Introduction
Glioma is the most common malignancy type in the
central neuronal system, according to prevalence studies conducted
in USA in 2010, while temozolomide (TMZ), is the first-choice
chemotherapy agent against glioma (1–3).
However, TMZ drug-resistance is a main cause of clinical treatment
failure (1). A number of studies
demonstrated that the abnormal transport of drugs attributes to
resistance (4,5). Furthermore, it was also demonstrated
that there are a number of mechanisms contributing to drug
resistance, including activation of DNA repair system, impairment
of apoptotic signaling and reduction in the drug uptake into cells,
but the precise mechanisms remain under investigation (6–8).
Therefore, understanding the precise mechanism underlying the drug
resistance of glioma cells is critical for developing novel
therapeutic strategies to overcome TMZ resistance.
Transient receptor potential cation channel
subfamily C member 5 (TRPC5) is a Ca2+-permeable channel
that is expressed in numerous types of cells and organs, including
endothelial and muscle cells, and the lungs and kidneys (9–11), and
attributes to a number of neuronal and vascular diseases, including
Huntington's disease and infantile hypertrophic pyloric stenosis
(12–14). Other studies demonstrated that TRPC5
is involved in cancer chemotherapy. For example, TRPC5 was
determined to mediate the Adriamycin resistance in breast carcinoma
via P-glycoprotein induction (15).
Additionally, it activated autophagy in chemotherapy-resistant
breast cells under Adriamycin exposure (16). Therefore, we hypothesized that TRPC5
may be a potential molecular target in glioma chemotherapy
treatment.
Macroautophagy (hereafter termed as autophagy) is a
catabolic process for the degradation and recycled use of cytosolic
excess proteins, and impaired or defective organelles in
autolysosomes (17). The hallmark
of autophagy is the formation of double- or multi-membrane vesicles
in the cytosol, termed autophagosomes. It encapsulates bulk
cytoplasm or cytoplasmic organelles, and then fuses with the
endocytic compartments, including early and late endosomes, and
multivesicular bodies (18).
Following maturation, it combines with the compartment of lysosomes
to form autolysosomes, where the cargo is degraded by acidic
lysosomal hydrolases (19,20). The contents of autolysosomes are
digested to recycle the fragment products and generate energy to
confer stress tolerance (21,22).
In cancer initiation and development, autophagy also serves a
controversial role and there is no precise and novel conclusion
(23–25). A number of studies demonstrated that
autophagy may support cancer survival (26–28);
while in contrast, other studies indicated that autophagy is
involved in programmed cell death (29–31).
Different tumor types, stages, genomic contexts and settings may
attribute to the different roles of autophagy in cancer. An
autophagic response simultaneously triggers the apoptotic cell
death induced by a number of anticancer drugs, including
Glychionide-A Flavonoid in pancreatic carcinoma and Thioridazine in
glioma (32,33). In cancer chemotherapy, the majority
of studies prefer autophagy as a protective pathway that postpones
or reverses apoptosis (34,35). However, the precise mechanism for
protective autophagy induced by chemotherapy and the potential
initiating factor remain unknown.
Based on previous research demonstrating that TRPC5
mediates drug resistance via autophagy in other cancer cells
(16), we hypothesized TRPC5 as an
autophagy initiator during glioma chemotherapy. In the present
study, the molecular mechanism of TRPC5 in autophagy and
chemotherapy was examined.
Materials and methods
Cell culture
U87 wild-type (U87/WT) cells were obtained from
Chinese Academy of Sciences (Shanghai, China), and then cultured in
Dulbecco's modified Eagle's medium (DMEM)/F12 culture medium
containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). TMZ-resistant human glioma cells (U87/TMZ)
were induced by exposing U87/WT cells to TMZ in high-dose therapy
at 400 nM for 6 months. TMZ was reconstituted with dimethyl
sulfoxide (DMSO) prior to use, resulting in an effective TMZ
concentration of 25 µM. When the cells were in the logarithmic
growth phase, TMZ was combined with DMEM to a final concentration
of 400 nM. Subsequently, at every 24-h incubation interval, the
medium was discarded and replaced with fresh medium with the
identical TMZ concentration. Dead cells were discarded with a wash
with PBS after 3 days and the remaining cells were diluted at
2×105 cells/ml by DMEM containing 10% FBS and replanted
in a 6 cm cell culture dish, and this procedure was repeated for 6
months. Finally, a cell line resistant to 400 nM TMZ (termed
U87/TMZ) was derived from U87/WT after 6 months. All cells were
incubated at 37°C in 5% CO2 humidified air.
For the inhibitor experiments, all inhibitors were
dissolved in DMSO and control experiments were performed with equal
volumes of DMSO. Cells were treated for 6 h at 37°C with
Bafilomycin A1 (BAF1; 400 nmol/l),
mTOR-inhibitor PP242 (400 nmol/l), autophagy activator chloroquine
(CQ; 20 µmol/l), CAMKK inhibitor KN−93 (10 µmol/l) and
AMPK inhibitor dorsomorphin (10 µmol/l) (all from MedChemExpress;
Monmouth Junction, NJ, USA) following the transfection of TRPC5
overexpression plasmid for 18 h, according to the subsequent
protocol.
Reagents and antibodies
The following antibodies were used: Anti-β-actin
(cat. no. sc-47778; 1:200) from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA); anti-TRPC5 (cat. no. ACC-020; 1:200) from
Alomone Labs (Jerusalem, Israel); anti-phospho-CaMKKβ (Ser286)
(cat. no. 12716; 1:500), anti-CaMKKβ (cat. no. 4436; 1:500),
anti-phospho-AMPKα (Ser172) (cat. no. 2535; 1:500), anti-AMPKα
(cat. no. 2532S; 1:500), anti-phospho-mTOR (Ser2448) (cat. no.
2971; 1:500), anti-mTOR (cat. no. 2972; 1:500) and anti-microtubule
associated protein 1 light chain 3 α (LC3; cat. no. 12741; 1:500)
from Cell Signaling Technology, Inc. (Danvers, MA, USA); and
AlexaFluor 488-conjugated goat anti-mouse IgG (cat. no. R37120;
1:2,000) and Alexa Fluor 555-conjugated goat anti-rabbit IgG (cat.
no. A21428; 1:2,000) from Thermo Fisher Scientific, Inc.
Plasmid and transfection
The pcDNA3.1-TRPC5 plasmid and control plasmid were
purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China). The
pcDNA3.1-GFP-LC3 plasmid was obtained from Suzhou University
(Suzhou, China). TRPC5 and/or LC3 plasmid transfection was
conducted by Lipofectamine® 3000 Transfection reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocols. The concentration of plasmids was 0.5
µg/ml for 6-well plates and 0.4 µg/ml for 24-well plates for both
cell lines. After transfection for 24 h, the subsequent experiments
were conducted.
Cell viability assay
U87/WT and U87/TMZ cells were added to 96-well
plates (5,000 cells/well) overnight at 37°C, and then treated by
400 nM TMZ for 48 h with or without TRPC5 transfection at 37°C,
according to the aforementioned protocol. cell viability was
measured by treating cells with MTT (20 µl; 5 mg/ml; Beijing
Solarbio Science & Technology Co., Ltd., Beijing, China) for 4
h. Subsequently, medium was replaced by 150 µl DMSO in each well
prior to measurement at 490 nm with a spectrophotometer.
Intracellular calcium level
measurement
U87/WT cells were added to 96-well plates (5,000
cells/well) overnight at 37°C and transfected with TRPC5 plasmid or
TRPC5-siRNA for 24 h. Subsequently Fluo-4 (2 mM/l; cat. no. F14201;
Thermo Fisher Scientific, Inc.) was added for 30 min at 37°C and
measured at 485 nm with a spectrophotometer.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cultured cells with the
use of TRIzol® reagent (Takara Bio, Inc., Otsu, Japan),
following the manufacturer's protocols. cDNA synthesis was
performed with a PrimeScript RT Reagent kit (Takara Bio, Inc.).
RT-qPCR was performed using the 7500 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) at conditions of 95°C
for 2 min, and 40 cycles of 95°C for 10 sec and 60°C for 40 sec.
