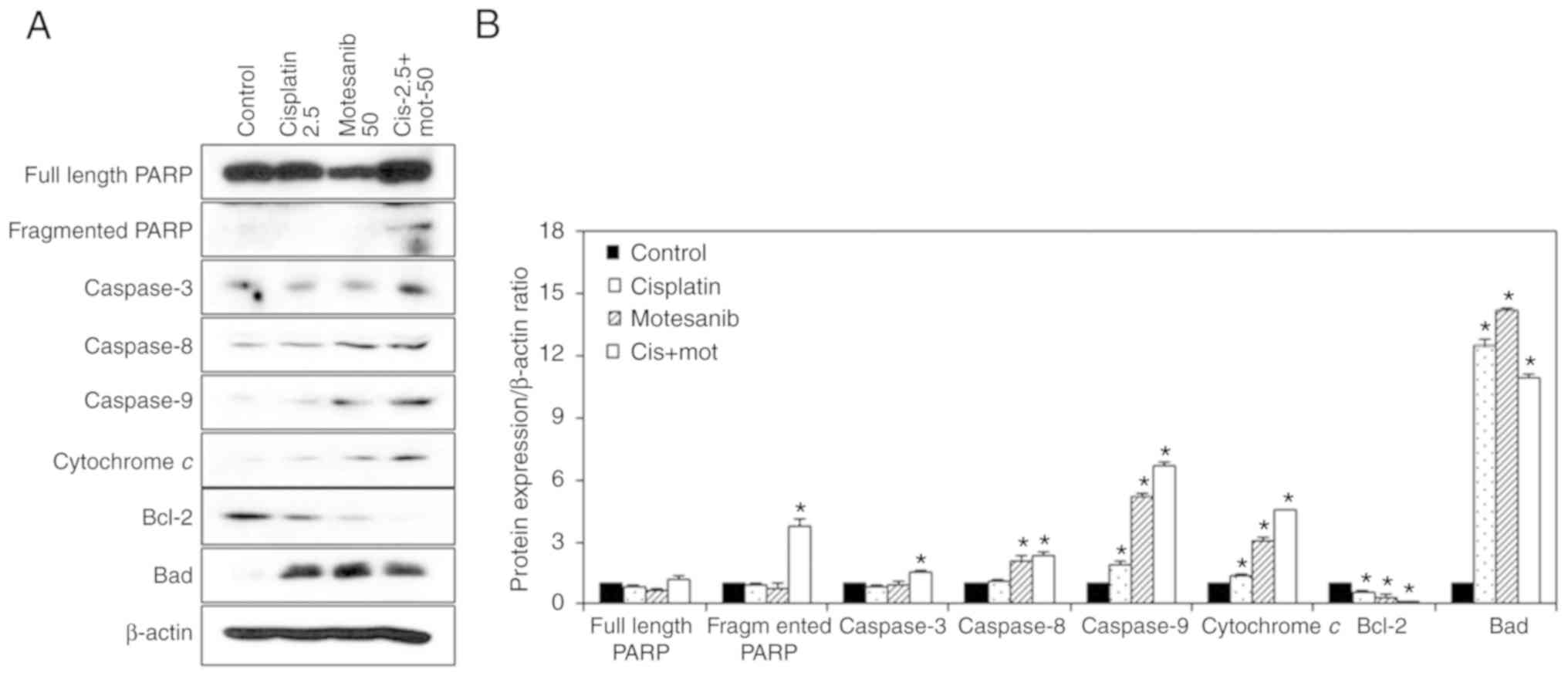
To confirm the antitumor effect of the combined
treatment of motesanib and cisplatin, western blot analysis was
performed. The expression levels of caspases-3, −8, and −9;
fragmented PARP; and cytochrome c were markedly enhanced by
the combined treatment of motesanib and cisplatin in T24R2 cells
(Fig. 8). Furthermore, the
expression of the anti-apoptotic protein Bcl-2 was markedly reduced
by combined treatment in T24R2 cells, whereas the expression of the
pro-apoptotic protein Bad was increased. In addition, the
expression of cyclins as an index of S-phase arrest was assessed
(21). The combined treatment of
motesanib and cisplatin markedly decreased cyclin E1 (Fig. 9). Phosphorylation of PI3K, Akt, and
Erk were significantly suppressed by the combined treatment in the
bladder cancer cells. Total PI3K, Akt, and Erk did not exhibit any
significant changes. In addition, both motesanib and cisplatin
individually resulted in lower levels of VEGF compared to those of
the non-treated control. These results indicated that the combined
treatment of motesanib and cisplatin inhibited the growth of
bladder cancer cells via an apoptosis-related mechanism, and
disrupted cell survival by inhibiting the PI3K/Akt signaling
pathway.
