Introduction
Small molecules that interact with specific targets
in signaling and epigenetic pathways have been demonstrated to be
useful agents for modulating cell differentiation. These agents can
enhance reprogramming of differentiated cells to pluripotent stem
cells (1–4) and also induce epithelial-mesenchymal
transition (EMT) in cancer cells in vitro (5–9). EMT
is a crucial trans-differentiation process that occurs during
embryogenesis and in adult tissue repair and also plays an
important role during cancer progression (10,11).
During EMT, epithelial cells acquire mesenchymal cell morphology,
leading to increased migration capacity and stemness (12–15).
Numerous studies have demonstrated the involvement
of epigenetic changes such as histone acetylation/deacetylation in
cancer development. Therefore, targeting histone deacetylase
complexes (HDACs) in cancer is a potential anticancer therapy
approach (16). The small molecule
valproic acid (VPA) inhibits HDACs, resulting in relief of
HDAC-dependent transcriptional repression. Previous studies
revealed that VPA regulated EMT-related gene expression in alveolar
epithelial cells and induced invasion of human melanoma cells in
vitro via regulation of N-cadherin expression and RhoA activity
(17,18).
TGF-β1 signaling plays a vital role in EMT. The
small molecule SB431542 inhibits activation of the activin
receptor-like kinase (ALK) receptors 4, 5 and 7, which are key
members of the TGF-β signaling pathway. Inhibition of TGF-β1/Smad
signaling was revealed to lead to repression of EMT in esophageal
squamous cell carcinoma by downregulating the expression of
N-cadherin and vimentin and upregulating the expression of
E-cadherin. TGF-β1 signaling blockade attenuated gastric cancer
cell-induced peritoneal mesothelial cell fibrosis and alleviated
peritoneal dissemination both in vitro and in vivo,
and sensitized drug-resistant pancreatic cancer cells to
gemcitabine (19–21).
Disrupted activation of
mitogen-activated/extracellular- signal-regulated protein kinases
(MEK) has been indicated in developmental malformation and
carcinogenesis. MEK inhibitor PD0325901 significantly reduced the
growth of papillary thyroid carcinoma cells in vitro and
in vivo (22). Combination
treatment using PD0325901 and SRC inhibitors synergistically
abrogated tumor cell growth and induced mesenchymal-epithelial
transition (MET) in non-small cell lung carcinoma. Combined
inhibition of PKC and MEK inhibited the proliferation of melanoma
cell lines (23).
Glycogen synthase kinase-3 (GSK3) is an important
regulator of cellular metabolism that plays complicated roles in
some types of cancer. The GSK3 inhibitor CHIR99021 dose-dependently
reduced the proliferation of three NSCLC cell lines (6). Inhibition of glycogen synthase
kinase-3 activity triggered an apoptotic response through
JNK-dependent mechanisms and induced prosurvival autophagic signals
in human pancreatic cancer cells (24).
We hypothesized that although some targeted
therapeutics initially were designed as antiproliferative agents,
some agents may also inhibit EMT initiation since proliferation and
EMT are regulated by similar signaling pathways. Therefore,
targeting EMT using inhibitors represents a promising therapeutic
strategy for treatment of cancer with an invasive phenotype.
It is recognized that a signaling pathway may
crosstalk with other pathways and lead to malignant cells mutating
quickly due to genome instability and DNA imprecise replication,
causing frequent failure of single-agent targeted therapy (25–27).
As a result, studies have evaluated the use of drug combinations in
abrogating signaling pathways crosstalk to regain treatment
sensitivity (28,29). If one inhibitor is not effective in
a particular cancer cell, other inhibitors may still be functional
leaving cancer cells treated with combination inhibitors less
chance for survival.
In the present study, we aimed to clarify the
potential effects of various inhibitors the HDAC inhibitor VPA (V),
the GSK3 inhibitor CHIR99021 (C), the MEK inhibitor PD0325901 (P)
and the ALK inhibitor SB431542 (S) and their combinations on
several cancer cell lines, including the uterine cervix carcinoma
cell line HeLa, the bladder cancer cell line 5637 and the squamous
cell carcinoma cell line SCC-15.
Materials and methods
Animals and cell lines
Thirty 4-week old female athymic Swiss nude mice
(weight range, 16–20 g) were purchased from the Medical
Experimental Animal Center of Guangdong Province China. They were
maintained in a specific pathogen-free (SPF) room at constant
temperature (20°C) and humidity (45%) on a 12-h light/dark cycle,
and had free access to sterile laboratory chow and tap water.
Animal experiment approval was obtained from the Ethics Committee
of Peking University Shenzhen Hospital (Shenzhen, China). Chinese
laws concerning animal caring and treatment were strictly followed
during the experiments, and the study complied with the ARRIVE
guidelines (https://www.nc3rs.org.uk/arrive-guidelines).
The HeLa uterine cervix carcinoma cell line
(CBP60232), the SCC-15 squamous cell carcinoma cell line (CBP61034)
and the 5637 bladder cancer cell line (CBP60309) were provided by
COBIOR Cell Bank (Nanjing, China; http://www.cobioer.com), and maintained in Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% heat inactivated
fetal bovine serum (FBS; HyClone; GE Healthcare, Chicago, IL, USA),
100 U/ml potassium penicillin, 100 U/ml streptomycin and 2 mM
glutamine. Cells were incubated at 37°C in a chamber with 5%
CO2 and 95% humidity.
Small molecules
SB431542, PD0325901 and CHIR99021 (Stemgent, Inc.,
Cambridge, MA, USA) and VPA (Sigma- Aldrich; Merck KGaA, Darmstadt,
Germany) were dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich;
Merck KGaA) and diluted in DMEM. Cells were treated with medium
containing a single inhibitor or inhibitor combinations, and medium
was changed every other day. The recommended concentration of the
single inhibitor (0.5 µM/l PD0325901, 2 µM/l SB431542, 3 µM/l
CHIR99021 and 0.5 mM/l VPA) in this experiment was safe to cell
viability (safe concentration) and previously used in reprogramming
of differentiated cells to pluripotent stem cells by promoting MET,
the reverse process of EMT, which is an important process in cell
dedifferentiation (1–4).
Quantitative real-time RT-PCR
Total RNA was isolated using the RNase Mini kit
(Qiagen, Inc., Valencia, CA, USA). A High Capacity RNA-to-cDNA kit
(Applied Biosystems; Thermo Fisher Scientific, Inc.) was used to
reverse-transcribe 1 µg of RNA into cDNA with MMLV Reverse
Transcriptase and random primers according to the manufacturer's
instructions (85°C, 5 sec and 37°C for 1 h). Real-time PCR was
performed using specific primers under optimal reaction conditions
(94°C, 30 sec; 60°C, 35 sec; 72°C, 40 sec; 30 cycles) to quantify
gene expression using SYBR-Green RT-PCR reagents (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The 2−ΔΔCq
method (30) was used to analyze
relative gene expression level. Table
I lists the primers used in the present study.
 | Table I.Primers used in quantitative
real-time RT-PCR. |
Table I.
Primers used in quantitative
real-time RT-PCR.
| Genes | PCR primers |
|---|
| E-cadherin | Sense primer:
5′-TTCTGCTGCTCTTGCTGTTT-3′ |
|
| Antisense primer:
5′-TGGCTCAAGTCAAAGTCCTG-3′ |
| N-cadherin | Sense primer:
5′-CCTGCGCGTGAAGGTTTGCC-3′ |
|
| Antisense primer:
5′-CCAAGCCCCGCACCCACAA-3′ |
| Vimentin | Sense primer:
5′-GCCCTTAAAGGAACCAATGA-3′ |
|
| Antisense primer:
5′-AGCTTCAACGGCAAAGTTCT-3′ |
| Oct3/4 | Sense primer:
5′-GACAGGGGGAGGGGAGGAGCTAGG-3′ |
|
| Antisense primer:
5′-CTTCCCTCCAACCAGTTGCCCCAAAC-3′ |
| Sox2 | Sense primer:
5′-GGGAAATGGGAGGGGTGCAAAAGAGG-3′ |
|
| Antisense primer:
5′-TTGCGTGAGTGTGGATGGGATTGGTG-3′ |
| Nanog | Sense primer:
5′-CAGCCCCGATTCTTCCACCAGTCCC-3′ |
|
| Antisense primer:
5′-CGGAAGATTCCCAGTCGGGTTCACC-3′ |
| GAPDH | Sense primer:
5′-CATCAATGGAAATCCCATCA-3′ |
|
| Antisense primer:
5′-TTCTCCATGGTGGTGAAGAC-3′ |
Cell viability assay
Cell viability was assessed using the Cell Counting
Kit-8 (CCK-8) assay (Roche Molecular Biochemicals, Mannheim,
Germany). Briefly, treated cells were seeded in 96-well plates at a
density of 1×103/well and cultured for another 24 h.