Relative expression of the miRNA was calculated using the
comparative Cq method (36). The
expression was normalized to GAPDH. The primer sequences used were:
TRPC5, forward, 5′-TGAACTCCCTCTACCTGGCAAC-3′, and reverse,
5′-CGAAGAGTGCTTCCGCAATCAGT-3′; LC3, forward,
5′-TACGAGCAGGAGAAAGACGAGG-3′, and reverse,
5′-GGCAGAGTARGGTGGGTTGGTG-3′; and GAPDH, forward,
5′-AAGGTCGGAGTCAACGGATTTGGT-3′, and reverse,
5′-AGTGATGGCATGGACTGTGGTCAT-3′.
Western blot analysis
Tissues from mice and cultured cells were lysed in
radio immunoprecipitation assay buffer containing protease
inhibitor and phosphatase inhibitor (Cell Signaling Technology,
Inc.). The protein concentration was quantified with the
Bicinchoninic Acid method using a Protein Assay kit (Beyotime
Institute of Biotechnology, Nanjing, China). The samples (15 µg)
were loaded onto 15 (for LC3 detection) or 8% (for other proteins)
SDS-PAGE. Subsequently, total protein was transferred to the
polyvinylidene fluoride membrane (350 mA for 2 h) and the membrane
was blocked at room temperature for 1 h with 5% bovine serum
albumin (BSA; Beijing Solarbio Science & Technology Co., Ltd.).
Following three washes with PBS for 10 min each, the membranes were
immunostained with primary and secondary antibodies at 4°C
overnight. The bands were detected by Enhanced Chemiluminescent
Western Blotting HRP Substrate (EMD Millipore, Billerica, MA, USA),
according to the manufacturer's protocols. The band intensity was
analyzed with ImageJ software 1.48 (National Institutes of Health,
Bethesda, MD, USA) and normalized to β-actin.
Small-interfering RNA (siRNA)
transfection
Human TRPC5 siRNA was synthesized by Invitrogen
(Thermo Fisher Scientific, Inc.). Briefly, cells (2×105
cells/well) were seeded in 6-well plates and transfected with 40 nM
siRNA (cat. no. 4392420; Thermo Fisher Scientific, Inc.) using
Lipofectamine® 3000, according to the manufacturer's
protocol. The sequences of TRPC5 are: Forward,
5′-CCAAUGGACUGAACCAGCUUUACUU-3′, and reverse,
5′-UGUCGUGGAAUGGAUGAUAUU-3′. After 24 h, the cells were lysed for
further treatments with PCR or western blot analysis.
Immunocytochemistry
U87/WT or U87/TMZ cells were plated at
1×104 cells/ml and co-transfected with GFP-LC3 and TRPC5
or control plasmid, according to the aforementioned protocol.
Subsequently, cells were fixed with 4% paraformaldehyde overnight
at room temperature, permeabilized with 0.1% Triton X-100 at room
temperature for 15 min, and blocked for 1 h at room temperature in
1% BSA. Primary antibodies (TRPC5; cat. no. ACC-020; 1:200; and
LC3, cat. no. 12741; 1:500) were added overnight at 4°C and
fluorescent secondary antibody were used for 2 h at room
temperature. Sections were counterstained with DAPI (Beijing
Solarbio Science & Technology Co., Ltd.) for 10 min at room
temperature. Images were acquired using a LSM 700 confocal
microscope (×400 magnification) and analyzed using Zeiss software
2011 and Image-Pro Plus 6.0 software (Media Cybernetics, Inc.,
Rockville, MD, USA). A total of 30–50 cells selected randomly from
3 or 4 replicated experiments were quantified.
Intracellular calcium measurement
U87/WT cells were plated at 5×103
cells/ml and co-transfected with TRPC5 or TRPC5-siRNA for 24 h,
according to the aforementioned protocol. Subsequently, cells were
incubated with the AM ester for 30 min at 37°C (Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocols.
Following this, each well was measured with a
spectrophotometer.
Mouse xenograft models
A total of 6 male mice (age, 6–8 weeks; weight,
20–25 g) were obtained from the Central Laboratorial Animal
Facility at the Jiangsu Institute of Parasitic Diseases (Wuxi,
China). Mice were housed in cages under controlled environmental
condition at 25°C with 55–65% humidity, a 12-h light/dark cycle and
had free access to food and water. To generate subcutaneous tumors,
U87/WT cells were first transfected with TRPC5-shRNA or
control-shRNA lentiviral particles for 48 h, according to the
aforementioned protocol, and then 5×106 cells were
injected into the nude mice. All mice were housed in air-filtered
pathogen-free condition and administered with TMZ (30
mg/m2). Tumor growth was measured after 5 weeks. Tumor
volumes were estimated using the formula: Volume (mm3) =
[(width)2 × length]/2. All experiments involving animals
were approved by the Animal Experimentation Ethics Committee of
Nanjing Medical University.
Statistical analysis
Each experiment was independently repeated at least
3 times. Two-tailed Student's test for two groups or one-way
analysis of variance with post hoc test least significant
difference test for more than two groups were performed using SPSS
software 16.0 (SPSS, Inc., Chicago, IL, USA). Values are presented
as the means ± standard error of the mean. P<0.05 was considered
to indicate a statistically significant difference.
Results
Chemotherapy upregulates the
expression of TRPC5 and autophagy level in glioma cells
Firstly, to confirm the effect of TMZ to U87/WT
cells, U87/WT cells were exposed to 400 nM TMZ for 48 h, and then
the cell viability was assessed by MTT, which demonstrated that it
was significantly reduced, compared with the control (Fig. S1A). Furthermore, the
transcriptional level of TRPC5 was assessed by RT-qPCR to determine
the influence of TMZ on TRPC5, and the results indicated that TRPC5
mRNA expression was also significantly increased following exposure
to TMZ, compared with the control (Fig. S1B). Additionally, TRPC5-siRNA was
applied to confirm the effect of TMZ on TPRC5, and the result
indicated that the mRNA expression of TRPC5 was also decreased
(Fig. S1C).
Subsequently whether TMZ increases TRPC5 protein
expression level and basal autophagy level in glioma cells was
investigated by analyzing TRPC5 and LC3 protein expression. LC3 is
a reliable marker of autophagy, and LC3-II is associated with the
amount of autophagosomes (16).
While, the TRPC5 and LC3 expression was significantly increased,
compared with the control (Fig.
1A). To further determine the autophagic flux, the LC3
expression in U87/WT cells exposed to TMZ combined with the
lysosomal protease inhibitor BAF1, a proton pump
inhibitor that raises lysosomal pH and blocks the activity of acid
hydrolases to restrict its proteolytic degradation and
autophagosome-lysosome fusion (37), was assessed. It was determined that
BAF1 significantly increased the LC3-II expression
level, indicating that increased LC3-II levels were attributed to
promotion of autophagy, rather than disruption of autophagic
degradation (Fig. 1B).
Subsequently, the autophagy level in TMZ-resistant glioma cells
(U87/TMZ), generated by high-dose concentrations of TMZ over 6
months, was detected. As depicted, U87/TMZ cells had enhanced
LC3-II levels, compared with U87/WT cells. Additionally,
BAF1 increased the LC3-II expression in U87/TMZ cells
(Fig. 1C). These results indicated
that U87/TMZ cells have increased basal levels of autophagy.
