Introduction
Neuroblastoma, one of the most aggressive
extracranial solid tumours occurring in childhood, remains a major
cause of cancer-related deaths in infancy (1). Despite therapeutic strategies based on
chemotherapy, radiotherapy, surgery, GD2-targeted immunotherapy,
stem cell transplant and treatment with 13-cis-retinoic acid,
high-risk neuroblastoma outcome remains poor, with a 5-year
event-free survival <40% (2–5).
Tumours show initial response to therapeutic interventions but
typically relapse into an incurable form of the disease. Moreover,
several drugs cause severe side effects, including cognitive
impairment and retarded growth (6).
Thus, to reduce drug toxicity and to improve the outcome and the
lifestyle of the patients affected by neuroblastoma, additional
therapeutic options are required.
Recently, cell-based approaches have been
increasingly investigated for the delivery of therapeutics agents.
Mesenchymal stem cells are multipotent adult stem cells isolated
from the umbilical cord, bone marrow and fat tissue and can
differentiate into multiple tissues including bone, cartilage,
muscle, fat cells and connective tissue (7–12).
Bone marrow-derived mesenchymal stem cells (MSCs) are ideally
suited for the delivery of anticancer agents to tumours, including
cytokines, interferons and prodrugs (13–15).
MSCs are particularly suitable for the role of
vectors for anticancer therapies for various reasons (16). MSCs are immunologically inert due to
their low expression of constitutive major histocompatibility
complex 1 (MHC1) and lack of MHC2 and co-stimulatory molecules
CD80, CD86 and CD40, meaning that allogeneic cells can be used in
immunocompetent patients abrogating the need of immunosuppressive
therapies (17). Furthermore, MSCs
are able to migrate to and incorporate into the tumour stroma when
administered in vivo and, if engineered with viral vectors, can
deliver therapeutic molecules that inhibit tumours or metastatic
growth (18,19).
In recent studies, MSCs have been engineered to
express IFN-γ, IL-12, IL-24 and tumour necrosis factor-related
apoptosis inducing ligand (TRAIL), to induce death of tumour cells
(15,20).
TRAIL is a member of the TNF superfamily and
interacts with fully functional death receptors DR4 and DR5, decoy
receptors DCR1 and DCR2, and osteoprotegerin (OPG), which lack
functional cytoplasmic signalling domains (21–23).
TRAIL is an interesting anticancer molecule, since it causes
apoptosis of cancer cells bearing DR4 and DR5 death receptors, but
it is unable to harm normal cells which express high levels of
TRAIL decoy receptors and low levels of TRAIL death receptors on
their surface. TRAIL-mediated killing is not dependent on a
specific molecular alteration and all molecular subtypes or
high-risk tumours are potentially amenable to TRAIL killing. Thus,
from a clinical point of view, TRAIL-based therapies are especially
attractive due to the extremely high therapeutic index.
Clinical trials in which cancer patients have been
treated with soluble, truncated forms of TRAIL have been
unsuccessful, due to the extremely short half-life of the molecule
in the blood stream and the emergence of resistance; therefore,
efficient TRAIL delivery is essential (24,25).
The major advantage of using MSCs to deliver TRAIL is that it is
continuously produced at the tumour site, overcoming the problem of
the short half-life of the protein. Furthermore, it has been
revealed that the full-length TRAIL protein secreted by MSCs
transduced with a lentiviral vector containing the TRAIL cDNA, can
resolve resistance in lung, colorectal and breast cancer (26–28).
These cells are also able to clear lung metastasis in mice injected
with extremely aggressive breast cancer cells or reduce the growth
of mesothelioma cells in mouse models (29).
Notably, a protease inhibitor currently used in the
clinic, Bortezomib, is able to enhance TRAIL-mediated killing of
neuroblastoma cells or render them sensitive to the molecule,
suggesting that in a TRAIL therapy setting, the problem of
resistance could be managed pharmacologically (30–32).
In the present study, we investigated the
tumour-homing ability and anticancer activity of TRAIL-MSCs in the
context of neuroblastoma.
Materials and methods
Cell lines
The primary human neuroblastoma cell lines A5, 2820,
0396 and 1043008 were isolated at the Institute of Child Health
(University College London, London, UK) by disaggregating surgical
resections. The patient characteristics are summarised in Table I. Consent for the isolation of cell
lines from patient material was obtained in accordance with the
Great Ormond Street Hospital (London, UK) Ethics Committee
regulations.
 | Table I.Primary cell lines and patient
information (Great Ormond Street Hospital cohort). |
Table I.
Primary cell lines and patient
information (Great Ormond Street Hospital cohort).
| Pathology
number | A5 | 2820 | 1043008 | 0396 |
|---|
| Procedure | Diagnostic
biopsy | Diagnostic
biopsy | Biopsy, second
relapse | Diagnostic
biopsy |
| Diagnosis | Poorly
differentiated neuroblastoma | Poorly
differentiated neuroblastoma | Poorly
differentiated neuroblastoma | Poorly
differentiated neuroblastoma |
| Site of origin | ‘Right neck mass’
(mediastinal into right supraclavicular fossa) | Abdominal mass | Supraclavicular
lymph node | Right inguinal
lymph node |
| MYCN status | Not amplified | Not amplified | Diagnostic biopsy:
not amplified | Not amplified |
| Chromosomal
abnormalities | 1p loss, 11q and
17q status inconclusive | 1p loss, 11q loss,
17 gain | Diagnostic biopsy:
1p loss, 11q loss, 17q gain | 11q loss, 17q gain,
no 1p/1q imbalance |
| Histopathology
immunostaining | CD56+,
NB84+ | CD56+,
NB84+ | Diagnostic biopsy:
CD56+, NB84+ | CD56+,
NB84+ |
The patient-derived xenograft (PDX) cells LU-NB-1,
LU-NB-2 and LU-NB-3 were established and characterized at the
laboratory of Dr Daniel Bexell (Lund University, Lund, Sweden) as
previously described (33). hNB
cells were isolated from a tumour metastasised in the neck of a
3-year-old male patient in 2011 (34). MSC-TRAIL was generated at the
laboratory of Dr Samuel Janes (University College London, London,
UK) as previously described (26).
