Introduction
Chronic myeloid leukemia (CML) is a common
hematologic malignancy in China. As a myeloproliferative disease,
CML results from the reciprocal translocation of chromosome 9 and
chromosome 22, which leads to the Bcr-Abl gene fusion and
increases constitutive tyrosine kinase activity (1).
Imatinib, a tyrosine kinase inhibitor (TKI),
directly inhibits constitutive tyrosine kinase activity, which
results in the modification of the functions of various genes
involved in the control of the cell cycle, cell adhesion,
cytoskeleton organization and ultimately in the apoptotic death of
Ph(+) cells (2). Imatinib-based
targeted treatment has become the standard therapy for CML, and
most patients in the chronic phase (CP) of CML achieve not only a
complete cytogenetic response (CCR) but also a major molecular
response (MMR) (3). A 10-year
follow-up of patients with CML who were treated with imatinib as
initial therapy showed that imatinib can improve the prognosis of
CML patients without unacceptable cumulative or late toxic effects
(4).
The major reason for the therapeutic success of
imatinib in CML is the well-defined molecular target toward the
Bcr-Abl gene and relatively selective therapies aimed at
this gene. However, drug resistance is a recurrent issue for
imatinib-based CML treatment. A clinical trial indicated that for
imatinib-based treatment of CML, there is a 15 to 25% rate of
primary cytogenetic resistance by 18 months of therapy, and the
secondary resistance rate was 7 to 15% (5). Imatinib resistance (IR) can be ascribed
to two major reasons: Bcr-Abl-dependent and -independent
resistance. The Bcr-Abl-dependent resistance includes
Bcr-Abl duplication and mutation. In vitro cell
experiments have demonstrated that continuous culture with
imatinib-containing medium elevated Abl kinase activity due to a
genetic duplication of the Bcr-Abl sequence (6,7). Gorre
et al demonstrated that the T315I mutation creates
steric hindrance to the bonding between imatinib and the Abl kinase
(8). Bcr-Abl-independent resistance
includes a decrease in drug influx and an increase in drug efflux
(9), drug sequestration in the plasma
(10), epigenetic modification
(11) and alternative signaling
pathway activation (12).
Ribonucleotide reductase regulatory subunit M2
(RRM2) plays a significant role in tumor progression and is
frequently overexpressed in cancer. It is involved in the
regulation of cell invasion, cell migration and tumor metastasis
(13). Elevated RRM2 expression has
been reported to be associated with a poor prognosis of several
types of cancer including gastric cancer, adrenocortical cancer,
and non-small cell lung cancer (13–15). RRM2
is also demonstrated to be significantly associated with drug
resistance. For example, Shah et al revealed that RRM2 is a
key contributor to AKT-induced tamoxifen resistance in breast
cancer treatment (16), and Tu et
al demonstrated that VASH2 reduced the chemosensitivity to
gemcitabine in pancreatic cancer cells via the JUN-dependent
transactivation of RRM2 (17).
In the present study, peripheral blood samples were
collected from 22 imatinib-sensitive (IS) and 17 imatinib-resistant
(IR) primary CML patients and the transcription profile of these
samples was analyzed using high-throughput sequencing (RNA-seq).
Numerous genes were found to be altered in IR patients. Four
significantly increased genes that may correlate with IR, aryl
hydrocarbon receptor nuclear translocator 2 (ARNT2), ATP
binding cassette subfamily A member 13 (ABCA13), RRM2
and secreted frizzled related protein 1 (SFRP1) were
screened (18–21), and three significantly decreased genes
were identified that may be correlated with IR, growth
differentiation factor 7 (GDF7 or BMP12), glutathione
S-transferase µ1 (GSTM1) and AP-1 transcription factor
subunit (c-Fos) (22–24). Among these genes, RRM2 was
observed to be elevated in both IR patients and an IR cell line. It
was also demonstrated that RRM2 is involved in the Bcl-2/caspase
and Akt cell signaling pathways and therefore affects the cell
survival in imatinib therapy.
Herein, for the first time, we report that RRM2 is
responsible for drug resistance in imatinib-based CML therapy. This
study evaluated RRM2 as a potential therapeutic target in the
clinical treatment of CML.
Materials and methods
Patients and peripheral blood
collection
Peripheral blood samples were collected from 20 IR
CML patients at the First Hospital of Jilin University (Changchun,
China) from April 2015 to August 2018. Among these IR patients, 11
were male and 9 were female, with a median age of 53 years (range
18–72). Two of the IR patients were in accelerated phase and 18
were in chronic phase. These patients had received imatinib as the
first line therapy for 8 months to 13 years. CML diagnosis and
resistance were defined on the basis of European Leukemia Net: ELN
Recommendations 2013 (https://www.leukemia-net.org/content/home/index_eng.html).
Peripheral blood samples were also collected from 17 IS CML
patients who had achieved a major molecular response (MMR). The
median age of the IS CML patients was 46 years (range 26–60).
Control peripheral blood samples were obtained from 15 healthy
people with a median age of 32 years (range 25–39). From these
blood samples, nucleated cells including lymphocytes, monocytes and
granulocytes were isolated. All of these samples were stored at
−80°C until use. Permission to use the clinical samples for
research purposes was obtained and approved by the Ethics Committee
of the First Hospital of Jilin University. Informed consents were
obtained from all patients.
RNA-seq
Two IR samples and two IS samples were randomly
selected and the RNA-seq was carried out. The distinct mRNAs
between IS and IR patients and their related pathways were
identified. Briefly, total RNA was isolated from the patient
peripheral blood samples using Qiazol (Qiagen, Shanghai, China).
Then the total RNA was reverse-transcribed to a cDNA library.
RNA-seq was carried out on a HiSeq4000 (Illumina, Inc.) and yielded
approximately 30 million reads with a length of 150 bp per sample
(Shanghai Biotechnology Corp., Shanghai, China). Gene counts were
normalized to the values of fragments per kilobase of transcript
per million mapped reads (FPKM).
Cell culture
The human myeloid leukemia K562 cell line and basic
IR type K562G cell line were purchased from the Chinese Academy of
Medical Sciences (Tianjin, China). K562 cells were grown in normal
RPMI-1640 media (Thermo Fisher Scientific, Inc, Beijing, China)
supplemented with 10% fetal bovine serum (HyClone, Beijing, China)
in 5% CO2 at 37°C. K562G cells were grown in the same
culturing condition except that 8 µM imatinib (Sigma-Aldrich; Merck
KGaA, Shanghai, China) was added to the media.
For the concentration-effect analysis of imatinib,
K562G cells were cultured in 8 µM imatinib as the initial
concentration for two weeks, then in 10 µM imatinib for another two
weeks, and finally in 12 µM imatinib for two weeks.