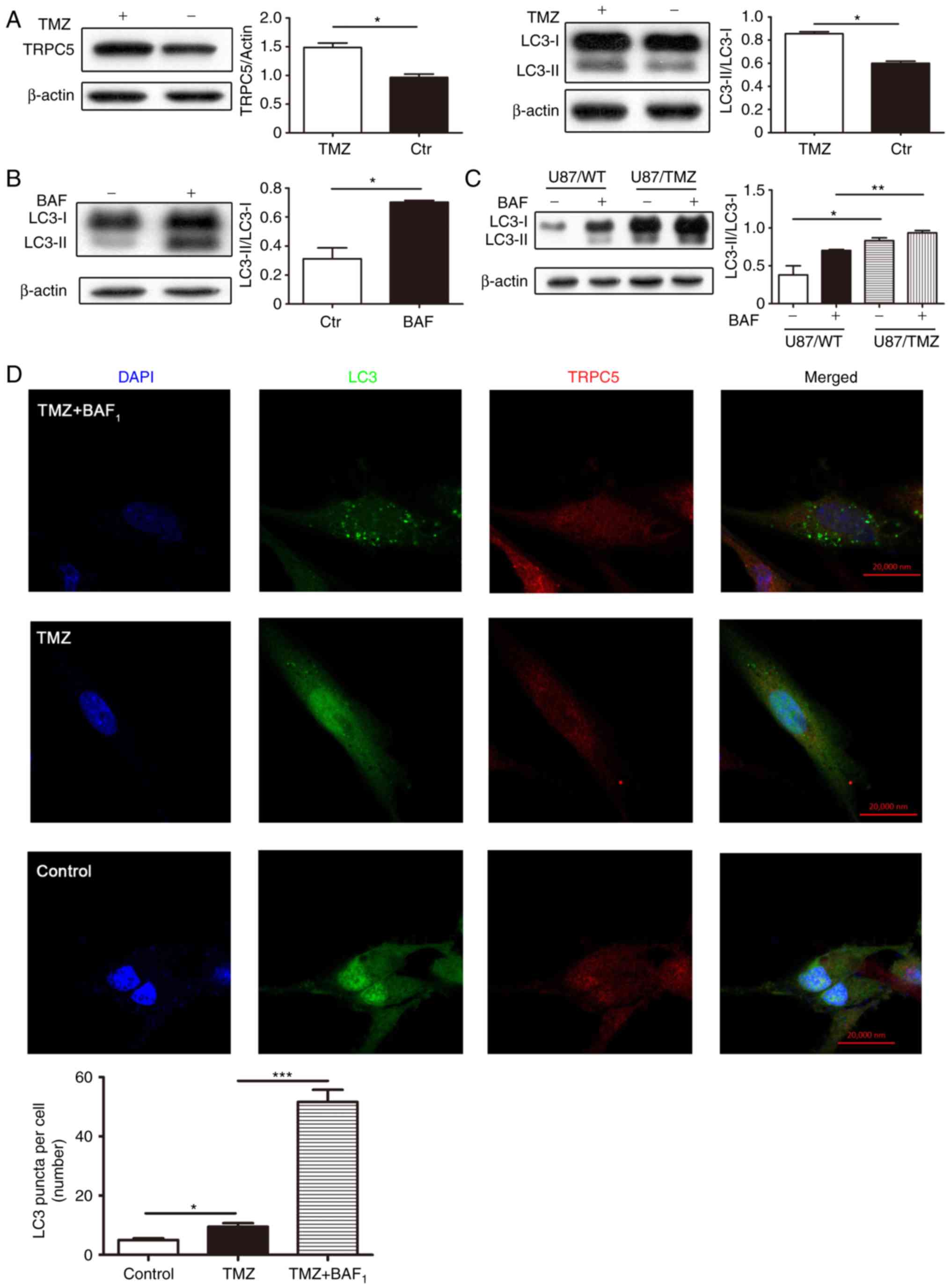 | Figure 1.Chemotherapy upregulates TRPC5
protein expression and autophagy level in glioma U87/WT cells. (A)
Glioma U87/WT cells were treated with 400 nM TMZ for 48 h, and the
protein expression of TRPC5 and LC3 were analyzed by western blot
analysis. It indicated that TMZ enhances TRPC5 and LC3-II
expression in U87/WT cells. Values are presented as the means ± SEM
of 4–6 experiments. (B) Glioma U87/WT cells were treated with 400
nM TMZ for 48 h and the lysosomal protease inhibitor 400 µM
BAF1 for 6 h, and the LC3 expression was analyzed by
western blot analysis. Representative western blotting indicated
that BAF1 accelerates TMZ-induced LC3-II accumulation.
Values are presented as the means ± SEM of 4–6 experiments. (C)
Glioma U87/TMZ cells were treated with 400 nM TMZ for 48 h and the
lysosomal protease inhibitor 400 µM BAF1 for 6 h, and
the LC3 expression was analyzed by western blot analysis. Values
are presented as the means ± SEM of 4–6 experiments. (D)
Representative immunofluorescence images of the accumulation of
autophagosomes in U87/WT cells, and the number of LC3 dots was
calculated in 40–50 cells. Scale bar, 20 µm. SEM, standard error of
the mean; BAF1, Bafilomycin A1; WT,
wild-type; TRPC5, transient receptor potential cation channel
subfamily C member 5; TMZ, temozolomide; Ctr, control; LC3,
microtubule associated protein 1 light chain 3 α. *P<0.05,
**P<0.01, ***P<0.001. |
To further determine the effect of TMZ on the
increase of the expression level of LC3-II, the basal level of
autophagy was visualized by using a LC3-GFP plasmid to immunostain
autophagosomes. Upon autophagy, LC3-II is localized on
autophagosomes and LC3 puncta is used as a marker for
autophagosomes. U87/WT cells were transfected with GFP-LC3 plasmid
for 24 h, and treated with or without TMZ for 24 h. As depicted,
compared with the number of LC3 dots contained in the control
group, a significantly increased number of LC3 dots were detected
in cells treated with TMZ. This indicated that TMZ treatment
accelerates the formation of LC3B and autophagosomes (Fig. 1D). Additionally, the present results
demonstrated that TRPC5 expression and basal autophagy level in
glioma cells are upregulated during TMZ exposure.
TRPC5 initiates TMZ-induced autophagy
in glioma cells
To confirm whether TRPC5 initiates autophagy under
exposure to TMZ, the LC3-II protein level and LC3 dots formation in
U87/WT cells following TRPC5-siRNA transfection were detected.
Knockdown of TRPC5 significantly decreased the intracellular
calcium level and the LC3-II expression in U87/WT cells under
exposure to TMZ, compared with the controls (Fig. 2A). The LC3 dots number per cell was
significantly decreased following TRPC5-knockdown and TMZ exposure,
compared with the control (Fig. 2B)
Furthermore, TRPC5-siRNA also significantly decreased the LC3-II
protein level in U87/TMZ cells. (Fig.
2C). To further confirm autophagy induced by TRPC5, U87/WT
cells were transfected with TRPC5 plasmid and it was determined
that TRPC5 overexpression significantly upregulated intracellular
calcium level, LC3-II expression and LC3 dots formation, compared
with the control (Fig. 2D and E).
Furthermore, the LC3 mRNA level was also determined following the
overexpression or silencing of TRPC5 and there was no significant
difference (data not shown). Collectively, the present data
indicated that chemotherapy may induce autophagy and TRPC5
initiated TMZ-induced autophagy in glioma cells.