SKNAS, IMR-32, Kelly, SHEP, LA-N-5 and SH-SY5Y were
obtained from the American Type Culture Collection (ATCC;
Teddington, Middlesex, UK). LA-N-1 and patient-derived
neuroblastoma cell lines were provided by Dr John Anderson
(Institute of Child Health). LA-N-1, SKNAS, SHEP and SH-SY5Y were
grown in Dulbeccos modified Eagles medium (DMEM) (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.),
1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.),
2 mM glutamine (Gibco; Thermo Fisher Scientific, Inc.) and 10 mM
sodium pyruvate (Gibco; Thermo Fisher Scientific, Inc.). IMR-32,
Kelly, LA-N-5 were cultured in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) containing 10% FBS, 2 mM glutamine
supplemented, 10 mM sodium pyruvate and 10 mM non-essential amino
acids (NEAA) (Gibco; Thermo Fisher Scientific, Inc.). hNB cells
were cultured in RPMI-1640 medium supplemented with 20% fetal calf
serum (FCS) (Gibco; Thermo Fisher Scientific, Inc.), 2 mM
glutamine, 10 µM 2-mercaptoethanol (Thermo Fisher Scientific,
Inc.), 1 mM sodium pyruvate, 1% penicillin/streptomycin and 10 mM
NEAA. TRAIL-MSCs were grown in α-MEM (Gibco; Thermo Fisher
Scientific, Inc.) with 16% FBS, 4 mM L-glutamine and 1%
penicillin/streptomycin.
Primary cell lines were grown in stem cell medium
(DMEM/F-12 medium with glutamine, 1% penicillin/streptomycin, 2%
B27 supplement (Thermo Fisher Scientific, Inc.), 40 ng/ml of basic
fibroblast growth factor (FGF) (PeproTech, Inc., Rocky Hill, NJ,
USA) and 20 ng/ml of epidermal growth factor (EGF) (PeproTech,
Inc.). All cell lines were incubated at 37°C and 5%
CO2.
Western blotting
Cells were lysed in Laemmli buffer (cat. no. NP0007;
Thermo Fisher Scientific, Inc.) supplemented with β-mercaptoethanol
(cat. no. 21985023; Thermo Fisher Scientific, Inc.) and protein
concentrations were determined using a Pierce BCA Protein Assay kit
(cat. no. 23227; Thermo Fisher Scientific, Inc.). The protein
extracts (20 µg) were resolved in 10% acrylamide gel and
transferred onto nitrocellulose membranes which were blocked in 5%
non-fat dry milk for 1 h at room temperature. The membranes were
incubated overnight at 4°C with the primary antibodies followed by
incubation with the goat-anti rabbit IgG HRP-conjugated secondary
antibody (dilution 1:3,000; cat. no. 1706515; Bio-Rad Laboratories,
Inc., Hercules, CA, USA) for 1 h at room temperature. Membranes
were exposed to ECL Western Blotting substrate (cat. no. 32106;
Thermo Fisher Scientific, Inc.) as described in the manufacturers
protocol.
The following antibodies were used: DR4 (dilution
1:1,000; cat. no. 42533), DR5 (dilution 1:1,000; cat. no. 8074),
α-tubulin (dilution 1:1,000; cat. no. 2144; all were from Cell
Signalling Technology, Danvers, MA, USA).
In vitro co-culture and cell death
assay
A total of 10,000 Dil-labeled (cat. no. V22885;
Thermo Fisher Scientific, Inc.) neuroblastoma cells were plated in
96-well plates, to which control medium, 50 ng/ml r-TRAIL (cat. no.
310–04; PeproTech, Inc.), 20 nM Bortezomib (cat. no. S-1013;
Selleck Chemicals, Houston, TX, USA), 10,000 TRAIL-MSCs or their
combinations were added after 24 h. Floating and adherent cells
were harvested after 48 h after plating. Apoptosis was quantified
by AF647-conjugated Annexin V (cat. no. A23204; Invitrogen; Thermo
Fisher Scientific, Inc.) and 2 µg/ml DAPI (cat. no. D9542;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) staining using flow
cytometry.
Primary patient-derived neuroblastoma cell lines
were grown as a monolayer in 96-well plates coated with human
recombinant laminin 521 (cat. no. LN-521; Biolamina AB, Sundbyberg,
Sweden) according to the manufacturers instructions.
Xenograft cancer models
All experimental procedures were non-retrospectively
approved by a the Brunel University London Ethics Committee and the
Home Office and were conducted under the Animal Scientific
Procedures Act, 1986 (UK). In addition, we confirm that the tumour
burden did not exceed the recommended dimensions.
Forty four-week-old female mice (initial weight
20–23 g), immunodeficient NOD/SCID (purchased from Charles River
Laboratories, Margate, UK) were injected subcutaneously into the
right flank with 5×106 neuroblastoma cells in a 1:1
mixture of Matrigel (Corning, Inc., Corning, NY, USA) and
phosphate-buffered saline (PBS). After tumours reached the size of
5 mm in diameter, the mice were randomly assigned to 4 groups (10
mice/group) and PBS, 5×106 TRAIL-MSCs labelled with DiR
(cat. no. D12731; Thermo Fisher Scientific, Inc.), Bortezomib (1
mg/kg body weight) and their combination were administered
intraperitoneally every 3 days for 3 weeks. Fluorescent TRAIL-MSCs
were tracked in vivo using the IVIS Lumina Imaging System (Caliper
Life Sciences, Hopkinton, MA, USA). Tumour size was monitored with
a calliper and calculated according to the formula: V = (length ×
width2)/2. Mice were housed under a 12-h light/dark
cycle in a specific pathogen-free facility with controlled
temperature and humidity (20–24°C, 45–65 % humidity) and allowed
access to food and water ad libitum. Body weight and general
physical status were recorded daily, and the mice were sacrificed
by cervical dislocation when the tumour reached 1.2 cm in
diameter.
Flow cytometry of
lentivirus-transduced cells
For the expression detection of TRAIL, MSC cells
were stained with a phycoerythrin (PE)-conjugated anti-TRAIL
antibody (dilution, 1:100; cat. no. 550516; BD Biosciences,
Franklin Lakes, NJ, USA) and analysed by flow cytometry.