Cell viability was evaluated using the kit according to the
manufacturer's instructions and results were detected using a Model
680 microplate reader (Bio-Rad Laboratories, Hercules, CA, USA).
All experiments were conducted in triplicate.
Cell invasion assays
Treated cells (2×104/well) were seeded in
a 24-well plate with a Millicell cell culture insert containing
polycarbonate filter membranes with 8-µm diameter pores (BD
Biosciences, Franklin Lakes, NJ, USA). Cells were maintained in
DMEM supplemented with 1% FBS. The lower chamber contained DMEM
supplemented with 10% FBS. The plates were incubated for 48 h at
37°C in a 5% CO2 humidified incubator. Invasive cells in
the lower chamber were stained with 0.1% (w/v) crystal violet.
Images were captured and visualized using QCapture Pro (version 7.0
software; QImaging, Surrey, BC, Canada).
Western blot analysis
Total protein was extracted using the EpiQuik Whole
Cell Extraction kit (AmyJet Scientific, Inc., Wuhan, China) and
protein concentration was measured by Bradford DC protein assay
(Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of protein
samples (30 µg) were separated on 12% Bis-Tris polyacrylamide gel
by electrophoresis and transferred to polyvinylidene difluoride
membranes (EMD Millipore, Temecula, CA, USA) which was pre-blocked
with 5% (w/v) skim milk (BD Biosciences) in a rotator for 2 h at
room temperature. The membranes were incubated with
anti-phosphorylated Smad1/2 (cat. no. 8828), Erk1/2 (cat. no. 9101)
(Cell Signaling Technology, Inc., Danvers, MA, USA) and GS (cat.
no. 07-817) primary antibodies (EMD Mililipore) (all at 1:1,000
dilution) at 4°C, for 12 h, followed by incubation with a goat
anti-rabbit polyclonal horseradish peroxidase (HRP)-conjugated
secondary antibody (cat. no. A0208) (Beyotime Institute of
Biotechnology, Shanghai, China) (1:10,000 dilution in 1% milk/TBS)
for 2 h at room temperature. Protein bands were visualized using a
chemiluminescence detection kit (Amersham Biosciences; GE
Healthcare, Piscataway, NJ, USA). The visualized bands were
analyzed by Tanon 5220S image analysis system (Tanon Image software
version 1.0; Tanon Science and Technology Co., Ltd., Shanghai,
China).
Cell cycle analysis
Cell cycle fractions were determined by propidium
iodide (PI) nuclear staining (Beyotime Institute of Biotechnology).
Briefly, cells were grown on 6-well plates and treated with
inhibitors or DMSO as control for 6 days. Cells were then
harvested, fixed with 70% cold ethanol, and stained with PI
staining buffer [0.03 mg/ml PI, 0.1 mg/ml RNAse A, 0.1% Triton
X-100 in phosphate-buffered saline (PBS)] for 30 min at 25°C. Flow
cytometry was performed using a BD LSR II flow cytometer (BD
Biosciences). At least 50,000 cell events were collected per sample
and cell cycle analysis was performed using FlowJo software
(version 10.0; FlowJo, LLC, Ashland, OR, USA).
Sphere formation and stemness-related
marker examination
Cells in single cell suspension were plated into
ultralow attachment 60 mm plates (Corning Inc., Corning, NY, USA)
at a density of 1×105 cells/plate in stem cell media:
DMEM supplemented with 20 ng/ml human basic fibroblast growth
factor, 20 ng/ml epidermal growth factor (Invitrogen; Thermo Fisher
Scientific, Inc.), 100 ng/ml insulin (Thermo Fisher Scientific,
Inc.) and 100 U/ml penicillin/streptomycin. Cells were then
subjected to different inhibitor treatments. Single cell suspension
of the spheres was prepared for assessment of
CD24−/CD24+ percentage by flow cytometry.
In vivo cancer cell clump formation
test
Cells (5×105 cells) isolated from VPA and
V+C+S+P-treated HeLa, SCC-15 and 5637 cancer cell spheres were
injected subcutaneously into the flank of female athymic Swiss nude
mice, respectively (total 30 mice were randomly separated into six
groups, 5 mice in each group; two groups were assigned to each cell
line, one group for injection of VPA-treated cells, and the other
