Introduction
Prostate cancer (PCa) is one of the most common
malignant tumors of the male urinary system. Statistical analysis
has shown that more than 250,000 men succumb to PCa worldwide, with
at least 900,000 new cases each year (1). In China, the incidence and mortality
rates of PCa are annually increasing and tend to affect younger
individuals (2,3). Current treatments for PCa include
surgical treatment and androgen deprivation therapy (ADT). However,
almost all patients become resistant to long-term treatment,
developing castration-resistant PCa (CRPC). CRPC is characterized
by the increased activation and/or overexpression of androgen
receptor (AR), resulting in the transcription of downstream target
genes and tumor progression, despite castrate levels of androgen in
the patient (4). Only a limited
number of drugs can be effective once the tumor progresses to CRPC.
Fortunately, enzalutamide is one of them. Enzalutamide is an AR
inhibitor that competitively inhibits the binding of androgens to
receptors and hinders the nuclear transport of the AR and the
interaction of the receptor with DNA. Although enzalutamide has
achieved some good clinical results (5–7), the
subsequent drug resistance remains a challenge. It is therefore,
necessary to determine the mechanism of CRPC resistance to
enzalutamide.
Phospholipase Cε (PLCε), a multifunctional signaling
protein harboring both PLC and guanine nucleotide exchange factor
activities, was discovered by Song et al in 2001 (8,9). As a
member of the human phospholipase C family, PLCε has been
identified as an oncogene involved in carcinogenesis, tumor
proliferation and migration (10,11).
Our previous study showed that PLCε knockdown inhibited PCa cell
proliferation via the PTEN/AKT signaling pathway (12). Furthermore, it was found that PLCε
inhibited the biological behavior of PCa cells by downregulating AR
(13). Nonetheless, the role of
PLCε in CRPC cells remains unknown. The aim of the present study
was to explore the effect of PLCε on the proliferation of CRPC
cells and determine whether PLCε can sensitize CRPC cells to the AR
axis inhibitor, enzalutamide.
The Hedgehog (Hh) signaling pathway plays a critical
role in the development and homeostasis of many organs and tissues.
It consists of the Hh ligand (Shh, Ihh and Dhh), two transmembrane
receptor complexes [patched (Ptch) and smoothened (Smo)], and the
downstream transcription factor glioma-associated homolog (Gli)
family (Gli-1, Gli-2 and Gli-3). Gli-1 and Gli-2 are responsible
for most transcriptional activator functions, whereas Gli-3 mainly
acts as a repressor. Gli-1 is a direct transcriptional target of
the Hh signaling and a marker for pathway activity (14). Vismodegib and cyclopamine are
classic Hh signaling pathway inhibitors. Vismodegib blocks the
biological activity of the Hh pathway. Since it binds to and
hinders Smo, thus, preventing the systemic activation of the
forward signaling, it has been used in the clinical treatment of
basal cell carcinoma (15).
Cyclopamine, a plant steroidal alkaloid that inhibits Smo, is a
therapeutic strategy for PCa (16,17)
and renal cell cancer (18).
GANT61, a small molecule antagonist directly acting on downstream
molecule Gli of the Hh signaling pathway, could interfere with
cellular DNA binding of Glis (19).
It has been reported that the Hh pathway is involved in PCa
development, progression, treatment resistance (20,21)
and epithelial-mesenchymal transition (17). An increasing number of studies have
reported that the Hh signaling pathway is associated with
chemotherapeutic drug resistance in pancreatic cancer and other
tumors (22–24). In addition, there is a crosstalk
between the Hh and AR signaling pathways in PCa cells (25,26).
Since, however, the role of the Hh signaling pathway in CRPC cells
is unclear, we hoped to determine whether it can regulate the drug
sensitivity of CRPC cells to enzalutamide by interacting with the
AR.
The aim of the present study was to assess whether
PLCε and/or GANT61 can increase the sensitivity of CRPC cells to
enzalutamide, and determine the interaction mechanism among PLCε,
Gli and AR, so as to provide a better strategy for the clinical
treatment of CRPC. In the present study, the expression of PLCε and
Gli-1/Gli-2 in benign prostatic hyperplasia (BPH), PCa and CRPC
tissues and cells was investigated. The correlation between the
PLCε and Gli-1/Gli-2 in CRPC tissues and cell lines was also
explored. Furthermore, the effect of PLCε on cell proliferation and
invasion was assessed in CRPC cell lines, and the sensitivity of
EN-R and 22RV1 cells to enzalutamide following the downregulation
of PLCε expression was determined using lentiviral-mediated shPLCε
and/or treatment with specific Gli inhibitor GANT61. The results
showed that the PLCε knockdown inhibits CRPC cell proliferation and
invasion and sensitizes CRPC cells to enzalutamide by suppressing
the AR expression and nuclear translocation. It was also shown that
GANT61 combined with PLCε knockdown significantly sensitized CRPC
cells to enzalutamide. These findings may provide a new therapeutic
approach for CRPC.
Materials and methods
Patients and tissue samples
A total of 30 BPH tissue samples, 64 PCa tissue
samples and 27 CRPC tissue samples were obtained from patients who
underwent needle biopsy, transurethral resection of the prostate or
radical prostatectomy at the Department of Urology of the First
Affiliated Hospital of Chongqing Medical University, Chongqing,
China between April 2010 and September 2015. Complete clinical data
were available for all patients. All patients met the EAU
guidelines for diagnostic criteria for BPH, PCa and CRPC. All
tissue samples were reviewed by a pathologist for the confirmation
of BPH or PCa. The study was approved by the Ethics Committee of
Chongqing Medical University, Chongqing, China. Informed consent
was obtained from the patients or their family members.
Immunohistochemistry assay
All formalin-fixed and paraffin-embedded tissue
samples were cut into 5-µm-thick sections. Immunohistochemical
staining was performed using a standard immunoperoxidase staining
procedure (anti-PLCε; dilution 1:50; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA; anti-Gli-1, dilution 1:200 and anti-Gli-2,
dilution 1:150; both were from Abcam, Cambridge, UK). The
expression status of immunostaining was reviewed and scored based
on the proportion of positive cells and staining intensity by a
pathologist. Staining intensity was scored as follows: 0 (no
staining); 1 (light yellow); 2 (light brown); 3 (brown); and 4
(deep brown). Immunoreactivity ratio was scored as follows: 0 (0%
immunoreactive cells), 1 (<5% immunoreactive cells), 2 (5–50%
immunoreactive cells), 3 (>50–75% immunoreactive cells) and 4
(>75% immunoreactive cells). The final immunoreactivity score
was defined as the sum of both parameters. Final scores of ≤1 were
regarded as negative expression, while scores of ≥2 were regarded
as positive.
Cell culture, treatment and
transfection
Human PCa cell lines LNCaP and 22RV1 were obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA), enzalutamide-resistant (EN-R) cells were generated as
previously described (27).
Briefly, the LNCaP cells, one of the androgen-dependent PCa cell
strains, were treated with enzalutamide (10 µM; Selleck Chemicals,
Houston, TX, USA) for at least 6 months. 22RV1 and EN-R cells
represent CRPC cells. All the cell lines were cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS; both from
Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1%
penicillin/streptomycin (Beyotime Institute of Biotechnology,
Haimen, China). Lentivirus-shRNA targeting human PLCε (LV-shPLCε,
5′-GGTTCTCTCCTAGAAGCAACC-3′) and the negative control (LV-shNC,
5′-TTCTCCGAACGTGTCACGT-3′) were purchased from Shanghai GenePharma
Co., Ltd. (Shanghai, China). A PLCε shRNA sequence was inserted
into a pGLV3/H1/GFP-Puro lentivirus vector. Puromycin (1 µg/ml) was
used to screen the stable cell lines. Fluorescence expression was
observed under a fluorescence microscope (Olympus Corp., Tokyo,
Japan) 3 days after lentiviral infection. The infected cells were
cultured for one week for subsequent experiments. The PLC knockdown
efficiency was assessed using RT-qPCR and western blot analysis.
The human Gli-1 and Gli-2 expression plasmids ad-Gli1 and ad-Gli2
containing full-length of Gli-1, Gli-2 and control vectors Gli1-NC,
Gli2-NC were purchased from Shanghai GenePharma Co., Ltd. Transient
transfection was performed using Lipofectamine 2000 transfection
reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Other reagents used in the present
study were as follows: Lipopolysaccharides (LPS; Merck KGaA,
Darmstadt, Germany), GANT61 (MedChemExpress Co., Ltd., Shanghai,
China).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
RNA was isolated from cells using TRIzol (Takara
Bio, Inc., Otsu, Japan) and reverse transcription was performed
using the PrimeScript RT reagent kit, according to the
manufacturer's instructions (Takara Bio, Inc.). RT-qPCR was
performed on a CFX Connect qPCR system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) with the SYBR Premix Ex Taq II kit (Takara Bio,
Inc.). The primer sequences used were as follows: PLCε (sense),
5′-GCAACTACAACGCTGTCATGGAG-3′ and PLCε (antisense),
5′-GCAACTACAACGCTGTCATGGAG-3′; Gli-1 (sense),
5′-ATCCTTACCTCCCAACCTCTGT-3′ and Gli-1 (antisense),
5′-AACTTCTGGCTCTTCCTGTAGC-3′; Gli-2 (sense),
5′-CGGTGTAGGCAGAGCTGATG-3′ and Gli-2 (antisense),
5′-CCACAAGGCAGAAACACCAA-3′; Smo (sense),
5′-CTCCTACTTCCACCTGCTCAC-3′ and Smo (antisense),
5′-CAAAACAAATCCCACTCACAGA-3′; β-actin (sense),
5′-TGACGTGGACATCCGCAAAG-3′ and β-actin (antisense),
5′-CTGGAAGGTGGACAGCGAGG-3′. The RT-qPCR comprised an initial
denaturation at 95°C for 15 sec, and then 45 cycles at 95°C for 5
sec and 60°C for 30 sec. The mRNA expression levels were calculated
using the comparative 2−ΔΔCq method (28), with β-actin as a calibrator. All
gene expression experiments were repeated at least 3 times.
