Introduction
Tumors develop as organ-like structures with a high
metabolic demand (1). In the early
stages, their growth depends on the passive diffusion of oxygen,
nutrients and metabolic waste (2).
When a tumor reaches 1–2 mm in diameter and its demand for oxygen
and nutrients surpasses the local supply, hypoxia and nutrient
starvation trigger an ‘angiogenic switch’ to allow for tumor
progression (3). The further
enlargement of these growths depends on angiogenesis initiated by
cancerous cells to enhance the supply of oxygen and nutrients
(4). Tumor angiogenesis involves a
complex series of events, starting with vessel membrane
degradation, followed by endothelial cell (EC) migration,
proliferation and re-organization, promoting vessel maturation,
indicating that ECs are essential in each step of angiogenesis
(5).
As ECs migrate towards a tumor, they can form loops
and branches to connect with it and supply it with nutrients
(6). This mechanism involves a
complex interaction between many factors, including adhesion
proteins, growth factors, junctional molecules, oxygen sensors and
endogenous inhibitors (7). The
crosstalk between tumor cells and ECs leads to modification of
their properties and facilitation of the tumor cell function
(8). It has been demonstrated that
the tumor microenvironment contains a variety of cytokines
regulating its angiogenesis (3).
Pro- and anti-angiogenic molecules, necessary for blood vessel
growth, are not only produced by tumor cells themselves, but can
also be products of ECs, pericytes, the extracellular matrix and
plasma clotting (9,10). This complex interplay between
stimulatory and inhibitory angiogenic factors is most likely
influenced by the tumor cells in order to initiate and further
support the vascular growth within the tumor (3). However, the underlying mechanisms and
any tumor type variations remain unclear.
In the present study, the influence of various tumor
types was analyzed, including that of breast cancer MCR86,
osteosarcoma ROS-1, colon cancer CC531 and rhabdomyosarcoma R1H
cells on EC52 cells in an in vitro rat cell model. Important
steps occurring during tumor vascularization were examined,
including EC proliferation, migration and tube formation, as well
as subsequent underlying changes in genetic expression patterns.
Investigation of tumor-induced angiogenesis may provide an improved
understanding of tumor vascularization and progression, which in
turn may lead to the development of novel anti-angiogenic
therapeutic strategies in the future.
Materials and methods
Cell lines and cell culture
Rat colon adenocarcinoma CC531 cells (CLS Cell Lines
Service GmbH, Eppelheim, Germany) were cultured in RPMI-1640 medium
(Thermo Fisher Scientific Inc., Waltham, MA, USA) supplemented with
10% fetal calf serum (FCS; Biochrom Ltd., Cambridge, UK) and 2 mM
L-glutamine (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Rat
rhabdomyosarcoma R1H cells were obtained from Dr A. Raabe
(Laboratory for Radiobiology and Experimental Radio-Oncology,
University Cancer Center Hamburg, Hamburg, Germany) and were
cultured in Dulbecco's modified Eagle's medium (Biochrom Ltd.)
supplemented with 5% FCS. Rat mammary carcinoma MCR86 cells were
provided by Professor P. J. K. Kuppen (Department of Surgery,
Leiden University Medical Center, Leiden, The Netherlands) and were
cultured in RPMI-1640 medium supplemented with 10% FCS. Rat
osteosarcoma ROS-1 cells were provided by Professor T. L. M. ten
Hagen (Laboratory of Experimental Surgical Oncology, Department of
Surgery, Erasmus MC, Rotterdam, The Netherlands) and were cultured
in minimum essential medium (Sigma-Aldrich; Merck KGaA)
supplemented with 10% FCS. Rat liver EC52 cells were cultured in
RPMI-1640 medium supplemented with 10% FCS, 2 mM L-glutamine and 1
µM dexamethasone (PeloBiotech GmbH, Planegg, Germany). All cell
lines were incubated at 37°C in a humidified atmosphere of 95% air
and 5% CO2. The culture medium was changed every 2–3
days and the cells were passaged when 80–90 % confluence was
reached.
Direct co-culture of EC52 and tumor
cells
A total of 2×104 tumor cells, including
colon cancer CC531, rhabdomyosarcoma R1H, breast cancer MCR86 and
osteosarcoma ROS-1 cells, were labeled with 25 µM CellTrace™ Oregon
Green® 488 (Thermo Fisher Scientific Inc.), and
2×104 EC52 cells were labeled with 2 µM PKH26
(Sigma-Aldrich; Merck KGaA), according to the manufacturers'
protocols. Equal numbers of tumor cells and EC52 cells were mixed
and seeded in 6-well plates. The co-cultured cells were incubated
in a 1:1 mixture of tumor cell and EC52 cell culture media. Images
were captured after 3–4 days using a fluorescent Olympus IX83
microscope and were analyzed with CellSens Dimensions 1.16 software
(both from Olympus Corp., Tokyo, Japan).
Preparation of tumor-conditioned
medium (CM)
For tumor-CM, tumor cells were grown in their normal
cell culture medium in cell culture flasks with an area of 75
cm2. When the cells reached ~80% confluence, the medium
was changed and the cells were cultured for another 48 h. The
supernatant was collected and filtered (pore size, 0.22 µm). The CM
was mixed with fresh EC culture medium in a 1:1 ratio and used for
the cell culture assays (termed tumor-CM). A 1:1 mixture of
non-conditioned tumor cell and EC culture media was used as a
negative control.
WST-8 cell proliferation and viability
assay
To assess the effect of tumor-CM on EC52
proliferation, the quantitative colorimetric WST-8 assay (PromoCell
GmbH, Heidelberg, Germany) was performed at 3 time-points. Briefly,
EC52 cells were seeded at a density of 1×103 cells/well
in 100 µl EC culture medium in 96-well culture plates in
triplicate. After 24 h of incubation, the medium was replaced with
tumor-CM or control medium. After 24, 36 and 48 h of incubation, 10
µl WST-8 tetrazolium salt was added to each well for 2 h at 37°C.
For each measurement time-point one culture well plate was
prepared. The absorption was measured at 450 nm with a reference
wavelength of 600 nm. After measurement, the culture well plate was
discarded. The data was analyzed using SkanIt™ software version 3.2
(Thermo Fisher Scientific Inc.). The assay was performed 3
times.
Transwell migration and invasion
assays
Transmigration and invasion assays were performed in
a 24-well Transwell system (BD Biosciences, Franklin Lakes, NJ,
USA) with 8-µm-pore polycarbonate membrane inserts. Transmigration
was performed 2 times in technical duplicates. For statistical
analyses the single technical duplicates were used. For the
invasion experiments, the Transwell chambers were coated with 40 µl
growth factor reduced Matrigel® (Growth Factor Reduced
Basement Membrane Matrix; Corning Inc., Corning, NY, USA) and
incubated for 60 min at 37°C. Invasion was performed 3 times. A
total of 1×105 EC52 cells/well were seeded into the
upper chambers in 200 µl EC culture medium and 700 µl tumor-CM or
control medium was added to the bottom chambers. Following
incubation at 37°C for 8 h, the cells remaining on the surface of
the membrane were wiped away using a cotton swab. The cells that
migrated through the membrane were fixed with ice-cold methanol and
stained with 1 µg/ml DAPI (Roche Molecular Diagnostics, Pleasanton,
CA, USA). The stained cells were scanned and quantified in 4 random
fields per membrane (1 image per quadrant) at an ×40 magnification
using an Olympus IX83 microscope. In a few wells only 3 regions of
interest (ROIs) could be evaluated due to technical problems such
as unclear images.
Wound-healing assay
A wound-healing assay was performed in 12-well
plates. EC52 cells were seeded at a density of 1×105
cells/well in EC culture medium. When the cells reached confluence,
a scratch was created using a 100-µl pipette tip. The cell layer
was washed with PBS and CM or control medium was added. Images were
captured at 0, 6, 12 and 24 h. The uncovered area of the scratch
was measured in ≤4 ROIs in each well and compared with the
measurement at 0 h, which was set to 1. In certain wells, not all
ROIs could be evaluated due to unequal cell growth at the borders
of the well. The assay was performed 3 times.