group for injection of V+C+S+P treated cells). Cancer cell clump
growth at the site of injection was monitored twice a week with
calipers. The mice were sacrificed 30 days after cancer cell
injection by cervical vertebra dissociation after pentobarbital
sodium anesthetization (40 mg/kg body weight). The cancer cell
clumps were excised and weighted.
Statistical analysis
All data are presented as the mean ± standard
deviation (SD) from three independent experiments (three
experiments were carried out under same conditions), except in the
in vivo cancer cell clump formation test. Data were analyzed
using SPSS software (version 11.0; SPSS, Inc., Chicago, IL, USA). A
Student's t-test was used in comparison between two groups, and
one-way analysis of variance (ANOVA) with Dunnett's test was used
in multiple comparisons to one control. P-values <0.05 were
considered to indicate a statistically significant difference.
Results
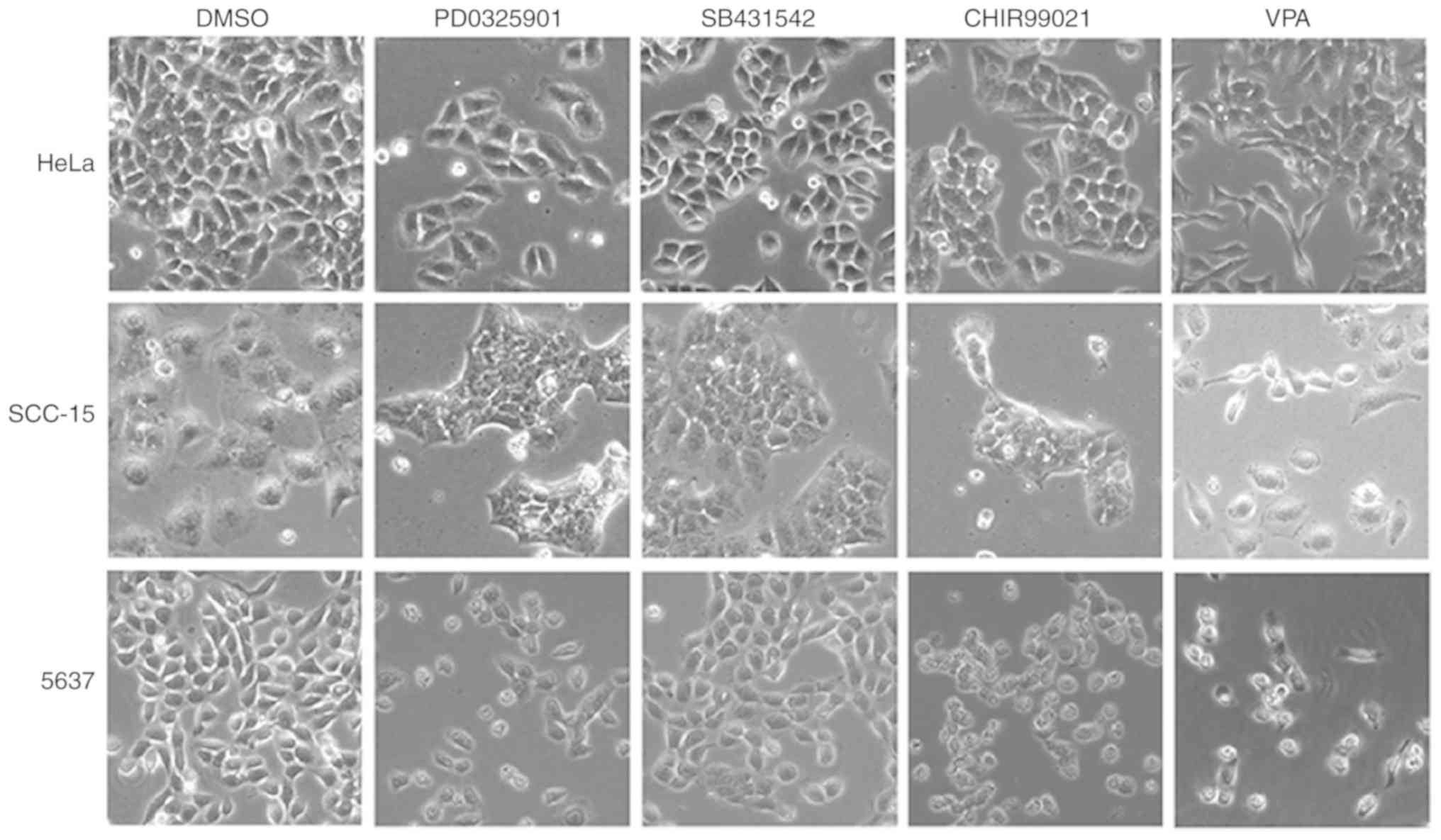
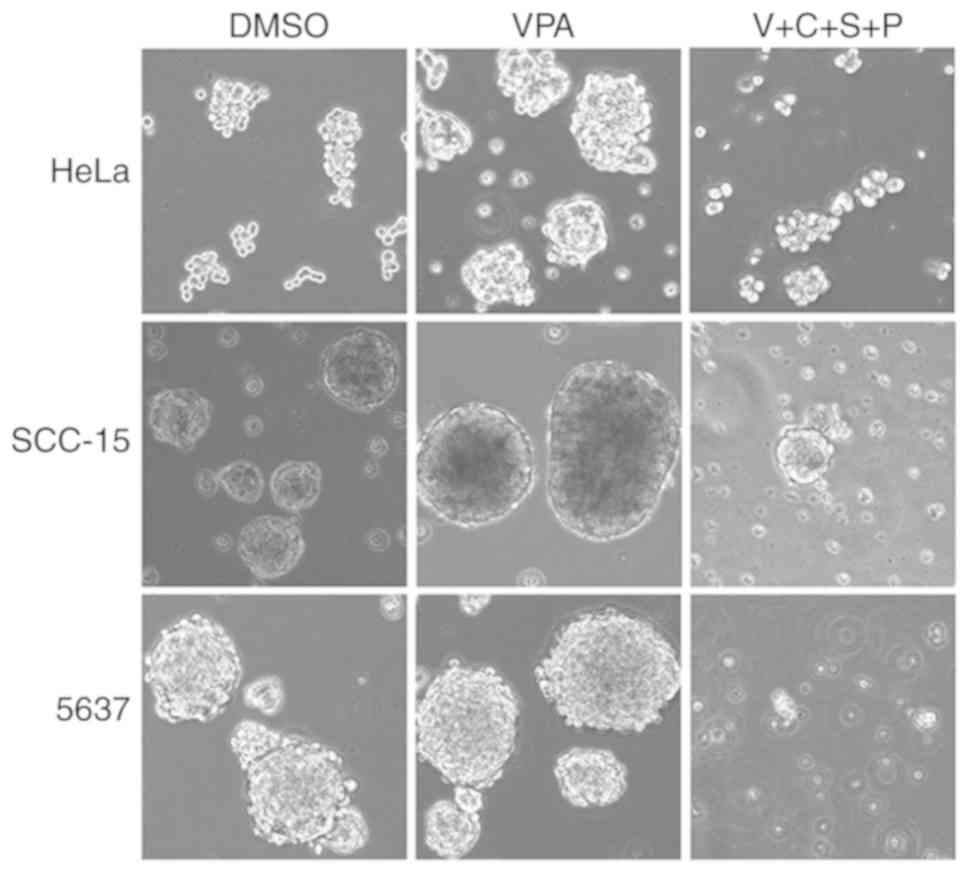
VPA induces EMT phenotype in cancer
cells
To investigate the influence of the small molecule
inhibitors on cancer cell morphology, HeLa, SCC-15 and 5637 cells
were treated with SB431542 (S; ALK inhibitor), PD0325901 (P; MEK
inhibitor), CHIR99021 (C; GSK3 inhibitor), and VPA (V; HDAC
inhibitor) at the recommended concentrations (0.5 µM/l PD0325901, 2
µM/l SB431542, 3 µM/l CHIR99021 and 0.5 mM/l VPA) for 4–6 days,
with medium changed every other day. Abundant spindle-shaped cells
were observed in all three cell lines treated with VPA, suggesting
that VPA treatment could lead to induction of EMT phenotype
(Fig. 1). In contrast, the other
three inhibitors did not exhibit similar effects in inducing
spindle cell morphology. Cells treated with the DMSO control
maintained their original morphology.
VPA promotes pluripotent transcription
factor and EMT-related gene expression
To examine whether VPA induced cancer cell EMT and
stemness, HeLa cells were treated with each of the inhibitors for 6
days and the gene expression levels of pluripotency-related
transcription factors and EMT-related factors were evaluated. In
HeLa cells treated with VPA, the gene expression of
pluripotency-related transcription factors Oct3/4, Sox2, Nanog and
EMT-related genes N-cadherin and vimentin were upregulated, while
the MET-related E-cadherin gene was downregulated compared with the
DMSO-treated control cells. In contrast, treatment with CHIR99021,
SB431542 or PD0325901 suppressed the gene expressions of
pluripotent-related transcription factors and EMT-related genes to
various levels (Fig. 2A).
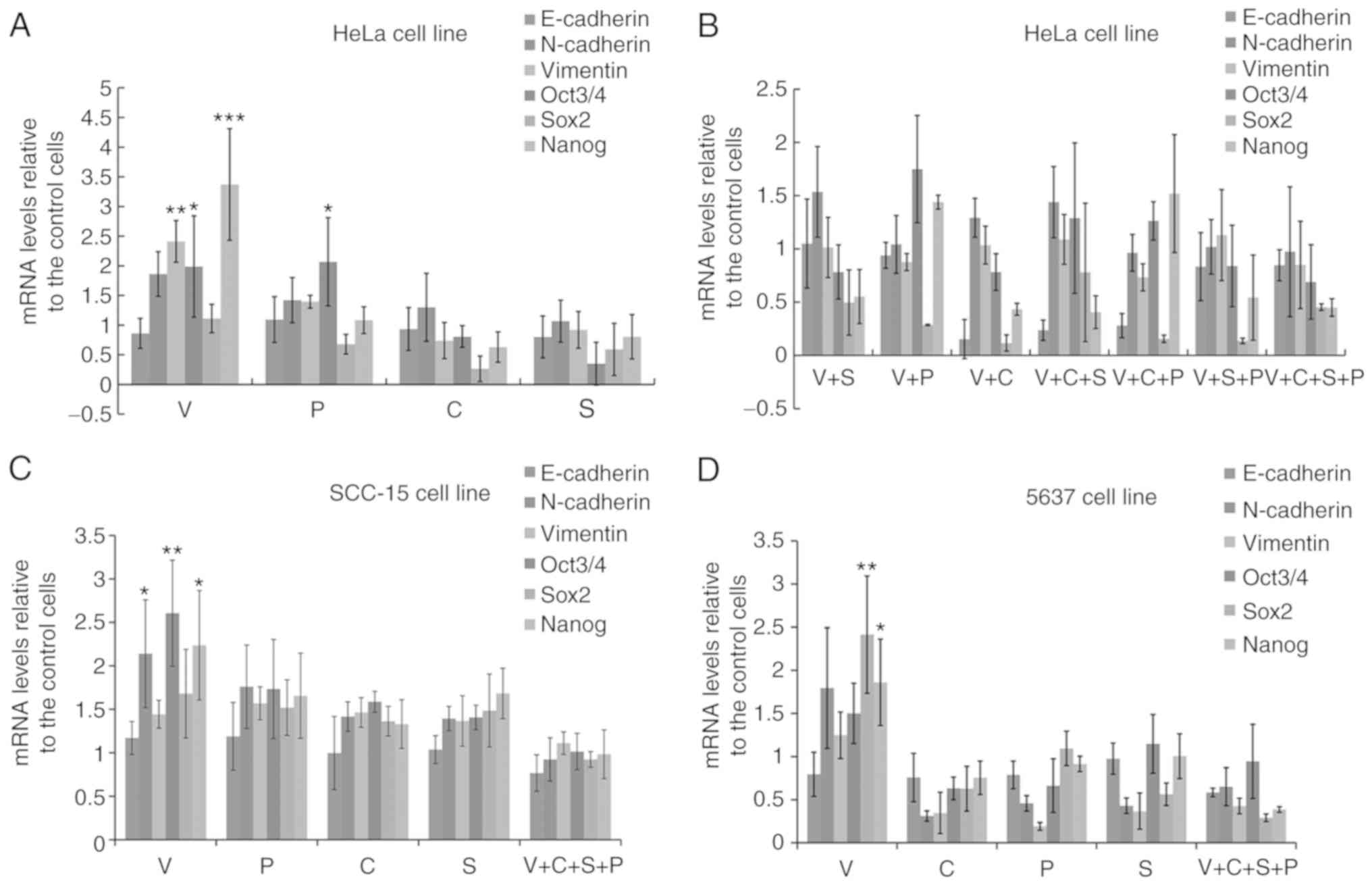 | Figure 2.VPA promotes pluripotent
transcription factor and EMT-related gene expression, and C+S+P
(CHIR99021+SB431542+PD0325901) combination abrogates the effect of
VPA. (A) Six-day VPA treatment upregulated the expression of
pluripotent-related transcription factors and EMT-related genes and
downregulated the MET-related E-cadherin gene in HeLa cells. (B)
Inhibitor and combination of inhibitors suppressed Oct3/4, Sox2,
Nanog, N-cadherin and vimentin expression down to differentiated
levels in HeLa cells. (C) C+S+P combination suppressed Oct3/4,
Sox2, Nanog, N-cadherin and vimentin expression of SCC-15 cells in
the presence of VPA. (D) C+S+P combination suppressed Oct3/4, Sox2,
Nanog, N-cadherin and vimentin expression of 5637 cells in the
presence of VPA. VPA, valproic acid; EMT, epithelial-mesenchymal
transition; MET, mesenchymal-epithelial transition. *P<0.05,
**P<0.01, ***P<0.001. |
Since single agent targeted therapy fails frequently
in cancer treatment, numerous researchers have conducted
investigation on a two-agent combination. Theoretically there is
still a risk that cancer cells could develop the ability to survive
with a two-agent combination. Thus, in the present study we
assessed three-agent combination on cancer cells trying to inhibit
signaling pathways in an extended range.