Reverse-transcription PCR (RT-PCR) and
real-time quantitative fluorescence PCR (qF-PCR)
Cells were collected and total RNA was extracted
using the Qiagen RNeasy Mini Kit (Qiagen, Shanghai, China). RNA (1
µg) was reverse-transcribed to cDNA using Moloney murine leukemia
virus reverse transcriptase (M-MLV, Invitrogen; Thermo Fisher
Scientific, Inc.). The reaction system for RT-PCR analysis
contained (6 µl): 2 µl of prepared sample cDNA as the template, 3
µl 2X Taq PCR StarMix buffer (GenStar, Beijing, China) and 1
µl forward and reverse primers. The reaction system for qF-PCR
analysis contained (20 µl): 2 µl of prepared sample cDNA as the
template, 10 µl 2X SYBR Premixed buffer (Roche, Shanghai, China), 2
µl forward and reverse primers and 6 µl ddH2O. The
primer sequences were as follows: ARNT2 forward primer,
5′-TGCATCGGAGAAGAAGATGATG-3′ and reverse primer,
5′-ATTCACTCCAGGCACATGAAC-3′ (136 bp); ABCA13 forward primer,
5′-CTGTGGAAGAATTGGCTCTGCA-3′ and reverse primer,
5′-TGTCTCTGTATCTGGGAGGTTC-3′ (127 bp); RRM2 forward primer,
5′-CTATGGCTTCCAAATTGCCATG-3′ and reverse primer,
5′-GACACAAGGCATCGTTTCAATG-3′ (127 bp); SFRP1 forward primer,
5′-TGTGCCACAACGTGGGCTAC-3′ and reverse primer,
5′-AGTTCTTGTTGAGCAGGGGCACC-3′ (114 bp); BMP12 forward
primer, 5′-CACTTCATGATGTCGCTTTACC-3′ and reverse primer,
5′-CGTTAAGGCTGGACACGTCGA-3′ (181 bp); GSTM1 forward primer,
5′-TTCCCAATCTGCCCTACTTG-3′ and reverse primer,
5′-CACGAATCTTCTCCTCTTCTG-3′ (120 bp); c-FOS forward primer,
5′-GATAGCCTCTCTTACTACCAC-3′ and reverse primer,
5′-GAATGAAGTTGGCACTGGAGAC-3′; Bcl-2 forward primer,
5′-ACCTGGATCCAGGATAACGGA-3′ and reverse primer,
5′-GATAGGCACCCAGGGTGATGC-3′ (148 bp); GAPDH forward primer,
5′-TGCACCACCAACTGCTTA-3′ and reverse primer,
5′-GGATGCAGGGATGATGTTC-3′ (178 bp). The RT-PCR amplification was
carried out on the Eppendorf Mastercycler Pro (Eppendorf, Shanghai,
China) and the PCR process was as follows: 10 min denaturation at
95°C, 26 to 32 cycles at 95°C for 20 sec, 62°C for 15 sec and 72°C
for 15 sec, and then at 72°C for 2 min. PCR productions were
visualized by 3% agarose gel electrophoresis. Densitometric
analysis was conducted using Quantity One software (version 4.6,
Bio-Rad). The qF-PCR amplification was carried out on the ABI
StepOnePlus (ABI, Beijing, China) and the PCR process was as
follows: 5 min denaturation at 95°C followed by 40 cycles at 95°C
for 20 sec, 60°C for 15 sec and 72°C for 15 sec. mRNA levels were
normalized to GAPDH levels within the same sample. Data analysis
was performed using the 2−ΔΔCq method (25).
siRNA and RNA interference
Control siRNA (siCT) and siRNA for RRM2 were
synthesized by Shanghai GenePharma Co. Ltd. (Shanghai, China). The
sequence of siCT is: 5′-UAGCGACUAAACACAUCAAUU-3′ and the sequence
of siRRM2 is: 5′-GCGAUUUAGCCAAGAAGUUCA-3′ (26). Amaxa® Cell Line
Nucleofector® Kit V (Lonza, Basel, Switzerland) was used
for the siRNA transfection. According to the manufacturer's
guidelines, 1×106 K562 or K562G cells were transfected
with 100 pmol siRNA. Twelve hours after the electrotransfection,
the cells were centrifuged at a low speed (90 × g) to exclude the
dead cells and the debris. The cells were incubated for another 12
h and the knockdown efficiency was measured by RT-PCR, qF-PCR and
western blotting. All the cell experiments were performed at 24 h
post-transfection.
Cell viability determination by the
CCK-8 assay
K562 and K562G cells (5×103) were plated
in 96-well plates and cultured for 24 h. For the cell viability
assay, 10 µl CCK-8 solution (Thermo Fisher Scientific, Inc,
Beijing, China) was added to each well and the plate was incubated
at 37°C for 2 h. The optical density (OD) at 450 nm (OD450) was
measured using a microplate reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Apoptosis assay
K562G cells were maintained in RPMI-1640 medium
supplemented with 8 µM imatinib. Approximately 1×106
cells were collected and washed with cold PBS. Then, the cells were
incubated with Annexin V-FITC/PI (BD Biosciences, Franklin Lakes,
NJ, USA) for 15 min in the dark and analyzed with
fluorescence-activated cell sorting (FACScan; BD Biosciences).
FlowJo software (FlowJo, LLC, Ashland, OR, USA) was used for
apoptosis analysis. Cells in the different proportions represent
the different cell states as follows: The dead cells are shown in
the upper left portion (Q1), the late-apoptotic cells are shown in
the upper right portion (Q2), the viable cells are shown in the
lower left portion (Q4), and the early apoptotic cells are the
cells present in the lower right portion (Q3).
Protein extraction and western
blotting
Cells were lysed in RIPA buffer (KeyGen Biotech.
Co., Ltd., Nanjing, China) compensated with a cocktail protease
inhibitor (Roche). Lysates were centrifuged at 20,000 × g for 30
min at 4°C. Protein concentration was determined by Bradford
Protein Assay Kit (Beyotime Institute of Biotechnology, Haimen,
Jiangsu, China). For protein expression assay, equal amount of 20
µg protein from each sample was separated by 12% sodium dodecyl
sulfate polyacrylamide gel electrophoresis and transferred to
polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA).
The blots were then blocked with 5% skim milk in TBST at 37°C for 1
h and incubated with primary antibodies including: RRM2 (dilution
1:1,000, cat. no. ab57653; Abcam, Shanghai, China), Bcl-2 (dilution
1:1,000; cat. no. ab59348; Abcam), Bax (dilution 1:1,000; cat. no.
ab32503; Abcam), cleaved caspase-3 (dilution 1:1,000; cat. no.