Tube formation assay
Matrigel® (10 µl) was applied to each
well of a µ-slide (a chambered coverslip) (ibidi GmbH, Martinsried,
Germany) and incubated at 37°C for 30 min for gel polymerization.
EC52 cells were seeded in technical triplicates at a density of
1.5×104 cells/well in 50 µl tumor-CM or control medium
on the surface of the gelled matrix. After 7 h, the cells were
stained with Calcein-AM (Sigma-Aldrich; Merck KGaA) and images were
captured with an Olympus IX83 microscope and processed using
CellSens software. Capillary-like tube formation was analyzed using
the WimTube analysis software (Wimasis GmbH, Munich, Germany)
regarding covered area, tube length, branching points and loops.
Tube formation was performed 3 times.
To detect the direct interaction of tumor cells and
EC52 cells, 3.9×103 CM DiI dye-labeled tumor cells
(staining solution 5 µg/ml; Thermo Fisher Scientific Inc.) and
1.3×104 Oregon Green-labeled EC52 cells were seeded in
direct co-cultures in Matrigel®-coated wells, as
described above, and incubated for 6 h in a 1:1 mixture of tumor-CM
and EC52 cell culture medium. A monoculture of EC52 cells was grown
as the control. The evaluation was performed as described above.
Direct tube formation was performed once with technical triplicates
that were used for statistical analysis.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the EC52 cells
incubated in tumor-CM for 4 days using the RNeasy Mini kit with the
corresponding QIAshredder Homogenizer (both from Qiagen GmbH,
Hilden, Germany). The RNA was converted to cDNA using the
QuantiTect Reverse Transcription kit with a DNase I incubation (15
min 42°C, 3 min 95°C) (Qiagen GmbH). The qPCR reactions were
performed using the SsoAdvanced Universal SYBR-Green SuperMix
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) with a Bio-Rad
CFX96 Touch™ Light Cycler (Bio-Rad Laboratories, Inc.) and the
following thermal cycling protocol: Polymerase activation and DNA
denaturation: 30 sec 95°C, amplification: 40 cycles with
denaturation at 95°C for 5 sec and annealing at 60°C for 30 sec,
melt-curve analysis 55–95 °C 0.5°C increment 5 sec/step. All kits
were used according to the manufacturers' protocols and
glucuronidase-β was used as the housekeeping control. The primer
sequences used are listed in Table
I. All reactions were performed in triplicate. The experiment
was performed 3 times. The expression of individual genes was
calculated and normalized by the 2−ΔΔCq method (11).
 | Table I.Primer sequences. |
Table I.
Primer sequences.
|
| Forward Primer
(5′→3′) | Reverse Primer
(3′→5′) |
|---|
| Tie1 |
AGGGTCCTGGAGACTGATCC |
AAGGTACTGCATCCCGTTGG |
| Flt1 |
ACAGCAATGTGTTCCACAGCGT |
TGGTTTCCTGCACCTGTTGCTT |
| Angpt2 |
AATGCACAGTAGCCCCTTCC |
GGTGCAGGCCTAAGTGATGT |
| Pecam1 |
GCAGACCCTTCCACGAAGAA |
GCTTCGGAGACTGGTCACAA |
| Cxcl12 |
TCCGTGGGCTCTGAGTTTTC |
GGAACCCAGAATCCCCACTG |
| Egf |
TCGAGTCAACAAAGGGCCTC |
GAGTACCAGATCTGCCGCTC |
| Egfr |
CAACATCCTGGAGGGGGAAC |
ATGTTCATGGTCTGGGGCAG |
| Vwf |
CCCGGGAAACTCCTTCTTCC |
CAAGCAAGTCACTGTGTGGC |
| Pdgfrb |
GCATTGGCTCCATTCTTCAT |
CCGTGGTCATTCACACTCAC |
| Fgf2 |
TCCATCAAGGGAGTGTGTGC |
GGACTCCAGGCGTTCAAAGA |
| Vegfa |
AATGATGAAGCCCTGGAGTG |
ATGCTGCAGGAAGCTCATCT |
| Gusb |
TGGCCTTGGCTTTGTGTACT |
CGTGGGTGCTAGGAATCGAA |
Statistical analysis
The data are expressed as the mean ± standard
deviation. Statistical analysis was performed using SPSS version
20.0 software (IBM Corp., Armonk, NY, USA). Due to the small sample
size with mostly n=3, non-parametric statistical tests were used
since testing of normal distribution was considered to be
negligible. In case of multiple comparisons, the Friedman test was
used for dependent samples and the Kruskal-Wallis test was used for
comparing independent samples. Mean differences between 2
independent samples were analyzed by the non-parametric
Mann-Whitney U test; the asymptotic significance was used. Values
of P≤0.05 were considered to indicate statistically significant
differences.
Results
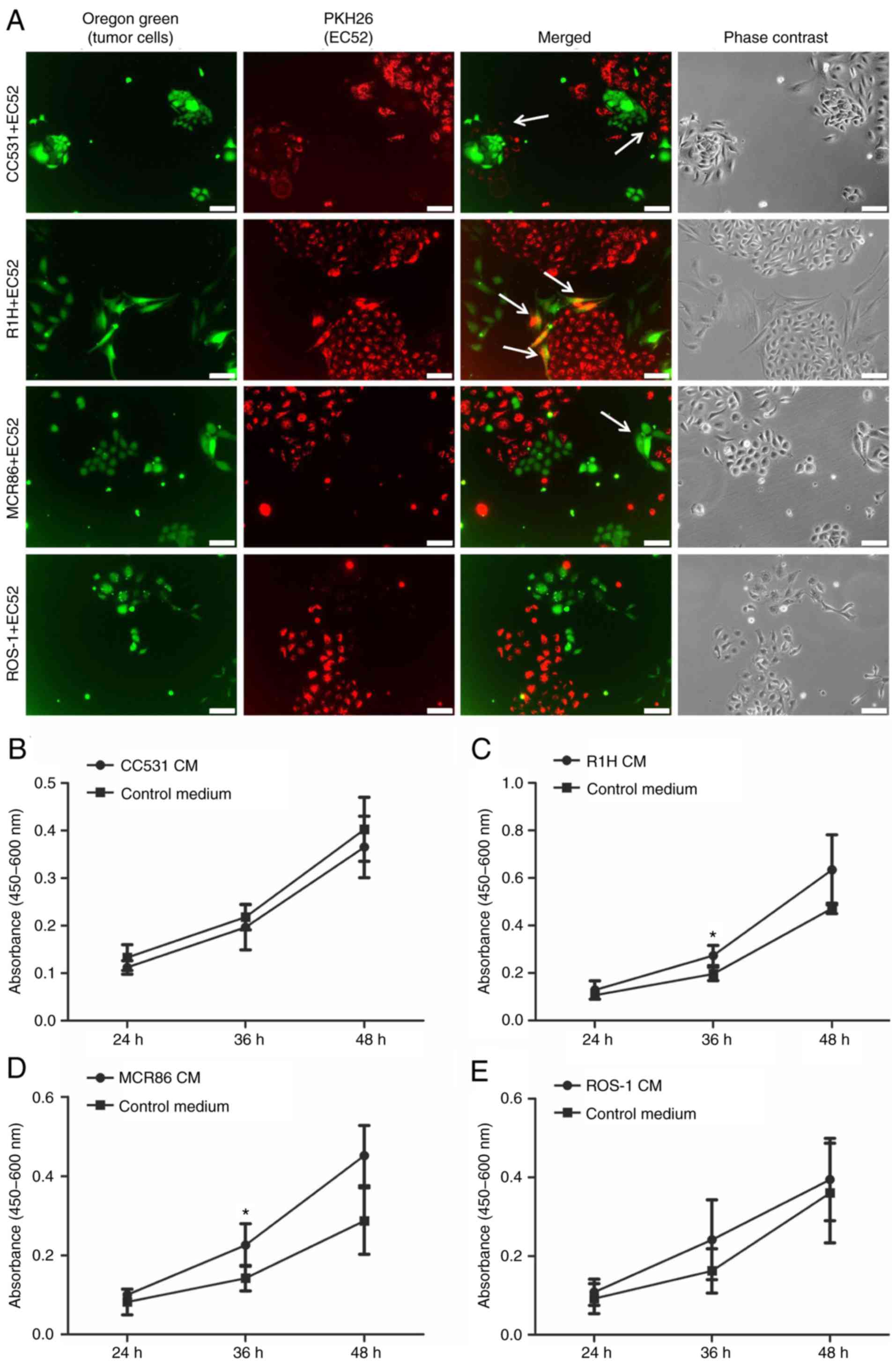
Effects of tumor-CM on EC52 morphology
and proliferation
EC52 cells in direct co-culture with various tumor
cells grew in colonies with uniform and characteristic cobblestone
appearance. No morphological changes were observed in the ECs. The
co-cultured cells grew evenly with each other with no signs of
overgrowth or inhibition of either one of the cell populations.