Statistical analysis
All data are expressed as the means ± standard
deviation (SD). Statistical significance between different test
conditions was determined using Students t-test. Probability values
<0.05 were considered to indicate a statistically significant
result. The statistical analysis of survival was carried out using
a log-rank test and the SPSS 16.0 software (SPSS, Inc., Chicago,
IL, USA).
Results
Expression of DR4 and DR5 death
receptors in neuroblastoma cells
The presence of death receptors in cancer cells is a
valuable biomarker to determine sensitivity to TRAIL. We therefore
quantified the expression of DR4 and DR5 in a panel of established
(classic) or patient-derived neuroblastoma cells and investigated
whether the anticancer drug Bortezomib could be used to enhance the
expression of TRAIL receptors.
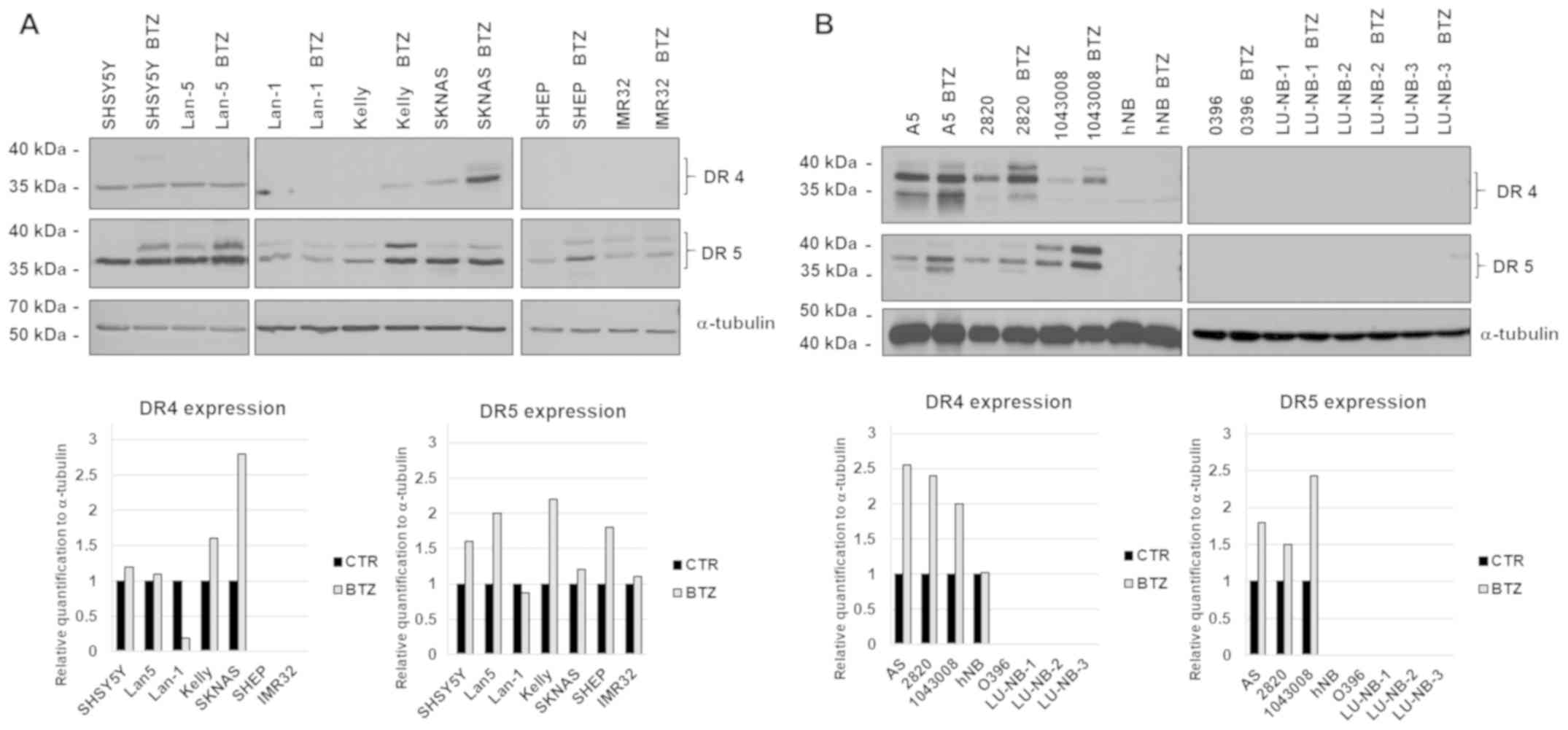
All classic neuroblastoma cells expressed DR5
receptor while the expression of DR4 was most prominent in a subset
of neuroblastoma cell lines. The expression of DR5 was markedly
increased after treatment with 20 nM Bortezomib for 24 h whereas
the DR4 receptor was increased by the drug only in the Kelly and
SKNAS cell lines (Fig. 1A).
There was a wide difference in expression of death
receptors in patient-derived cell lines, ranging from strong to
undetectable. In keeping with the results obtained with classic
neuroblastoma cell lines, Bortezomib enhanced the expression of
DR4-5 also in primary cells (Fig.
1B).
TRAIL-MSCs induce neuroblastoma cell
death in vitro
To examine whether the TRAIL-death receptor system
could be exploited for therapeutic purposes in neuroblastoma, the
different cell lines were subjected to in vitro killing assays.