9664; Cell Signaling Technology, Shanghai, China), cleaved
caspase-9 (dilution 1:1,000; cat. no. 9505; Cell Signaling
Technology), Akt (dilution 1:1,000. cat. no. 2920; Cell Signaling
Technology), phospho-Akt (Ser473, dilution 1:1,000; cat. no. 4060;
Cell Signaling Technology) and β-actin (dilution 1:1,000; cat. no.
sc47778; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Then the
membranes were incubated with horseradish peroxidase-coupled goat
anti-rabbit or goat anti-mouse secondary antibody (dilution
1:3,000; cat. nos. sc-2004 and sc-2005; Santa Cruz Biotechnology)
at room temperature for 1 h. The chemiluminescence signals were
detected with a chemiluminescence system (ECL, Thermo Fisher
Scientific, Inc.). Densitometric analysis was conducted using
Quantity One software, version 4.6 (Bio-Rad Laboratories).
Statistical analysis
All experimental results represent the average of at
least three independent experiments. Data were analyzed using SPSS
16.0 software (SPSS Inc., Chicago, IL, USA) and expressed as mean ±
SD. Statistical analysis was performed using t-test (between two
groups) or one-way analysis of variance followed by an LSD post hoc
test (more than two groups). *P<0.05, **P<0.01,
#P<0.05 or ##P<0.01 was considered to
indicate a statistically significant difference (relevant symbols
are shown in the figures and legends).
Results
Identification of IR-related genes
(IRGs) using RNA-seq and qF-PCR
The relative mRNA levels from the RNA-seq are
expressed as FPKM value and are listed in Fig. 1A. Referring to the KEGG Pathway
Database (https://www.kegg.jp/kegg/pathway.html), 7 genes were
screened that may be involved in the IR process. The expression
levels of ARNT2, ABCA13, RRM2 and SFRP1 were
significantly increased and the expression levels of GDF7,
GSTM1 and c-FOS were significantly decreased.
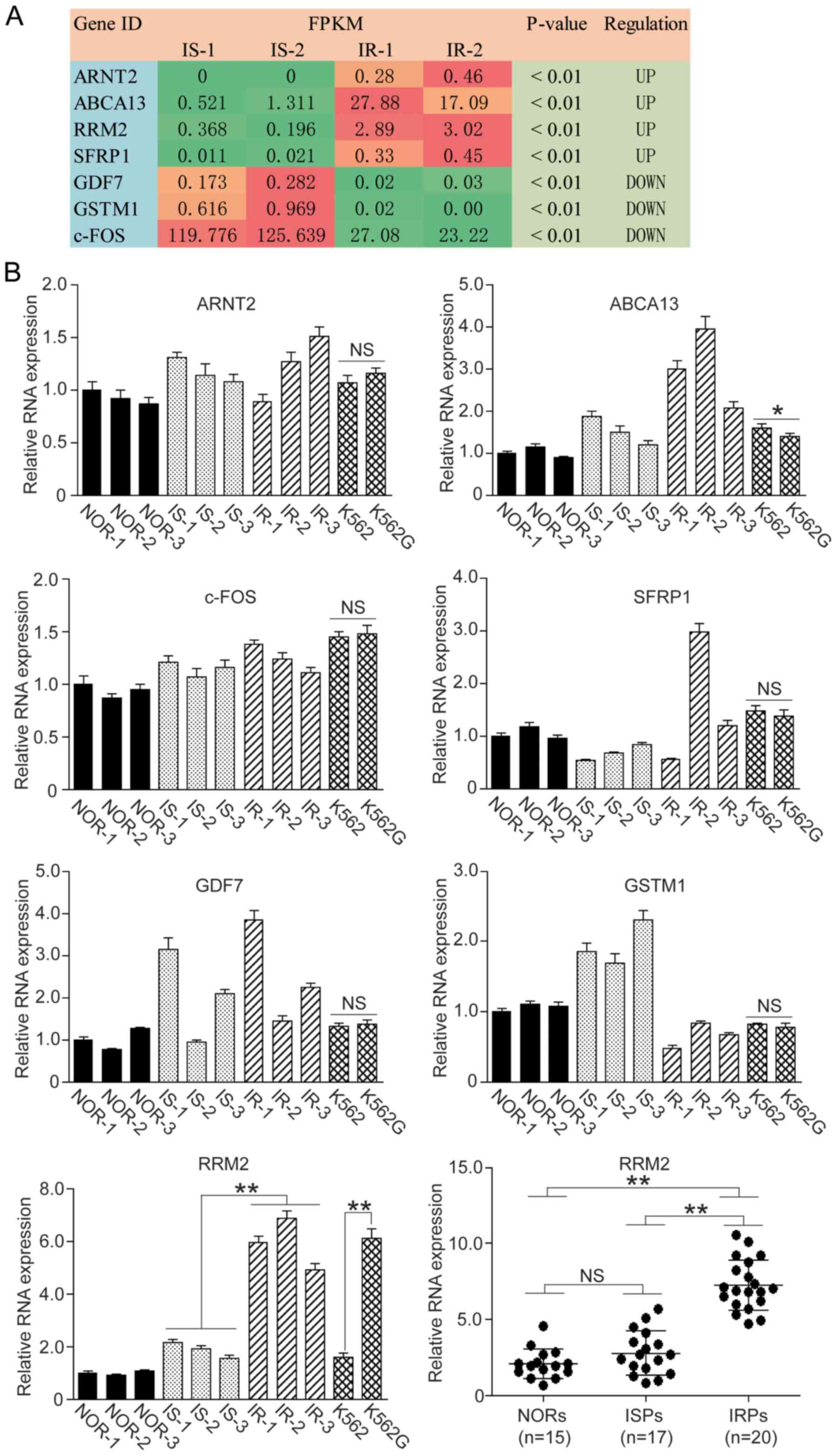 | Figure 1.Screening of IRGs in CML patients.
RNA-seq was employed to screen specific IRGs in imatinib-sensitive
(IS) and imatinib-resistant (IR) CML patients and 7 genes were
identified. Among these genes, 4 genes were upregulated and 3 genes
were downregulated in the IR patients. We carried out qF-PCR to
confirm the levels of these IRGs in peripheral blood samples and
cell lines. (A) FPKM values are presented in RNA-seq. Green box,
downregulation; orange and red box, upregulation. (B) qF-PCR assay
of 7 IRGs in blood samples and cell lines. IRGs, IR-related genes;
CML, chronic myeloid leukemia; NORs, normal patients; ISPs,
imatinib-sensitive patients; IRPs, imatinib-resistant patients.
One-way ANOVA and t-test, *P<0.05; **P<0.01; NS, not
significant. |
We carried out qF-PCR to confirm whether the
variation trends of these genes were consistent with the RNA-seq
result, and we found that the variation trends of ABCA13,
GSTM1 and RRM2 were consistent with the RNA-seq result,
whereas ARNT2 and c-FOS were not consistent with the
RNA-seq result. As for SFRP1 and GDF7, the individual
variation in the gene transcription was significant (Fig. 1B).