Where direct cell-cell contact occurred, different interaction
patterns were observed. In the rhabdomyosarcoma R1H co-culture,
double-labeled cells were visible, indicating close cell-cell
interactions. In many areas of the colon cancer CC531 co-culture,
the ECs formed a border around the tumor cell colonies. In the
culture with the breast cancer MCR86 cells, the tumor cells began
to grow in close contact with the ECs and grew around EC colonies
in certain areas (Fig. 1A).
The effect of tumor-CM on the viability and
proliferation of EC52 cells was investigated using a WST-8 assay at
24, 36 and 48 h of culture. The ECs proliferated over time in the
tumor-CM as well as the control medium, mostly with a tendency of
higher proliferation being observed in the tumor-CM (Fig. 1B-E). A significant increase in
absorbance was observed when the ECs were cultured in R1H CM
compared to control for 36 h (P≤0.05) (Fig. 1C), as well as in MCR86 CM compared
to control at 36 h (P≤0.05; Fig.
1D).
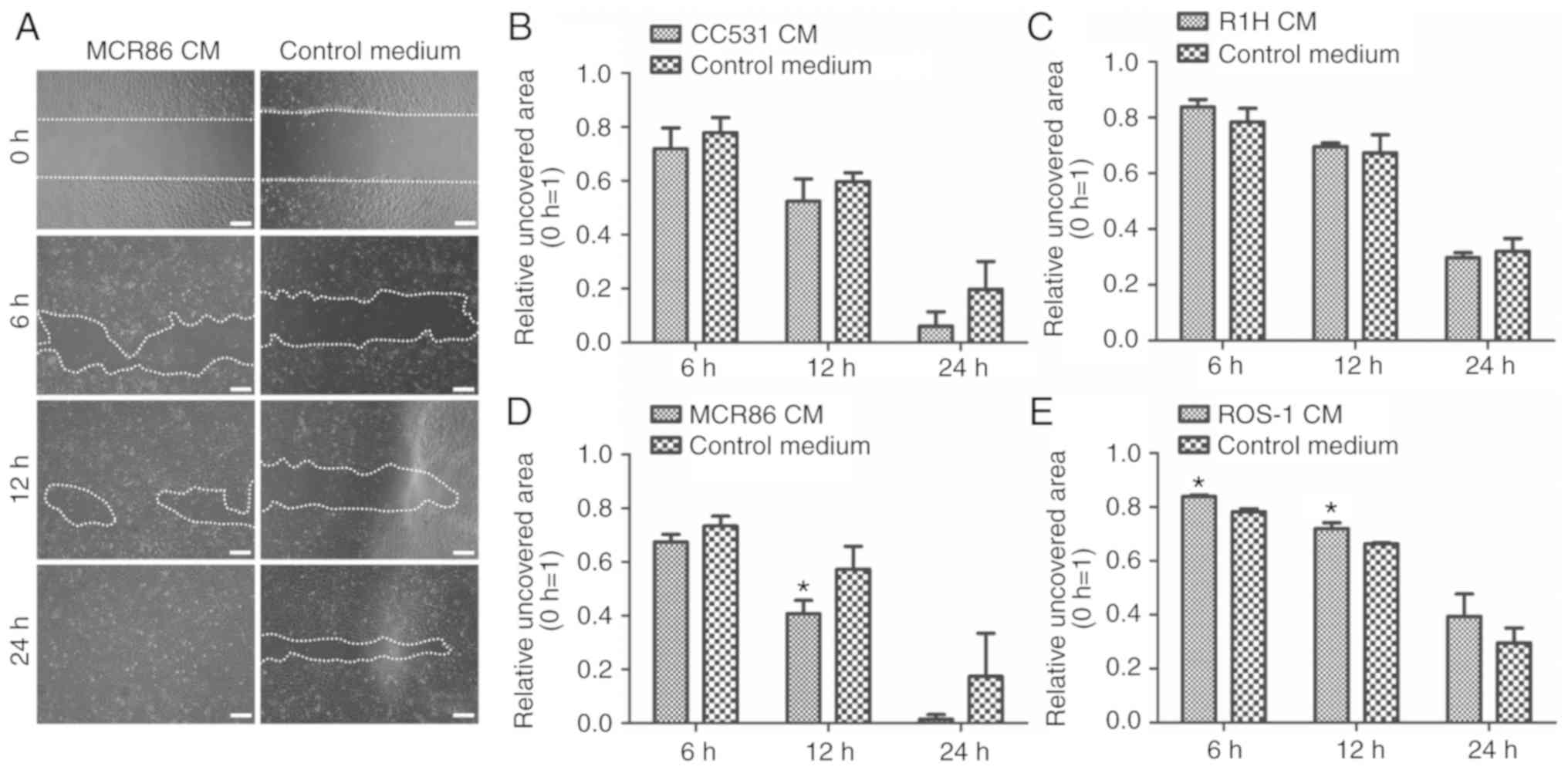
Tumor-CM promotes the migratory
capacity of ECs
To investigate the effect of tumor-CM on the
migratory ability of ECs, 3 distinct cell culture assays were
performed. First, a wound-healing assay was conducted. Compared
with that of the control, the migration of EC52 cells into the
scratched area was increased in the CC531 and MCR86 CM. This
difference became significant in the MCR86 CM compared to control
at time-point 12 h (P≤0.05; Fig.
2).