Discussion
Angiogenesis plays a critical role in tumor growth
and metastasis and involves several growth factors and their
receptors, particularly VEGF and VEGFRs (22). Therefore, multiple agents that
inhibit VEGFR and PDGFR have proved to be beneficial in cancer
treatment (23). Recently, new
targeted agents, i.e., monoclonal antibodies, fusion proteins and
small-molecule inhibitors, have been developed and are being used
clinically (24). In our study, we
determined the antitumor effects of motesanib, a small-molecule
multikinase inhibitor, alone or in combination with cisplatin by
evaluating cell proliferation, colony formation, cell cycle, and by
expression of mRNA and proteins associated with angiogenesis,
apoptosis, and cell survival in human bladder cancer cells.
VEGF is the primary proangiogenic mediator. VEGF
mRNA and protein are overexpressed in advanced bladder cancer
compared with those in normal bladder epithelium (25). Wang et al reported that
inhibition of VEGF expression reduced the development of metastasis
(26). Blocking VEGF or its
receptors reduced tumor growth in animal models (27,28).
VEGF acts on two principal tyrosine kinase receptors, VEGFR-1 and
VEGFR-2, both of which are overexpressed in most vascular tumors
and therefore are attractive therapeutic targets (25). As anticipated, motesanib
significantly reduced the mRNA expression of VEGFR-1, VEGFR-2, and
PDGFR-α and the protein expression of VEGF, VEGFR-1 and VEGFR-2.
Furthermore, the combined treatment of motesanib and cisplatin
significantly decreased the mRNA expression of VEGFR-1, VEGFR-3 and
PDGFR-α and the protein expression of VEGF, VEGFR-1, VEGFR-2 and
VEGFR-3.
The targeting and induction of apoptosis are
particularly interesting strategies in cancer therapy, as the
occurrence of apoptosis shifts the treatment effect from a
cytostatic to cytotoxic state (29). Cisplatin primarily induces cell
death by apoptosis, and a defect in apoptotic signaling could
confer cisplatin resistance (5). To
better understand the effect of combination treatment on apoptosis,
a western blot analysis was performed. Treatment with a combination
of 50 µM motesanib and 2.5 µg/ml cisplatin led to a synergistic
increase in the expression of apoptosis-related proteins
(fragmented PARP, cleaved caspase-3, −8, and −9 and cytochrome
c). Furthermore, combination treatment of motesanib and
cisplatin markedly reduced the expression of the anti-apoptotic
protein Bcl-2, whereas Bad expression was increased, confirming the
promotion of apoptosis. Since Bcl-2 overexpression is responsible
for cisplatin resistance (30),
combination treatment with motesanib could have resulted in
comprehensive downregulation of anti-apoptotic proteins in T24R2
cells, lowering the hurdle to apoptosis induction. Treatment with
motesanib alone or motesanib plus cisplatin revealed significant
reduction in cell proliferation and colony formation compared with
those of non-treated control cells. The effect on cell
proliferation reduction observed with the combination of motesanib
and cisplatin may be due, at least in part, to the induction of
apoptosis, which may increase the antitumor activity of cisplatin
(9). Consistent with our findings,
Kaya et al, reported that the main mechanism of action of
motesanib is the inhibition of tumor angiogenesis, but it also has
antiproliferative and apoptotic effects on HT29 colorectal cancer
cells (24).
The PI3K signaling pathway is involved in the
regulation of cancer cell growth, motility, survival and metabolism
(31). Several studies have
demonstrated that the PI3K signaling pathway is excessively
activated in muscle-invasive or metastatic bladder cancer (32). Akt is a family of serine/threonine
kinases that acts downstream of PI3K and plays a critical role in
cell survival and growth (5,28). Akt
activity is dependent on phosphorylation at two sites: T308 and
S473 (33). Phosphorylated Akt can
inhibit apoptosis resulting in the degradation of the p53 protein,
and inactivating pro-apoptotic proteins Bad, Bax or caspase-3
(34). In our results, PI3K/Akt
phosphorylation levels were markedly decreased by combination
treatment of motesanib and cisplatin. Therefore, motesanib and
cisplatin synergistically suppressed T24R2 bladder cancer cell
growth through the promotion of apoptosis and reduction of survival
related proteins. Collectively, these results emphasized the
superior cisplatin sensitizing effect of motesanib in
cisplatin-resistant bladder cancer cells.