We then detected the mRNA level of ABCA13,
GSTM1 and RRM2 in normal K562 cells and the IR type
K562G cells. Among these three genes, RRM2 showed the most
significant difference between K562 and K562G cells (Fig. 1B, P<0.01). We went on to detect the
RRM2 level in 15 normal individuals (NORs), 17 IS patients (ISPs)
and 20 IR patients (IRPs), and we observed that the RRM2 level in
the IR group was significantly higher than that of the IS and NOR
group (Fig. 1B, P<0.01).
Therefore, we selected RRM2 as our target gene and performed the
following cell experiments.
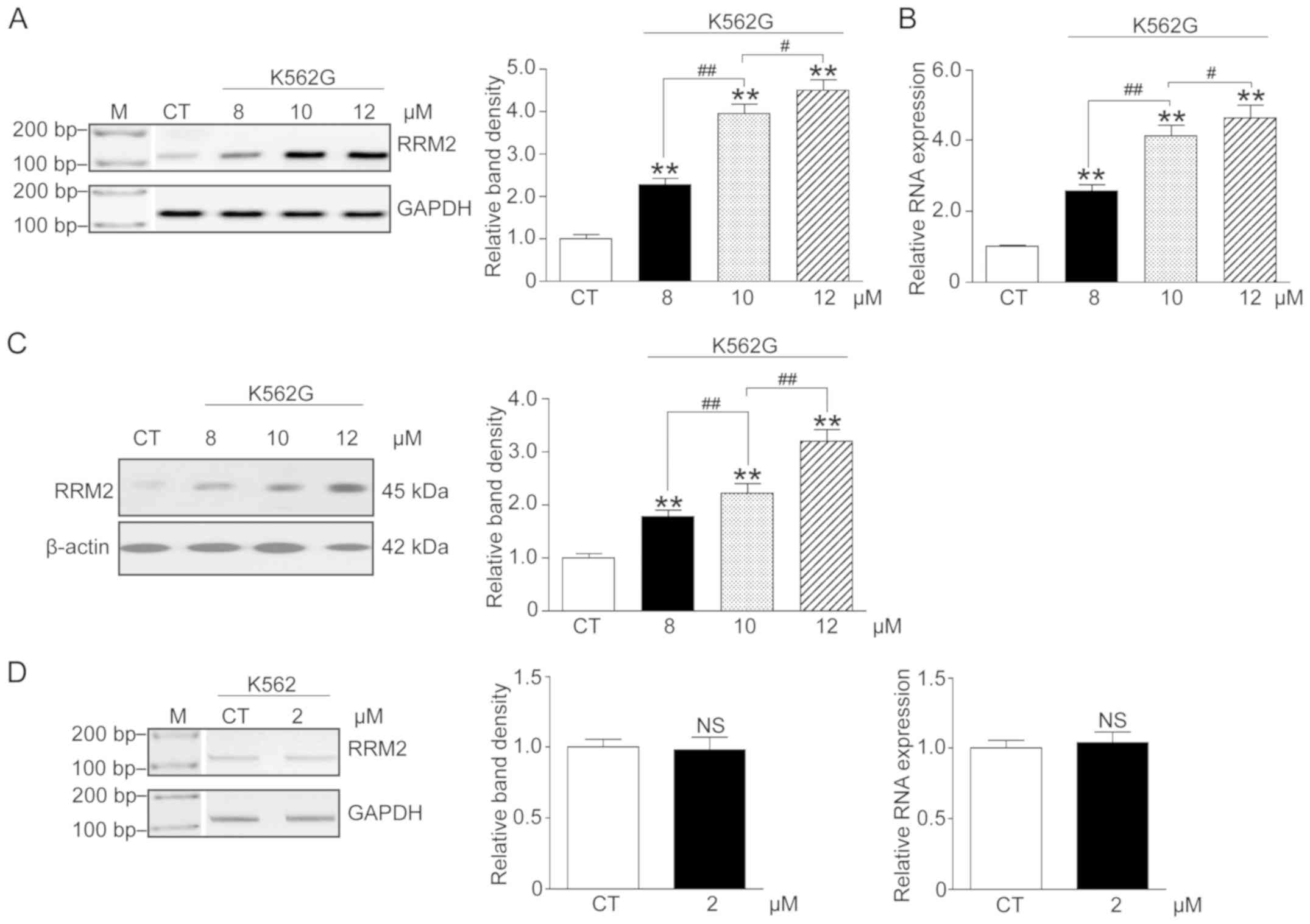
Imatinib increases the RRM2 level in a
dose-dependent manner in K562G cells
In order to confirm the correlation between imatinib
therapy and RRM2 expression, K562G cells were treated with three
doses of imatinib (8, 10 and 12 µM), and K562 cells were treated
with 2 µM imatinib. We analyzed the RRM2 mRNA and protein levels by
RT-PCR, qF-PCR and western blot analysis. The results indicated
that following increasing concentrations of imatinib, both the mRNA
and protein levels of RRM2 in the K562G cells were significantly
elevated when compared with the CT group (Fig. 2A-C, P<0.01). However, the mRNA
level in K562 cells was not significantly altered (Fig. 2D, P>0.05). These data showed an
apparent concentration-effect relationship of imatinib and RRM2 in
IR cells.