Neuroblastoma cells were cultured in the presence of soluble,
recombinant TRAIL (r-TRAIL), Bortezomib, TRAIL-MSCs and their
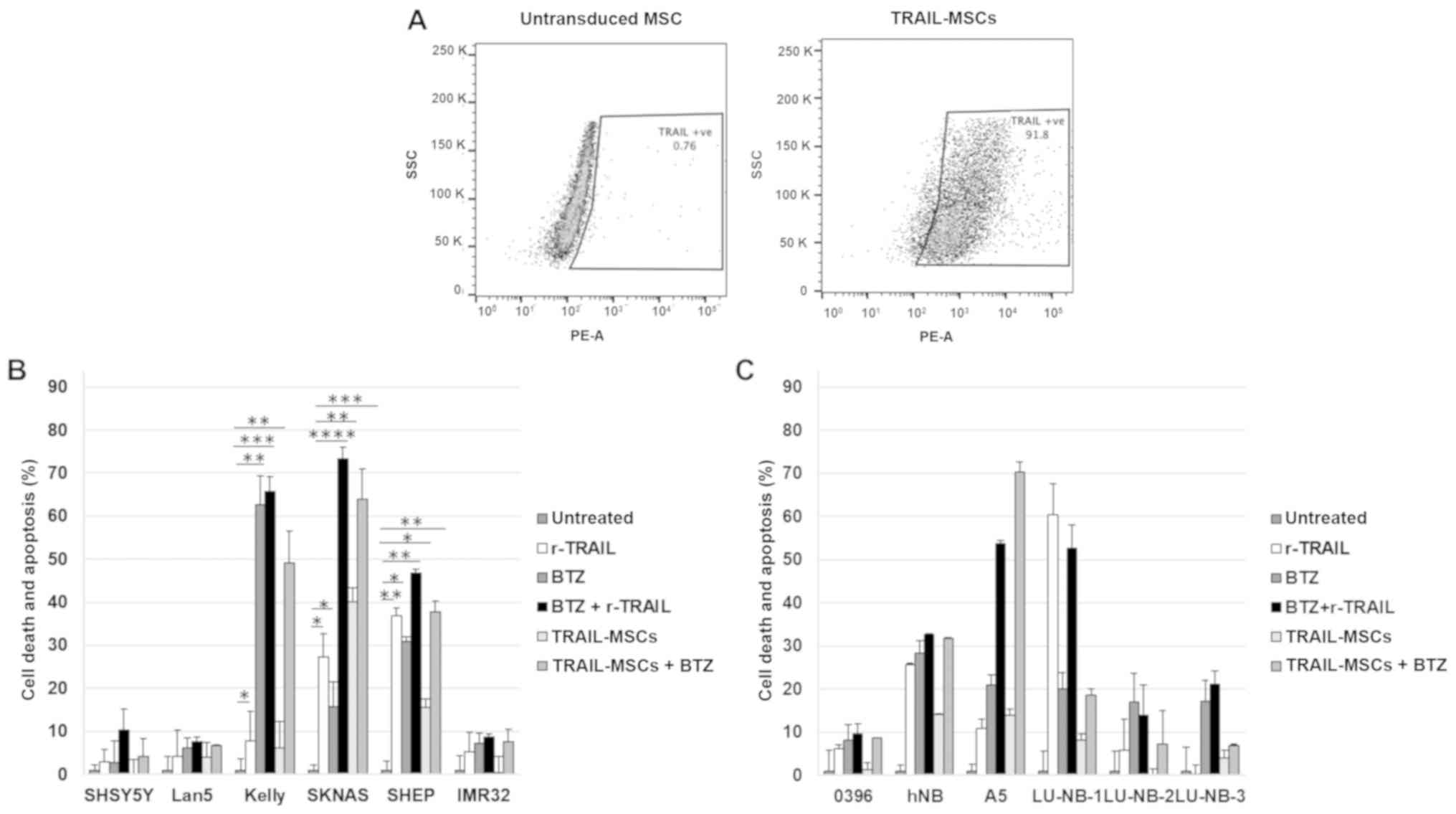
combinations. Before performing the experiments, TRAIL expression
on the surface by the modified MSCs was verified by flow cytometry
(Fig. 2A). The results revealed
that soluble TRAIL and TRAIL-MSCs could induce killing of death
receptor-positive cell lines that was increased by Bortezomib. The
classic neuroblastoma cell line SKNAS was particularly sensitive to
the combination, with 60–70 % of cells undergoing apoptosis
(Fig. 2B). Primary cells were
generally less sensitive to soluble TRAIL or TRAIL-MSCs, and, as
anticipated, the degree of cell death was in most instances
proportional to the expression levels of the TRAIL receptors
(Fig. 2C). Intraperitoneally
injected TRAIL-MSCs migrate to neuroblastoma xenotransplants. Since
TRAIL-MSCs and the combined treatment with Bortezomib enhanced
TRAIL-induced apoptosis in vitro, we evaluated the therapeutic
efficacy of this treatment in vivo. SKNAS were injected
subcutaneously into the flank of NOD/SCID mice to establish a
xenograft tumour model.
Tumours were allowed to grow until they reached a
volume of 50 mm3. Mice were then randomly assigned into
4 groups and treated with vehicle (PBS), TRAIL-MSCs labelled with
the fluorescent lipophilic dye DiR, Bortezomib or their
combination.