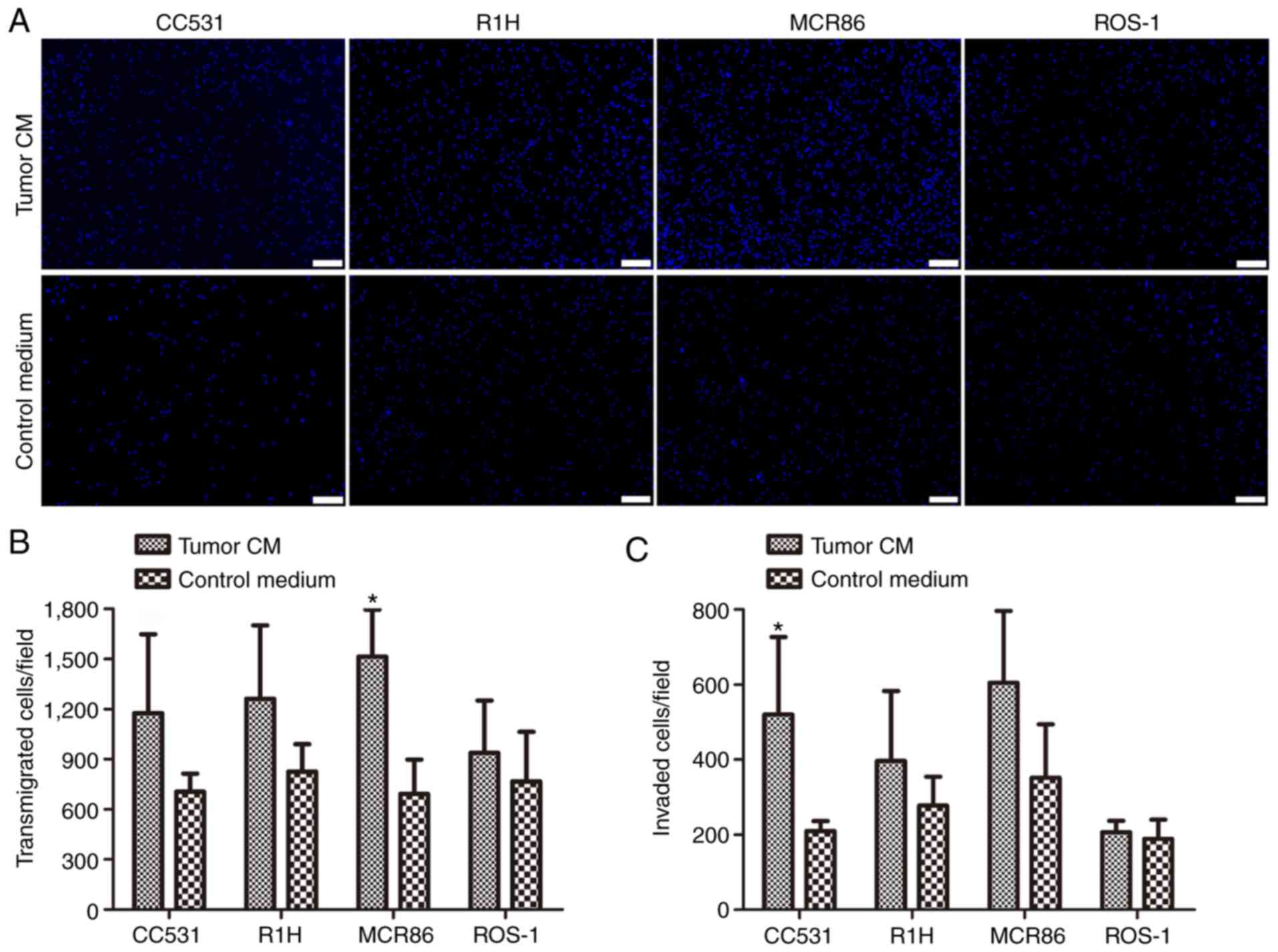
Subsequently, the influence of tumor-CM on the
transmigration and invasion capacities of ECs was analyzed using
Transwell assays. The transmigration was significantly stimulated
in tumor-CM breast cancer MCR86 groups (P≤0.05; Fig. 3A and B). The invasion of the ECs
through the Matrigel®-coated Boyden chambers was
significantly stimulated by the colon cancer CC531 CM compared with
the effect of the control (P≤0.05; Fig.
3C).
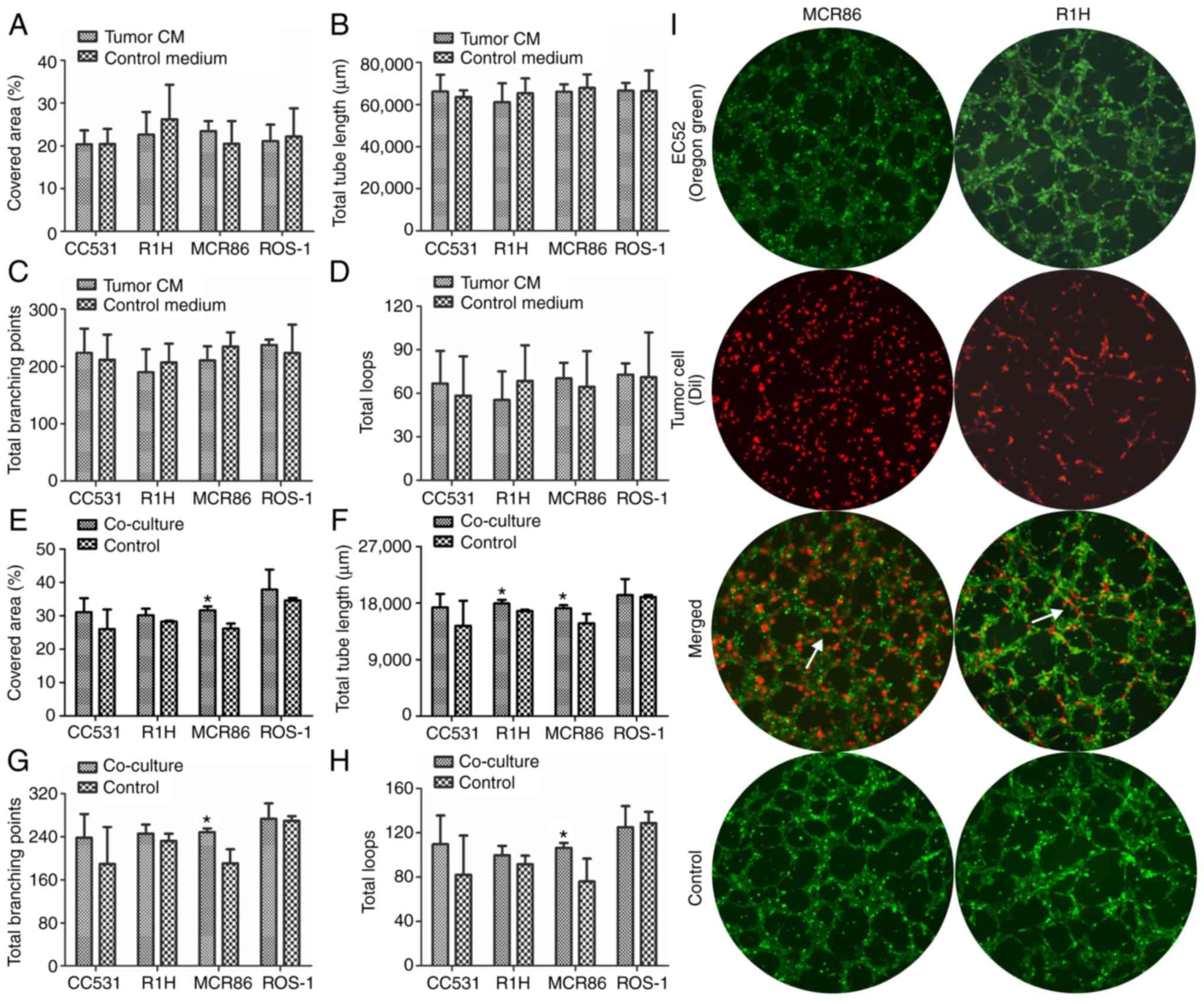
Stimulation of tube formation depends
on direct cell-cell contact
To evaluate the angiogenic capabilities of ECs in a
quantifiable manner, a tube-formation assay was performed.
Following seeding on Matrigel®, the EC52 cells formed
typical endothelial networks in the tumor-CM and control medium
with no significant differences (Fig.
4A-D).
To further analyze whether the stimulation of tube
formation depends on direct cell-cell interactions, ECs and tumor
cells were seeded in direct co-culture on Matrigel®. By
labeling tumor cells and ECs with different fluorescence dyes, the
contribution of the different cell populations to tube formation
could be visualized. The ECs directly co-cultured with breast
cancer MCR86 cells displayed a significantly higher area, total
tube length, number of branching points and total loops (P≤0.05)
compared with the mono-cultured ECs (Fig. 4E-H). In addition, the total tube
length was significantly longer in the rhabdomyosarcoma R1H group
compared with that of the control (P≤0.05; Fig. 4F). Tumor cells were observed in
close contact with the tubes formed by the ECs in all groups.
Furthermore, the rhabdomyosarcoma R1H cells participated in forming
tube-like structures by cell elongation and cell-cell interactions
in close contact with the tubes formed by the ECs (Fig. 4I).