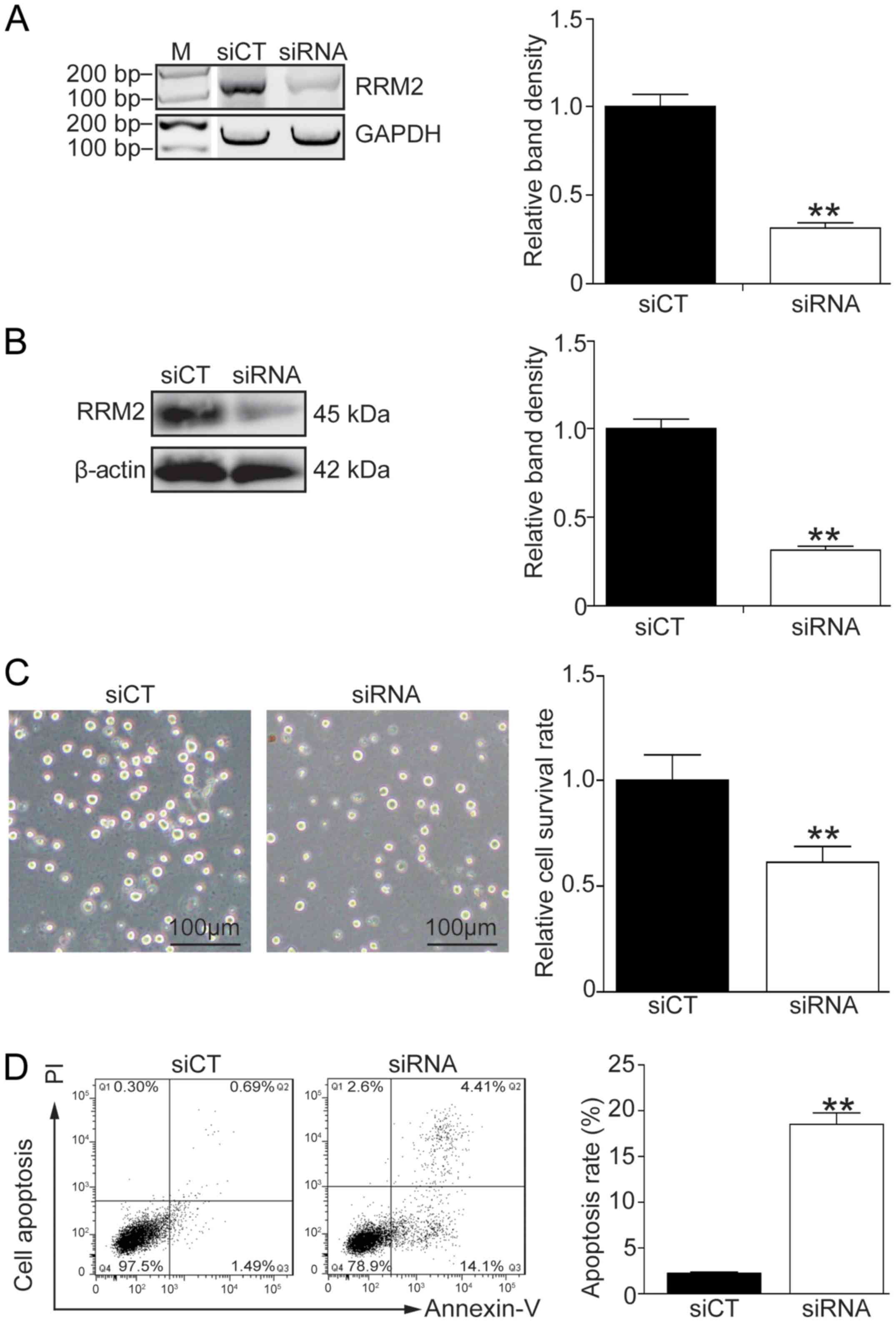
Knockdown of RRM2 enhances the
apoptosis of K562G cells
In order to address the correlation between the RRM2
level and imatinib-induced cell apoptosis, endogenous RRM2 was
effectively knocked down using siRNA (Fig. 3A and B, P<0.01). Cell morphology
showed that K562G cells became sensitive to 8 µM imatinib treatment
after RRM2 was knocked down (Fig. 3C,
left panel). Cell survival experiment and flow cytometry also
demonstrated that knockdown of RRM2 inhibited the cell growth and
induced a high rate of cell death following imatinib-based
treatment (Fig. 3C, right panel,
P<0.01; Fig. 3D, P<0.01).
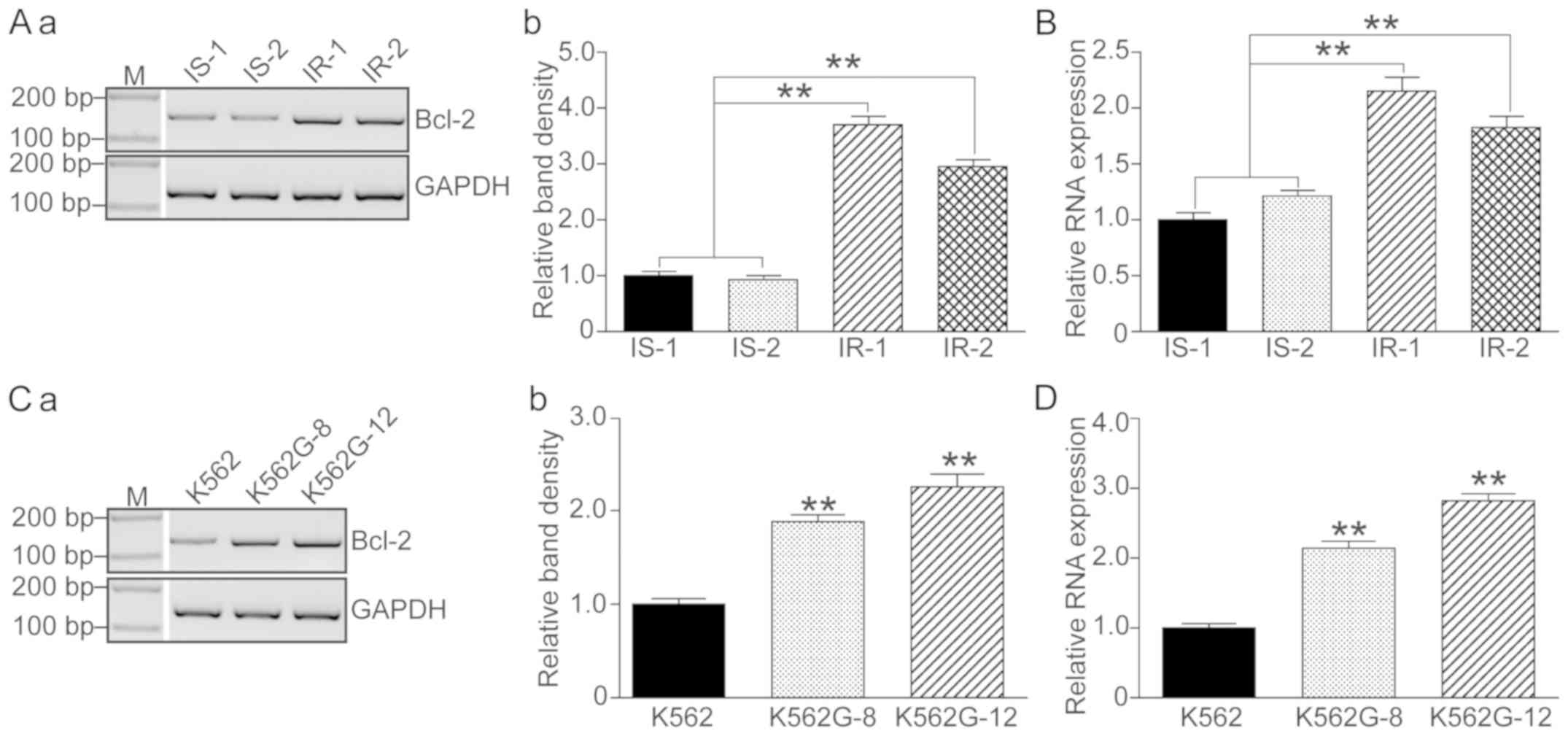
Bcl-2 is upregulated in IR patients
and in K562G cells
Since it was demonstrated that a high level of RRM2
induced imatinib resistance and that knockdown of RRM2 increased
drug sensitivity of K562G cells to imatinib, we then focused on the
variations in apoptosis-related genes. Bcl-2 levels were detected
in 2 IS patients (IS-1 and IS-2), 2 IR patients (IR-1 and IR-2) and
the imatinib-treated K562G cells. As shown in Fig. 4, both the IR patients and the K562G
cells showed significant higher levels of Bcl-2 compared to the IS
patients and the normal K562 cells (P<0.01).