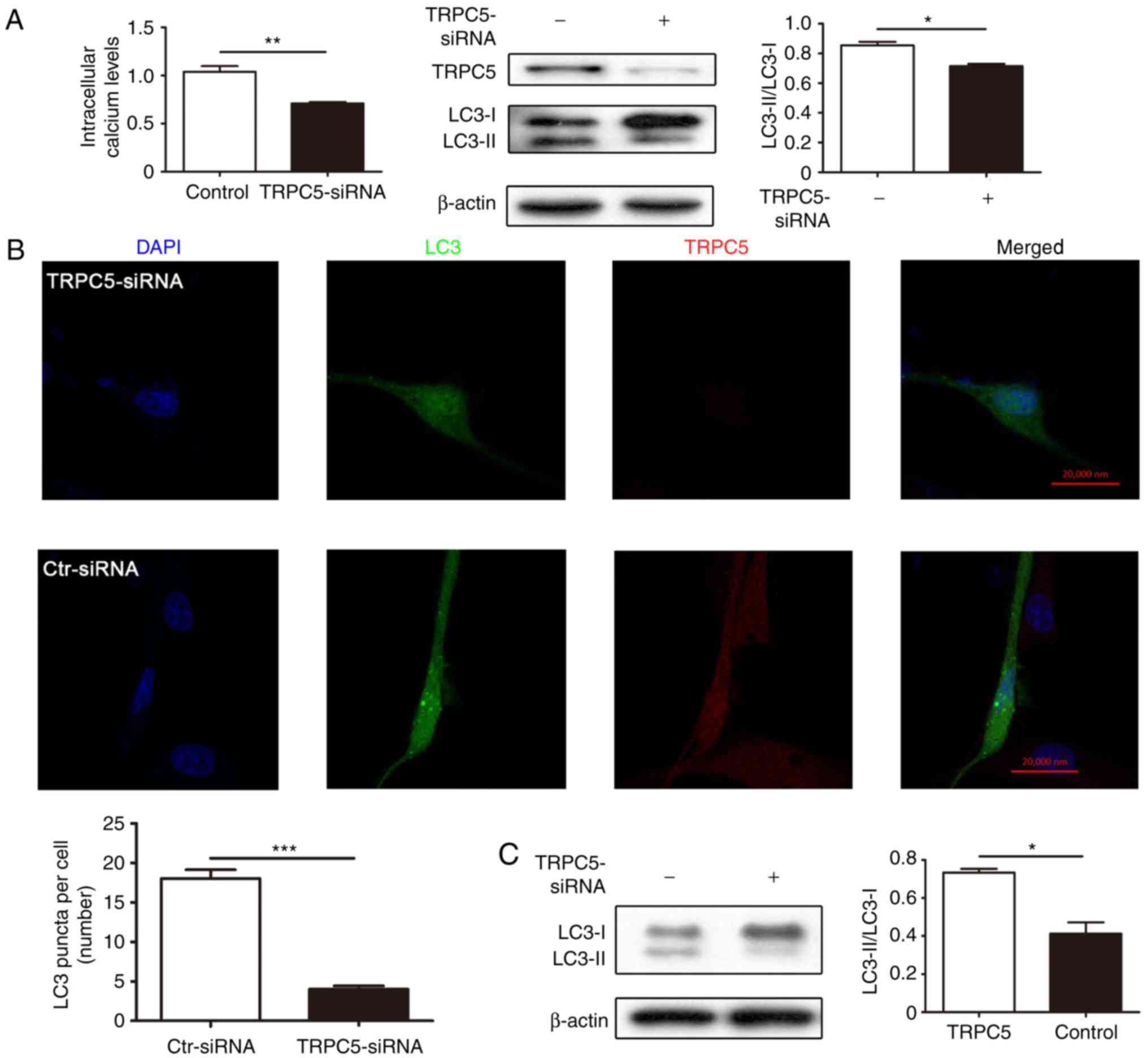 | Figure 2.TRPC5 regulates autophagy induced by
chemotherapy in glioma cells. (A) U87/WT cells were transfected
with TRPC5-siRNA or scramble-siRNA for 24 h, and then exposed to
TMZ for 48 h. The intracellular calcium levels were first measured
and the intracellular calcium levels were decreased following
TRPC5-siRNA transfection. The protein expression of TRPC5 and LC3
were analyzed by western blot analysis. Representative images
indicated that TRPC5-siRNA decreased TRPC5 and LC3-II protein
expression. (B) Representative immunofluorescence images
demonstrated that TRPC5-siRNA decreased the mean number of LC3 dots
per cell in U87/WT cells. (C) U87/TMZ cells were transfected with
TRPC5-siRNA or scramble-siRNA for 24 h, and then exposed to TMZ for
48 h. The knockdown of TRPC5 restricted the accumulation of LC3-II
in U87/TMZ cells. (D) The intracellular calcium levels were
measured and the intracellular calcium levels were increased
following TRPC5-plasmid transfection. Representative western blot
and densitometric analyses normalized to β-actin demonstrated the
effect of TRPC5 overexpression on the accumulation of LC3-II in
indicated cells. (E) Representative immunofluorescence images of
the accumulation of autophagosomes in indicated cells transfected
by TRPC5 plasmid, and the mean number of LC3 dots was calculated in
40–50 cells. Scale bar, 20 µm. Values are presented as the mean ±
standard error of the mean of 3–6 experiments. WT, wild-type;
TRPC5, transient receptor potential cation channel subfamily C
member 5; TMZ, temozolomide; LC3, microtubule associated protein 1
light chain 3 α; siRNA, small interfering RNA. *P<0.05,
**P<0.01, ***P<0.001. |
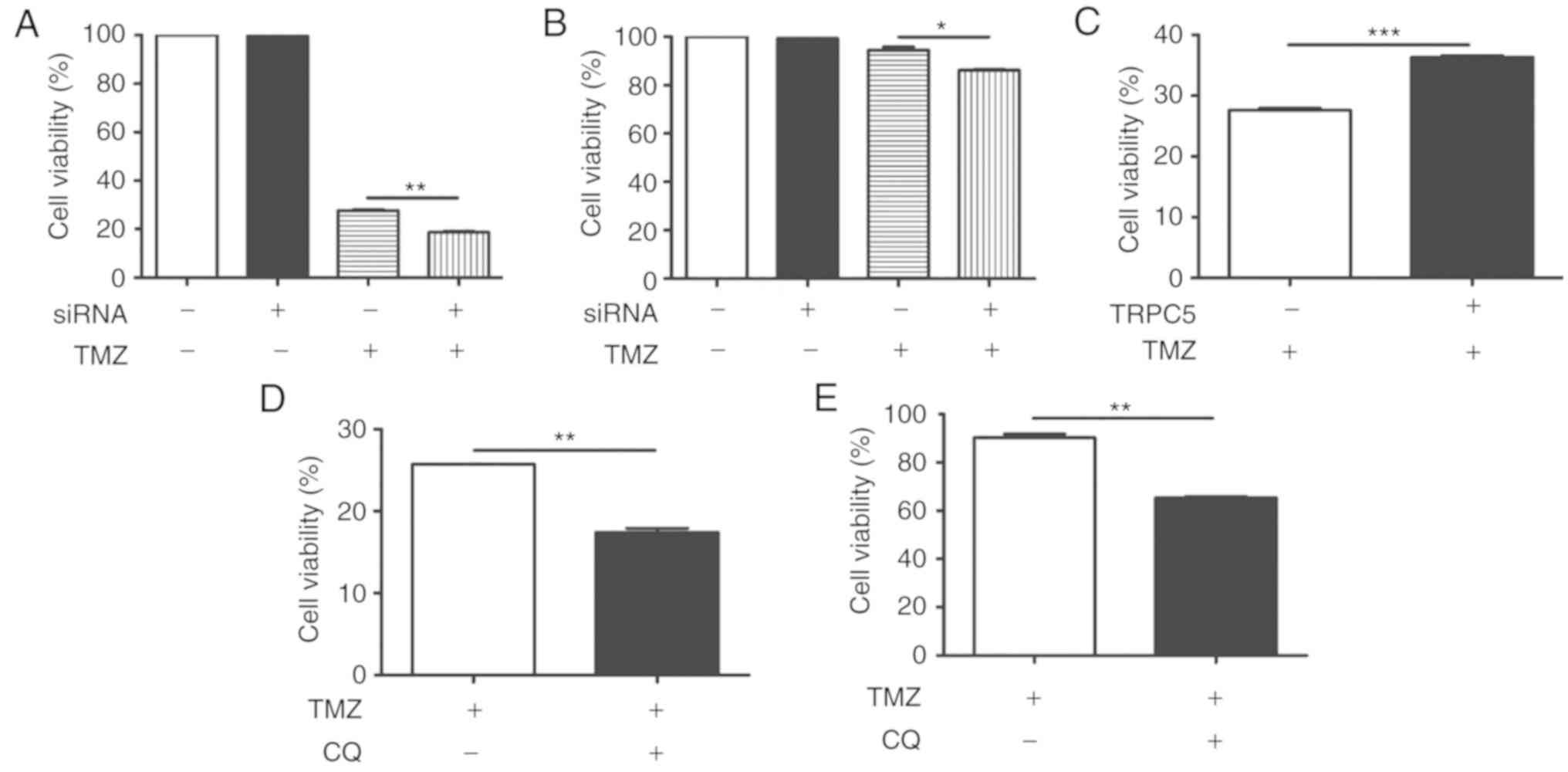
TRPC5 silencing or autophagy blockage
enhances glioma cell chemotherapy sensitivity to TMZ
TRPC5-siRNA was transfected into U87/WT cells to
determine whether autophagy induced by TRPC5 is involved in cell
survival under exposure to TMZ. The results demonstrated that cell
viability reduced to 28% in 400 nmol/l TMZ, compared with untreated
cells (100%). Additionally, downregulation of TRPC5 caused TMZ to
decrease proliferation of U87/WT cells to 18%, relative to the
siRNA control (Fig. 3A).