Cell Counting Kit-8 (CCK-8) assay
Cell proliferation was evaluated using the CCK-8
assay. The cells were seeded in 96-well plates (2,000 cells/well),
and incubated at 37°C for 12 h, and then cultured with the
different treatment agents in each 5 replicate wells. CCK-8 reagent
(10 µl; Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) was added into each well and incubated at 37°C for
2 h. Optical density was detected using a microplate reader at the
absorbance of 450 nm. Half maximal inhibitory concentration
(IC50) of enzalutamide in the cells was calculated by
the CCK-8 assay. The pretreated cells were seeded into 96-well
plates (4,000 cells/well) and incubated at 37°C for 12 h. Different
concentrations (0.2–640 µM) of enzalutamide were added to the cells
in each 3 replicate wells for 24 h, dimethyl sulfoxide (DMSO)
(Merck KGaA) was used as the control. According to the research of
Gonnissen et al (29),
GANT61 inhibited the survival rate of 22RV1 cells in a
dose-dependent manner (from 1 to 50 µM), but the survival rate was
significantly inhibited at a concentration of 10 µM. Considering
the cytotoxicity, the concentration of 10 µM was chosen for the
present study.
Transwell invasion assay
For the Transwell invasion assay, 1×104
cells were plated in serum-free medium in the upper chamber with a
Matrigel-coated membrane, and the lower chamber was filled with
medium supplemented with 10% FBS. Following 48 h of incubation, the
cells at the lower chamber inserts were stained with 0.1% crystal
violet and 4% formaldehyde (Beyotime Institute of Biotechnology) at
room temperature for 15 min. After removing the membrane, the
number of cells was counted under a fluorescence microscope (Nikon,
Tokyo, Japan).
Colony formation assay
The cells were plated in 6-well plates (400
cells/well), and the medium was refreshed every 3 days. Following
culture for 14 days, the cells were fixed in 70% ethanol and then
stained with 0.05% crystal violet solution for 20 min. The number
of colonies was counted under a light microscope.
Western blot assay
The total protein of cells was extracted using RIPA
buffer supplemented with protease inhibitor PMSF and phosphatase
inhibitors NaF and Na3VO4 (Roche Diagnostics,
Basel, Switzerland). The protein concentration was determined using
the BCA protein assay kit. The isolated proteins (50 µg per lane)
were separated by sodium dodecyl sulfate-polyacrylamide gel (10 or
12%) electrophoresis (SDS-PAGE) and transferred to polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes was immersed in Tris-buffered saline (TBS) with Tween-20
blocking solution containing 5% non-fat milk for 2 h, and then
incubated with the primary antibodies at 4°C overnight. The
membranes were then incubated with a goat anti-rabbit IgG secondary
antibody (dilution 1:2,000; cat. no. TA130015; OriGene
Technologies, Inc., Rockville, MD, USA) or a goat anti-mouse IgG
secondary antibody (dilution 1:2,000; cat. no. TA130001; OriGene
Technologies, Inc.) for 1 h at 37°C. The enhanced chemiluminescent
(ECL) kit was purchased from Merck Millipore. The intensity level
of each protein band was quantified using Image-Pro plus 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA). The following antibodies
were used: PLCε (dilution 1:300; cat. no. sc28402; Santa Cruz
Biotechnology, Inc.), STAT3 (dilution 1:1,000; cat. no. ab119352;
Abcam), p-STAT3 (dilution 1:1,000; cat. no. ab32143; Abcam), Gli-1
(dilution 1:500; cat. no. ab49314; Abcam), Gli-2 (dilution 1:500;
cat. no. ab26056; Abcam), Smo (dilution 1:500; cat. no. ab113438;
Abcam) and AR (dilution 1:500; cat. no. sc7305; Santa Cruz
Biotechnology, Inc.).
Immunofluorescence
EN-R cells (1.0×105 cells/well) were
seeded on sterile glass coverslips and incubated for 48 h, fixed in
4% paraformaldehyde for 20 min, and incubated with anti-AR primary
antibody overnight at 4°C. They were then cultured with a goat
anti-rabbit secondary antibody (dilution 1:2,000; cat. no.
TA130015; OriGene Technologies, Inc., Rockville, MD, USA) for 45
min in the dark at room temperature, and nuclei were stained with
4′6-diamidino-2-phenylindole (DAPI). Immunofluorescent images were
acquired using a fluorescence microscope (Nikon, Tokyo, Japan) at a
magnification of ×400.
Animal xenograft model
All animal experiments were approved by the Ethics
Committee of Chongqing Medical University (Chongqing, China).
Twelve male castrated athymic nude mice aged 4 weeks (BALB/c;
Beijing Huafukang Bioscience Co., Ltd., Beijing, China) weighing
18–20 g were raised in a cabinet with laminar air flow under
pathogen-free conditions in a humidity- and temperature-controlled
environment with a 12 h light/dark schedule. The mice had ad
libitum access to food and water. Prior to the study
initiation, the mice were allowed to acclimatize for 1 week. Then
the mice were injected subcutaneously with 2×106 EN-R
(Cont group) or LV-shPLCε EN-R (LV-shPLCε group) cells (suspended
in 0.1 ml Matrigel) into the right flank area. The animals were
then divided into four groups: The Cont+PBS, the LV-shPLCε+PBS, the
Cont+Enz and the LV-shPLCε+Enz (3 mice per group). A week later,
the mice were treated with vehicle phosphate-buffered saline (PBS)
or enzalumatide at 10 mg/kg by oral gavage (5 days on and 2 days
off) up to 32 days treatment. The tumor growth was monitored every
5 days and the tumor volume was calculated using the following
formula: Volume (mm3) = 1/2 × length ×
width2. At the end of the experiment or when the tumor
volume exceeded 1,000 mm3, the mice were euthanized with
cervical dislocation and the tumors were collected and measured.
The following animal humane endpoints were established: tumors
exceeding 10% of the body weight of mice, tumors that became
festered and infected, and mice that could not eat and drink water
on their own. Mice were euthanized when these humane endpoints
occurred.
Statistical analysis
SPSS 20.0 (IBM Corp, Armonk, NY, USA) was used to
process data. Data are presented as the mean ± standard deviation
(SD). Data from two groups were analyzed by unpaired t-test and
>2 groups were analyzed by one-way and two-way ANOVA with post
hoc contrasts by the Student-Newman-Keuls (SNK) method. For the
data of characteristics of the patient groups, Mann-Whitney test
for 2 independent variables was used; Chi-square test was used for
trend for the number of rows or columns >2; McNemar's test was
used to compare the differences between the matched categorical
variables; Pearson's Chi-square test was used for 2 groups of
independent variables. Progressive-free survival (PFS) and overall
survival (OS) curves were estimated using the Kaplan-Meier method.
Correlation curve analysis for PLCε protein vs. corresponding Gli1
and Gli2 protein in CRPC specimens was conducted using Pearson's
linear correlation analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
Inhibition of PLCε and Gli-1/Gli-2
suppresses the proliferation and invasion of CRPC cells
Western blot analysis and RT-qPCR were performed to
investigate the protein expression and mRNA levels of PLCε and
Gli-1/Gli-2 in the LNCaP, 22RV1 and EN-R cells. As shown in
Fig. 1A and B, PLCε was highly
expressed in the three cell lines. Gli-1/Gli-2 was highly expressed
in 22RV1 and ENR cells, but slightly expressed in the LNCaP cells.
The expression of AR was also found to be higher in the 22RV1 and
EN-R cells, as compared with the LNCaP cells.
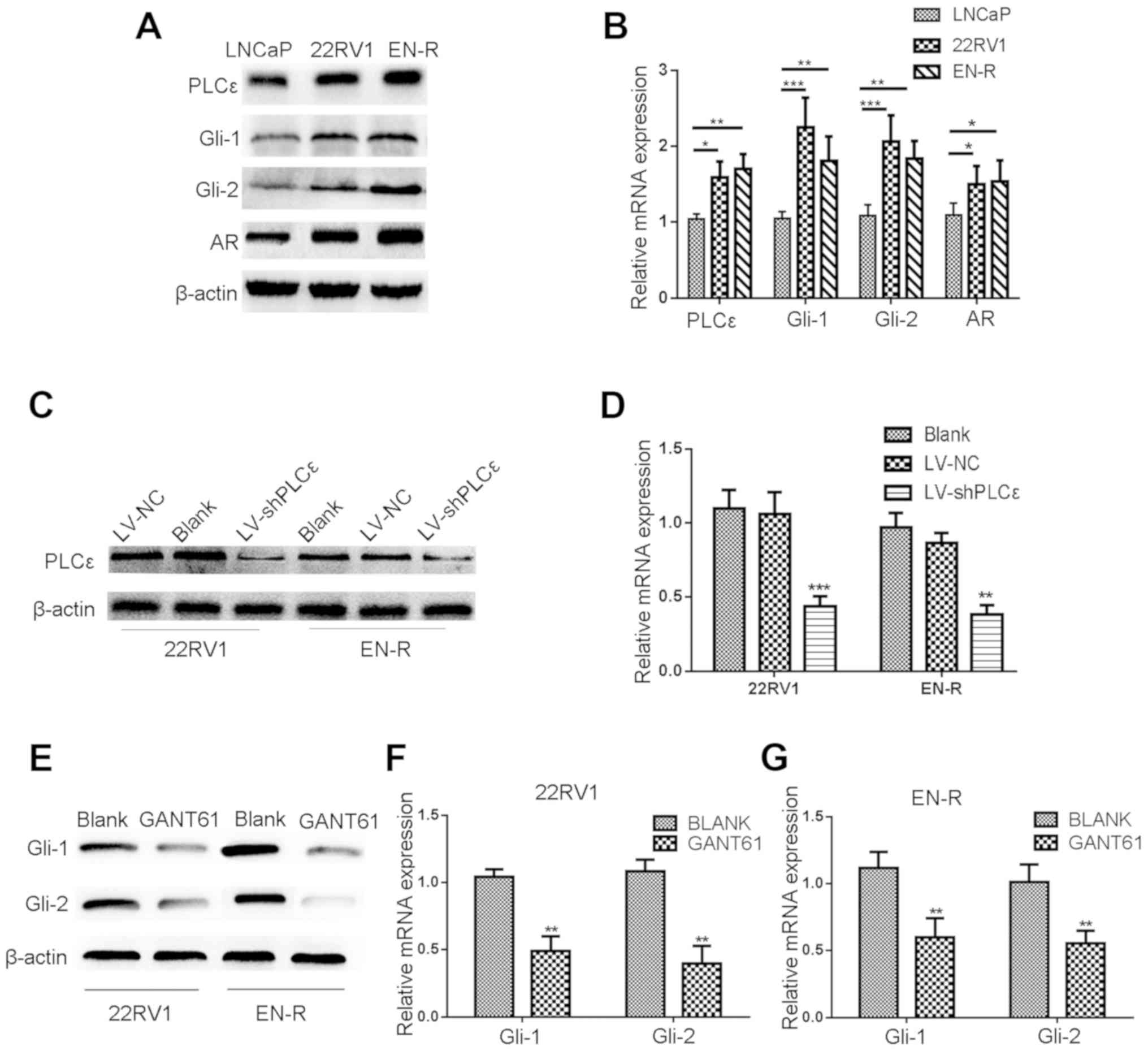 | Figure 1.Lentivirus-shPLCε and GANT61 inhibit
the protein and mRNA expression level of PLCε and Gli-1/Gli-2. (A)
PLCε was highly expressed in the PCa (LNCaP) and CRPC (22RV1 and
EN-R) cell lines. Gli-1/Gli-2 and AR expression levels were higher
in CRPC (22RV1 and EN-R) cells compared to PCa (LNCaP) cells as
shown by western blotting. (B) The mRNA expression levels of PLCε,
Gli-1/Gli-2 and AR in LNCap, 22RV1 and EN-R cells were detected by
RT-qPCR (*P<0.05, **P<0.01 and ***P<0.001). (C) PLCε
knockdown was induced by transfecting lentivirus (LV)-shPLCε into
CRPC cell line. Total cellular proteins were detected by western
blotting. (D) Relative PLCε mRNA expression level was determined by
RT-qPCR. β-actin was used as an internal control (**P<0.01 and
***P<0.001 compared with the blank control). (E-G) GANT61
inhibited the protein and mRNA expression level of Gli-1/Gli-2.