Since VPA upregulated EMT and cell
pluripotency-related gene expression, and CHIR99021, SB431542 or
PD0325901 suppressed the expression of these genes to various
extents, different combinations of inhibitors (V+S, V+P, V+C,
V+C+S, V+C+P, V+S+P, or V+C+S+P) were then used to treat HeLa cells
for 6 days to examine whether any of the inhibitors or inhibitor
combinations could counteract the effects of VPA. Single and
combination treatments could counteract the effect of VPA to
various extents. The C+S+P combination exerted the strongest
suppressive effect on pluripotency and EMT-related gene expression
in the presence of VPA compared with single and two inhibitor
combinations (Fig. 2B).
Since VPA upregulated EMT and cell
pluripotency-related gene expression in HeLa cells, and the C+S+P
combination suppressed the parameters to ultra-low levels in the
presence of VPA. In order to demonstrate this result, single
inhibitors and the C+S+P combination were assessed in the SCC-15
and 5637 cell lines and similar results were obtained (Fig. 2C and D). It was observed that under
C+S+P treatment, cells quickly lost cell viability, thus, 6 days
was set as the termination time-point in the following
experiments.
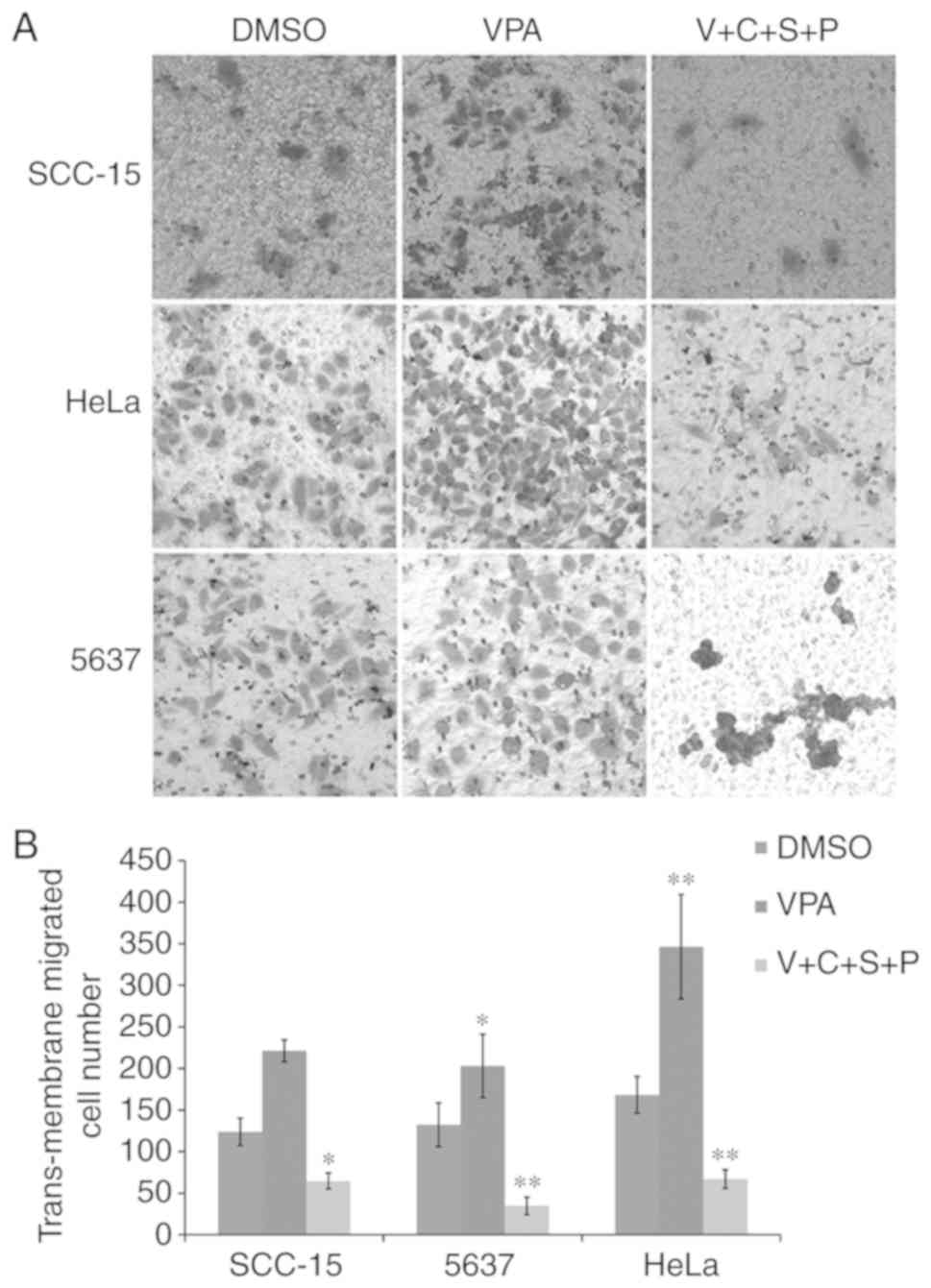
Inhibitor combination reduces
VPA-induced cell migration
The present results revealed that VPA could induce
EMT in cultured cancer cells, and C+S+P could effectively inhibit
VPA-induced EMT-related gene expression. Therefore, the three cell
lines were then treated with VPA or V+C+S+P for 6 days and the
migration ability of these cells was evaluated. In VPA-treated
cells, the number of migrated cells was increased compared with the
DMSO-treated control cells, while the C+S+P combination effectively
inhibited migration compared with the DMSO-treated control cells in
the presence of VPA (SCC-15 cells: VPA/DMSO, 221.3±13.05/124±16.37,
P<0.01; C+S+P/DMSO, 64.66±9.50/124±16.37, P<0.05; 5637 cells:
VPA/DMSO, 203±38.052/132.3±26.24, P<0.05; C+S+P/DMSO,
34.6±10.6/132.3±26.24, P<0.01; HeLa cells: VPA/DMSO,
346.3±62.7/168.3±22, P<0.01; C+S+P/DMSO, 67±11.2/168.3±22,
P<0.01) (Fig. 3A and B).
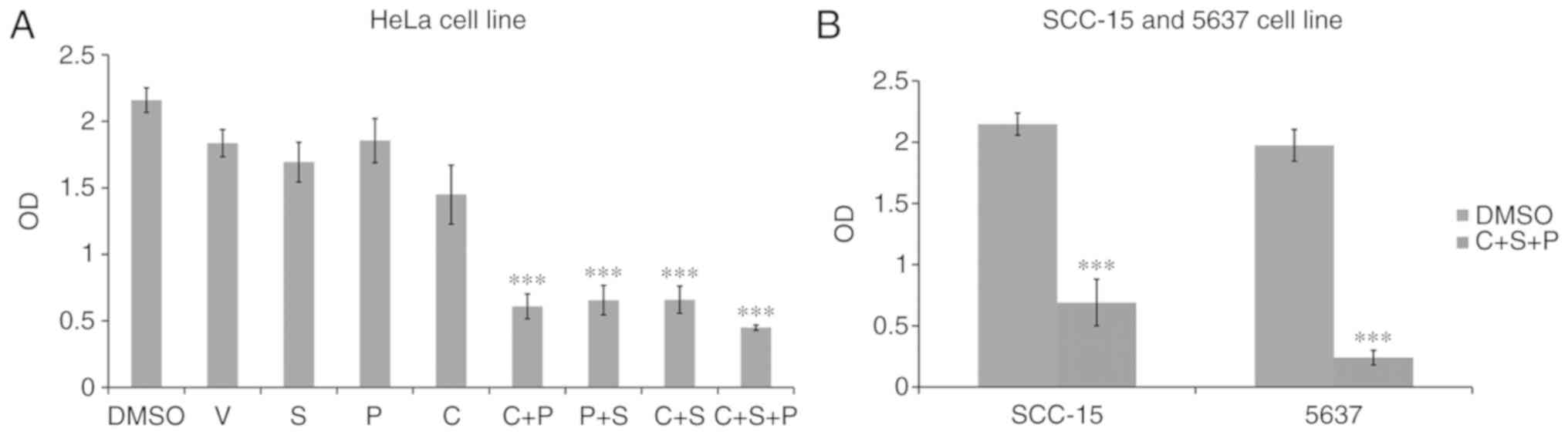
Inhibitor combination synergizes
proliferation inhibition
Proliferation assays were next performed in cells
treated with single inhibitor or combination treatments for 6 days.