In conclusion, the combined treatment of motesanib
and cisplatin can actively induce tumor cell growth inhibition,
cell cycle arrest at the S phase, reduced VEGFR and PDGFR mRNA
expression and enhanced apoptosis with increasing levels of cleaved
PARP, caspases, and Bad in cisplatin-resistant human bladder cancer
cells. Our results indicated that motesanib is a promising agent
for the treatment of bladder cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the grant nos.
13-2015-014 and 03-2010-013 from the Seoul National University
Bundang Hospital Research Fund.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SL designed the study, assembled and interpreted the
data, wrote the manuscript and managed the project. JNH performed
the experiments, analyzed the results and wrote the manuscript. SSB
and SEL were responsible for the collection and data analysis and
revised the manuscript. JIY analyzed the data and approved the
final version of the manuscript. All authors read and approved the
final manuscript and agree to be accountable for all aspects of the
research in ensuring that questions related to the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Antoni S, Ferlay J, Soerjomataram I, Znaor
A, Jemal A and Bray F: Bladder cancer incidence and motality: A
Global overview and recent trends. Eur Urol. 71:96–108. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kamat AM, Hahn NM, Efstathiou JA, Lerner
SP, Malmström PU, Choi W, Guo CC, Lotan Y and Kassouf W: Bladder
cancer. Lancet. 388:2796–2810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Godwin JL, Hoffman-Censits J and Plimack
E: Recent developments in the treatment of advanced bladder cancer.
Urol Oncol. 36:109–114. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McHugh LA, Kriajevska M, Mellon JK and
Griffiths TR: Combined treatment of bladder cancer cell lines with
lapatinib and varying chemotherapy regimens-evidence of
schedule-dependent synergy. Urology. 69:390–394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao Y and Adjei AA: Targeting
angiogenesis in cancer therapy: Moving beyond vascular endothelial
growth factor. Oncologist. 20:660–673. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fus ŁP and Górnicka B: Role of
angiogenesis in urothelial bladder carcinoma. Cent Eurpean J Urol.
69:258–263. 2016.
|
|
8
|
Bellmunt J, Hussain M and Dinney CP: Novel
approaches with targeted therapies in bladder cancer therapy of
bladder cancer by blockade of the epidermal growth factor receptor
family. Crit Rev Oncol Hematol. 46 (Suppl 46):S85–S104. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Coxon A, Bush T, Saffran D, Kaufman S,
Belmontes B, Rex K, Hughes P, Caenepeel S, Rottman JB, Tasker A, et
al: Broad antitumor activity in breast cancer xenografts by
motesanib, a highly selective, oral inhibitor of vascular
endothelial growth factor, platelet-derived growth factor, and Kit
receptors. Clin Cancer Res. 15:110–118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Yang X, Su LJ and Flaig TW:
Pazopanib synergizes with docetaxel in the treatment of bladder
cancer cells. Urology. 78:233.e7–233.e13. 2011. View Article : Google Scholar
|
|
11
|
Black PC, Agarwal PK and Dinney CP:
Targeted therapies in bladder cancer-an update. Urol Oncol.
25:433–438. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jordan EJ and Iyer G: Targeted therapy in
advanced bladder cancer: What have we learned? Urol Clin North Am.
42:253–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Polverino A, Coxon A, Starnes C, Diaz Z,
DeMelfi T, Wang L, Bready J, Estrada J, Cattley R, Kaufman S, et
al: AMG 706, an oral, multikinase inhibitor that selectively
targets vascular endothelial growth factor, platelet-derived growth
factor, and kit receptors, potently inhibits angiogenesis and
induces regression in tumor xenografts. Cancer Res. 66:8715–8721.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rosen LS, Kurzrock R, Mulay M, Van Vugt A,
Purdom M, Ng C, Silverman J, Koutsoukos A, Sun YN, Bass MB, et al:
Safety, pharmacokinetics, and efficacy of AMG 706, an oral
multikinase inhibitor, in patients with advanced solid tumors. J
Clin Oncol. 25:2369–2376. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Coxon A, Bready J, Kaufman S, Estrada J,
Osgood T, Canon J, Wang L, Radinsky R, Kendall R, Hughes P, et al:
Anti-tumor activity of motesanib in a medullary thyroid cancer
model. J Endocrinol Invest. 35:181–190. 2012.PubMed/NCBI
|
|
16
|
Coxon A, Ziegler B, Kaufman S, Xu M, Wang
H, Weishuhn D, Schmidt J, Sweet H, Starnes C, Saffran D and
Polverino A: Antitumor activity of motesanib alone and in
combination with cisplatin or docetaxel in multiple human
non-small-cell lung cancer xenograft models. Mol Cancer. 11:702012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tebbutt N, Kotasek D, Burris HA,
Schwartzberg LS, Hurwitz H, Stephenson J, Warner DJ, Chen L, Hsu CP
and Goldstein D: Motesanib with or without panitumumab plus FOLFIRI
or FOLFOX for the treatment of metastatic colorectal cancer. Cancer
Chemother Pharmacol. 75:993–1004. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu JF, Claret L, Sutjandra L, Kuchimanchi
M, Melara R, Bruno R and Sun YN: Population
pharmacokinetic/pharmacodynamic modeling for the time course of
tumor shrinkage by motesanib in thyroid cancer patients. Cancer
Chemother Pharmacol. 66:1151–1158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schilder RJ, Sill MW, Lankes HA, Gold MA,
Mannel RS, Modesitt SC, Hanjani P, Bonebrake AJ, Sood AK, Godwin
AK, et al: A phase II evaluation of motesanib (AMG 706) in the
treatment of persistent or recurrent ovarian, fallopian tube and
primary peritoneal carcinomas: A Gynecologic Oncology Group study.