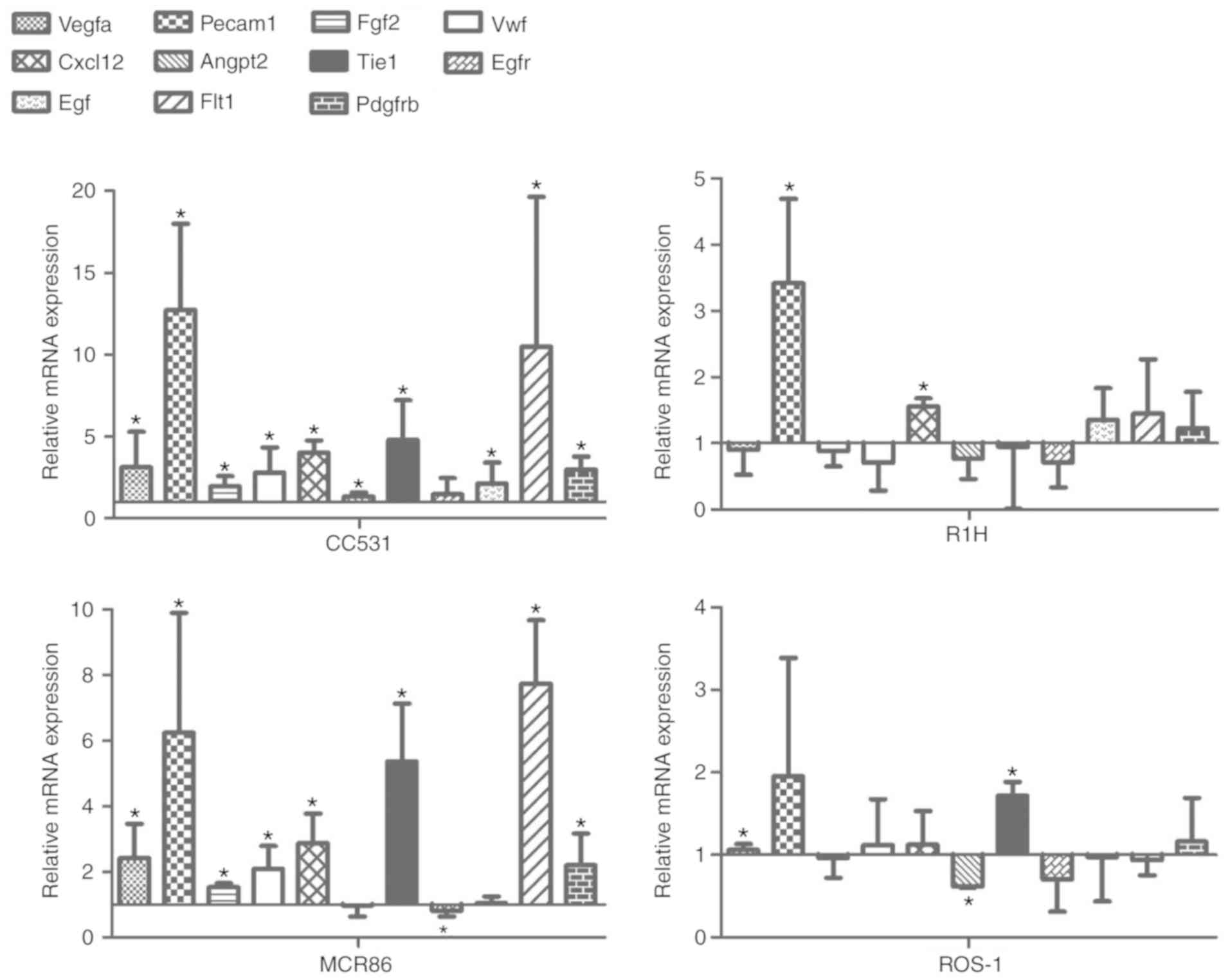
Tumor-CM upregulates
angiogenesis-associated genes
Following incubation in tumor-CM for 4 days, the
expression of 11 angiogenesis-associated genes in EC52 cells was
determined by RT-qPCR (Fig. 5). In
each group, the expression levels of angiogenic gene platelet
endothelial cell adhesion molecule-1 (PECAM1) were
significantly higher (except for ROS-1) in the tumor-CM compared
with those in the control. The expression levels of other
endothelial markers, including vascular endothelial growth factor A
(VEGFA), fibroblast growth factor 2 (FGF2), Von
Willebrand factor (VWF), C-X-C motif chemokine ligand 12
(CXCL12), tyrosine kinase with immunoglobulin-like and
EGF-like domains 1 (TIE1), VEGF receptor 1 (VEGFR1) and
platelet-derived growth factor receptor β (PDGFRB) were
significantly upregulated in groups stimulated with CC531 and MCR86
CM. PECAM1 exhibited a ~2-6-fold higher expression
level in the colon cancer CC531 and breast cancer MCR86 groups
compared with the rhabdomyosarcoma R1H and osteosarcoma ROS-1
groups. Whereas numerous angiogenesis-associated genes were
upregulated in ECs that were cultured in the CC531 or MCR86 CM,
only PECAM1 and CXCL12 in the R1H CM and VEGFA
and TIE1 in the ROS-1 CM were significantly upregulated
compared with the control.
Discussion
Angiogenesis is necessary for tumor growth,
development and metastasis (8).
Tumor vascularization is a complex multistep process. Important
components of this process are EC proliferation and migration,
followed by the formation of mostly abnormal capillaries (12,13).
ECs are stimulated by tumor-secreted factors to migrate and divide
at the tumor sites, ultimately forming capillary structures
stabilized by smooth muscle cells (14). Previous studies have demonstrated
that. The tumor microenvironment serves a crucial role in its
survival and development, and various factors influence its
vascularization, however, the mechanism remains unclear (3,15). As
part of the tumor microenvironment, direct interaction of cancer
cells with ECs may be of great importance for tumor metastasis, but
also for tumor angiogenesis (16).
Considering the growing awareness of the role of the tumor
microenvironment in its progression, the present study aimed at
deciphering its effect on tumor vascularization by studying the
changes in phenotype and gene expression. Furthermore, the
functional properties of ECs under the influence of various tumor
types were analyzed. The design of the present tumor/EC in
vitro model was based on previous studies (17–19).
In the direct co-culture, signs of cell-cell
interactions between the tumor cells and ECs were observed. It is
widely known that ECs in tumors have a unique activated phenotype
compared with those under normal conditions (3). In the colon and breast cancer groups,
the cells began to surround each other, perhaps due to tumor
cell-secreted soluble factors, such as tumor necrosis factor α,
which can rearrange F-actin cytoskeleton filaments influencing the
shape and motility of the cell (20), as well as transforming growth
factor-β, which can alter the content of collagen type IV and
provide a supporting structure for ECs (21,22).
Although no distinct change in the morphology of the ECs could be
detected, in certain areas they began to align with tumor cells in
a manner similar to that reported by a study on the interaction
between umbilical vein ECs and human glioma cells (22). Similar findings revealed that the CM
derived from breast cancer cells affected the phenotype and
behavior of normal cells (21).
After a number of days of co-culture, double-labeled cells became
visible in the rhabdomyosarcoma group. This could be an indication
of cell fusion, although loss of dye or it being absorbed up by
other cells cannot be excluded. Tumor cells have the ability to
fuse spontaneously with normal host cells, including ECs, which may
alter the biological behavior of tumors (23). Cell fusion with macrophages or bone
marrow-derived cells is quite a common feature of tumor cells to
enhance metastasis, drug resistance and resistance to apoptosis
(24). Similar findings were
described in a study reporting on the direct co-culture of
adipose-derived stem cells with breast cancer cells, indicating an
exchange of cellular vesicles and fusion of cells, which led to a
more malignant phenotype of the cancer cells (25). Hybrid cells derived from lung cancer
cells and mesenchymal stem cells underwent an
epithelial-mesenchymal transition (EMT), which resulted in a higher
metastatic capacity and a higher expression of matrix
metalloproteinase (MMP)2, MMP9 and typical mesenchymal markers,
including vimentin (26). The
authors suggest that this could be an explanation of the origin of
lung cancer stem cells. A possible mechanism of the tumor-EC fusion
was proposed by Mortensen et al (27), who demonstrated that, following
intravascular dissemination, breast cancer cells fused with ECs in
an in vivo mouse model. Hybrid cells exhibited markers of
the two cell types and underwent mitosis. Although the present
study did not further analyze cells in direct co-culture and
drawing a distinct conclusion is therefore not possible, it was
demonstrated that tumor cells not only interact with ECs in a
paracrine way, but also via direct cell-cell contact.