The single inhibitors had no effect on HeLa cell growth (Fig. 4A). The C+S, C+P and P+S treatments
had various levels of inhibition (C+S, 70±10%, P<0.001; C+P,
72±9% P<0.001; P+S, 70±11%, P<0.001), with the C+S+P group
exerting the strongest inhibitory effect compared with the
DMSO-treated control cells (C+S+P, 80±2% P<0.001). Strong growth
inhibition was also revealed in the C+S+P-treated SCC-15 (69±7%,
P<0.001) and 5637 cells (88±2%, P<0.001) (Fig. 4B).
Inhibitor combination inhibits signal
transduction
To evaluate the effects of the small molecules on
inhibition of ALK, MEK and GSK3, western blot analysis of the
phosphorylation levels of their respective targets Smad1/2, ERK1/2
and glycogen synthase (GS) was performed.
In all three cell lines, compared with the
DMSO-treated cells, VPA treatment did not influence phosphorylation
levels of any of the examined proteins. In 5637 and HeLa cell
lines, C+S+P combination treatment significantly decreased
phosphorylation levels of GS, ERK1/2 and Smad1/2, (P<0.05,
P<0.01 and P<0.001, respectively). In single treatment of
PD0325901, SB431542 or CHIR99021, inhibition to no self-target
could be found. In SCC-15 cells, C+S+P treatment significantly
decreased phosphorylation levels of ERK1/2 and Smad1/2 (P<0.05
and P<0.01, respectively), without influencing GS, and these
were cross-inhibitions to their targets when SB431542 and CHIR99021
were used as single agents (Fig.
5).
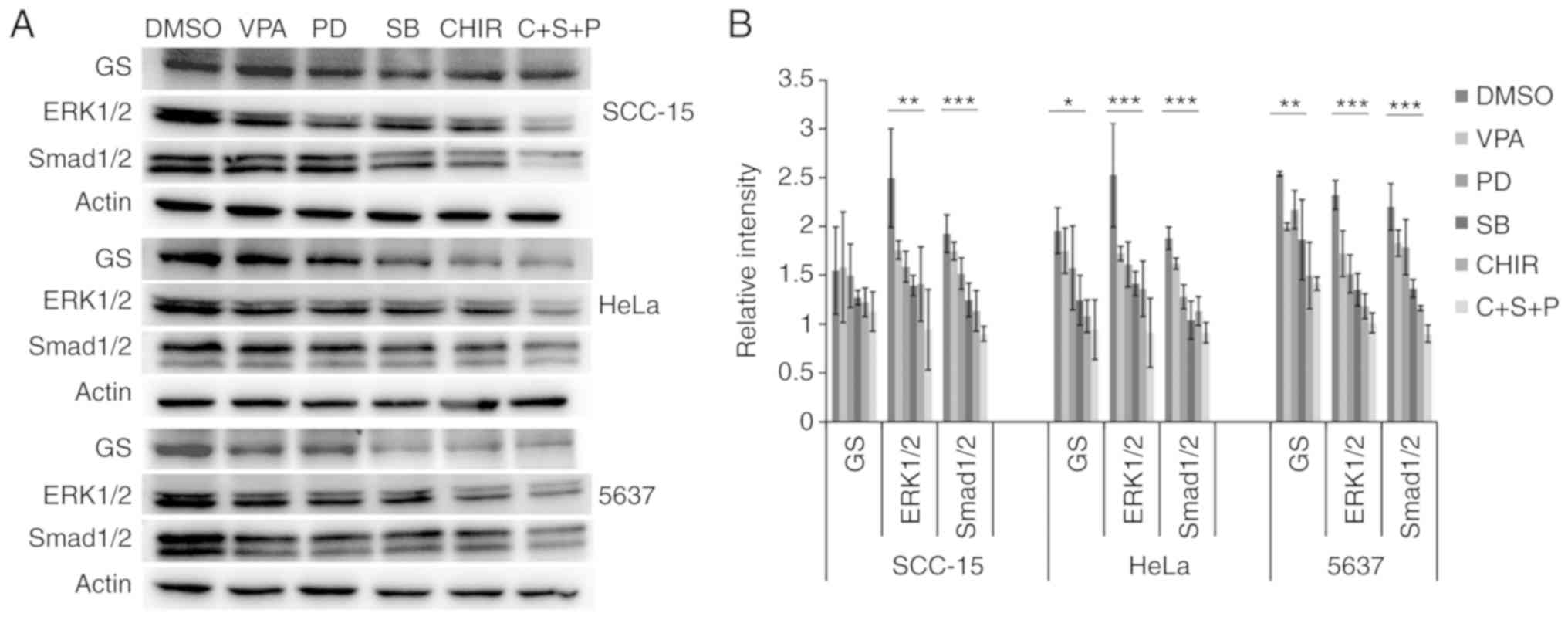 | Figure 5.Inhibitor and combination of
inhibitors suppresses phosphorylation levels of GS, ERK1/2 and
Smad1/2 in treated cells. (A and B) Phosphorylation level detection
of GS, ERK1/2 and Smad1/2 in treated cells. In HeLa, 5637 and
SCC-15 cells, compared with DMSO-treated control cells VPA
treatment did not significantly influence phosphorylation levels of
GS, ERK1/2 and Smad1/2. In 5637 and HeLa cell lines, C+S+P
(CHIR99021+SB431542+PD0325901) combination treatment markedly
decreased phosphorylation levels of GS, ERK1/2 and Smad1/2. When
PD0325901, SB431542 and CHIR99021 were used as single agents,
significant inhibition to no self-target could be found. In the
SCC-15 cell line, the C+S+P combination treatment significantly
decreased phosphorylation levels of ERK1/2 and Smad1/2, without
influencing GS, and there were significant cross-inhibitions to
their targets when SB431542 and CHIR99021 were used as single
agents. *P<0.05, **P<0.01, ***P<0.001. VPA, valproic acid;
DMSO, dimethyl sulfoxide. |
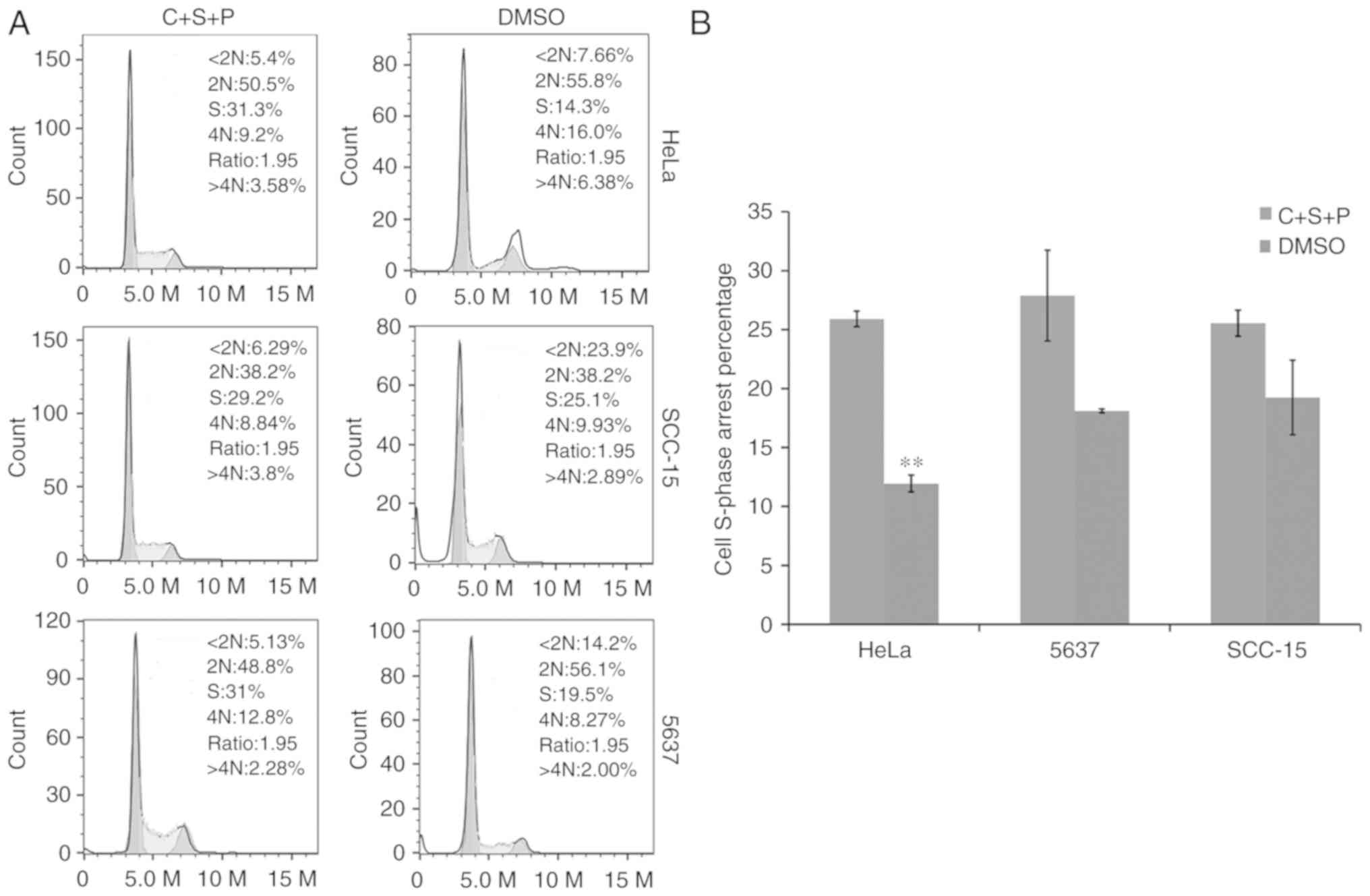
Inhibitor combination causes cell
cycle arrest
The cell cycle was next investigated in cells
treated with C+S+P. PI and flow cytometry revealed that C+S+P
exerted an increased cytostatic effect and induced S-phase
accumulation in cells compared with DMSO-treated control cells
(S-phase in C+S+P/DMSO HeLa cells: 25.9±0.66%/11.9±0.718%,
P<0.01; 5637 cells: 27.83±3.8%/18.1±0.16%; and SCC-15 cells:
25.33±1.1%/19.23±3.16%) (Fig.