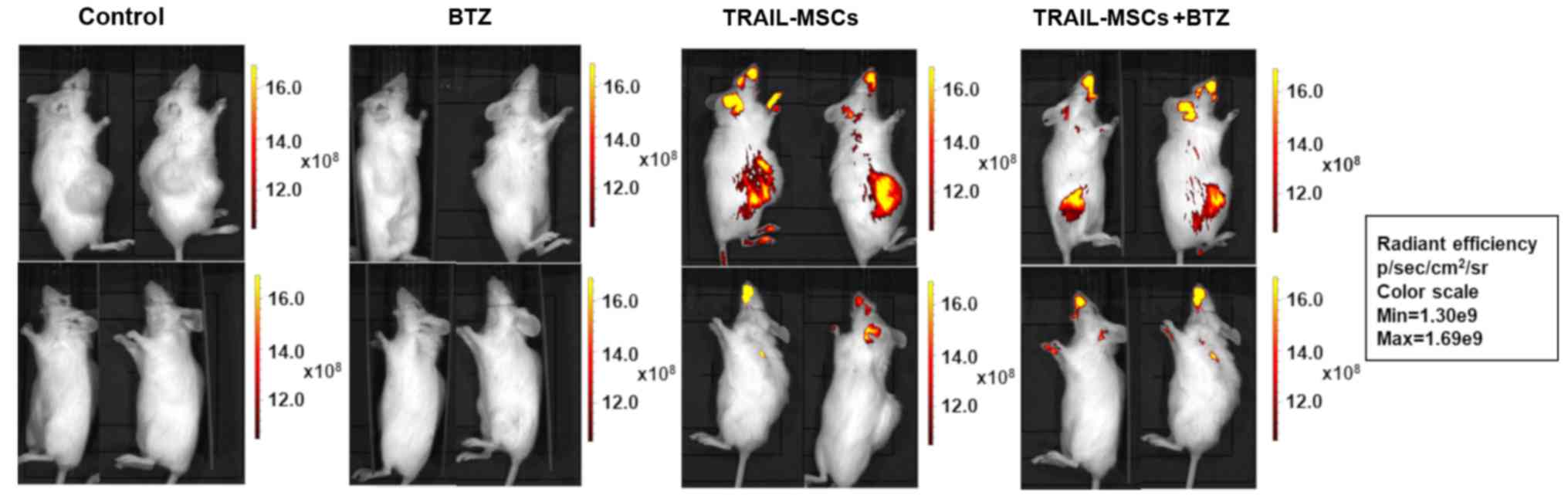
Fluorescent TRAIL-MSCs were tracked in vivo using
the IVIS Lumina Imaging System (Caliper Life Sciences). Animals
were imaged 24 h after injection to determine the localisation of
TRAIL-MSCs in vivo. A strong signal from DiR-labeled TRAIL-MSCs was
detected in the flanks containing the tumour masses whereas a weak
signal was detected in other anatomical locations (Fig. 3). Control animals receiving no MSC
injections were negative for fluorescence. To evaluate the effect
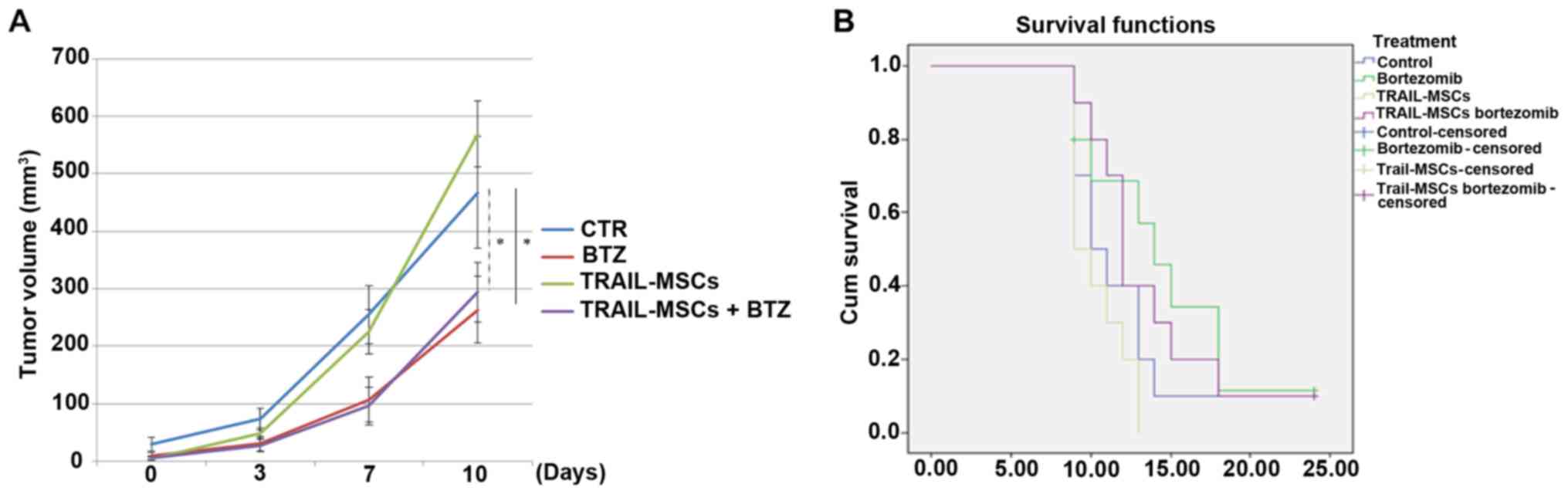
of TRAIL-MSCs in tumour growth, mice were injected with TRAIL-MSCs,
Bortezomib or TRAIL-MSCs+Bortezomib every 3 days for 3 weeks and
monitored until tumour masses reached a diameter of 1.2 cm or mice
lost 20% of their weight or showed signs of distress. There were no
significant changes in tumour volumes in mice injected with the
TRAIL-MSCs compared with the control group, but a significant
anticancer effect was noted in the Bortezomib groups (Fig. 4A). We did not observe statistically
significant differences in survival between different treatment
groups, but there was a trend of increased survival in the
Bortezomib-treated mice, consistent with previous observations
(32) (Fig. 4B).
Discussion
Gene rearrangements and mutation of key oncogenes
such as MYCN, ALK, ATRX, TERT and chromosomal losses or reduced
expression of CLU, CHD5, PHOX2B, CASZ1, PML and other tumour
suppressor genes are thought to play a crucial role in the
pathogenesis of neuroblastoma (35–39).
Despite the significant advances in the understanding of its
molecular causes, the life expectancy of patients bearing high-risk
neuroblastoma is still very poor, suggesting that new therapeutic
approaches are urgently needed. MYCN, the main neuroblastoma
oncogene, is essentially undruggable (40). Small molecule inhibitors against ALK
have been developed, however, monotherapies with small molecule
inhibitors are prone to the problem of resistance and new therapies
based on the inhibition of molecular drivers, for example by small
interfering RNAs or CRISPR, are still far from being translated
into clinical practice.
One of the many challenges of developing a therapy
based on biomolecules is the delivery of these to the tumour site.
It has been recently revealed that MSCs can specifically home in
the tumour stroma, and this property can be exploited for cancer
therapy. In this context, the death ligand TRAIL was reported to
induce apoptosis selectively in tumour cells while sparing normal
cells in vitro and in vivo (41,42).
Recently, several research groups have explored the possibility of
using TRAIL as an anticancer molecule that could be delivered in
the tumour microenvironment by mesenchymal stem cells. Loebinger
et al demonstrated that TRAIL-MSCs were able to home and
kill tumour cells, and significantly induce cancer regression in a
lung metastatic cancer model of breast cancer cells (20). In addition, TRAIL-expressing MSCs
exhibited anticancer activity in other experimental tumour models,
such as glioma and sarcoma (43–45).