The presence of actively proliferating and
functional ECs is essential for tumor angiogenesis (16). To clarify if and to which extent the
various tumor cell lines contribute to the proliferation of ECs,
EC52 cells were incubated in the CM of tumor cell lines and their
viability was analyzed by a WST-8 assay. The breast cancer MCR86
and rhabdomyosarcoma R1H cells significantly stimulated the
viability of the ECs. A previous study also detected a relatively
low influence of tumor cells on the proliferation of endothelial
progenitor cells (EPC), but a significant contribution to the
recruitment and tube-like formation (28). Therefore, further experiments were
performed focusing on the migration of ECs. As demonstrated by the
WST-8 assay, the ECs were significantly affected by
rhabdomyosarcoma and breast cancer cells. Their migration in the
scratch test and the transmigration and invasion in the Boyden
chamber assay were notably stimulated especially in the mammary
carcinoma CM. Ding et al (29) reported a positive correlation
between increasing invasion and migration capacities and enhanced
proliferation ability. This effect of the breast cancer cells
appears not to be limited only to ECs, as they have also been
demonstrated to activate an alteration in the morphology and
migration properties of normal breast epithelial cells to trigger
the EMT (21), and the chemotaxis
of bone marrow-derived mesenchymal stromal cells (30).
The communication between ECs and other cell types
is not only based on paracrine, but also on direct cell-cell
interactions (28,31). Therefore, a co-culture system with
the incubation of ECs in tumor-CM was assessed in a tube-formation
assay. Tube formation is an in vitro model for evaluating
the differentiation of ECs into tube-like structures, occurring
subsequent to proliferation and migration (32). As anticipated from the
aforementioned findings, the breast cancer cell line significantly
stimulated the ECs to form tubes in the direct co-culture system.
Furthermore, the tumor cells participated in the formation of
tube-like structures (Fig. 4E-I).
These findings support the hypothesis that tumor cells can modify
their properties to form tumor blood vessels in terms of
vasculogenic mimicry (28,33,34).
However, the tumor-CM did not result in stimulating the EC52 cells
to form capillary-like structures in the present study (Fig. 4A-D). This observation strengthens
the hypothesis that tumor cells themselves contribute to tube
formation rather than further stimulating the tube-formation
capability of ECs.
Contrary to the present findings, the CM of
hepatocellular carcinoma and esophageal squamous cell carcinoma
cells enhanced the tube-formation abilities of ECs (8,35). In
another study, the CM from colon and breast cancer cells supported
EC tube formation (36). On the one
hand, these inconsistencies could be due to variances in cell
lines, however they could also be explained by the different
conditions of tumor-CM production. Possibly the concentration of
tumor-derived pro-angiogenic factors in the CM produced in the
present study is too low or the accumulation of metabolic waste
products by tumor cells during the preparation of the CM is too
high. On the other hand, since the effects of the tumor-CM in the
other functional cell assays were notable, it is hypothesized that
the tube formation in the present controls is itself high to the
extent that a detectable increase is not possible.
Collectively, the present results demonstrated that
breast cancer MCR86 cells exhibited the strongest potential to
stimulate the angiogenic functions of ECs compared with the other
assessed cancer types, supporting the clinical findings that
invasive breast cancer belongs to the group of highly vascularized
tumors and produces various angiogenic factors (37). The different effects observed among
the tumor types can likely be linked to the soluble factors
secreted by the tumor cells. In a previous study, the effect of
these tumor cell lines on EPCs was analyzed and the results
suggested that tumor-derived cytokines, such as monocyte
chemoattractant protein-1 (MCP-1), macrophage inflammatory protein
2-α (MIP-2) and TNF-related apoptosis-inducing ligand (TRAIL), may
serve a vital role in the cell-cell communications of tumor cells
with their neighbors (28).
Given the contribution of tumor cells in changing
the functional properties of ECs, the question of implicated gene
expression changes arose. Genes associated with the phenotype of
tumor-activated ECs may include the expression of the
tumor-specific/induced markers of neovascularization (22). ECs isolated from human tumors
exhibit enhanced angiogenic capabilities, survival, adhesion to
tumor cells, chemotaxis and motility by expressing distinct markers
to modulate their functions (3,38).
PECAM1 was upregulated in each group. This gene serves a key
role in the initiation of vasculogenesis and angiogenesis,
involving EC interactions on a cell-cell basis, but also with
extracellular matrix components (39). CXCL12 was significantly
upregulated in the colon cancer CC531-, rhabdomyosarcoma R1H- and
breast cancer MCR86-CM groups. This factor is typically expressed
in stromal cells in various tissues and organs (40). However, it can interact
synergistically with VEGF to promote the functions of vascular ECs,
including cell migration, cell survival and changes in gene
expression (41). TIE1
levels were highest in the breast cancer MCR86-CM. Upregulation of
TIE1 has been observed in a variety of human solid tumors
and tumor ECs, and is associated with tumor progression (42). The increase in the angiogenic
capabilities of ECs following culture in tumor-CM could be partly
attributed to these genes, based on the consistent upregulation in
all the assessed groups.
VEGFA, VWF, FGF2 and PDGFRB were
revealed to be upregulated in the breast cancer MCR86- and colon
cancer CC531-CM groups, and downregulated or unaffected in the
osteosarcoma ROS-1- and rhabdomyosarcoma R1H-CM groups. The
expression of VWF, an EC marker, is regulated by angiogenesis
factors, such as FGF2 and VEGF. High VWF mRNA levels in
tumors may be an early sign of activation of the endothelium
(43). PDGFR serves a key
role in regulating the formation and function of blood vessels
(44). These gene expression
results reflect the findings of the present functional assays. In
nearly all the angiogenesis assays a stimulating effect of the
breast cancer MCR86 cells was observed on the ECs. Furthermore,
colon cancer CC531- and breast cancer MCR86-CM significantly
induced EC migration. Conversely, CM derived from rhabdomyosarcoma
R1H and osteosarcoma ROS-1, both categorized as sarcoma, induced
nearly no significant changes in the functional assays or the
expression of the majority of the tested genes, suggesting that the
secretion of pro-angiogenic factors is likely dependent on the
origin of the tumor cells.
Since it is widely reported that there is an
association between vascularization density and disease prognosis
for a number of carcinomas, but not sarcomas, this was evaluated by
West et al (45) in 42
patients with high grade soft tissue sarcomas. The authors revealed
no correlation between vascularization and metastasis or survival.
In contrast, in invasive breast carcinoma, microvessel counts and
density were associated with metastasis (46). Notably, Tomlinson et al
(47) analyzed the angiogenesis
patterns in sarcoma and carcinoma samples and demonstrated that, in
breast carcinoma tumors, the vessels were clustered within the
stroma, whereas the vessels in the sarcoma stroma were
homogeneously distributed. This could be due to the fibroblasts and
myoblasts within the carcinoma stroma leading to a
compartmentalization of the tumor, and thus of the angiogenic
factors (47). In a review by
Rocchi et al (48), the
authors concluded that the angiogenesis in sarcomas could also
serve an important role, but that there is an urgent requirement to
analyze sarcoma neovascularization in greater detail. Based on the
findings of the present study it can be concluded that the effect
of carcinoma cells on angiogenesis is stronger than that of sarcoma
cells. Whether this is associated with enhanced tumor progression
or disease prognosis is to be evaluated in further in vivo
studies. In the present study, rat tumor cell lines originating
from tumors grown in the WAG/Rij rat were used. This allows using
the same tumor cell lines in an established in vivo rat
model for future studies making the translation from in
vitro to in vivo much simpler. Further, using the
WAG/Rij rat we can be certain that tumor cell lines used in the
present study will grow in vivo. This would only be possible
with human tumor cell lines when using an immunodeficient rat model
with the disadvantage of having no immune system which is described
to have an important effect for tumor growth and development.