Furthermore, the effect of TRPC5 knockdown in drug-resistant
U87/TMZ cells was measured. It also demonstrated a significant
proliferation reduction, compared with TMZ exposure alone (Fig. 3B). This indicates that knockdown of
TRPC5 sensitizes U87/WT cells to TMZ-induced damage. The
TRPC5-plasmid was also transfected into U87/WT cells and it became
significantly more resistant to TMZ-induced injury, compared with
the control (Fig. 3C).
Additionally, CQ, an inhibitor of autophagy by inhibiting lysosomal
acidification (37), significantly
reduced cell viability with TMZ in U87/WT cells, compared with the
control (Fig. 3D). CQ also
significantly restricted the proliferation of drug-resistant
U87/TMZ cells, compared with TMZ alone. (Fig. 3E) These results indicated that TRPC5
knockdown or autophagy inhibition increases the chemotherapy
sensitivity of glioma cells.
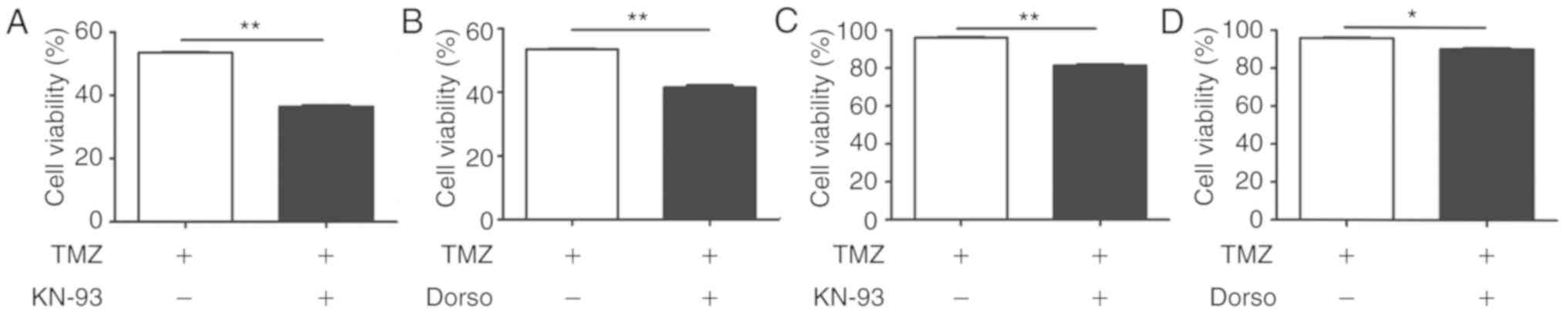
CaMKKβ/AMPKα/mTOR pathway activated by
TRPC5 mediates autophagy under chemotherapy
To confirm the potential mechanism of TRPC5
activation in autophagy, the autophagy-associated kinases
downstream of TRPC5 were investigated. Previous studies reported
that overexpression of TRPC5 activated CaMKKβ in breast cells
(16). After exposing U87/WT cells
to TMZ, the phospho-CaMKKβ level was determined to be significantly
increased, compared with the control, which is a downstream kinase
of TRPC5. Phosphorylation of CaMKKβ may activate AMPKα, therefore,
AMPKα activity was also detected and it was demonstrated to be
significantly increased in the U87/WT cell line under TMZ exposure,
compared with the control (Fig.
4A). Previous research indicated that phospho-AMPKα may inhibit
the expression of mTOR to activate autophagy (38). The present data demonstrated that
phospho-mTOR is significantly downregulated under exposure to TMZ,
compared with the control (Fig.
4A). All of these results indicated that the CaMKKβ/AMPKα/mTOR
pathway contributed to activation of autophagy when exposed to TMZ.
To further investigate the function of TRPC5, whether the
CaMKKβ/AMPKα/mTOR pathway was activated was investigated, and
TRPC5-siRNA was used and the key proteins of this pathway were
assessed. It was determined that knockdown of TRPC5 significantly
downregulated the phospho-CaMKKβ and phospho-AMPKα levels, and
upregulated the phospho-mTOR levels under TMZ exposure, compared
with the controls (Fig. 4B). Thus,
TRPC5 may be attributed to the initiation of autophagy via the
CaMKKβ/AMPKα/mTOR pathway. Subsequently, CaMKKβ was significantly
inhibited using KN−93, an inhibitor of CaMK (39), in U87/WT cells, and AMPKα activity
was significantly inhibited and phospho-mTOR expression was
significantly enhanced on exposure to TMZ (Fig. 4C). Furthermore, AMPKα was
significantly inhibited by dorsomorphin, an inhibitor of AMPK
(40), and it also significantly
upregulated the phospho-mTOR levels and significantly attenuated
the LC3-II levels (Fig. 4D).
Additionally, inhibition of the CaMKKβ/AMPKα/mTOR pathway also
significantly increased the TMZ sensitivity of U87/TMZ cells
(Fig. 5A-D). The present data
indicated that TRPC5 upregulated the CaMKKβ/AMPKα/mTOR pathway to
activate cytoprotective autophagy during TMZ exposure.
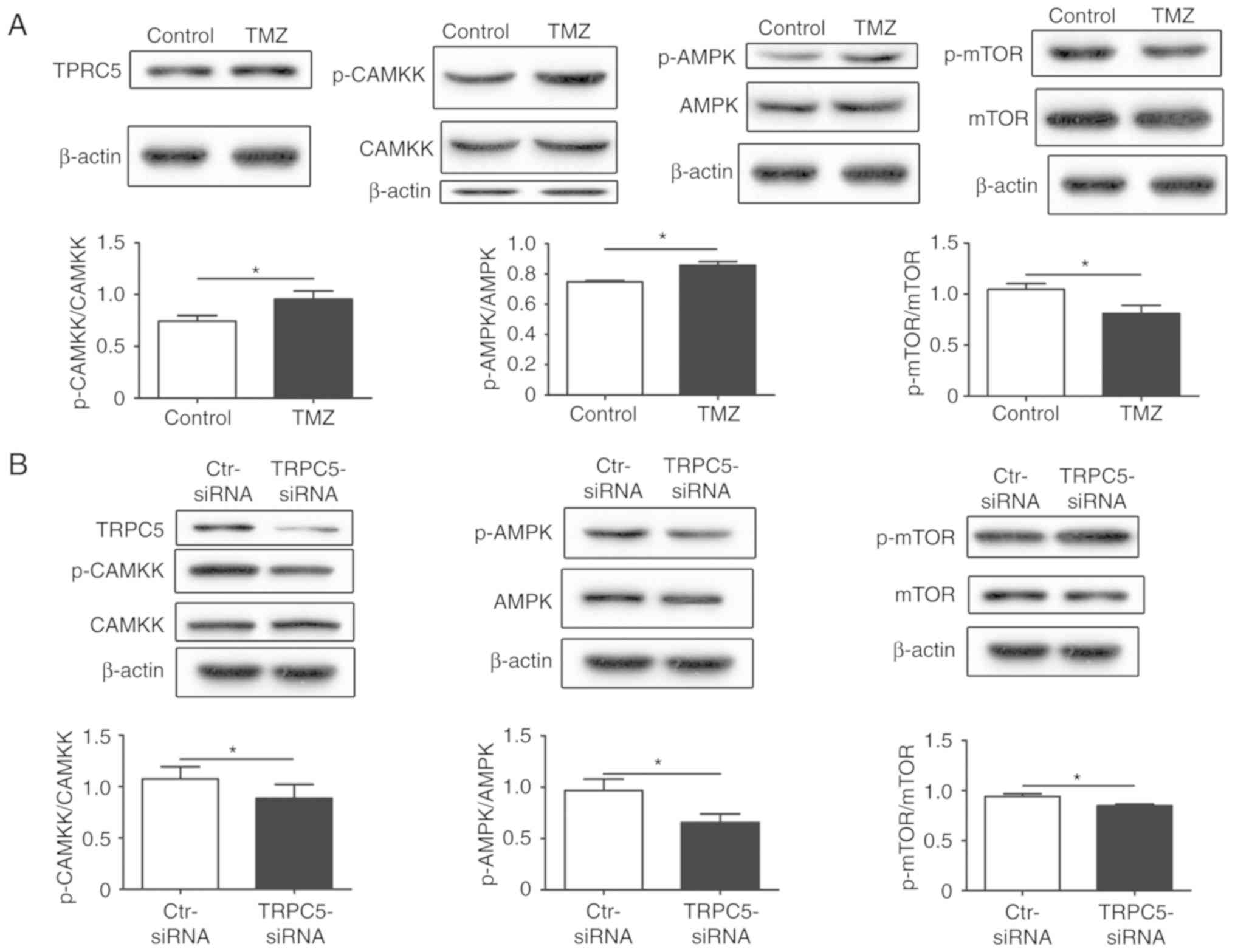 | Figure 4.TRPC5 initiates autophagy via the
CaMKKβ/AMPKα/mTOR pathway during chemotherapy. (A) U87/WT cells
were exposed to TMZ for 48 h and the protein levels of the
CaMKKβ/AMPKα/mTOR pathway were analyzed by western blot analysis.