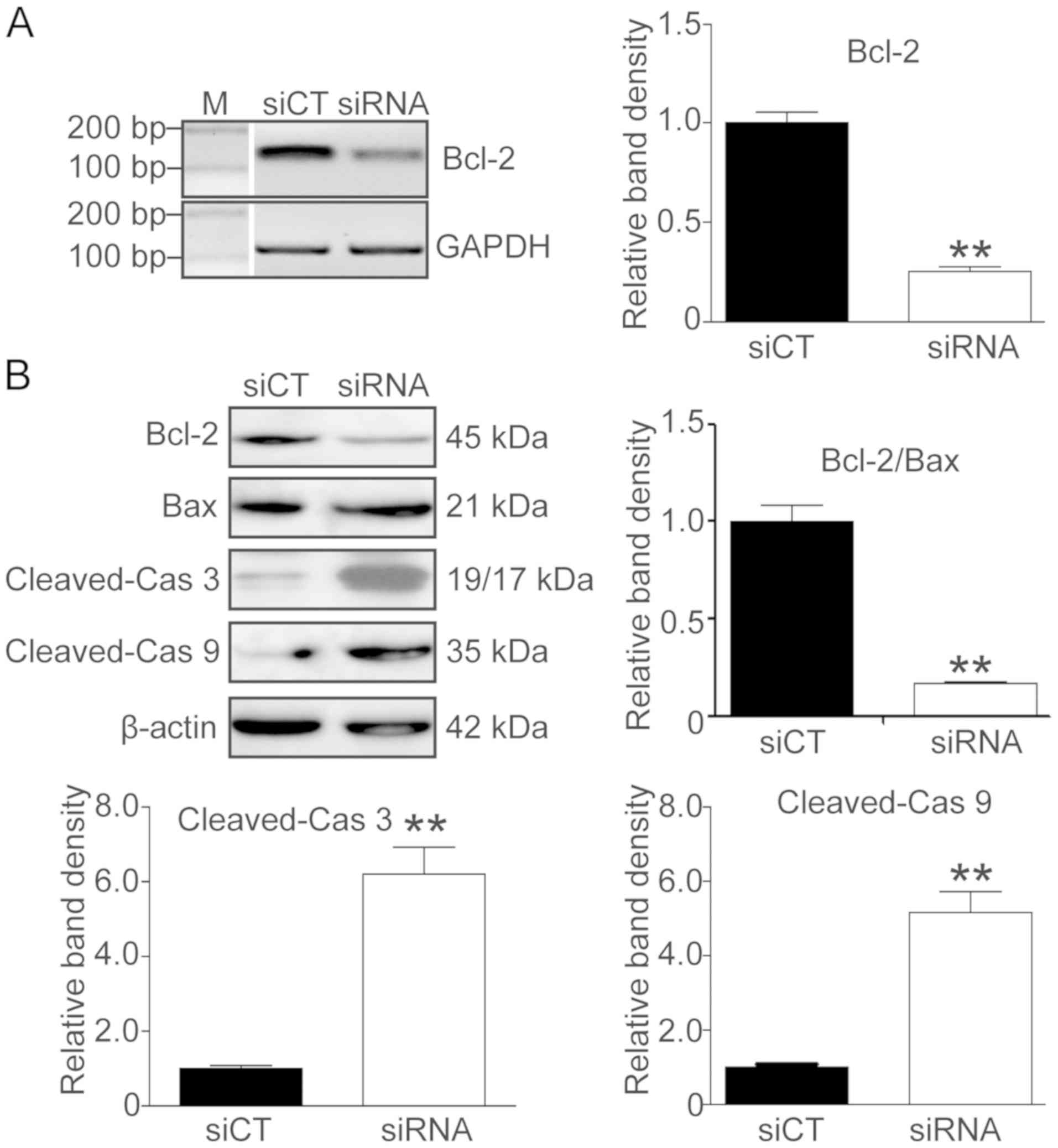
RRM2-induced IR is associated with the
Bcl-2/caspase cell apoptotic pathway
To confirm the role of Bcl-2/Bax and the caspase
apoptotic pathway in the RRM2-induced imatinib resistance, RRM2
siRNA was used to knock down RRM2 and RT-PCR, qF-PCR and western
blot analysis were utilized to detect the expression of Bcl-2, Bax,
cleaved caspase-3 and −9. As shown in Fig. 5, after RRM2 was knocked down, the
Bcl-2 level was decreased (Fig. 5A and
B, P<0.01) and the Bax level was increased while the
Bcl-2/Bax ratio was decreased (Fig.
5B, P<0.01). Simultaneously, the levels of cleaved caspase-3
and −9 were significantly elevated (Fig.
5B, P<0.01).