Despite expressing death receptors, cancer cells can become
resistant to TRAIL-induced apoptosis (46,47).
Several studies have indicated that Bortezomib can reverse
resistance of cancer cells to TRAIL killing by increasing the
expression of DR4 and DR5 (48).
Notably, Naumann et al reported that Bortezomib synergised
with TRAIL in inducing apoptosis of neuroblastoma cells (49).
A key question for the potential exploitation of
MSCs in neuroblastoma therapeutics is whether stromal cells
injected systemically can reach tumour sites. Contradicting
information has been published that still leaves the question open.
For example, a previous study suggested that neuroblastoma tumours
are unable to attract systemically injected MSCs, whereas a recent
study revealed imaging of MSCs infiltrating human neuroblastomas
growing in vivo (50,51).
To further address this issue, we investigated
whether MSCs engineered to express TRAIL were attracted by and able
to kill neuroblastoma cells transplanted into immunocompromised
mice. Firstly, MSCs expressing TRAIL were co-cultured with
classical and patient-derived neuroblastoma cell lines and
subjected to cell killing assays. The protease inhibitor and
anticancer drug Bortezomib was used to increase the expression of
death receptors and sensitise cells to TRAIL killing. Once we had
identified the cell line more susceptible to TRAIL-MSCs killing, we
carried out xenotransplantation experiments to assess whether the
engineered stem cells were attracted by neuroblastomas.
Bioluminescent imaging (BLI) clearly indicated that TRAIL-MSCs
infiltrated neuroblastomas hours after intraperitoneal injections.
Disappointingly, despite the fact that TRAIL-MSCs were able to kill
SKNAS cells in vitro, they were not able to do so in vivo and the
marginal therapeutic effect that we observed was caused by
Bortezomib. Resistance to TRAIL killing may have different causes,
including high expression of decoy receptors and downregulation or
upregulation of apoptotic proteins. Expression of the intracellular
apoptotic inhibitor c-FLIP can confer TRAIL resistance in different
types of cancer cell lines and it may be involved in the protection
of neuroblastoma cells from the cytotoxic effect of TRAIL (52). Deficient expression of caspases, in
particular caspase-8, essential with FADD to form the death
receptor complex DISC, may contribute to TRAIL resistance (53). Also, overexpression of Bcl-2 or
Bcl-x or loss of Bax and Bad function or high expression of an
inhibitor of apoptotic proteins could lead to TRAIL resistance
(54). Furthermore, high expression
of inhibitors that act downstream of the receptors, which includes
XIAP, c-IAP1, c-IAP2 and survivin could block the activation and
activity of caspase-9, −3 and −7 (55). It is possible that one of these
mechanisms of resistance is activated and selected in tumours
developing in vivo and could be responsible for the failure of
TRAIL-MSCs to kill their targets. An alternative explanation of the
failure of the MSCs to induce cancer regression could be that the
cells have lost their ability to express TRAIL in the tumour
microenvironment. Although we cannot exclude this possibility, it
is unlikely since the same TRAIL-modified MSCs have been previously
used to successfully inhibit breast cancer growth in
immunodeficient mice (20,26).
Nonetheless, the present study suggests that
mesenchymal stem cells are suitable for neuroblastoma cell and gene
therapy but should be loaded with biomolecules more effective than
TRAIL in further preclinical studies. For example, MSCs producing
interferon gamma (IFNγ) have shown promise in reducing
neuroblastoma growth when injected intratumorally (56). In light of this study, it is likely
that systemic injections of MSCs producing IFNγ or other
neuroblastoma-specific drugs could be successfully developed for
neuroblastoma therapeutics.
Acknowledgements
Not applicable.
Funding
The present study was supported by a Niamhs Next
Step/SPARKS and Associazione Italiana per la Lotta al Neuroblastoma
awards to AS and NIHR GOSH BRC to JA and NS.
Availability of data and materials
The datasets and cell lines used in the study are
available from the corresponding author upon reasonable
request.
Authors contributions
The study was conceived and written by AS and VN. VN
performed all the experiments with the assistance of RP and KK.
SMJ, JB, JA and DB provided the patient-derived materials, the cell
lines and the key reagents, and were involved in the data analysis
and the interpretation of the results. NS carried out the pathology
analyses. EK assisted with the interpretation, revision and
analysis of the data. All authors read and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Consent for the isolation of cell lines from patient
material was obtained in accordance with the Great Ormond Street
Hospital (London, UK) Ethics Committee regulations. All
experimental procedures were non-retrospectively approved by the
Brunel University London Ethics Committee and the Home Office and
were conducted under the Animal Scientific Procedures Act, 1986
(UK).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Matthay KK, Maris JM, Schleiermacher G,
Nakagawara A, Mackall CL, Diller L and Weiss WA: Neuroblastoma. Nat
Rev Dis Primers. 2:160782016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang RK and Sondel PM: Anti-GD2 strategy
in the treatment of neuroblastoma. Drugs Future. 35:6652010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maris JM: Recent advances in
neuroblastoma. N Engl J Med. 362:2202–2211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fish JD and Grupp SA: Stem cell
transplantation for neuroblastoma. Bone Marrow Transplant.
41:159–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wagner LM and Danks MK: New therapeutic
targets for the treatment of high-risk neuroblastoma. J Cell
Biochem. 107:46–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ren N, Atyah M, Chen WY and Zhou CH: The
various aspects of genetic and epigenetic toxicology: Testing
methods and clinical applications. J Transl Med. 15:1102017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bianco P, Robey PG and Simmons PJ:
Mesenchymal stem cells: Revisiting history, concepts, and assays.
Cell Stem Cell. 2:313–319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hwang NS, Zhang C, Hwang YS and Varghese
S: Mesenchymal stem cell differentiation and roles in regenerative
medicine. Wiley Interdiscip Rev Syst Biol Med. 1:97–106. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pittenger MF, Mackay AM, Beck SC, Jaiswal
RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S and
Marshak DR: Multilineage potential of adult human mesenchymal stem
cells. Science. 284:143–147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Granero-Molto F, Weis JA, Longobardi L and
Spagnoli A: Role of mesenchymal stem cells in regenerative
medicine: Application to bone and cartilage repair. Expert Opin
Biol Ther. 8:255–268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Savkovic V, Li H, Seon JK, Hacker M, Franz
S and Simon JC: Mesenchymal stem cells in cartilage regeneration.