β-actin was used as an internal control (**P<0.01 compared with
the blank control). PLCε, phospholipase Cε; Gli, glioma-associated
homolog; PCa, prostate cancer; CRPC, castration-resistant PCa;
EN-R, enzalutamide-resistant cell line; AR, androgen receptor;
RT-qPCR, reverse transcription quantitative polymerase chain
reaction. |
As previously described (12,13),
PLCε knockdown inhibits the proliferation and invasion of PCa
cells, but whether it has the same inhibitory effect on CRPC cells
remains unknown. GANT61, a specific Gli inhibitor, has been
reported to suppress the growth of tumor cells by inhibiting the
expression of Gli-1/Gli-2 (29,30).
To determine the effect of targeting PLCε and Gli-1/Gli-2 on the
proliferative and invasive capacity of 22RV1 and EN-R cells,
lentivirus-shPLCε (LV-shPLCε) was used to knock down PLCε (Fig. 1C and D). Meanwhile, GANT61 (10 µM)
was used to inhibit the expression of Gli-1/Gli-2 in 22RV1 and EN-R
cells (Fig. 1E-G). CCK-8 and colony
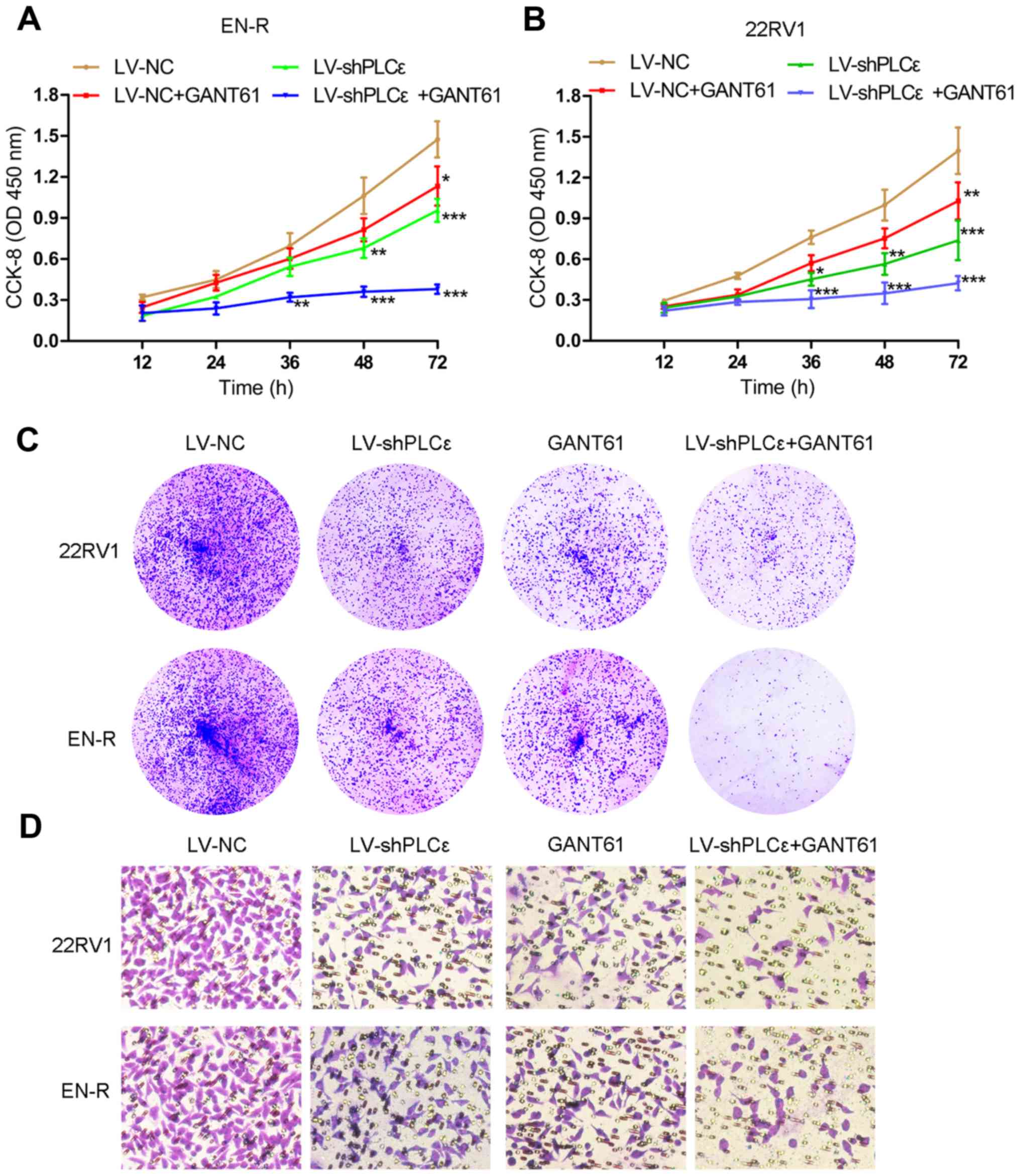
formation assays showed that PLCε knockdown and GANT61 treatment
suppressed the proliferation of the 22RV1 and EN-R cells. The
combination of PLCε knockdown and GANT61 enhanced the inhibitory
effect (Fig. 2A-C). Accordingly,
Transwell assay showed that PLCε knockdown and GANT61 treatment
inhibited the invasion of the 22RV1 and EN-R cells. The combination
of PLCε knockdown and GANT61 enhanced the inhibitory effect
(Fig. 2D). These findings indicated
that PLCε knockdown and Gli-1/Gli-2 inhibition suppressed cell
proliferation and invasion in CRPC cells.