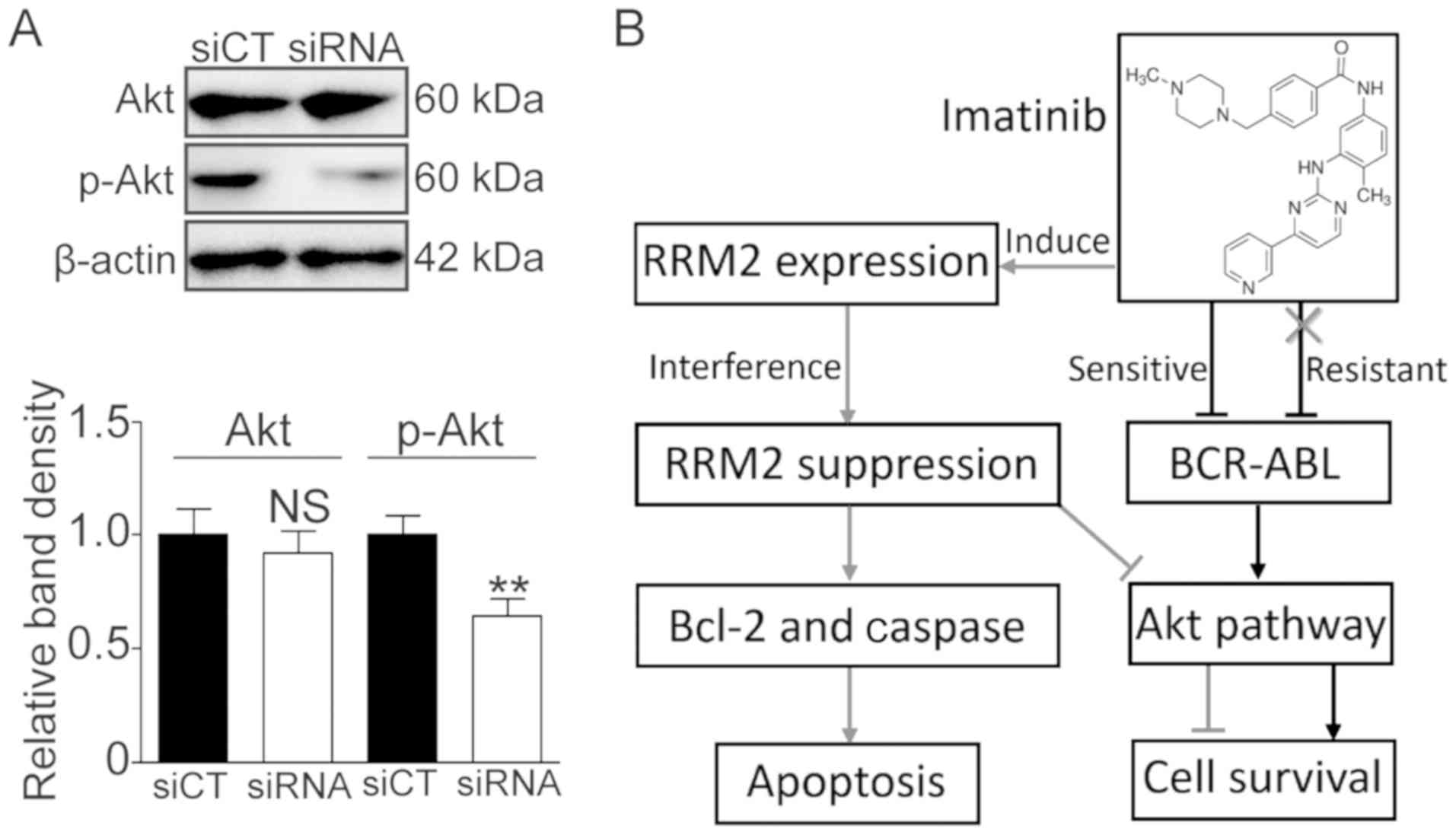
Knockdown of RRM2 suppresses
activation of the Akt pathway
It was reported that RRM2 can activate Akt and
promote cell invasion in gastric cancer (13). Herein, we also investigated whether or
not RRM2 activates the Akt pathway. As shown in Fig. 6, the phosphorylated Akt (p-Akt) level
was significantly decreased following RRM2 knockdown (Fig. 6A, P<0.01). These data suggest that
RRM2 suppression may enhance the sensitivity of CML cells to
imatinib via the Akt pathway.
Discussion
Ribonucleotide reductase (RNR) is a multimeric
enzyme that catalyzes the conversion of ribonucleoside diphosphates
to deoxyribonucleoside diphosphates. It contains two subunits, a
large subunit RRM1 and a small subunit RRM2. RRM1 and RRM2 form an
active enzyme (27). RRM1 is a dimer
of Mr 170,000 that contains nucleotide binding
sites and is responsible for the complex allosteric regulation of
the enzyme. RRM2 is a dimer of Mr 88,000 that
contains stoichiometric amounts of non-heme iron and a unique
tyrosyl free radical, which is essential for its activity (28).
RRM2 is a pivotal molecule that modulates the
activity of RNR, which is essential for DNA synthesis. It was
reported that RRM2 is a determinant of malignant cellular behavior
in a variety of human cancers, such as breast cancer (29), gastric cancer (30), cervical cancer (31), glioblastoma (32), and head and neck cancer (33). RRM2 also plays a role in the
chemoresistance process. Duxbury et al indicated that RRM2
overexpression is associated with gemcitabine chemoresistance in
pancreatic adenocarcinoma cells, and that the suppression of RRM2
enhances gemcitabine-induced cytotoxicity both in vitro and
in vivo (34). Huang et
al used an integrative bioinformatics approach and found that
RRM2 was overexpressed in tamoxifen-resistant breast tumors
(35).
The present study was designed to identify various
novel genes that are responsible for imatinib resistance (IR) in
chronic myeloid leukemia (CML) treatment. The RNA-seq approach was
employed, and we observed that the transcription profiles of many
genes were altered in IR patients. KEGG pathway analysis revealed 7
genes that may be involved in the IR process. After confirming
these genes by qF-PCR in both the patient samples and cell lines,
we finally selected RRM2 as the optimal gene and
investigated its molecular function in the IR process.
The concentration effect experiment showed that
imatinib can induce a dose-dependent increase in RRM2 levels in IR
cells, and knockdown of RRM2 enhanced the sensitivity of K562G
cells to imatinib therapy (Figs. 2
and 3). Further investigation
revealed that the overexpression of RRM2 may induce imatinib drug
resistance and antiapoptosis through the Bcl-2/caspase pathway
(Figs. 4 and 5). Bcl-2 family members regulate the
mitochondrial pathway of apoptosis (36). The ratio between Bcl-2 and Bax is
important for regulating the release of cytochrome c from
mitochondria, which activates caspase-3 and induces apoptosis
(37). Rahman et al
demonstrated that RRM2 depletion significantly reduced Bcl-2
protein expression in head and neck squamous cell carcinoma (HNSCC)
and non-small cell lung cancer (NSCLC) cells. They observed that
RRM2 regulates Bcl-2 protein stability, with RRM2 suppression
leading to increased Bcl-2 degradation, and demonstrated their
colocalization (26). Our results are
consistent with those of Rahman et al.
Activation of the alternative signaling pathway is
an important mechanism in the Bcr-Abl-independent resistance of
imatinib. The crosstalk between RRM2 and the Akt pathway may be a
novel alternative signaling pathway for IR. Zhong et al
found that the overexpression of RRM2 promotes gastric cancer cell
invasion via the Akt/NF-κB signaling pathway (13). Shah et al found that inhibition
of RRM2 significantly reversed Akt-induced tamoxifen-resistant cell
growth and inhibited breast cancer cell motility. They also
indicated that Akt-expression upregulated RRM2 levels, leading to
increased DNA repair and protection from tamoxifen-induced
apoptosis (16). The Akt signaling
pathway plays an important role in the response to extracellular
stimuli to regulate cell growth, apoptosis and survival (38). In our experimental model, we observed
that the knockdown of RRM2 suppressed the activation of Akt
(Fig. 6A). We speculate that there
exists feedback regulation between RRM2 and Akt, and therefore RRM2
affects IR through the Akt signaling pathway (Fig. 6B).
To conclude, in the present study, RRM2 was found to
be elevated in both the IR CML patients and an IR cell line. It was
also demonstrated that RRM2 may affect the cell survival via the
Bcl-2/caspase cell apoptotic pathway and the Akt cell signaling
pathway in imatinib therapy. It is noteworthy that different CML
phases (chronic phase or accelerated phase) probably affect the
level of a certain carcinoma biomarker (39), however, this was not taken into
consideration in the present study. Therefore, to gain deeper
insight into the predictive value of RRM2 for IR, a larger sample
size and a more detailed sample classification are still needed in
subsequent investigations.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by grants
from the Provincial Science Fund of Jilin Provincial Department of
Science and Technology (no. 20190201041JC to CL) and the Ability
Improvement Fund of Health Commission of Jilin Province (no.