6).
Inhibitor combination suppresses tumor
cell sphere formation
The impact of inhibitors on sphere formation ability
of these cells was next evaluated by culturing cells in cancer stem
cell induction media containing VPA or V+C+S+P. In 4–6 days, small,
bright, typical cell spheres were observed in SCC-15 and 5637 cell
cultures, while spheres appeared in HeLa cell culture at days 8–10.
In VPA-containing medium, cells formed compact, fast growing and
large sized cell spheres compared with DMSO-treated control
spheres. In V+C+S+P-containing medium, cells initially formed
small-sized, atypical cell spheres that broke down into tiny lumps
or single cells within 10 to 11 days (Fig. 7).
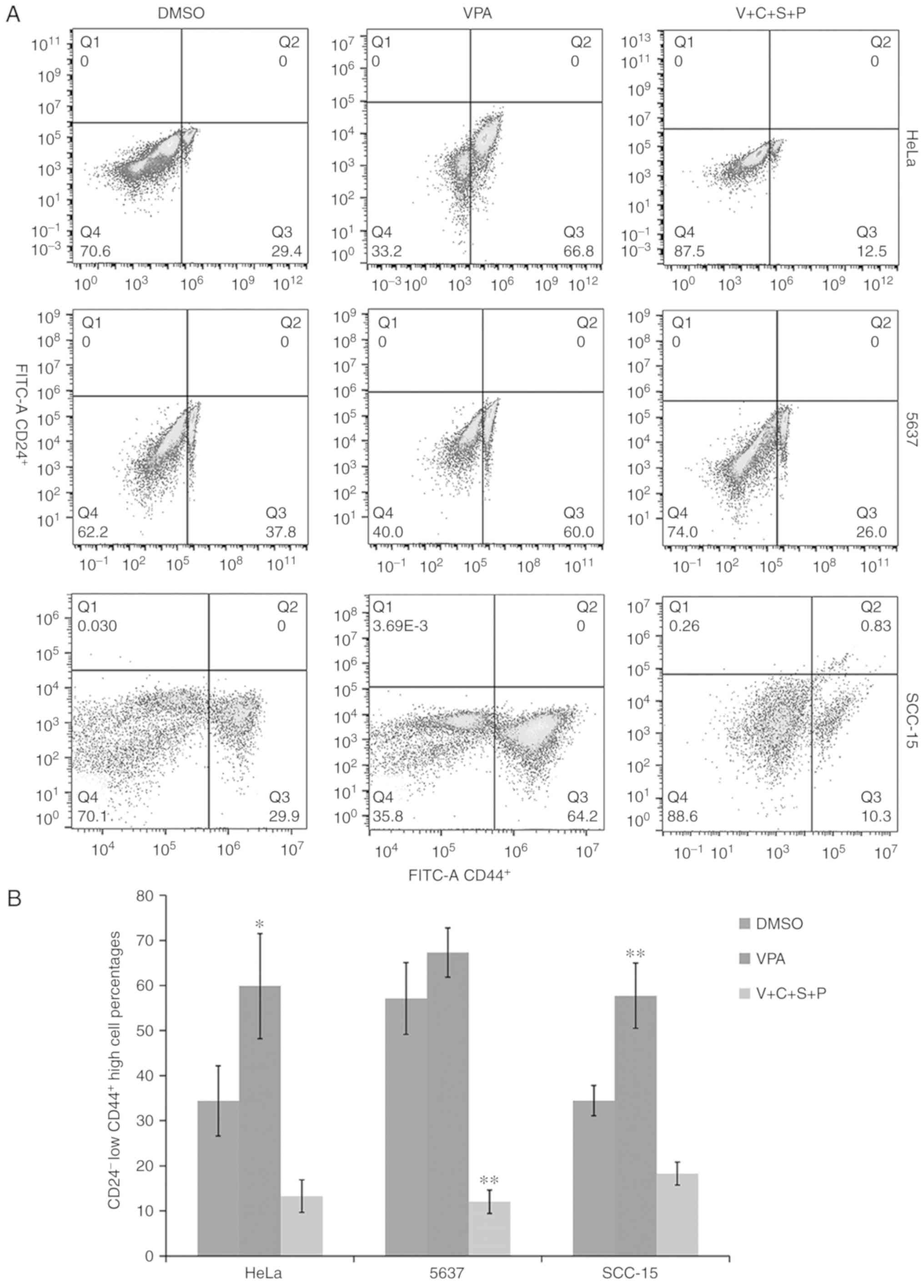
Inhibitor combination reduces
CD24−/CD44+ cell percentages in cancer cell
spheres
The cells with CD24−/CD44+
phenotype are regarded as cells that have cancer stem cell
potential (31). Thus, the relative
abundance of CD24−/CD44+ cells in cell
spheres was compared using fluorescence-activated cell sorting
analysis. Compared with the DMSO-treated control cells, the
CD24−/CD44+ cell percentages in VPA-treated
cells were increased, while the percentages in the V+C+S+P-treated
cells were decreased (HeLa: VPA/DMSO, 59.8±11.6/34.4±7.78,
P<0.05; V+C+S+P/DMSO, 13.28±3.6/34.4±7.78; 5637: VPA/DMSO,
67.3±5.46/57.1±7.919; V+C+S+P/DMSO, 12.03±2.6/57.1±7.9, P<0.001;
SCC-15: VPA/DMSO, 57.72±7.1/34.46 3.35, P<0.01; V+C+S+P/DMSO,
10.26±2.5/34.46±3.35, P<0.05) (Fig.
8).