Inhibition of PLCε sensitizes CRPC
cells to enzalutamide in vitro
It is well known that AR amplification is
responsible for CRPC. As previously mentioned, the knockdown of
PLCε inhibited the expression of AR in PCa cells. The inhibition of
Gli has been shown to regulate the activity of AR, rather than its
expression (25). However, whether
PLCε knockdown or Gli inhibition regulates the sensitivity of CRPC
cells to enzalutamide by interacting with the AR signaling pathway
remains unclear. We therefore hypothesized that the knockdown of
PLCε and inhibition of Gli-1/Gli-2 could sensitize CRPC cells to
enzalutamide. To test this, the IC50 value of
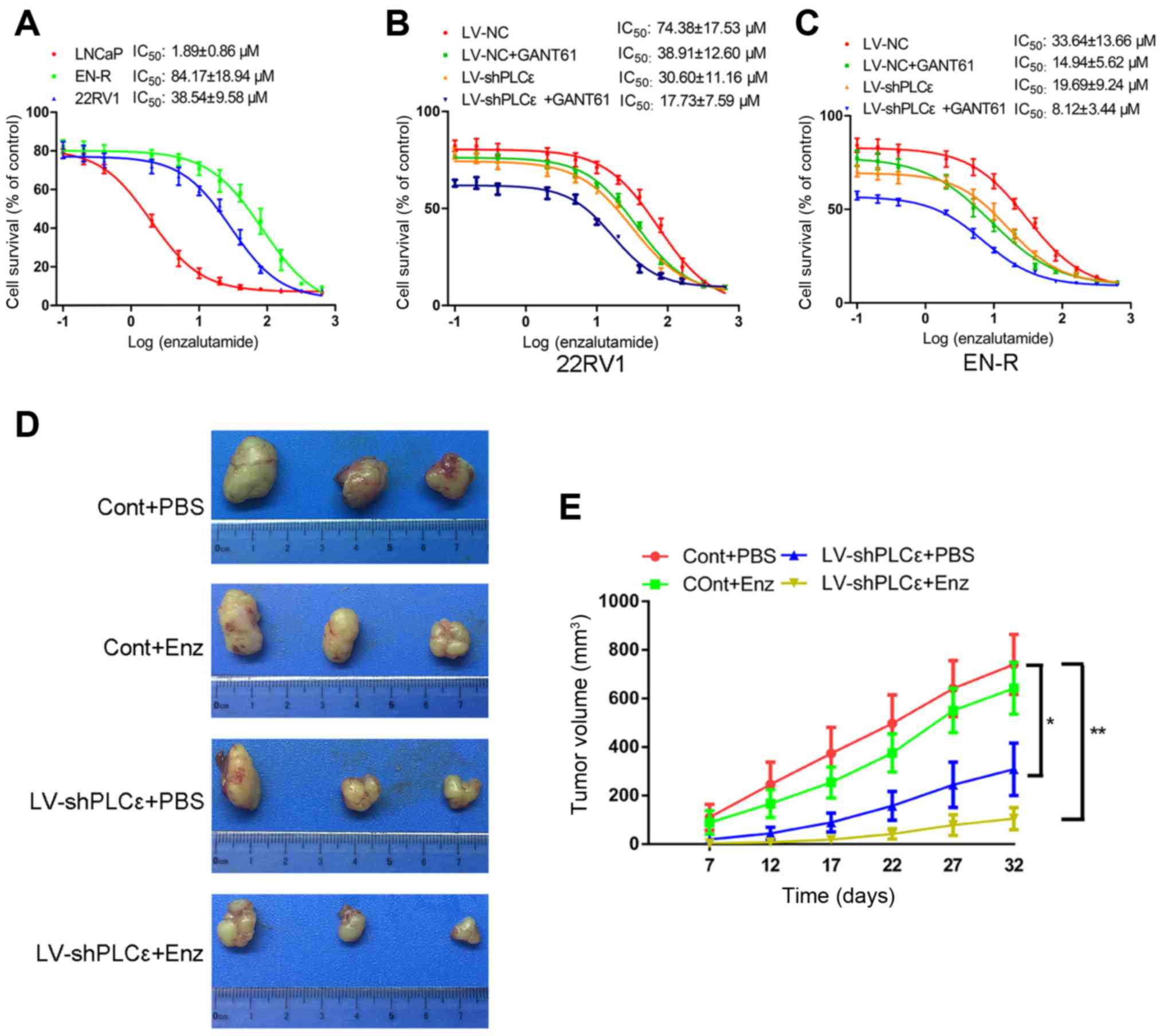
enzalutamide for the LNCaP, 22RV1and EN-R cells was determined by
CCK-8 assay, as 1.89±0.86, 38.54±9.58 and 84.17±18.94 µM,
respectively (Fig. 3A). The
IC50 value was detected again in 22RV1and EN-R cells
following treatment with LV-shPLCε, GANT61 10 µM or combination
treatment. The data showed that, although both LV-shPLCε and GANT61
reduced the IC50 value of enzalutamide to varying
degrees, the combination treatment significantly decreased the
IC50 value of 22RV1 and EN-R cells, as compared to
either single treatment (Fig. 3B and
C). These data indicated that PLCε knockdown and Gli-1/Gli-2
inhibition could sensitize CRPC cells to enzalutamide in
vitro.
PLCε knockdown inhibits the expression
of Gli-1/Gli-2 through the non-canonical Hh signaling pathway in
CRPC cells
As described above, the increase in PLCε expression
was positively correlated with an increase in Gli-1/Gli-2
expression in PCa and CRPC tissue samples. In addition, PLCε
knockdown or inhibition of Gli-1/Gli-2 expression had a similar
inhibitory effect on cell proliferation and invasion in the 22RV1
and EN-R cells. Both helped sensitize 22RV1 and EN-R cells to
enzalutamide in vitro. We therefore, hypothesized that there
is an interaction between PLCε and Gli-1/Gli-2. In order to
determine that interaction, western blot analysis and RT-qPCR were
performed. PLCε knockdown inhibited the mRNA and protein expression
levels of Gli-1/Gli-2, while their inhibition did not affect the
expression of PLCε in 22RV1 and EN-R cells, suggesting that PLCε is
the upstream regulator of Gli-1/Gli-2 (Fig. 4). However, following PLCε knockdown,
Smo (upstream regulatory gene of Gli in the classical Hh signaling
pathway) showed no significant mRNA and protein expression change
(Fig. 4). These data indicated that
PLCε knockdown inhibited the expression of Gli-1/Gli-2 through the
non-canonical Hh signaling pathway.
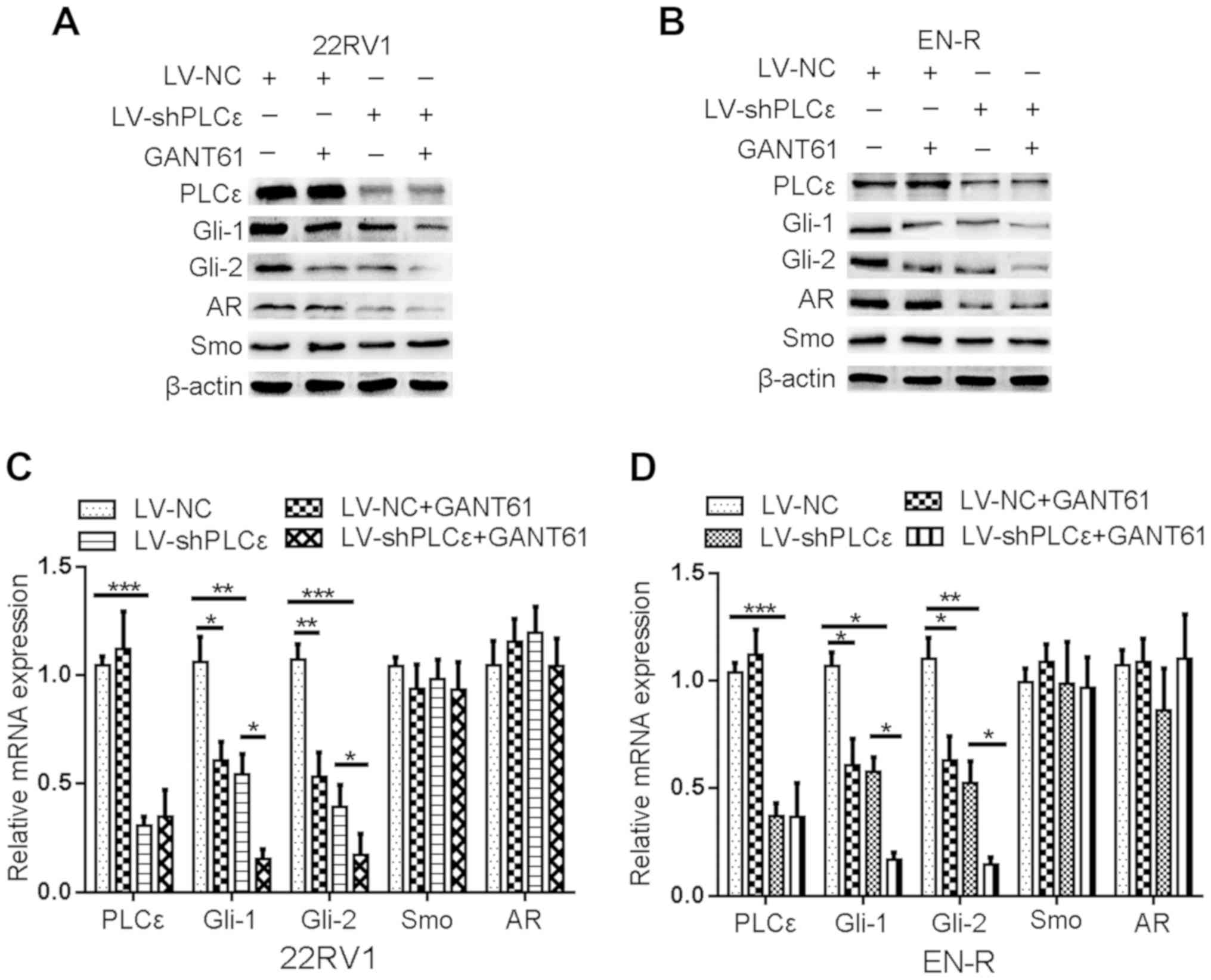 | Figure 4.PLCε knockdown inhibits the
expression of Gli-1/Gli-2. (A and B) The expression of PLCε, Gli-1,
Gli-2, AR and Smo in 22RV1 and EN-R cells was examined by western
blotting. The cells were treated with LV-shPLCε and/or GANT61 10 µM
for 72 h. β-actin served as a loading control. (C and D) The
relative mRNA expression of PLCε, Gli-1, Gli-2, AR and Smo in 22RV1
and EN-R cells was examined by RT-qPCR (*P<0.05, **P<0.01 and
***P<0.001). PLCε, phospholipase Cε; Gli, glioma-associated
homolog; AR, androgen receptor; Smo, smoothened; EN-R,
enzalutamide-resistant cell line; RT-qPCR, reverse transcription
quantitative polymerase chain reaction. |
PLCε knockdown increases the
sensitivity of CRPC cells to enzalutamide by suppressing AR
expression and nuclear translocation via different signaling
pathways
To further investigate the mechanism by which PLCε
increases the sensitivity of CRPC cells to enzalutamide, the effect
of PLCε on AR expression and subcellular localization in EN-R cells
was assessed using immunofluorescence and western blotting. It was
found that PLCε knockdown inhibited AR translocation from the
cytoplasm to the nucleus (Fig. 5A).
Next, the overexpression of plasmid ad-Gli1and ad-Gli2 was used to
overexpress Gli-1 and Gli-2 in EN-R cells (Fig. 5B-D). Of note, it was found that the
AR nuclear translocation was significantly increased in the
LV-shPLCε+ad-Gli2 group compared to the LV-shPLCε+ad-Gli1 group
(Fig. 5A). In addition, the
cytoplasmic vs. nuclear distribution of AR in EN-R cells was
examined using western blot analysis. The data showed that AR
expression was decreased in the nucleus following PLCε knockdown,
while the overexpression of Gli-2 reversed this effect (Fig. 5E), indicating that PLCε knockdown
suppressed AR nuclear translocation via the Gli-2/AR signaling
pathway.