Gynecol Oncol. 129:86–91. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Byun SS, Kim SW, Choi H, Lee C and Lee E:
Augmentation of cisplatin sensitivity in cisplatin-resistant human
bladder cancer cells by modulating glutathione concentrations and
glutathione-related enzyme activities. BJU Int. 95:1086–1090. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yeo EJ, Ryu JH, Chun YS, Cho YS, Jang IJ,
Cho H, Kim J, Kim MS and Park JW: YC-1 induces S cell cycle arrest
and apoptosis by activating checkpoint kinases. Cancer Res.
66:6345–6352. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carmeliet P: VEGF as a key mediator of
angiogenesis in cancer. Oncology. 69 (Suppl 3):S4–S10. 2005.
View Article : Google Scholar
|
|
23
|
Appelmann I, Liersch R, Kessler T, Mesters
RM and Berdel WE: Angiogenesis inhibition in cancer therapy:
Platelet-derived growth factor (PDGF) and vascular endothelial
growth factor (VEGF) and their receptors: Biological functions and
role in malignancy. Recent Results Cancer Res. 180:51–81. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kaya TT, Altun A, Turgut NH, Ataseven H
and Koyluoglu G: Effects of a multikinase inhibitor motesanib (AMG
706) alone and combined with the selective DuP-697 COX-2 inhibitor
on colorectal cancer cells. Asian Pac J Cancer Prev. 17:1103–1110.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elfiky AA and Rosenberg JE: Targeting
angiogenesis in bladder cancer. Curr Oncol Rep. 11:244–249. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang F, Li HM, Wang HP, Ma JL, Chen XF,
Wei F, Yi MY and Huang Q: siRNA-mediated knockdown of VEGF-A,
VEGF-C and VEGFR-3 suppresses the growth and metastasis of mouse
bladder carcinoma in vivo. Exp Ther Med. 1:899–904. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu CL, Ping SY, Yu CP and Yu DS: Tyrosine
kinase receptor inhibitor-targeted combined chemotherapy for
metastatic bladder cancer. Kaohsiung J Med Sci. 28:194–203. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Van Kessel KE, Zuiverloon TC, Alberts AR,
Boormans JL and Zwarthoff EC: Targeted therapies in bladder cancer:
An overview of in vivo research. Nat Rev Urol. 12:681–694. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Madka V, Zhang Y, Li Q, Mohammed A,
Sindhwani P, Lightfoot S, Wu XR, Kopelovich L and Rao CV:
P53-stabilizing agent CP-31398 prevents growth and invasion of
urothelial cancer of the bladder in transgenic UPII-SV40T mice.
Neoplasia. 15:966–974. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim SH, Ho JN, Jin H, Lee SC, Lee SE, Hong
SK, Lee JW, Lee ES and Byun SS: Upregulated expression of BCL2,
MCM7, and CCNE1 indicate cisplatin-resistance in the set of two
human bladder cancer cell lines: T24 cisplatin sensitive and T24R2
cisplatin resistant bladder cancer cell lines. Investig Clin Urol.
57:63–72. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bartholomeusz C and Gonzalez-Angulo AM:
Targeting the PI3K signaling pathway in cancer therapy. Expert Opin
Ther Targets. 16:121–130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sathe A and Nawroth R: Targeting the
PI3K/AKT/mTOR pathway in bladder cancer. methods Mol Biol.
1655:335–350. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dienstmann R, Rodon J, Serra V and
Tabernero J: Picking the point of inhibition: A comparative review
of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 13:1021–1031.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fayard ETL, Baudry A and Hemmings BA:
Protein kinase B/Akt at a glance. J Cell Sci. 118:5675–5678. 2005.
View Article : Google Scholar : PubMed/NCBI
|