Collectively, these results demonstrated that
tumor-CM exerts a significant influence on the gene expression in
ECs, leading to a modulation of signaling pathways that mediate the
EC response. Significant upregulation of pro-angiogenic genes in
ECs following culture in various tumor-CM is in agreement with the
findings of enhanced angiogenic capacities of ECs in certain
groups. The activation of quiescent ECs, which in turn secret
pro-angiogenic factors such as VEGF and FGF, can further lead to an
autocrine and paracrine loop of angiogenesis stimulation (1). Therefore, it is reasonable to assume
that tumor-CM exerts an effect on tumor angiogenesis by inducing
the expression of pro-angiogenic genes in ECs. Previous findings
demonstrated that CM derived from these various tumor cell types
contain diverse secreted growth factors in varying amounts, capable
of affecting the functional behavior of mature, but also of EPCs to
different extents (28). Future
studies should focus on which distinct factors, most likely among
MCP-1, MIP-2, and TRAIL, serve a significant role in the
interaction of tumor cells and mature ECs. Based on the present
findings, the development of antibodies against these tumor
cell-secreted molecules could provide a novel therapeutic option
for treatment of highly vascularized tumors. Recently, a study by
Kami Reddy et al (49)
proposed the use of a novel inhibitor against dimethylarginine
dimethylaminohydrolase 1 (DDAH1) for the treatment of prostate
cancer. Inhibition of DDAH1 abrogated the secretion of angiogenic
growth factors and reduced the vascularization of tumors, which led
to an inhibition of xenograft tumors (49). Another approach to block the
tumor-EC interaction is the inhibition of VEGFR1, which serves a
role in tumor-associated, but not in adult physiological
angiogenesis (50). Selective
inhibition of this receptor would open up a new perspective in the
treatment of highly vascularized tumors.
In summary, the present study demonstrated that
tumor cells can significantly enhance the pro-angiogenic properties
of ECs, including proliferation, migration, invasion and tube
formation, in a tumor-type-dependent manner, by influencing the
genetic expression in ECs. Comparing the 4 distinct tumor types
analyzed within this study, the breast cancer MCR86 cell line
exhibited the greatest influence on the behavior of ECs. It is now
the aim of future studies to evaluate the effectiveness of
inhibiting tumor cell-secreted molecules to block the tumor-EC
interaction in highly vascularized tumors.
Acknowledgements
The authors would like to thank Dr A. Raabe
(University Cancer Center Hamburg, Hamburg, Germany) for providing
the R1H cells; Professor P.J.K. Kuppen (Leiden University Medical
Center, Leiden, The Netherlands) for providing the MCR86 cells;
Professor T.L.M. ten Hagen (Erasmus Medical Center, Rotterdam, The
Netherlands) for providing the ROS-1 cells; and Mr. S. Fleischer
and Mrs. I. Arnold-Herberth (University Hospital of Erlangen,
Erlangen, Germany) for their excellent technical support.
Funding
The present study was funded by the Staedtler
Foundation, ELAN-Fonds (grant no. 13-03-18-1-Brandl/Boos),
University of Erlangen-Nürnberg, The Interdisciplinary Center for
Clinical Research (IZKF, Faculty of Medicine, Friedrich-Alexander
University of Erlangen-Nürnberg), the Medicine Research Foundation,
University of Erlangen-Nürnberg and the Xue Hong and Hans Georg
Geis Foundation.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MAA, RA, AMB, REH and AKW designed the study
research and participated in the manuscript preparation, revision
and data analysis. RS performed the quantitative polymerase chain
reaction assays and data analysis. MW and MAA performed the
proliferation and tube formation assays and the data analysis. AK
assisted with the migration, transmigration and invasion assays and
data analysis. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EC
|
endothelial cell
|
|
FCS
|
fetal calf serum
|
|
CM
|
conditioned medium
|
|
PECAM-1
|
platelet endothelial cell adhesion
molecule-1
|
|
VEGF
|
vascular endothelial growth factor
|
|
FGF2
|
fibroblast growth factor 2
|
|
VWF
|
Von Willebrand factor
|
|
CXCL12
|
C-X-C motif chemokine ligand 12
|
|
TIE1
|
tyrosine kinase with
immunoglobulin-like and EGF-like domains 1
|
|
PDGFRB
|
platelet-derived growth factor
receptor β
|
|
EPC
|
endothelial progenitor cell
|
|
MCP-1
|
monocyte chemoattractant protein-1
|
|
MIP-2
|
macrophage inflammatory protein
2-α
|
|
TRAIL
|
TNF-related apoptosis-inducing
ligand
|
References
|
1
|
Ronca R, Van Ginderachter JA and Turtoi A:
Paracrine interactions of cancer-associated fibroblasts,
macrophages and endothelial cells: Tumor allies and foes. Curr Opin
Oncol. 30:45–53. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carmeliet P and Jain RK: Angiogenesis in
cancer and other diseases. Nature. 407:249–257. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weis SM and Cheresh DA: Tumor
angiogenesis: Molecular pathways and therapeutic targets. Nat Med.
17:1359–1370. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Billy F, Ribba B, Saut O, Morre-Trouilhet
H, Colin T, Bresch D, Boissel JP, Grenier E and Flandrois JP: A
pharmacologically based multiscale mathematical model of
angiogenesis and its use in investigating the efficacy of a new
cancer treatment strategy. J Theor Biol. 260:545–562. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bai Y, Bai L, Zhou J, Chen H and Zhang L:
Sequential delivery of VEGF, FGF-2 and PDGF from the polymeric
system enhance HUVECs angiogenesis in vitro and CAM angiogenesis.
Cell Immunol. 323:19–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hosseini F and Naghavi N: Modelling
tumor-induced angiogenesis: combination of stochastic sprout
spacing and sprout progression. J Biomed Phys Eng. 7:233–256.
2017.PubMed/NCBI
|
|
7
|
Laschke MW, Harder Y, Amon M, Martin I,
Farhadi J, Ring A, Torio-Padron N, Schramm R, Rücker M, Junker D,
et al: Angiogenesis in tissue engineering: Breathing life into
constructed tissue substitutes. Tissue Eng. 12:2093–2104. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feng T, Yu H, Xia Q, Ma Y, Yin H, Shen Y
and Liu X: Cross-talk mechanism between endothelial cells and
hepatocellular carcinoma cells via growth factors and integrin
pathway promotes tumor angiogenesis and cell migration. Oncotarget.
8:69577–69593. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Addison-Smith B, McElwain DL and Maini PK:
A simple mechanistic model of sprout spacing in tumour-associated
angiogenesis. J Theor Biol. 250:1–15. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chaplain MA, McDougall SR and Anderson AR:
Mathematical modeling of tumor-induced angiogenesis. Annu Rev
Biomed Eng. 8:233–257. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lamalice L, Le Boeuf F and Huot J:
Endothelial cell migration during angiogenesis. Circ Res.
100:782–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gao D, Nolan DJ, Mellick AS, Bambino K,
McDonnell K and Mittal V: Endothelial progenitor cells control the
angiogenic switch in mouse lung metastasis. Science. 319:195–198.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiarugi V, Ruggiero M and Magnelli L:
Molecular polarity in endothelial cells and tumor-induced
angiogenesis. Oncol Res. 12:1–4. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tei K, Kawakami-Kimura N, Taguchi O,
Kumamoto K, Higashiyama S, Taniguchi N, Toda K, Kawata R, Hisa Y
and Kannagi R: Roles of cell adhesion molecules in tumor
angiogenesis induced by cotransplantation of cancer and endothelial
cells to nude rats. Cancer Res. 62:6289–6296. 2002.PubMed/NCBI
|
|
17
|
Watnick RS: The role of the tumor
microenvironment in regulating angiogenesis. Cold Spring Harb
Perspect Med. 2:a0066762012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Samples J, Willis M and Klauber-DeMore N:
Targeting angiogenesis and the tumor microenvironment. Surg Oncol
Clin N Am. 22:629–639. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chouaib S, Kieda C, Benlalam H, Noman MZ,
Mami-Chouaib F and Rüegg C: Endothelial cells as key determinants
of the tumor microenvironment: Interaction with tumor cells,
extracellular matrix and immune killer cells. Crit Rev Immunol.