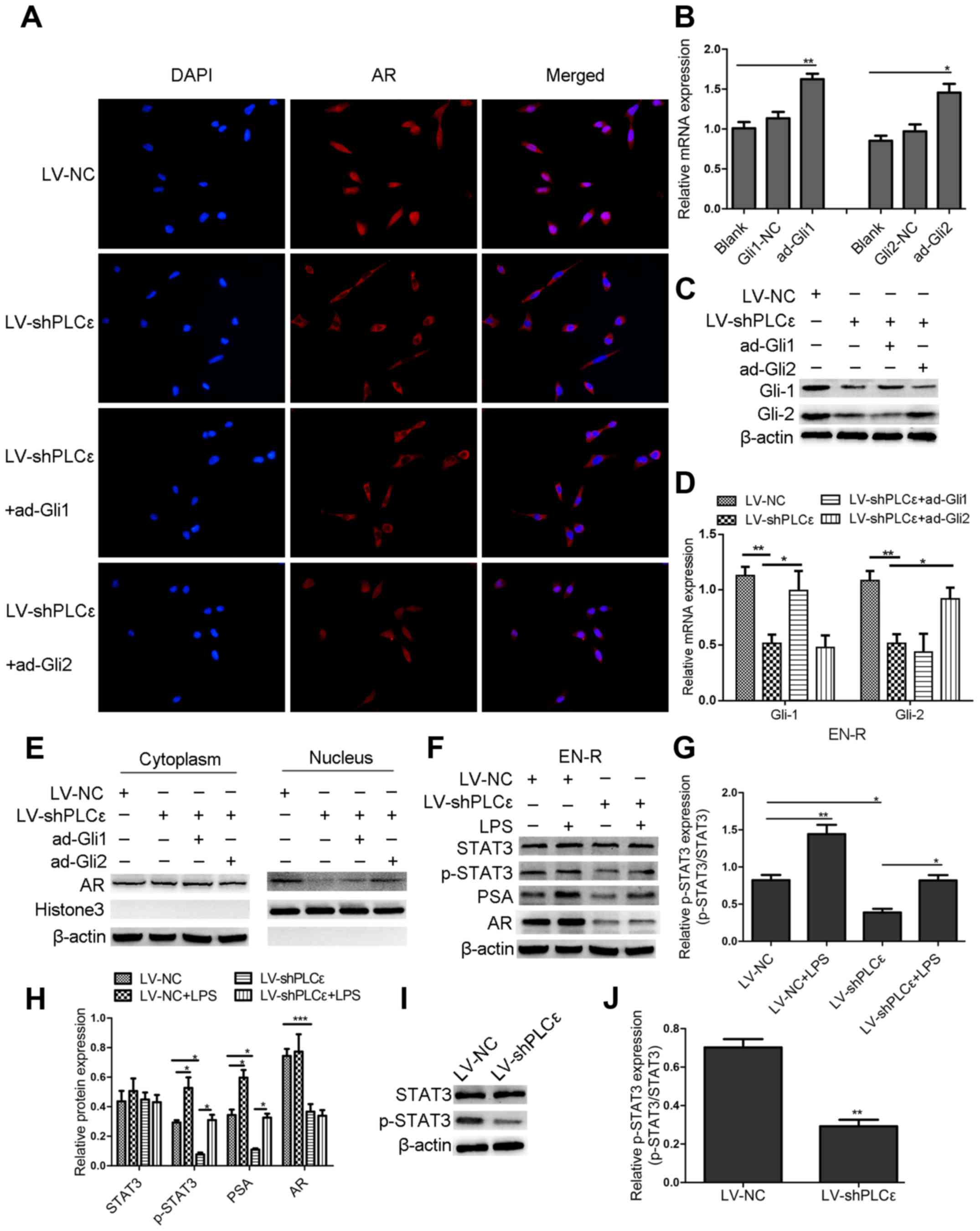 | Figure 5.PLCε knockdown suppresses AR
expression and nuclear translocation via different signaling
pathways. (A) Immunofluorescence demonstrated AR intracellular
distribution at 48 h following infection with LV-shPLCε and
ad-Gli1/ad-Gli2 in EN-R cells. Magnification, ×400. PLCε knockdown
inhibited AR nuclear translocation in EN-R cells. However, the
overexpression of Gli-2 reversed the inhibitory effect produced by
PLCε. (B) The relative mRNA expression level of Gli-1 and Gli-2
following treatment with an overexpression plasmid of ad-Gli1,
ad-Gli2 was examined by RT-qPCR and β-actin served as loading
control (NC stands for empty vector plasmid group (*P<0.05,
**P<0.01). (C) Protein expression level of Gli-1 and Gli-2
following treatment with the overexpression plasmid of ad-Gli1,
ad-Gli2 and PLCε knockdown. (D) The mRNA expression level of Gli-1
and Gli-2 following treatment with overexpression plasmid of
ad-Gli1, ad-Gli2 and PLCε knockdown (*P<0.05, **P<0.01). (E)
Western blotting showed that PLCε knockdown significantly decreased
the AR expression in the nucleus. However, the expression of AR in
the nucleus increased following Gli-2 overexpression. (F-H) Western
blotting indicated that PLCε knockdown inhibited the PSA expression
via the p-STAT3 signaling pathway (The results are represented as
the mean ± SD; *P<0.05, **P<0.01 and ***P<0.001). (I and
J) Western blotting showed that the downregulation of PLCε
decreased the p-STAT3 protein expression in EN-R cells (The results
are represented as the mean ± SD; **P<0.01). PLCε, phospholipase
Cε; AR, androgen receptor; Gli, glioma-associated homolog; EN-R,
enzalutamide-resistant cell line. |
The data also showed that the knockdown of PLCε
inhibited the protein expression of AR but not its mRNA expression.
However, the inhibition of the Gli-1/Gli-2 expression by GANT61 did
not lead to any significant change in the AR mRNA and protein
expression (Fig. 4). We also found
that the protein expression of phosphorylated STAT3 (p-STAT3) was
decreased following PLCε knockdown in EN-R cells, but there was no
obvious change in the total STAT3 (t-STAT3) protein expression
(Fig. 5I and J). We therefore
hypothesized that PLCε affects the expression of AR through p-STAT3
signaling. To verify the role of the p-STAT3 signaling pathway and
in the regulation of AR, LPS a p-STAT3 agonist, were used to
activate p-STAT3. The data showed that the expression of p-STAT3
was activated by LPS, and that the activated p-STAT3 increased the
expression of AR target gene PSA, rather than that of AR. PLCε
knockdown inhibited the expression of p-STAT3 and PSA protein
(Fig. 5F-H). These findings
indicated that the knockdown of PLCε inhibited the PSA expression
via the p-STAT3 signaling pathway.
PLCε inhibition sensitizes CRPC cells
to enzalutamide in vivo
The role of LV-shPLCε in the sensitivity to
enzalutamide in EN-R-derived tumors was also examined in
vivo. Castrated nude mice were injected subcutaneously with
2×106 EN-R (Cont group) or LV-shPLCε EN-R cells
(LV-shPLCε group) into the right flank area and then divided into
four groups: Cont+PBS, LV-shPLCε+PBS, Cont+Enz and LV-shPLCε+Enz
(n=3 per group). According to the research of Guerrero et al
(31), 10 and 50 mg/kg of
enzalutamide significantly inhibited the volume in a mouse LNCaP-AR
xenograft model. Considering the toxicity of drugs, the dose of 10
mg/kg enzalutamide was chosen for animal experiments. The mice were
treated with vehicle PBS or enzalutamide at 10 mg/kg by oral gavage
(5 days on, 2 days off). The mice were then sacrificed, and tumors
were collected and measured. No animal was sacrificed due to
reaching humane endpoints. No mice were found to exhibit multiple
tumors. The largest diameter of the tumors was 1.5 cm. As shown in
Fig. 3D and E, no significant
difference was identified in the tumor volume between the Cont+PBS
and the Cont+Enz groups. The tumor volume in the LV-shPLCε+PBS
group was smaller than that of the Cont+PBS and Cont+Enz groups. In
addition, the LV-shPLCε+Enz group exhibited the smallest tumor
volume of the four groups. These results suggested that PLCε
knockdown inhibited the tumor growth of the EN-R cell xenografts
and contributed to the sensitization of CRPC cells to enzalutamide
in vivo.
Increased PLCε expression contributes
to unfavorable disease phenotype and poor outcome of patients with
PCa
A total of 30 BPH samples, 64 PCa and 27 CRPC
samples were collected (Table I).
The expression of PLCε was detected in the BPH, PCa and CRPC
tissues by immunohistochemistry. A high expression of PLCε was
identified in most PCa (49/64) and CRPC (21/27) tissue samples. By
contrast, none of the BPH tissues stained positive for PLCε
(Fig. 6A). Noticeably, the
expression of PLCε in the CRPC tissues was significantly higher
than that in the PCa tissues (P=0.0447; Fig. 6B). It was also observed that the
expression of Gli-1 and Gli-2 in the tumor (PCa and CRPC) tissues
was significantly higher than that in the benign (BPH) tissues
(Fig. 6A, C and D). Interestingly,
the expression of Gli-1, but not Gli-2, in the CRPC tissues was
significantly upregulated, as compared to that in the PCa tissues
(P=0.152; Fig. 6C). In addition,
Pearson's linear correlation results showed that PLCε expression
was positively correlated with Gli-1 (r=0.581, P=0.01; Fig. 6E) and Gli-2 expression (r=0.409,
P=0.034; Fig. 6F).