p-CaMKKβ and p-AMPKα levels were increased, while p-mTOR/mTOR
levels decreased, compared with the control group. (B) U87/WT cells
were treated with TRPC5-siRNA and the protein levels of the
CaMKKβ/AMPKα/mTOR pathway were analyzed in U87/WT cells exposed to
TMZ. The p-CaMKKβ and p-AMPKα levels were decreased, while p-mTOR
levels were increased, compared with the control group. (C) CaMKKβ
inhibited by KN−93 decreased p-CaMKKβ and p-AMPKα
levels, and increased p-mTOR levels in U87/WT cells treated with
TMZ. (D) AMPKα silencing by dorsomorphin decreased p-AMPKα levels,
increased p-mTOR levels and downregulated LC3-II levels in U87/WT
cells exposed to TMZ. Values are presented as the mean ± standard
error of the mean of 3–6 experiments. CaMKKβ,
Ca2+/calmodulin dependent protein kinase β; AMPKα,
AMP-activated protein kinase α; p-, phospho-; mTOR, mechanistic
target of rapamycin kinase; TRPC5, transient receptor potential
cation channel subfamily C member 5; TMZ, temozolomide; siRNA,
small interfering RNA; Ctr, control. *P<0.05, **P<0.01,
***P<0.001. |
Downregulation of TRPC5 to suppress
autophagy increases TMZ sensitivity in vivo
To determine whether inhibition of autophagy induced
by TRPC5 also upregulates sensitivity to TMZ in vivo, nude
mice were injected subcutaneously with U87/WT cells previously
treated with TRPC5 short hairpin (sh)RNA or control shRNA
lentiviral particles. The tumor size of TRPC5 shRNA-treated cancer
cells was significantly reduced, compared with cells transfected
with control shRNA following TMZ exposure (Fig. 6A). Additionally, it was determined
that tumors treated with TRPC5 shRNA exhibited reduced autophagy
following TMZ exposure. Furthermore, the LC3 level was
significantly upregulated in TRPC5-shRNA treated U87/WT xenografts,
compared with in control-shRNA treated U87/WT xenografts (Fig. 6B).
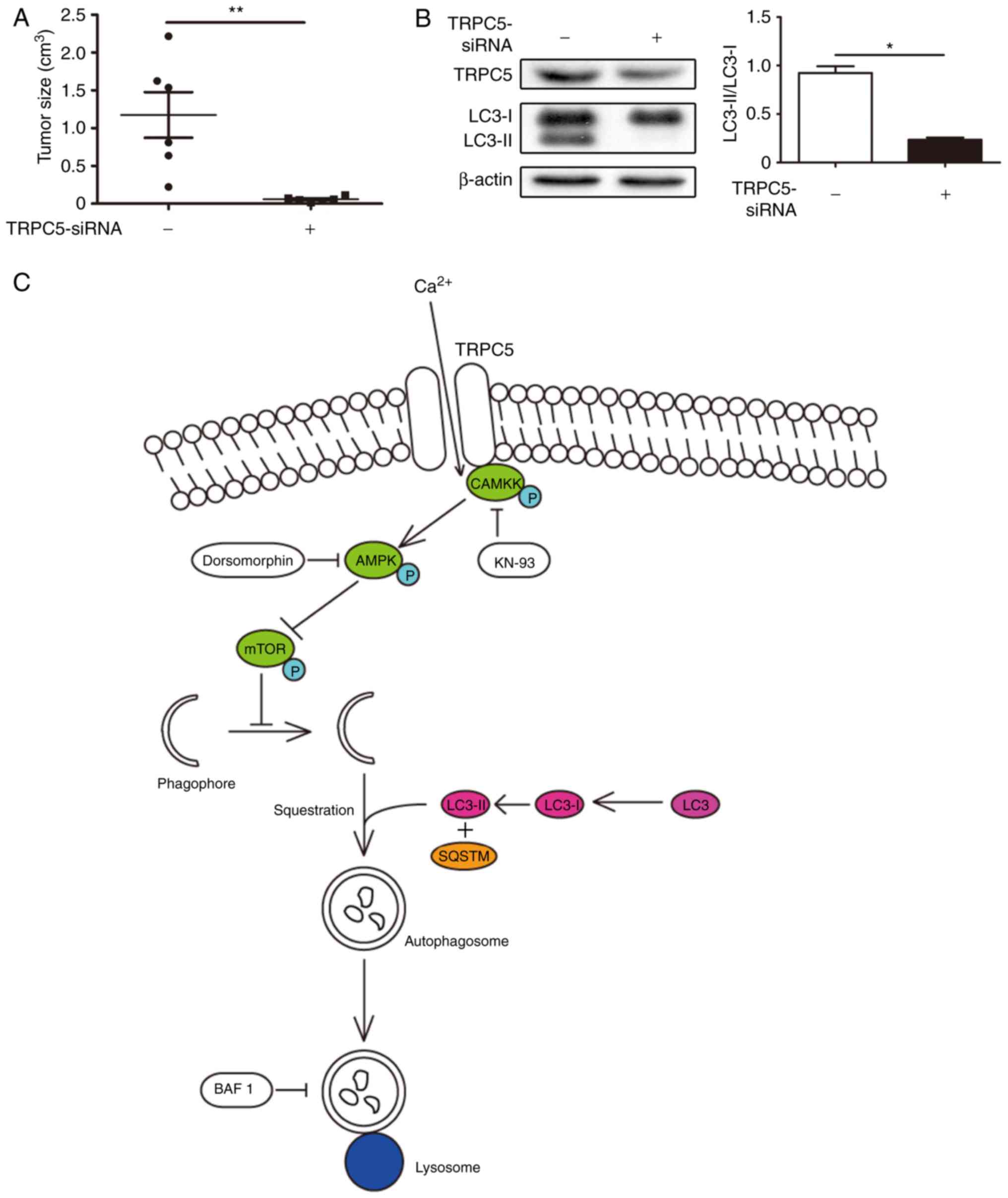 | Figure 6.Inhibition of autophagy by TRPC5
knockdown enhances sensitivity to TMZ in vivo. (A) Nude mice
were inoculated with U87/WT cells pre-transfected with TRPC5 or
control shRNA lentivirus and administered with TMZ (30
mg/m2) after 5 weeks (n=3 in each group). The tumor size
was measured after 5 weeks. (B) TRPC5 and LC3-II protein expression
were measured by western blot analysis. Values are presented as the
mean ± standard error of the mean. (C) Signal pathway involved in
TRPC5-activated autophagy in glioma cells under exposure to TMZ.
TMZ increases TRPC5 expression and activates phospho-CaMKKβ, and
then activates phospho-AMPKα. AMPKα activation negatively regulates
phospho-mTOR, resulting in upregulated basic autophagy level. TRPC5
induces autophagy during chemotherapy and accelerates glioma cell
survival. Arrows represent upregulation events, blunt arrows
represent downregulation events. TRPC5, transient receptor
potential cation channel subfamily C member 5; siRNA, small
interfering RNA; LC3, microtubule associated protein 1 light chain
3 α; CaMKK, Ca2+/calmodulin dependent protein kinase;
AMPK, AMP-activated protein kinase; P, phosphate; mTOR, mechanistic
target of rapamycin kinase; BAF1, Bafilomycin
A1; SQSTM, sequestosome; TMZ, temozolomide. *P<0.05,
**P<0.01. |
Discussion
Numerous patients with glioma acquire resistance to
the first-choice drug TMZ and chemotherapy resistance is the major
cause for recurrence and mortality. Autophagy induced by
chemotherapy is considered as a novel participant in drug
resistance in cancer cells (16),
but the precise mechanism and initiator of autophagy remains
unknown. In the present study, it was determined that
TRPC5-activated autophagy serves as a novel participant in the
occurrence and development of TMZ resistance in glioma cells.