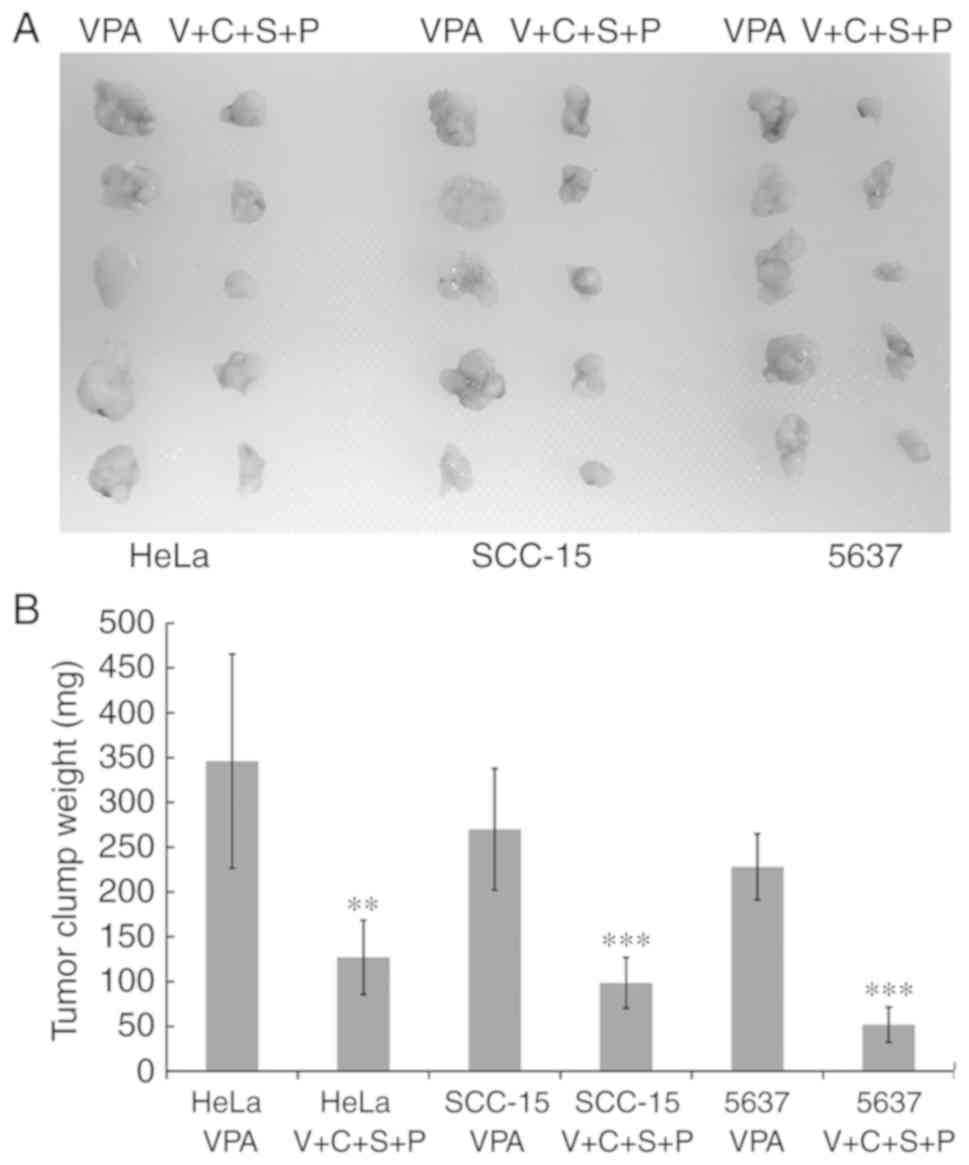
Inhibitor combination delays cancer
cell clump formation in vivo
Cancer stem cells exhibit enhanced tumor-initiating
capacity upon transplantation into a permissive host. Cells
isolated from VPA and V+C+S+P-treated spheres were subcutaneously
transplanted into the flanks of female athymic Swiss nude mice. The
transplanted cells isolated from VPA-treated spheres formed a
single visible solid cell clump beneath the injection site within 6
to 10 days. In mice that received cells from V+C+S+P-treated
spheres, the single cell clump was invisible until 15 to 20 days
after the injection. Cancer cell clumps were excised at day 30
after injection and weighed. Cell clumps from V+C+S+P-treated cells
exhibited growth retardation compared with VPA-treated cells in all
cell lines (VPA/V+C+S+P, HeLa: 346±119.53 mg/127±41.33 mg,
P<0.01; SCC-15: 270±67.82 mg/98.4±28.33 mg, P<0.001; 5637:
228±19.63 mg/51.8±19.63 mg, P<0.001) (Fig. 9).
Discussion
A small subpopulation of cancer cells within a
heterogeneous tumor mass, termed CSCs, have the capacity to
propagate, relapse, metastasize, and resist treatment. EMT plays a
critical role in cancer progression, in which in situ
carcinoma cells acquire mesenchymal characteristics and become
mobile, leading to localized and metastatic invasion (15–21).
While many cancer therapies may effectively kill the
bulk of tumor cells, failure to eliminate CSCs would enable the
remaining CSCs to regenerate new tumors after treatment ends.
Therefore, the complete elimination of CSCs is an issue of primary
importance in cancer treatment (32,33).
Since CSCs account for only a small subpopulation of the entire
cancer cell mass, direct isolation of these cells from tumor tissue
for subsequent in vitro propagation is difficult. Without a
sufficient number of CSCs, it is impossible to set up a screening
system, with which to identify agents that are effective in
eliminating CSCs.
In the present study, the results revealed that VPA
induced EMT morphology in all three cell lines, as well as
increased pluripotency and EMT-related gene expression, promoted
cell migration and accelerated cell sphere formation. When the
concentration was decreased to 0.25 mM, no effects were observed,
and tumor cells treated with 1 mM did not survive more than 5 days
(data not shown). These observations indicated that different
concentrations of VPA exhibit different effects on cell fate.
The other three inhibitors, PD0325901, SB431542 and
CHIR99021, had no significant influence on EMT- and
pluripotency-related gene expression, and cell proliferation rate.
Proliferation inhibition was only revealed at high concentrations
(4–6 µm/l) (data not shown). The combination treatments exhibited
significant inhibition on cell growth and migration of all three
cell lines, even in the presence of the EMT-promoting reagent VPA.
This result indicated the effectiveness of the combination
treatment in reversing VPA promotion of cancer cell invasiveness
and stemness. It is possible that modulating the EMT status of
cancer cells towards a more epithelial state may reduce the
metastatic aggressiveness of cells and render cells more
susceptible to conventional chemotherapy.
Western blot analyses demonstrated that in
PD0325901, SB431542 and CHIR99021-treated 5637 and HeLa cell lines,
there were cross reactions among their targets. In SCC-15 cells,
treatment with a single inhibitor or C+S+P for 6 days had no impact
on the phosphorylation levels of GS, and cross reactions occurred
among the ERK1/2 and Smad1/2 pathways. These results indicated that
inhibitors have different cell-type specific effects. Therefore, we
surmised that a multi-inhibitor treatment would be more
advantageous in treating cancer cells, and this notion was
demonstrated by our results revealing that the combination of
PD0325901, SB431542 and CHIR99021 had significant synergized
inhibition on phosphorylation of GS, ERK1/2 and Smad1/2. This
synergized signaling pathway inhibition may cause cancer cell cycle
arrest and result in cell growth suppression, as observed in the
cell cycle and growth inhibition experiments of the present
study.
Cancer stem cells form cell spheres in cancer stem
cell induction medium and in the present study, the results
revealed that VPA induced quick formation of cell spheres in all
three cell lines, and the CD24−/CD44+ cell
percentages were increased compared with the control group. This
result indicated that 0.5 mM VPA treatment could amplify the number
of CSCs in these three cell lines, indicating that VPA may be a
potent CSC stimulator for establishing a screening system to select
potential anti-CSC agents. When cells were treated with all four
inhibitors, the percentages of CD24−/CD44+,
the cell sphere forming ability and the in vivo
tumor-initiating potential of tumor cells greatly decreased,
demonstrating the effectiveness of this combination in eliminating
CSCs in tumor cells, even in the presence of a CSC-promoting agent.
These results were consistent with the qPCR results, in which C+S+P
significantly downregulated VPA-mediated promotion of EMT and
stemness-related gene expression.
VPA and the combination treatments were also
assessed in other cell lines (CNE1, CNE2, MCF-7, CRL-2073 and J82;
data not shown) and the same results were obtained with CNE2 and
MCF-7 cells. CNE1, CRL-2073 and J82 cell lines were markedly
sensitive to VPA and the combination treatment and could not
survive more than 3 days. This sensitivity may be due to their
slow-growing rate and less invasive and relatively higher
epithelial-like differentiation characteristics (data not shown).
Since CNE2 and MCF-7 cells are fast-growing and more invasive,
similar to HeLa, SCC-15 and 5637 cells, we consider this CSC
screening and inhibitory system more suitable in dealing with the
highly invasive and fast-growing advanced malignant cancer
cells.
In conclusion, in the present study, it was
demonstrated that a specific concentration of VPA could be used as
an agent to promote EMT and CSCs and establishing a screening
system to identify agents for eliminating CSCs for advanced stage
cancer. PD0325901, SB431542 and CHIR99021 co-treatment could act as
a CSC eliminator by downregulating pluripotency and EMT-related
gene expression and intervention with signal transduction
pathways.
Acknowledgements
We thank Dr Zeng Zhang for technical assistance in
the flow cytometric and cell cycle analysis.
Funding
The present study was supported by the Shenzhen
Science and Technology Innovation Committee (grant nos.
JCYJ20160429090753103 and JCYJ20170816105345191); and the Guangdong
Bureau of Traditional Chinese Medicine Project (grant no.