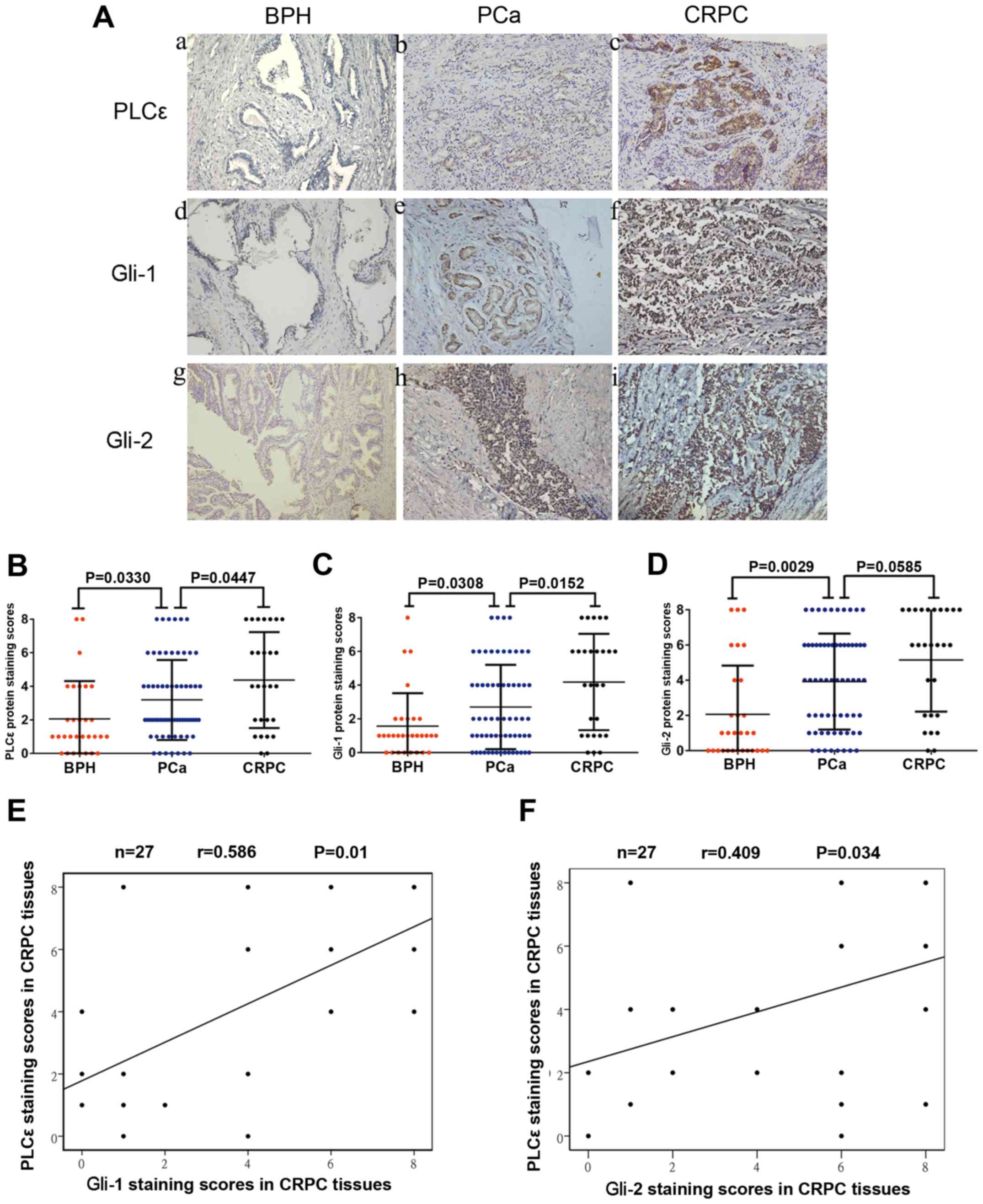 | Figure 6.Increased PLCε expression in PCa and
CRPC tissues is associated with Gli-1/Gli-2 expression. (A)
Immunohistochemical staining in prostate tissues. Magnification,
×200. (a, d and g) BPH tissues. (b, e and h) PCa tissues. (c, f and
i) CRPC tissues. (B-D) Average staining scores for PLCε, Gli-1 and
Gli-2 in BPH, PCa and CRPC tissues. (E and F) Correlation curve
analysis for PLCε vs. Gli-1/Gli-2 staining scores in CRPC tissues.
P<0.05 was considered statistically significant. PLCε,
phospholipase Cε; BPH, benign prostatic hyperplasia; PCa, prostate
cancer; CRPC, castration-resistant PCa; Gli, glioma-associated
homolog. |
 | Table I.Demographic and clinical
characteristics of the PCa patients. |
Table I.
Demographic and clinical
characteristics of the PCa patients.
|
|
| PLCε |
|
|---|
|
|
|
|
|
|---|
| Patient
characteristics Total number | Overall n=64 | Negative 15/64
(23%) | Positive 49/64
(77%) | P-value |
|---|
| Age, years |
|
|
| 0.405 |
|
Median | 69 | 65 | 70 |
|
|
Quartiles 25–75 | 59–74 | 58–74 | 60–75 |
|
| PSA ng/ml |
|
|
| 0.108 |
|
Median | 47.41 | 82.14 | 38.46 |
|
|
Quartiles 25–75 | 23.70–89.89 | 31.18–144.35 | 21.44–86.39 |
|
| Histological stage,
n (%) |
|
|
| 0.028a |
|
Ta-T1 | 27 (42%) | 10 (67%) | 17 (35%) |
|
|
T2-T4 | 37 (58%) | 5 (33%) | 32 (65%) |
|
| Gleason score, n
(%) |
|
|
| 0.053 |
|
<7 | 21 (33%) | 8 (53%) | 13 (27%) |
|
| ≥7 | 43 (67%) | 7 (47%) | 36 (73%) |
|
| Bone metastases, n
(%) |
|
|
| 0.147 |
|
Yes | 25 (39%) | 6 (40%) | 19 (39%) |
|
| No | 39 (61%) | 9 (60%) | 30 (61%) |
|
To investigate the effects of PLCε on the disease
phenotype and patient clinical outcome in PCa and CRPC, the
association between various clinical parameters and the PLCε
expression in PCa and CRPC tissues was analyzed. On the one hand,
it was found that the PLCε expression was positively correlated
with the tumor histological stage of the PCa patients (P=0.028;
Table I) and bone metastasis of the
CRPC patients (P=0.030; Table II).
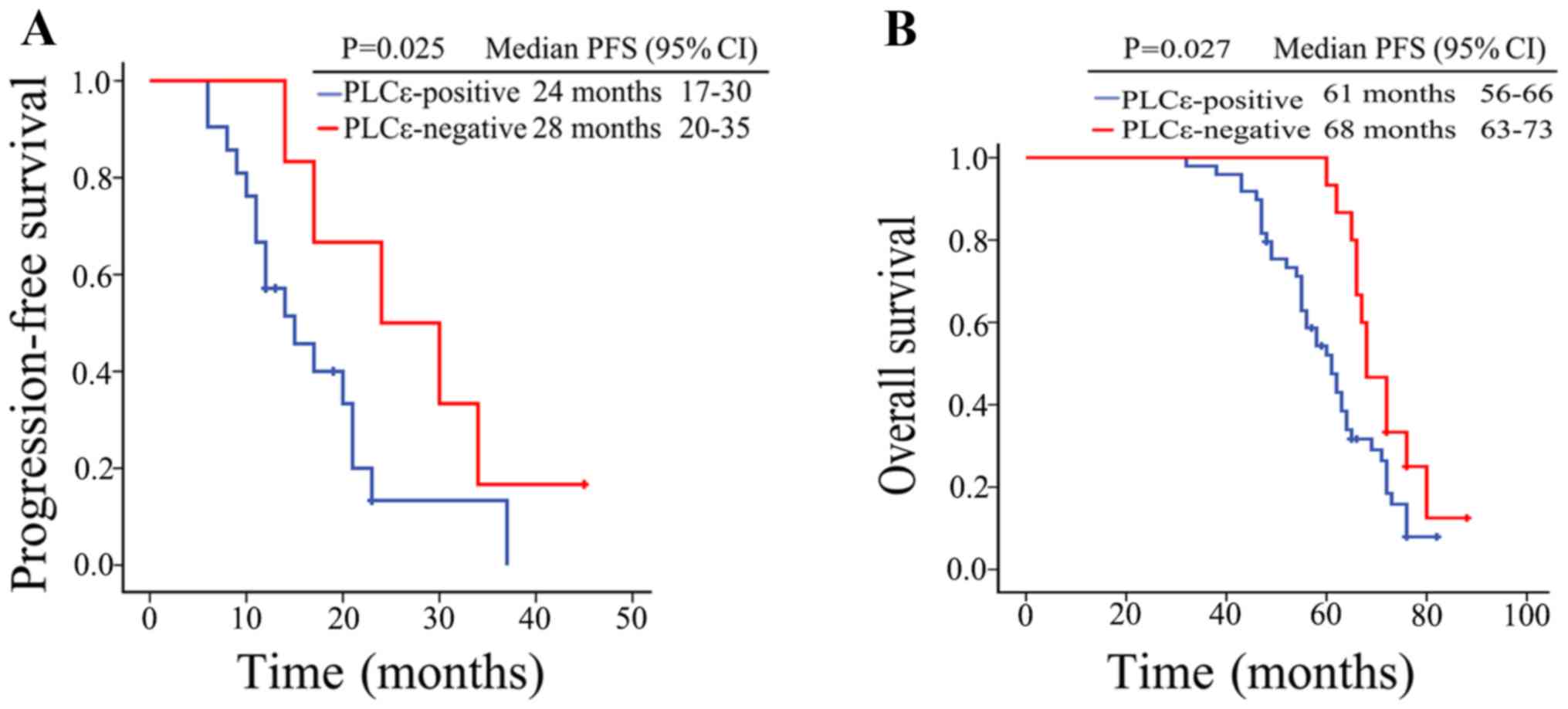
On the other hand, Kaplan-Meier survival analysis results showed
that the median progression-free survival (PFS) was 24 months (95%
CI, 17–30 months) in the PLCε-positive CRPC patients, while the
median PFS was 28 months (95% CI, 21–35 months) in the
PLCε-negative patients; PLCε positivity in the CRPC tissues was
associated with a shorter PFS in the CRPC patients (P=0.025;
Fig. 7A). Similarly, the overall
survival (OS) of PCa patients with a negative PLCε expression was
significantly longer than that of PCa patients with a positive PLCε
expression (P=0.027; Fig. 7B).