Blockage of TRPC5 or autophagy accelerated glioma cell death under
exposure to TMZ. The present results also indicated significant
inhibition of autophagy and xenografts treated with TRPC5 shRNA in
response to TMZ in vivo.
A number of mechanisms, including abnormal
ex-transport of drug, inhibition of cell death pathways and
activation of the DNA repair system, are considered to contribute
to chemotherapy resistance (15,41,42).
In the present research, TMZ exposure enhanced TRPC5 protein
expression in U87/WT cells. Furthermore, silencing TRPC5 expression
enhances drug sensitivity and restricts resistance in glioma cells
exposed to TMZ. Similar results were also determined in U87/TMZ
cells. Collectively, it was demonstrated that TRPC5 acts as a
positive regulator against TMZ-induced cell death in glioma cells.
Chemotherapy destroys cancer cells by resulting in cell death via a
number of mechanisms, including apoptosis and damage to DNA
duplication (42,43); however, autophagy is considered to
accelerate the degradation of and recycle damaged or excess
components to maintain survival (19,44,45).
Previous research demonstrated that a number of types of TRP
channels, including TRP mucolipin 1 (TRPML1), TRPML3, TRPV1, TRPC1
and TRPM7, are involved in the regulation of autophagy via
different mechanisms (46). TRPML1
impairs lysosomal pH, and accumulates autophagosomes, abnormal
mitochondria, p62 and ubiquitin proteins to regulate autophagy
(47–50). TRPV1 activates autophagy through the
reactive oxygen species-associated AMPK and autophagy related 4C
cysteine peptidase pathway (51).
TRPC1 serves as a key regulator in hypoxia and nutrient depletion
dependent autophagy (52), while
TRPM7 regulates basal autophagy (53). Autophagy is also exhibited in a
number of cancer cells, including glioma and lung cells, to
maintain cell survival under chemotherapy. In gastric cancer cells,
TRPM2 downregulation inhibited the c-Jun N-terminal kinase signal
pathway, accumulated the damaged mitochondria and upregulated the
chemosensitivity to paclitaxel and doxorubicin (54). In the present study, increased
autophagy occurs under chemotherapy in glioma cells. In line with
these results, TMZ exposure was determined to increase the LC3-II
protein expression and accelerate LC3 dots formation in glioma
cells. U87/TMZ cells acquired an increased basic autophagy level,
compared with U87/WT cells. The combination of CQ and TMZ
facilitates the cell death of sensitive or drug-resistant glioma
cells, compared with TMZ alone, indicating that autophagy may be a
main mechanism of cell survival. Subsequently, the association
between TRPC5 and autophagy was determined. The present results
indicated that overexpression of TRPC5 significantly upregulates
LC3-II levels and accelerates LC3 dots formation in glioma cells
under exposure to TMZ. Knockdown of TRPC5 reduced LC3-II expression
and facilitated cell death of glioma cells in response to TMZ.
Furthermore, U87/WT cells treated with TRPC5 shRNA lentiviral
particles acquired decreased autophagy level and restricted tumor
size with TMZ chemotherapy. In conclusion, TRPC5 mediates glioma
cell survival to TMZ via autophagy activation.
Autophagy is negatively regulated by mTOR via
regulating the binding of the ULK1-ATG13-FIP200 complex (55). Therefore, the function of mTOR may
depend on a number of upstream components. AMPK, the upstream
molecule of mTOR, functions as a positive regulator in the
activation of autophagy (56,57).
AMPKα is phosphorylated and activated by CaMKKβ (58). The activation of Ca2+
influx via TRPC5 in response to numerous physiological stimuli to
activate CaMKKβ has been determined (59). Thus, to confirm whether autophagy
activation by TRPC5 exposed to chemotherapy via the
CaMKKβ/AMPKα/mTOR pathway, the effects of TRPC5-siRNA and
pharmacological agents on this pathway were examined.
Downregulation of TRPC5 inhibited the phosphorylated activity of
CaMKKβ and AMPKα, and increased the phosphorylated activity of mTOR
under TMZ exposure. Furthermore, KN−93 to silence
CaMKKβ, and dorsomorphin to silence AMPKα, restricted the autophagy
activation and accelerated cell death under expose to TMZ. In line
with previous research (16), the
present results indicated that TRPC5-induced autophagy is
mTOR-dependent in glioma cells mediating chemotherapy. Therefore,
the present data indicated that the CaMKKβ/AMPKα/mTOR pathway is
involved in TRPC5-induced autophagy in chemotherapy.
In conclusion, the present results indicated that
TRPC5-mediated autophagy facilitated the cell viability of glioma
cells via the CaMKKβ/AMPKα/mTOR pathway under exposure to TMZ
(Fig. 6C). TRPC5 expression has a
positive correlation with autophagy in vivo prior to and
following TMZ chemotherapy. This research confirmed TRPC5 to be an
initiator of autophagy, and revealed a novel mechanism for drug
resistance in chemotherapy for glioma.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr De'en Xu
(Department of Neurology, The Affiliated Wuxi No. 2 Hospital of
Nanjing Medical University, Wuxi, China), Mr. Xiu'yun Zhao
(Institute of Neuroscience and Jiangsu Key Laboratory of
Translational Research and Therapy for Neuro-psycho-Diseases,
Soochow University, Suzhou, China), Mr Pei'pei Yao (Institute of
Neuroscience & Jiangsu Key Laboratory of Translational Research
and Therapy for Neuro-psycho-Diseases, Soochow University, Suzhou,
China) and Mr De'juan Yuan (Institute of Neuroscience and Jiangsu
Key Laboratory of Translational Research and Therapy for
Neuro-psycho-Diseases, Soochow University, Suzhou, China) for their
advice and assistance.
Funding
No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
XJL and YZ conceived and designed the experiments.
YZ, JW, SZ and MC performed the experiments. YZ, XDZ and ZLM
analyzed the data. YZ and XJL wrote the paper. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All animals were kept in a pathogen-free environment
and fed ad libitum. The procedures for care and use of
animals were approved by the Ethics Committee of the Affiliated No.
2 Hospital of Nanjing Medical University (Wuxi, China) and all
applicable institutional and governmental regulations concerning
the ethical use of animals were followed.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Towner RA, Smith N, Saunders D, Brown CA,
Cai X, Ziegler J, Mallory S, Dozmorov MG, Coutinho De Souza P, et
al: OKN-007 Increases temozolomide (TMZ) sensitivity and suppresses
TMZ-resistant glioblastoma (GBM) tumor growth. Transl Onco.
12:320–335. 2019. View Article : Google Scholar
|
|
2
|
Ostrom QT, Gittleman H, Liao P,
Vecchione-Koval T, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and other central nervous
system tumors diagnosed in the United States in 2010–2014. Neuro
Oncol. 19 (Suppl 5):v1–v88. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lai SW, Huang BR, Liu YS, Lin HY, Chen CC,
Tsai CF, Lu DY and Lin C: Differential characterization of
temozolomide-resistant human glioma cells. Int J Mol Sci. 19(pii):
E1272018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kunjachan S, Rychlik B, Storm G, Kiessling
F and Lammers T: Multidrug resistance: Physiological principles and
nanomedical solutions. Adv Drug Deliv Rev. 65:1852–1865. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen C, Hanson E, Watson JW and Lee JS:
P-glycoprotein limits the brain penetration of nonsedating but not
sedating H1-antagonists. Drug Metab Dispos. 31:312–318. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Minchinton AI and Tannock IF: Drug
penetration in solid tumours. Nat Rev Cancer. 6:583–592. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rebucci M and Michiels C: Molecular
aspects of cancer cell resistance to chemotherapy. Biochem
Pharmacol. 85:1219–1226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kiselyov K, van Rossum DB and Patterson
RL: TRPC channels in pheromone sensing. Vitam Horm. 83:197–213.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lehen'kyi V and Prevarskaya N: Oncogenic
TRP channels. Adv Exp Med Biol. 704:929–945. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zholos AV: TRPC5. Handb Exp Pharmacol.