Curr Stem Cell Res Ther. 9:469–488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dezawa M, Ishikawa H, Itokazu Y, Yoshihara
T, Hoshino M, Takeda S, Ide C and Nabeshima Y: Bone marrow stromal
cells generate muscle cells and repair muscle degeneration.
Science. 309:314–317. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakamizo A, Marini F, Amano T, Khan A,
Studeny M, Gumin J, Chen J, Hentschel S, Vecil G, Dembinski J, et
al: Human bone marrow-derived mesenchymal stem cells in the
treatment of gliomas. Cancer Res. 65:3307–3318. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Duan X, Guan H, Cao Y and Kleinerman ES:
Murine bone marrow-derived mesenchymal stem cells as vehicles for
interleukin-12 gene delivery into Ewing sarcoma tumors. Cancer.
115:13–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Studeny M, Marini FC, Champlin RE,
Zompetta C, Fidler IJ and Andreeff M: Bone marrow-derived
mesenchymal stem cells as vehicles for interferon-beta delivery
into tumors. Cancer Res. 62:3603–3608. 2002.PubMed/NCBI
|
|
16
|
Ozawa K, Sato K, Oh I, Ozaki K, Uchibori
R, Obara Y, Kikuchi Y, Ito T, Okada T, Urabe M, et al: Cell and
gene therapy using mesenchymal stem cells (MSCs). J Autoimmun.
30:121–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
1Javazon EH, Beggs KJ and Flake AW:
Mesenchymal stem cells: Paradoxes of passaging. Exp Hematol.
32:414–425. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xin H, Kanehira M, Mizuguchi H, Hayakawa
T, Kikuchi T, Nukiwa T and Saijo Y: Targeted delivery of CX3CL1 to
multiple lung tumors by mesenchymal stem cells. Stem Cells.
25:1618–1626. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Loebinger MR and Janes SM: Stem cells as
vectors for antitumour therapy. Thorax. 65:362–369. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Loebinger MR, Eddaoudi A, Davies D and
Janes SM: Mesenchymal stem cell delivery of TRAIL can eliminate
metastatic cancer. Cancer Res. 69:4134–4142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pitti RM, Marsters SA, Ruppert S, Donahue
CJ, Moore A and Ashkenazi A: Induction of apoptosis by Apo-2
ligand, a new member of the tumor necrosis factor cytokine family.
J Biol Chem. 271:12687–12690. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kelley SK and Ashkenazi A: Targeting death
receptors in cancer with Apo2L/TRAIL. Curr Opin Pharmacol.
4:333–339. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wiley SR, Schooley K, Smolak PJ, Din WS,
Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA,
et al: Identification and characterization of a new member of the
TNF family that induces apoptosis. Immunity. 3:673–682. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Herbst RS, Eckhardt SG, Kurzrock R,
Ebbinghaus S, ODwyer PJ, Gordon MS, Novotny W, Goldwasser MA,
Tohnya TM, Lum BL, et al: Phase I dose-escalation study of
recombinant human Apo2L/TRAIL, a dual proapoptotic receptor
agonist, in patients with advanced cancer. J Clin Oncol.
28:2839–2846. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Soria JC, Smit E, Khayat D, Besse B, Yang
X, Hsu CP, Reese D, Wiezorek J and Blackhall F: Phase 1b study of
dulanermin (recombinant human Apo2L/TRAIL) in combination with
paclitaxel, carboplatin, and bevacizumab in patients with advanced
non-squamous non-small-cell lung cancer. J Clin Oncol.
28:1527–1533. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yuan Z, Kolluri KK, Sage EK, Gowers KH and
Janes SM: Mesenchymal stromal cell delivery of full-length tumor
necrosis factor-related apoptosis-inducing ligand is superior to
soluble type for cancer therapy. Cytotherapy. 17:885–896. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sheard MA, Asgharzadeh S, Liu Y, Lin TY,
Wu HW, Ji L, Groshen S, Lee DA and Seeger RC: Membrane-bound TRAIL
supplements natural killer cell cytotoxicity against neuroblastoma
cells. J Immunother. 36:319–329. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mueller LP, Luetzkendorf J, Widder M,
Nerger K, Caysa H and Mueller T: TRAIL-transduced multipotent
mesenchymal stromal cells (TRAIL-MSC) overcome TRAIL resistance in
selected CRC cell lines in vitro and in vivo. Cancer Gene Ther.
18:229–239. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sage EK, Kolluri KK, McNulty K, Lourenco
Sda S, Kalber TL, Ordidge KL, Davies D, Gary Lee YC, Giangreco A
and Janes SM: Systemic but not topical TRAIL-expressing mesenchymal
stem cells reduce tumour growth in malignant mesothelioma. Thorax.
69:638–647. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Loi M, Becherini P, Emionite L, Giacomini
A, Cossu I, Destefanis E, Brignole C, Di Paolo D, Piaggio F, Perri
P, et al: sTRAIL coupled to liposomes improves its pharmacokinetic
profile and overcomes neuroblastoma tumour resistance in
combination with Bortezomib. J Control Release. 192:157–166. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tong HX, Lu CW, Wang QS and Ma LY:
Combination of IFNgamma and chemotherapeutic agents increase TRAIL
sensitivity of neuroblastoma cell lines. Eur J Pediatr Surg.
21:304–309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brignole C, Marimpietri D, Pastorino F,
Nico B, Di Paolo D, Cioni M, Piccardi F, Cilli M, Pezzolo A,
Corrias MV, et al: Effect of bortezomib on human neuroblastoma cell
growth, apoptosis, and angiogenesis. J Natl Cancer Inst.