20171228).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YaZ performed the western blot analyses,
quantitative real-time RT-PCR, tumor cell invasion assay and the
cell cycle analysis; YuZ conducted the flow cytometric analysis,
the cell growth inhibition test and the cell sphere formation; ML
was responsible for the cell culture and cell sample preparation;
FM carried out the cancer cell inoculation, the nude mice
anesthetization and the cell clumps measurement; ZY assisted with
the interpretation of the data, revision of the project and the
statistical analysis; YC and GC contributed to the design and
revision of the project, acquisition and analysis of the data,
drafting, revising and publication of the manuscript, and were also
responsible for all parts of the work being appropriately
investigated. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Animal experiment approval was obtained from the
Ethics Committee of Peking University Shenzhen Hospital (Shenzhen,
China). Chinese laws concerning animal caring and treatment were
strictly followed during the experiments, and the study complied
with ARRIVE guidelines (https://www.nc3rs.org.uk/arrive-guidelines).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shi Y, Do JT, Desponts C, Hahm HS, Scholer
HR and Ding S: A combined chemical and genetic approach for the
generation of induced pluripotent stem cells. Cell Stem Cell.
2:525–528. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin TX, Ambasudhan R, Yuan X, Li WL,
Hilcove S, Abujarour R, Lin XY, Hahm HS, Hao E, Hayek A, et al: A
chemical platform forimproved induction of human iPscs. Nat
Methods. 6:805–808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huangfu DW, Osafune KJ, Maehr R, Guo WJ,
Eijkelenboom A, Chen SB, Muhlestein W and Melton DA: Induction of
pluripotent stem cells from primary human fibroblasts with only
Oct4 and Sox2. Nat Biotechnol. 26:1269–1275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li YQ, Zhang Q, Yin XL, Yang WF, Du YY,
Hou PP, Ge J, Liu C, Zhang WQ, Zhang X, et al: Generation of iPSCs
from mouse fibroblasts with a single gene, Oct4, and small
molecules. Cell Res. 21:196–204. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ji MY, Lee EJ, Kim KB, Kim YM, Sung RY,
Lee SJ, Kim DS and Park SM: HDAC inhibitors induce
epithelial-mesenchymal transition in colon carcinoma cells. Oncol
Rep. 33:2299–2308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vincent EE, Elder DJ, O'Flaherty L, Pardo
OE, Dzien P, Phillips L, Morgan C, Pawade J, May MT, Sohail M, et
al: Glycogen synthase kinase 3 protein kinase activity is
frequently elevated in human non-small cell lung carcinoma and
supports tumour cell proliferation. PLoS One. 9:e1147252014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chua KN, Kong LR, Sim WJ, Ng HC, Ong WR,
Thiery JP, Huynh H and Goh BC: Combinatorial treatment using
targeted MEK and SRC inhibitors synergistically abrogates tumor
cell growth and induces mesenchymal-epithelial transition in
non-small-cell lung carcinoma. Oncotarget. 6:29991–30005. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu Y, Jiang F, Zheng X, Katakowski M,
Buller B, To SS and Chopp M: TGF-β1 promotes motility and
invasiveness of glioma cells through activation of ADAM17. Oncol
Rep. 25:1329–1335. 2011.PubMed/NCBI
|
|
9
|
Noguchi S, Eitoku M, Moriya S, Kondo S,
Kiyosawa H, Watanabe T and Suganuma N: Regulation of gene
expression by sodium valproate in epithelial-to-mesenchymal
transition. Lung. 193:691–700. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial-mesenchymal
and mesenchymal-epithelial transitions in carcinoma progression. J
Cell Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kong D, Li Y, Wang Z, Banerjee S, Ahmad A,
Kim HR and Sarkar FH: miR-200 Regulates PDGF-D-mediated
epithelial-mesenchymal transition, adhesion, and invasion of
prostate cancer cells. Stem Cells. 27:1712–1721. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kong D, Banerjee S, Ahmad A, Li Y, Wang Z,
Sethi S and Sarkar FH: Epithelial to mesenchymal transition is
mechanistically linked with stem cell signatures in prostate cancer
cells. PLoS One. 5:e124452010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kong D, Li Y, Wang Z and Sarkar FH: Cancer
stem cells and epithelial-to mesenchymal transition
(EMT)-phenotypic cells: Are they cousins or twins? Cancers.
3:716–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, VandenBoom TG II, Kong D, Wang Z,
Ali S, Philip PA and Sarkar FH: Up-regulation of miR-200 and let-7
by natural agents leads to the reversal of epithelial-tomesenchymal
transition in gemcitabine-resistant pancreatic cancer cells. Cancer
Res. 69:6704–6712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Robey RW, Chakraborty AR, Basseville A,
Luchenko V, Bahr J, Zhan Z and Bates SE: Histone deacetylase
inhibitors: Emerging mechanisms of resistance. Mol Pharm.
8:2021–2031. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Díaz-Núñez M, Díez-Torre A, Wever D,
Andrade R, Arluzea J, Silió M and Aréchaga J: Histone deacetylase
inhibitors induce invasion of human melanoma cells in vitro via
differential regulation of N-cadherin expression and RhoA activity.
BMC Cancer. 22:6672016. View Article : Google Scholar
|
|
19
|
Pang L, Li Q, Wei C, Zou H, Li S, Cao W,
He J, Zhou Y, Ju X, Lan J, et al: TGF-β1/Smad signaling pathway
regulates epithelial-to-mesenchymal transition in esophageal
squamous cell carcinoma: In vitro and clinical analyses of cell
lines and nomadic Kazakh patients from northwest Xinjiang, China.
PLoS One. 9:e1123002014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miao ZF, Zhao TT, Wang ZN, Miao F, Xu YY,
Mao XY, Gao J, Wu Jh, Liu XY, You Y, et al: Transforming growth
factor-beta1 signaling blockade attenuates gastric cancer
cell-induced peritoneal mesothelial cell fibrosis and alleviates
peritoneal dissemination both in vitro and in vivo. Tumour Biol.
35:3575–3583. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim YJ, Hwang JS, Hong YB, Bae I and Seong
YS: Transforming growth factor beta receptor I inhibitor sensitizes
drug-resistant pancreatic cancer cells to gemcitabine. Anticancer
Res. 32:799–806. 2012.PubMed/NCBI
|
|
22
|
Henderson YC, Chen Y, Frederick MJ, Lai SY
and Clayman GL: MEK inhibitor PD0325901 significantly reduces the
growth of papillary thyroid carcinoma cells in vitro and in vivo.
Mol Cancer Ther. 9:1968–1976. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Wu Q, Tan L, Porter D, Jager MJ,
Emery C and Bastian BC: Combined PKC and MEK inhibition in uveal
melanoma with GNAQ and GNA11 mutations. Oncogene. 33:4724–4734.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marchand B, Tremblay I, Cagnol S and
Boucher MJ: Inhibition of glycogen synthase kinase-3 activity
triggers an apoptotic response in pancreatic cancer cells through
JNK-dependent mechanisms. Carcinogenesis. 33:529–537. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lackner MR, Wilson TR and Settleman J:
Mechanisms of acquired resistance to targeted cancer therapies.
Future Oncol. 8:999–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yun CH, Mengwasser KE, Toms AV, Woo MS,
Greulich H, Wong KK, Meyerson M and Eck MJ: The T790M mutation in
EGFR kinase causes drug resistance by increasing the affnity for
ATP. Proc Natl Acad Sci USA. 105:2070–2075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Buck E, Eyzaguirre A, Rosenfeld-Franklin
M, Thomson S, Mulvihill M, Barr S, Brown E, O'Connor M, Yao Y,
Pachter J, et al: Feedback mechanisms promote cooperativity for
small molecule inhibitors of epidermal and insulin-like growth
factor receptors. Cancer Res. 68:8322–8332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fukumoto S, Kanbara K and Neo M:
Synergistic anti-proliferative effects of mTOR and MEK inhibitors
in high-grade chondrosarcoma cell line OUMS-27. Acta Histochem.
120:142–150. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Langlands AJ, Carroll TD, Chen Y and
Näthke I: Chir99021 and Valproic acid reduce the proliferative
advantage of Apc mutant cells. Cell Death Dis. 9:2552018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee YJ, Wu CC, Li J, Ou CC, Hsu SC, Tseng
HH, Kao MC and Liu JY: A rational approach for cancer stem-like
cell isolation and characterization using CD44 andprominin-1(CD133)
as selection markers. Oncotarget. 7:78499–78515. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Blake RA, Broome MA, Liu X, Wu J, Gishizky
M, Sun L and Courtneidge SA: SU6656, a selective src family kinase
inhibitor, used to probe growth factor signaling. Mol Cell Biol.
20:9018–9027. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marchion D and Munster P: Development of
histone deacetylase inhibitors for cancer treatment. Expert Rev
Anticancer Ther. 7:583–598. 2007. View Article : Google Scholar : PubMed/NCBI
|