222:129–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hong C, Seo H, Kwak M, Jeon J, Jang J,
Jeong EM, Myeong J, Hwang YJ, Ha K, Kang MJ, et al: Increased TRPC5
glutathionylation contributes to striatal neuron loss in
Huntington's disease. Brain. 138:3030–3047. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Y, Xu Y, Thilo F, Friis UG, Jensen BL,
Scholze A, Zheng J and Tepel M: Erythropoietin increases expression
and function of transient receptor potential canonical 5 channels.
Hypertension. 58:317–324. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Everett KV, Chioza BA, Georgoula C, Reece
A, Gardiner RM and Chung EM: Infantile hypertrophic pyloric
stenosis: Evaluation of three positional candidate genes, TRPC1,
TRPC5 and TRPC6, by association analysis and re-sequen.
Hum Genet. 126:819–831. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma X, Cai Y, He D, Zou C, Zhang P, Lo CY,
Xu Z, Chan FL, Yu S, Chen Y, et al: Transient receptor potential
channel TRPC5 is essential for P-glycoprotein induction in
drug-resistant cancer cells. Proc Natl Acad Sci USA.
109:16282–16287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang P, Liu X, Li H, Chen Z, Yao X, Jin J
and Ma X: TRPC5-induced autophagy promotes drug resistance in
breast carcinoma via CaMKKβ/AMPKα/mTOR pathway. Sci Rep.
7:31582017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lamark T, Svenning S and Johansen T:
Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays
Biochem. 61:609–624. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kroemer G, Mariño G and Levine B:
Autophagy and the integrated stress response. Mol Cell. 40:280–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yorimitsu T and Klionsky DJ: Autophagy:
Molecular machinery for self-eating. Cell Death Differ. 12 (Suppl
2):S1542–S1552. 2005. View Article : Google Scholar
|
|
22
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jin S and White E: Role of autophagy in
cancer: Management of metabolic stress. Autophagy. 3:28–31. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Levine B: Unraveling the role of autophagy
in cancer. Autophagy. 2:65–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin S and White E: Tumor suppression by
autophagy through the management of metabolic stress. Autophagy.
4:563–566. 2008. View Article : Google Scholar :
|
|
28
|
Xie CM, Liu XY, Sham KW, Lai JM and Cheng
CH: Silencing of EEF2K (eukaryotic elongation factor-2 kinase)
reveals AMPK-ULK1-dependent autophagy in colon cancer cells.
Autophagy. 10:1495–1508. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cao L, Walker MP, Vaidya NK, Fu M, Kumar S
and Kumar A: Cocaine-mediated autophagy in astrocytes involves
sigma 1 receptor, PI3K, mTOR, Atg5/7, Beclin-1 and induces type II
programed cell death. Mol Neurobiol. 53:4417–4430. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gozuacik D and Kimchi A: Autophagy and
cell death. Curr Top Dev Biol. 78:217–245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li W, Zhou Y, Yang J, Li H, Zhang H and
Zheng P: Curcumin induces apoptotic cell death and protective
autophagy in human gastric cancer cells. Oncol Rep. 37:3459–3466.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo Y and Pei X: Tetrandrine-induced
autophagy in MDA- MB-231 triple-negative breast cancer cell through
the inhibition of PI3K/AKT/mTOR signaling. Evid Based Complement
Alternat Med. 2019:75174312019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sun WL, Chen J, Wang YP and Zheng H:
Autophagy protects breast cancer cells from epirubicin-induced
apoptosis and facilitates epirubicin-resistance development.
Autophagy. 7:1035–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chittaranjan S, Bortnik S, Dragowska WH,
Xu J, Abeysundara N, Leung A, Go NE, DeVorkin L, Weppler SA, Gelmon
K, et al: Autophagy inhibition augments the anticancer effects of
epirubicin treatment in anthracycline-sensitive and -resistant
triple-negative breast cancer. Clin Cancer Res. 20:3159–3173. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Takeuchi M and Yamamoto T: Apoptosis
induced by NAD depletion is inhibited by KN-93 in a
CaMKII-independent manner. Exp Cell Res. 335:62–67. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dasgupta B and Seibel W: Compound
C/dorsomorphin: Its use and misuse as an AMPK inhibitor. Methods
Mol Biol. 1732:195–202. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen X, Zhang M, Gan H, Lee JH, Fang D,
Kitange GJ, He L, Hu Z, Parney IF, Meyer FB, et al: A novel
enhancer regulates MGMT expression and promotes temozolomide
resistance in glioblastoma. Nat Commun. 9:29492018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Goldar S, Khaniani MS, Derakhshan SM and
Baradaran B: Molecular mechanisms of apoptosis and roles in cancer
development and treatment. Asian Pac J Cancer Prev. 16:2129–2144.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Si W, Shen J, Zheng H and Fan W: The role
and mechanisms of action of microRNAs in cancer drug resistance.
Clin Epigenetics. 11:252019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xie Z and Klionsky DJ: Autophagosome
formation: Core machinery and adaptations. Nat Cell Biol.
9:1102–1109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Düzen IV, Yavuz F, Vuruskan E, Saracoglu
E, Poyraz F, Göksülük H, Candemir B and Demiryürek S: Leukocyte TRP
channel gene expressions in patients with non-valvular atrial
fibrillation. Sci Rep. 7:92722017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jennings JJ Jr, Zhu JH, Rbaibi Y, Luo X,
Chu CT and Kiselyov K: Mitochondrial aberrations in mucolipidosis
Type IV. J Biol Chem. 281:39041–39050. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Curcio-Morelli C, Charles FA, Micsenyi MC,
Cao Y, Venugopal B, Browning MF, Dobrenis K, Cotman SL, Walkley SU
and Slaugenhaupt SA: Macroautophagy is defective in
mucolipin-1-deficient mouse neurons. Neurobiol Dis. 40:370–377.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vergarajauregui S, Connelly PS, Daniels MP
and Puertollano R: Autophagic dysfunction in mucolipidosis type IV
patients. Hum Mol Genet. 17:2723–2737. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y,
Bashllari E, Bisceglia J, Muallem S and Kiselyov K: TRP-ML1
regulates lysosomal pH and acidic lysosomal lipid hydrolytic
activity. J Biol Chem. 281:7294–7301. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Farfariello V, Amantini C and Santoni G:
Transient receptor potential vanilloid 1 activation induces
autophagy in thymocytes through ROS-regulated AMPK and Atg4C
pathways. J Leukoc Biol. 92:421–431. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sukumaran P, Sun Y, Vyas M and Singh BB:
TRPC1-mediated Ca2+ entry is essential for the
regulation of hypoxia and nutrient depletion-dependent autophagy.
Cell Death Dis. 6:e16742015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Oh HG, Chun YS, Park CS, Kim TW, Park MK
and Chung S: Regulation of basal autophagy by transient receptor
potential melastatin 7 (TRPM7) channel. Biochem Biophys Res Commun.
463:7–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Almasi S, Kennedy BE, El-Aghil M, Sterea
AM, Gujar S, Partida-Sánchez S and El Hiani Y: TRPM2
channel-mediated regulation of autophagy maintains mitochondrial
function and promotes gastric cancer cell survival via the
JNK-signaling pathway. J Biol Chem. 293:3637–3650. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et
al: Nutrient- dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell.
20:1981–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Behrends C, Sowa ME, Gygi SP and Harper
JW: Network organization of the human autophagy system. Nature.
466:68–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shaw RJ, Bardeesy N, Manning BD, Lopez L,
Kosmatka M, DePinho RA and Cantley LC: The LKB1 tumor suppressor
negatively regulates mTOR signaling. Cancer cell. 6:91–99. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bort A, Sánchez BG, Spínola E,
Mateos-Gómez PA, Rodriguez- Henche N and Diaz-Laviada I: The red
pepper's spicy ingredient capsaicin activates AMPK in HepG2 cells
through CaMKKβ. PLoS One. 14:e02114202019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yoshida T, Inoue R, Morii T, Takahashi N,
Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y and Mori Y:
Nitric oxide activates TRP channels by cysteine S-nitrosylation.
Nat Chem Biol. 2:596–607. 2006. View Article : Google Scholar : PubMed/NCBI
|