98:1142–1157. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Braekeveldt N, Wigerup C, Gisselsson D,
Mohlin S, Merselius M, Beckman S, Jonson T, Börjesson A, Backman T,
Tadeo I, et al: Neuroblastoma patient-derived orthotopic xenografts
retain metastatic patterns and geno- and phenotypes of patient
tumours. Int J Cancer. 136:E252–E261. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chaiwatanasirikul KA and Sala A: The
tumour-suppressive function of CLU is explained by its localisation
and interaction with HSP60. Cell Death Dis. 2:e2192011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fujita T, Igarashi J, Okawa ER, Gotoh T,
Manne J, Kolla V, Kim J, Zhao H, Pawel BR, London WB, et al: CHD5,
a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. J
Natl Cancer Inst. 100:940–949. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang C, Liu Z, Woo CW, Li Z, Wang L, Wei
JS, Marquez VE, Bates SE, Jin Q, Khan J, et al: EZH2 Mediates
epigenetic silencing of neuroblastoma suppressor genes CASZ1, CLU,
RUNX3, and NGFR. Cancer Res. 72:315–324. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Corvetta D, Chayka O, Gherardi S, DAcunto
CW, Cantilena S, Valli E, Piotrowska I, Perini G and Sala A:
Physical interaction between MYCN oncogene and polycomb repressive
complex 2 (PRC2) in neuroblastoma: Functional and therapeutic
implications. J Biol Chem. 288:8332–8341. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dvorkina M, Nieddu V, Chakelam S, Pezzolo
A, Cantilena S, Leite AP, Chayka O, Regad T, Pistorio A, Sementa
AR, et al: A promyelocytic leukemia protein-thrombospondin-2 axis
and the risk of relapse in neuroblastoma. Clin Cancer Res.
22:3398–3409. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Valentijn LJ, Koster J, Zwijnenburg DA,
Hasselt NE, van Sluis P, Volckmann R, van Noesel MM, George RE,
Tytgat GA, Molenaar JJ, et al: TERT rearrangements are frequent in
neuroblastoma and identify aggressive tumors. Nat Genet.
47:1411–1414. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shalaby T and Grotzer MA: MYC as
therapeutic target for embryonal tumors: Potential and challenges.
Curr Cancer Drug Targets. 16:2–21. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Walczak H, Miller RE, Ariail K, Gliniak B,
Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, et al:
Tumoricidal activity of tumor necrosis factor-related
apoptosis-inducing ligand in vivo. Nat Med. 5:157–163. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ashkenazi A, Pai RC, Fong S, Leung S,
Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert
A, et al: Safety and antitumor activity of recombinant soluble Apo2
ligand. J Clin Invest. 104:155–162. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mohr A, Lyons M, Deedigan L, Harte T, Shaw
G, Howard L, Barry F, O'Brien T and Zwacka R: Mesenchymal stem
cells expressing TRAIL lead to tumour growth inhibition in an
experimental lung cancer model. J Cell Mol Med. 12:2628–2643. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Menon LG, Kelly K, Yang HW, Kim SK, Black
PM and Carroll RS: Human bone marrow-derived mesenchymal stromal
cells expressing S-TRAIL as a cellular delivery vehicle for human
glioma therapy. Stem Cells. 27:2320–2330. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Grisendi G, Spano C, D'souza N, Rasini V,
Veronesi E, Prapa M, Petrachi T, Piccinno S, Rossignoli F, Burns
JS, et al: Mesenchymal progenitors expressing TRAIL induce
apoptosis in sarcomas. Stem Cells. 33:859–869. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dyer MJ, MacFarlane M and Cohen GM:
Barriers to effective TRAIL-targeted therapy of malignancy. J Clin
Oncol. 25:4505–4506. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rieger J, Frank B, Weller M and Wick W:
Mechanisms of resistance of human glioma cells to Apo2
ligand/TNF-related apoptosis-inducing ligand. Cell Physiol Biochem.
20:23–34. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mahalingam D, Szegezdi E, Keane M, de Jong
S and Samali A: TRAIL receptor signalling and modulation: Are we on
the right TRAIL? Cancer Treat Rev. 35:280–288. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Naumann I, Kappler R, von Schweinitz D,
Debatin KM and Fulda S: Bortezomib primes neuroblastoma cells for
TRAIL-induced apoptosis by linking the death receptor to the
mitochondrial pathway. Clin Cancer Res. 17:3204–3218. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bianchi G, Morandi F, Cilli M, Daga A,
Bocelli-Tyndall C, Gambini C, Pistoia V and Raffaghello L: Close
interactions between mesenchymal stem cells and neuroblastoma cell
lines lead to tumor growth inhibition. PLoS One. 7:e486542012.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cussó L, Mirones I, Peña-Zalbidea S,
García-Vázquez V, García-Castro J and Desco M: Combination of
single-photon emission computed tomography and magnetic resonance
imaging to track 111in-oxine-labeled human mesenchymal stem cells
in neuroblastoma-bearing mice. Mol Imaging. 13:132014. View Article : Google Scholar
|
|
52
|
French R, Hayward O, Jones S, Yang W and
Clarkson R: Cytoplasmic levels of cFLIP determine a broad
susceptibility of breast cancer stem/progenitor-like cells to
TRAIL. Mol Cancer. 14:2092015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schneider P, Thome M, Burns K, Bodmer JL,
Hofmann K, Kataoka T, Holler N and Tschopp J: TRAIL receptors 1
(DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate
NF-kappaB. Immunity. 7:831–836. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
LeBlanc H, Lawrence D, Varfolomeev E,
Totpal K, Morlan J, Schow P, Fong S, Schwall R, Sinicropi D and
Ashkenazi A: Tumor-cell resistance to death receptor - induced
apoptosis through mutational inactivation of the proapoptotic Bcl-2
homolog Bax. Nat Med. 8:274–281. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Deveraux QL, Takahashi R, Salvesen GS and
Reed JC: X-linked IAP is a direct inhibitor of cell-death
proteases. Nature. 388:300–304. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
56
|
Relation T, Yi T, Guess AJ, La Perle K,
Otsuru S, Hasgur S, Dominici M, Breuer C and Horwitz EM:
Intratumoral delivery of interferon γ-secreting mesenchymal stromal
cells repolarizes tumor-associated macrophages and suppresses
neuroblastoma proliferation in vivo. Stem Cells. 36:915–924. 2018.
View Article : Google Scholar : PubMed/NCBI
|