30:529–545. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stroka KM, Vaitkus JA and Aranda-Espinoza
H: Endothelial cells undergo morphological, biomechanical, and
dynamic changes in response to tumor necrosis factor-α. Eur Biophys
J. 41:939–947. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo J, Liu C, Zhou X, Xu X, Deng L, Li X
and Guan F: Conditioned medium from malignant breast cancer cells
induces an EMT-Like Phenotype and an Altered N-Glycan profile in
Normal Epithelial MCF10A cells. Int J Mol Sci. 18(pii): E15282017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Khodarev NN, Yu J, Labay E, Darga T, Brown
CK, Mauceri HJ, Yassari R, Gupta N and Weichselbaum RR:
Tumour-endothelium interactions in co-culture: Coordinated changes
of gene expression profiles and phenotypic properties of
endothelial cells. J Cell Sci. 116:1013–1022. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bjerregaard B, Holck S, Christensen IJ and
Larsson LI: Syncytin is involved in breast cancer-endothelial cell
fusions. Cell Mol Life Sci. 63:1906–1911. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dittmar T, Nagler C, Niggemann B and
Zanker KS: The dark side of stem cells: Triggering cancer
progression by cell fusion. Curr Mol Med. 13:735–750. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kuhbier JW, Bucan V, Reimers K, Strauss S,
Lazaridis A, Jahn S, Radtke C and Vogt PM: Observed changes in the
morphology and phenotype of breast cancer cells in direct
co-culture with adipose-derived stem cells. Plast Reconstr Surg.
134:414–423. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang LN, Kong CF, Zhao D, Cong XL, Wang
SS, Ma L and Huang YH: Fusion with mesenchymal stem cells
differentially affects tumorigenic and metastatic abilities of lung
cancer cells. J Cell Physiol. 234:3570–3582. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mortensen K, Lichtenberg J, Thomsen PD and
Larsson LI: Spontaneous fusion between cancer cells and endothelial
cells. Cell Mol Life Sci. 61:2125–2131. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
An R, Schmid R, Klausing A, Robering JW,
Weber M, Bäuerle T, Detsch R, Boccaccini AR, Horch RE, Boos AM and
Weigand A: Proangiogenic effects of tumor cells on endothelial
progenitor cells vary with tumor type in an in vitro and in vivo
rat model. Faseb J. 32:5587–5601. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ding Q, Xia Y, Ding S, Lu P, Sun L, Fan Y,
Li X, Wang Y, Tian DA and Liu M: Potential role of CXCL9 induced by
endothelial cells/CD133+ liver cancer cells co-culture
system in tumor transendothelial migration. Genes Cancer.
7:254–259. 2016.PubMed/NCBI
|
|
30
|
Lin SY, Yang J, Everett AD, Clevenger CV,
Koneru M, Mishra PJ, Kamen B, Banerjee D and Glod J: The isolation
of novel mesenchymal stromal cell chemotactic factors from the
conditioned medium of tumor cells. Exp Cell Res. 314:3107–3117.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Novosel EC, Kleinhans C and Kluger PJ:
Vascularization is the key challenge in tissue engineering. Adv
Drug Deliv Rev. 63:300–311. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cartland SP, Genner SW, Zahoor A and
Kavurma MM: Comparative evaluation of TRAIL, FGF-2 and
VEGF-A-induced angiogenesis in vitro and in vivo. Int J Mol
Sci. 17(pii): E20252016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wei B, Han XY, Qi CL, Zhang S, Zheng ZH,
Huang Y, Chen TF and Wei HB: Coaction of spheroid-derived stem-like
cells and endothelial progenitor cells promotes development of
colon cancer. PLoS One. 7:e390692012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Maniotis AJ, Folberg R, Hess A, Seftor EA,
Gardner LM, Pe'er J, Trent JM, Meltzer PS and Hendrix MJ: Vascular
channel formation by human melanoma cells in vivo and in vitro:
Vasculogenic mimicry. Am J Pathol. 155:739–752. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang Y, Jin G, Liu H, Liu K, Zhao J, Chen
X, Wang D, Bai R, Li X and Jang Y: Metformin inhibits esophageal
squamous cell carcinoma-induced angiogenesis by suppressing
JAK/STAT3 signaling pathway. Oncotarget. 8:74673–74687.
2017.PubMed/NCBI
|
|
36
|
Katkoori VR, Basson MD, Bond VC, Manne U
and Bumpers HL: Nef-M1, a peptide antagonist of CXCR4, inhibits
tumor angiogenesis and epithelialtomesenchymal transition in colon
and breast cancers. Oncotarget. 6:27763–27777. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gasparini G and Harris AL: Clinical
importance of the determination of tumor angiogenesis in breast
carcinoma: much more than a new prognostic tool. J Clin Oncol.
13:765–782. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xiong YQ, Sun HC, Zhang W, Zhu XD, Zhuang
PY, Zhang JB, Wang L, Wu WZ, Qin LX and Tang ZY: Human
hepatocellular carcinoma tumor-derived endothelial cells manifest
increased angiogenesis capability and drug resistance compared with
normal endothelial cells. Clin Cancer Res. 15:4838–4846. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Duncan GS, Andrew DP, Takimoto H, Kaufman
SA, Yoshida H, Spellberg J, de la Pompa JL, Elia A, Wakeham A,
Karan-Tamir B, et al: Genetic evidence for functional redundancy of
platelet/endothelial cell adhesion molecule-1 (PECAM-1):
CD31-deficient mice reveal PECAM-1-dependent and
PECAM-1-independent functions. J Immunol. 162:3022–3030.
1999.PubMed/NCBI
|
|
40
|
Yu Y, Huang X, Di Y, Qu L and Fan N:
Effect of CXCL12/CXCR4 signaling on neuropathic pain after chronic
compression of dorsal root ganglion. Sci Rep. 7:57072017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Okada H, Tsuzuki T, Shindoh H, Nishigaki
A, Yasuda K and Kanzaki H: Regulation of decidualization and
angiogenesis in the human endometrium: Mini review. J Obstet
Gynaecol Res. 40:1180–1187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang P, Chen N, Jia JH, Gao XJ, Li SH, Cai
J and Wang Z: Tie-1: A potential target for anti-angiogenesis
therapy. J Huazhong Univ Sci Technolog Med Sci. 35:615–622. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zanetta L, Marcus SG, Vasile J, Dobryansky
M, Cohen H, Eng K, Shamamian P and Mignatti P: Expression of Von
Willebrand factor, an endothelial cell marker, is up-regulated by
angiogenesis factors: A potential method for objective assessment
of tumor angiogenesis. Int J Cancer. 85:281–288. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liang T, Zhu L, Gao W, Gong M, Ren J, Yao
H, Wang K and Shi D: Coculture of endothelial progenitor cells and
mesenchymal stem cells enhanced their proliferation and
angiogenesis through PDGF and Notch signaling. FEBS Open Bio.
7:1722–1736. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
West CC, Brown NJ, Mangham DC, Grimer RJ
and Reed MW: Microvessel density does not predict outcome in high
grade soft tissue sarcoma. Eur J Surg Oncol. 31:1198–1205. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Weidner N, Semple JP, Welch WR and Folkman
J: Tumor angiogenesis and metastasis-correlation in invasive breast
carcinoma. N Engl J Med. 324:1–8. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tomlinson J, Barsky SH, Nelson S, Singer
S, Pezeshki B, Lee MC, Eilber F and Nguyen M: Different patterns of
angiogenesis in sarcomas and carcinomas. Clin Cancer Res.
5:3516–3522. 1999.PubMed/NCBI
|
|
48
|
Rocchi L, Caraffi S, Perris R and Mangieri
D: The angiogenic asset of soft tissue sarcomas: A new tool to
discover new therapeutic targets. Biosci Rep. 34:e001472014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kami Reddy KR, Dasari C, Vandavasi S,
Natani S, Supriya B, Jadav SS, Sai Ram N, Kumar JM and Ummanni R:
Novel cellularly active inhibitor regresses DDAH1 induced prostate
tumor growth by restraining tumor angiogenesis through targeting
DDAH1/ADMA/NOS pathway. ACS Comb Sci. 2019.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lacal PM and Graziani G: Therapeutic
implication of vascular endothelial growth factor receptor-1
(VEGFR-1) targeting in cancer cells and tumor microenvironment by
competitive and non-competitive inhibitors. Pharmacol Res.
136:97–107. 2018. View Article : Google Scholar : PubMed/NCBI
|