These findings revealed that high PLCε expression contributes to
unfavorable disease phenotype and poor outcomes in patients with
PCa.
| Table II.Demographic and clinical
characteristics of the CRPC patients. |
Table II.
Demographic and clinical
characteristics of the CRPC patients.
|
|
| PLCε |
|
|---|
|
|
|
|
|
|---|
| Characteristics
Total number | Overall n=27 | Negative 6/27
(22%) | Positive 21/27
(78%) | P-value |
|---|
| Age, years |
|
|
| 0.884 |
|
Median | 71 | 73 | 70 |
|
|
Quartiles 25–75 | 65–77 | 65–78 | 66–79 |
|
| PSA ng/ml |
|
|
| 0.726 |
|
Median | 32.23 | 28.26 | 32.24 |
|
|
Quartiles 25–75 | 16.76–56.18 | 16.13–58.32 | 24.90–58.12 |
|
| Bone metastases, n
(%) |
|
|
| 0.030a |
|
Yes | 15 (56%) | 1 (17%) | 14 (67%) |
|
| No | 12 (44%) | 5 (83%) | 7
(33%) |
|
Discussion
As a member of the PLC family, PLCε does not only
have typical catalytic domains XY and C2, but also a
carboxyl-terminated Ras domain RA and amino acid guanylate exchange
factor domain CDC25 (32,33). These special domains can activate
multiple signaling pathways and promote the development of
malignant tumors. Current experimental and clinical data suggest
that PLCε may play a pivotal role in the regulation of the
development and progression of different types of cancer, such as
skin cancer (10), lung cancer
(34), gastric cancer and
esophageal squamous cell carcinoma (35,36).
We previously investigated the expression level of PLCε in bladder
cancer and renal cell carcinoma using immunohistochemistry. It was
found to be significantly higher than that in normal tissue,
suggesting that PLCε is closely associated with the occurrence and
development of renal cell carcinoma and bladder cancer (37,38).
Recently, our research (12,13)
has shown that the high expression of PLCε is associated with cell
proliferation and invasion in PCa. In the present study, it was
found that the expression of PLCε was significantly increased in
PCa and CRPC, as compared to BPH tissues, and that this high
expression was linked to poor prognosis in patients with PCa. PLCε
knockdown inhibited the proliferation and invasion of CRPC cells.
These results revealed that PLCε is involved in the progression of
CRPC and further demonstrated that PLCε is an oncogene, which is
consistent with previous literature reports.
The activity of Hh/Gli signaling has been shown to
be elevated in PCa (20), and is
more intensive in those of metastatic PCa than in specimens of
localized-PCa (21). Consistent
with these reports, it was found in the present study that Hh/Gli
signaling was markedly increased in CRPC, as compared to PCa
tissues, suggesting that Hh/Gli signaling plays an important role
in the emergence and development of CRPC. In addition, it was found
that the increase of PLCε was positively correlated with the
increase in Gli-1/Gli-2 expression, and that PLCε knockdown and/or
inhibition of Gli-1/Gli-2 using GANT61 inhibited the proliferation
and invasion of CRPC cells. PLCε knockdown also interacted with the
Hh/Gli signaling pathway by decreasing the mRNA and protein
expression of the Hh target genes Gli-1 and Gli-2. However, Smo,
the upstream regulatory gene of Gli did not decrease, suggesting
that PLCε could be involved in the progression of CRPC cells
through the non-canonical Hh/Gli signaling pathway.
Androgen-deprivation therapy (ADT) remains the main
treatment strategy for PCa patients (39). However, the transition from
hormone-sensitive PCa to CRPC is inevitable during treatment. AR
amplification is a major mechanism of resistance to ADT (40). Enzalutamide is one of the first-line
drugs for the treatment of CRPC; it has not only been shown to
potently inhibit the binding of androgens to AR, but also the
nuclear translocation and subsequent binding of the AR-ligand
complex to DNA, thereby inhibiting the transcription of AR target
genes (41). However, enzalutamide
resistance can occur within a few months. In the present study, it
was found that PLCε knockdown increased the sensitivity of CRPC
cells to enzalutamide, which might be achieved by PLCε knockdown
suppressing AR nuclear translocation via the Gli-2/AR signaling
pathway, or PLCε knockdown inhibiting the expression of the AR
target gene PSA via the p-STAT3 signaling pathway. Although PLCε
knockdown decreased the protein expression of AR, we failed to
determine the exact mechanism in this study.
GANT61, a specific Gli-1/Gli-2 inhibitor, has been
reported to significantly decrease cell survival of PC3 and 22Rv1
cells (29). As a small molecule
that blocks the transcription of essential Hh proteins, GANT61 has
a broad spectrum of potential mechanisms (either through the
specific inhibition of Hh signaling or not) by which it can elicit
anticancer effects, as it targets many of the ‘classical hallmarks
of cancer’ (42). In present study,
GANT61 was shown to not only suppress proliferation and invasion in
CRPC cells, but also sensitize CRPC cells to enzalutamide,
suggesting that GANT61 may be a novel adjuvant drug for use in the
treatment of CRPC. However, the potential mechanism of
sensitization is unclear. Although it was demonstrated herein that
downregulation of Gli-2, rather than Gli-1, inhibited AR
translocation, that inhibition did not affect AR expression, which
was consistent with a previous study on androgen-independent PCa
cells (25). It was therefore
deduced that GANT61 restored the sensitivity of CRPC cells to
enzalutamide not only via the AR pathway, but also other non-AR
pathways, such as cell autophagy. Autophagy is an important
mechanism of resistance to enzalutamide in CRPC; GANT61 induces
autophagy in cancer cells, and the enhancement of autophagy
increases the sensitivity of enzalutamide in CRPC (43,44).
Taken together, the signal transduction relationship among PLCε,
Gli and AR was that: GANT61 inhibited AR nuclear translocation by
inhibiting Gli2, GANT61 also increased the drug sensitivity of
enzalutamide through other non-AR signaling pathways. PLCε
inhibited the activity of AR axis in three ways. On the one hand,
PLCε inhibited AR nuclear translocation by inhibiting the
non-classical Hh/Gli-2 signaling pathway. On the other hand, PLCε
inhibited the expression of AR target gene PSA by inhibiting the
expression of p-STAT3. In addition PLCε inhibited the expression of
AR protein, but the mechanism has not been clarified. Certain
issues were encountered in this study, including the failure to
define the mechanism through which PLCε affects AR protein
expression and GANT61 affects drug sensitivity to enzalutamide in
CRPC through non-AR pathways. Further research is required to
confirm these. In immunohistochemical experiments, the expression
status of immunostaining was reviewed and scored based on the
proportion of positive cells and staining intensity by pathologist,
and the staining intensity was defined as follows: 0 (no staining),
1 (light yellow), 2 (light brown), 3 (brown) and 4 (deep brown) and
the immunoreactivity ratio was 0 (0% immunoreactive cells), 1
(<5% immunoreactive cells), 2 (5–50% immunoreactive cells), 3
(>50–75% immunoreactive cells) and 4 (>75% immunoreactive
cells). The final immunoreactivity score was defined as the sum of
both parameters. However, the lack of positive control is still a
limitation of this study.
In combination, the present results showed that PLCε
expression and Hh/Gli signaling were excessively activated in the
majority of CRPC tissues. PLCε positivity was linked to poor
prognosis in patients with PCa. The knockdown of PLCε significantly
suppressed CRPC cell proliferation and invasion by reducing
Gli-1/Gli-2 expression. More importantly, PLCε knockdown increased
the sensitivity of CRPC cells to enzalutamide by suppressing AR
nuclear translocation via the Gli-2/AR signaling pathway and
inhibiting PSA expression via the p-STAT3 signaling pathway. In
addition, the combination of PLCε knockdown and GANT61
significantly sensitized CRPC cells to enzalutamide. Targeting PLCε
may therefore serve as a potential treatment strategy for CRPC, and
GANT61 may prove to be a promising agent for use in CRPC
treatment.
Acknowledgements
The authors would like to thank Dr Dingyun Liu,
Department of Pathology, Fuling Center Hospital of Chongqing City,
Chongqing, China, for providing technical assistance with the
immunohistochemistry assays.
Funding
The present study was supported by a grant from the
Natural Science Foundation of China (no. 81802543).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
WS, XW and CL designed the experiments. WS, LL, ZD
and MY collected the specimens and analyzed the clinical data. WS,
LL, MY, YG, HC and ZQ conducted the experiments and collected the
data. WS and LL co-wrote the manuscript. CL and XW provided
technical support of this research project and supervised the
progress of the experiments. ZD, ZQ and MY analyzed the statistical
data. WS, LL and ZD assembled and designed the figures. All authors
read and approved the final manuscript and agree to be accountable
for all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
Chongqing Medical University (Chongqing, China). Informed consent
was obtained from the patients or their family members. All animal
experiments were approved as well by the Ethics Committee of
Chongqing Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Du LB, Li HZ, Wang XH, Zhu C, Liu QM, Li
QL, Li XQ, Shen YZ, Zhang XP, Ying JW, et al: Analysis of cancer
incidence in Zhejiang cancer registry in China during 2000 to 2009.
Asian Pac J Cancer Prev. 15:5839–5843. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qu M, Ren SC and Sun YH: Current early
diagnostic biomarkers of prostate cancer. Asian J Androl.
16:549–554. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu C, Armstrong C, Zhu Y, Lou W and Gao
AC: Niclosamide enhances abiraterone treatment via inhibition of
androgen receptor variants in castration resistant prostate cancer.
Oncotarget. 7:32210–32220. 2016.PubMed/NCBI
|
|
5
|
Scher HI, Fizazi K, Saad F, Taplin ME,
Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et
al: Increased survival with enzalutamide in prostate cancer after
chemotherapy. N Engl J Med. 367:1187–1197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tombal B, Borre M, Rathenborg P, Werbrouck
P, Van Poppel H, Heidenreich A, Iversen P, Braeckman J, Heracek J,
Baskin-Bey E, et al: Long-term efficacy and safety of enzalutamide
monotherapy in hormone-naïve prostate cancer: 1- and 2-year
open-label follow-up results. Eur Urol. 68:787–794. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Beer TM, Armstrong AJ, Rathkopf DE, Loriot
Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J,
Chowdhury S, et al: Enzalutamide in metastatic prostate cancer
before chemotherapy. N Engl J Med. 371:424–433. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song C, Hu CD, Masago M, Kariyai K,
Yamawaki-Kataoka Y, Shibatohge M, Wu D, Satoh T and Kataoka T:
Regulation of a novel human phospholipase C, PLCepsilon, through
membrane targeting by Ras. J Biol Chem. 276:2752–2757. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bunney TD and Katan M: PLC regulation:
Emerging pictures for molecular mechanisms. Trends Biochem Sci.
36:88–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bai Y, Edamatsu H, Maeda S, Saito H,
Suzuki N, Satoh T and Kataoka T: Crucial role of phospholipase
Cepsilon in chemical carcinogen-induced skin tumor development.
Cancer Res. 64:8808–8810. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang LD, Zhou FY, Li XM, Sun LD, Song X,
Jin Y, Li JM, Kong GQ, Qi H, Cui J, et al: Genome-wide association
study of esophageal squamous cell carcinoma in Chinese subjects
identifies susceptibility loci at PLCE1 and C20 or f54. Nat Genet.
42:759–763. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang X, Fan Y, Du Z, Fan J, Hao Y, Wang J,
Wu X and Luo C: Knockdown of phospholipase Cε (PLCε) inhibits cell
proliferation via phosphatase and tensin homolog deleted on
chromosome 10 (PTEN)/AKT signaling pathway in human prostate
cancer. Med Sci Monit. 24:254–263. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Wu X, Ou L, Yang X, Wang X, Tang
M, Chen E and Luo C: PLCε knockdown inhibits prostate cancer cell
proliferation via suppression of Notch signalling and nuclear
translocation of the androgen receptor. Cancer Lett. 362:61–69.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Buonamici S, Williams J, Morrissey M, Wang
A, Guo R, Vattay A, Hsiao K, Yuan J, Green J, Ospina B, et al:
Interfering with resistance to smoothened antagonists by inhibition
of the PI3K pathway in medulloblastoma. Sci Transl Med.
2:51ra702010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wahid M, Jawed A, Mandal RK, Dar SA, Khan
S, Akhter N and Haque S: Vismodegib, itraconazole and sonidegib as
hedgehog pathway inhibitors and their relative competencies in the
treatment of basal cell carcinomas. Crit Rev Oncol Hematol.
98:235–241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shigemura K, Huang WC, Li X, Zhau HE, Zhu
G, Gotoh A, Fujisawa M, Xie J, Marshall FF and Chung LW: Active
sonic hedgehog signaling between androgen independent human
prostate cancer cells and normal/benign but not cancer-associated
prostate stromal cells. Prostate. 71:1711–1722. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamamichi F, Shigemura K, Behnsawy HM,
Meligy FY, Huang WC, Li X, Yamanaka K, Hanioka K, Miyake H, Tanaka
K, et al: Sonic hedgehog and androgen signaling in tumor and
stromal compartments drives epithelial-mesenchymal transition in
prostate cancer. Scand J Urol. 48:523–532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Behnsawy HM, Shigemura K, Meligy FY,
Yamamichi F, Yamashita M, Haung WC, Li X, Miyake H, Tanaka K,
Kawabata M, et al: Possible role of sonic hedgehog and
epithelial-mesenchymal transition in renal cell cancer progression.
Korean J Urol. 54:547–554. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lauth M, Bergstrom A, Shimokawa T and
Toftgard R: Inhibition of GLI-mediated transcription and tumor cell
growth by small-molecule antagonists. Proc Natl Acad Sci USA.
104:8455–8460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gonnissen A, Isebaert S and Haustermans K:
Hedgehog signaling in prostate cancer and its therapeutic
implication. Int J Mol Sci. 14:13979–14007. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Karhadkar SS, Bova GS, Abdallah N, Dhara
S, Gardner D, Maitra A, Isaacs JT, Berman DM and Beachy PA:
Hedgehog signalling in prostate regeneration, neoplasia and
metastasis. Nature. 431:707–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Y, Bieber MM and Teng NN: Hedgehog
signaling regulates drug sensitivity by targeting ABC transporters
ABCB1 and ABCG2 in epithelial ovarian cancer. Mol Carcinog.
53:625–634. 2014.PubMed/NCBI
|
|
23
|
Sims-Mourtada J, Izzo JG, Ajani J and Chao
KS: Sonic Hedgehog promotes multiple drug resistance by regulation
of drug transport. Oncogene. 26:5674–5679. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Singh S, Chitkara D, Mehrazin R, Behrman
SW, Wake RW and Mahato RI: Chemoresistance in prostate cancer cells
is regulated by miRNAs and Hedgehog pathway. PLoS One.
7:e400212012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen M, Feuerstein MA, Levina E, Baghel
PS, Carkner RD, Tanner MJ, Shtutman M, Vacherot F, Terry S, de la
Taille A and Buttyan R: Hedgehog/Gli supports androgen signaling in
androgen deprived and androgen independent prostate cancer cells.
Mol Cancer. 9:892010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen G, Goto Y, Sakamoto R, Tanaka K,
Matsubara E, Nakamura M, Zheng H, Lu J, Takayanagi R and Nomura M:
GLI1, a crucial mediator of sonic hedgehog signaling in prostate
cancer, functions as a negative modulator for androgen receptor.
Biochem Biophys Res Commun. 404:809–815. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Du Z, Li L, Sun W, Wang X, Zhang Y, Chen
Z, Yuan M, Quan Z, Liu N, Hao Y, et al: HepaCAM inhibits the
malignant behavior of castration-resistant prostate cancer cells by
downregulating Notch signaling and PF-3084014 (a gamma-secretase
inhibitor) partly reverses the resistance of refractory prostate
cancer to docetaxel and enzalutamide in vitro. Int J Oncol.
53:99–112. 2018.PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gonnissen A, Isebaert S, McKee CM, Dok R,
Haustermans K and Muschel R: The hedgehog inhibitor GANT61
sensitizes prostate cancer cells to ionizing radiation both in
vitro and in vivo. Oncotarget. 7:84286–84298. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Q, Xu R, Zeng C, Lu Q, Huang D, Shi
C, Zhang W, Deng L, Yan R, Rao H, et al: Down-regulation of Gli
transcription factor leads to the inhibition of migration and
invasion of ovarian cancer cells via integrin β4-mediated FAK
signaling. PLoS One. 9:e883862014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guerrero J, Alfaro IE, Gomez F, Protter AA
and Bernales S: Enzalutamide, an androgen receptor signaling
inhibitor, induces tumor regression in a mouse model of
castration-resistant prostate cancer. Prostate. 73:1291–1305. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hicks SN, Jezyk MR, Gershburg S, Seifert
JP, Harden TK and Sondek J: General and versatile autoinhibition of
PLC isozymes. Mol Cell. 31:383–394. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wing MR, Snyder JT, Sondek J and Harden
TK: Direct activation of phospholipase C-epsilon by Rho. J Biol
Chem. 278:41253–41258. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Luo XP: Phospholipase C epsilon-1 inhibits
p53 expression in lung cancer. Cell Biochem Funct. 32:294–298.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Abnet CC, Freedman ND, Hu N, Wang Z, Yu K,
Shu XO, Yuan JM, Zheng W, Dawsey SM, Dong LM, et al: A shared
susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma
and esophageal squamous cell carcinoma. Nat Genet. 42:764–767.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Smrcka AV, Brown JH and Holz GG: Role of
phospholipase Cε in physiological phosphoinositide signaling
networks. Cell Signal. 24:1333–1343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Du HF, Ou LP, Song XD, Fan YR, Yang X, Tan
B, Quan Z, Luo CL and Wu XH: Nuclear factor-kappa B signaling
pathway is involved in phospholipase Cepsilon-regulated
proliferation in human renal cell carcinoma cells. Mol Cell
Biochem. 389:265–275. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang X, Ou L, Tang M, Wang Y, Wang X, Chen
E, Diao J, Wu X and Luo C: Knockdown of PLCepsilon inhibits
inflammatory cytokine release via STAT3 phosphorylation in human
bladder cancer cells. Tumour Biol. 36:9723–9732. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sharp A, Welti J, Blagg J and de Bono JS:
Targeting androgen receptor aberrations in castration-resistant
prostate cancer. Clin Cancer Res. 22:4280–4282. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ardiani A, Gameiro SR, Kwilas AR, Donahue
RN and Hodge JW: Androgen deprivation therapy sensitizes prostate
cancer cells to T-cell killing through androgen receptor dependent
modulation of the apoptotic pathway. Oncotarget. 5:9335–9348. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tran C, Ouk S, Clegg NJ, Chen Y, Watson
PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, et al:
Development of a second-generation antiandrogen for treatment of
advanced prostate cancer. Science. 324:787–790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gonnissen A, Isebaert S and Haustermans K:
Targeting the Hedgehog signaling pathway in cancer: Beyond
smoothened. Oncotarget. 6:13899–13913. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tang X, Deng L, Chen Q, Wang Y, Xu R, Shi
C, Shao J, Hu G, Gao M, Rao H, et al: Inhibition of Hedgehog
signaling pathway impedes cancer cell proliferation by promotion of
autophagy. Eur J Cell Biol. 94:223–233. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nguyen HG, Yang JC, Kung HJ, Shi XB, Tilki
D, Lara PN Jr, DeVere White RW, Gao AC and Evans CP: Targeting
autophagy overcomes enzalutamide resistance in castration-resistant
prostate cancer cells and improves therapeutic response in a
xenograft model. Oncogene. 33:4521–4530. 2014. View Article : Google Scholar : PubMed/NCBI
|