2018J065).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SG contributed to the design of the study and wrote
the manuscript. CL and YL performed the experiments and analyzed
the data. RH was involved in conducting the experiments and
analyzing of the data. WH provided constructive comments and
discussions. All authors have read and approved this manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Permission to use the clinical samples for research
purposes was obtained and approved by the Ethics Committee of the
First Hospital of Jilin University. Informed consents were obtained
from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
References
|
1
|
Bartram CR, de Klein A, Hagemeijer A, van
Agthoven T, Geurts van Kessel A, Bootsma D, Grosveld G,
Ferguson-Smith MA, Davies T, Stone M, et al: Translocation of c-ab1
oncogene correlates with the presence of a Philadelphia chromosome
in chronic myelocytic leukaemia. Nature. 306:277–280. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deininger MW: Milestones and monitoring in
patients with CML treated with imatinib. Hematology Am Soc Hematol
Educ Program. 419–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Quintás-Cardama A, Cortes JE and
Kantarjian HM: Early cytogenetic and molecular response during
first-line treatment of chronic myeloid leukemia in chronic phase:
Long-term implications. Cancer. 117:5261–5270. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hochhaus A, Larson RA, Guilhot F, Radich
JP, Branford S, Hughes TP, Baccarani M, Deininger MW, Cervantes F,
Fujihara S, et al: Long-term outcomes of imatinib treatment for
chronic myeloid leukemia. N Engl J Med. 376:917–927. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bixby D and Talpaz M: Mechanisms of
resistance to tyrosine kinase inhibitors in chronic myeloid
leukemia and recent therapeutic strategies to overcome resistance.
Hematology. Hematology. Am Soc Hematol Educ Program. 461–476. 2009.
View Article : Google Scholar
|
|
6
|
Weisberg E and Griffin JD: Mechanism of
resistance to the ABL tyrosine kinase inhibitor STI571 in
BCR/ABL-transformed hematopoietic cell lines. Blood. 95:3498–3505.
2000.PubMed/NCBI
|
|
7
|
le Coutre P, Tassi E, Varella-Garcia M,
Barni R, Mologni L, Cabrita G, Marchesi E, Supino R and
Gambacorti-Passerini C: Induction of resistance to the Abelson
inhibitor STI571 in human leukemic cells through gene
amplification. Blood. 95:1758–1766. 2000.PubMed/NCBI
|
|
8
|
Gorre ME, Mohammed M, Ellwood K, Hsu N,
Paquette R, Rao PN and Sawyers CL: Clinical resistance to STI-571
cancer therapy caused by BCR-ABL gene mutation or amplification.
Science. 293:876–880. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alves R, Fonseca AR, Goncalves AC,
Ferreira-Teixeira M, Lima J, Abrantes AM, Alves V, Rodrigues-Santos
P, Jorge L, Matoso E, et al: Drug transporters play a key role in
the complex process of Imatinib resistance in vitro. Leuk Res.
39:355–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gambacorti-Passerini C, Zucchetti M, Russo
D, Frapolli R, Verga M, Bungaro S, Tornaghi L, Rossi F, Pioltelli
P, Pogliani E, et al: Alpha1 acid glycoprotein binds to imatinib
(STI571) and substantially alters its pharmacokinetics in chronic
myeloid leukemia patients. Clin Cancer Res. 9:625–632.
2003.PubMed/NCBI
|
|
11
|
Bozkurt S, Özkan T, Özmen F, Baran Y,
Sunguroğlu A and Kansu E: The roles of epigenetic modifications of
proapoptotic BID and BIM genes in imatinib-resistant chronic
myeloid leukemia cells. Hematology. 18:217–223. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hentschel J, Rubio I, Eberhart M, Hipler
C, Schiefner J, Schubert K, Loncarevic IF, Wittig U, Baniahmad A
and von Eggeling F: BCR-ABL- and Ras-independent activation of Raf
as a novel mechanism of Imatinib resistance in CML. Int J Oncol.
39:585–591. 2011.PubMed/NCBI
|
|
13
|
Zhong Z, Cao Y, Yang S and Zhang S:
Overexpression of RRM2 in gastric cancer cell promotes their
invasiveness via AKT/NF-κB signaling pathway. Pharmazie.
71:280–284. 2016.PubMed/NCBI
|
|
14
|
Grolmusz VK, Karászi K, Micsik T, Tóth EA,
Mészáros K, Karvaly G, Barna G, Szabó PM, Baghy K, Matkó J, et al:
Cell cycle dependent RRM2 may serve as proliferation marker and
pharmaceutical target in adrenocortical cancer. Am J Cancer Res.
6:2041–2053. 2016.PubMed/NCBI
|
|
15
|
Wang L, Meng L, Wang XW, Ma GY and Chen
JH: Expression of RRM1 and RRM2 as a novel prognostic marker in
advanced non-small cell lung cancer receiving chemotherapy. Tumour
Biol. 35:1899–1906. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shah KN, Mehta KR, Peterson D, Evangelista
M, Livesey JC and Faridi JS: AKT-induced tamoxifen resistance is
overturned by RRM2 inhibition. Mol Cancer Res. 12:394–407. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tu M, Li H, Lv N, Xi C, Lu Z, Wei J, Chen
J, Guo F, Jiang K, Song G, et al: Vasohibin 2 reduces
chemosensitivity to gemcitabine in pancreatic cancer cells via Jun
proto-oncogene dependent transactivation of ribonucleotide
reductase regulatory subunit M2. Mol Cancer. 16:662017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kimura Y, Kasamatsu A, Nakashima D,
Yamatoji M, Minakawa Y, Koike K, Fushimi K, Higo M, Endo-Sakamoto
Y, Shiiba M, et al: ARNT2 regulates tumoral growth in oral squamous
cell carcinoma. J Cancer. 7:702–710. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Joha S, Dauphin V, Leprêtre F, Corm S,
Nicolini FE, Roumier C, Nibourel O, Grardel N, Maguer-Satta V,
Idziorek T, et al: Genomic characterization of Imatinib resistance
in CD34+ cell populations from chronic myeloid leukaemia
patients. Leuk Res. 35:448–458. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun H, Yang B, Zhang H, Song J, Zhang Y,
Xing J, Yang Z, Wei C, Xu T, Yu Z, et al: RRM2 is a potential
prognostic biomarker with functional significance in glioma. Int J
Biol Sci. 15:533–543. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
An C, Guo H, Wen XM, Tang CY, Yang J, Zhu
XW, Yin JY, Liu Q, Ma JC, Deng ZQ, et al: Clinical significance of
reduced SFRP1 expression in acute myeloid leukemia. Leuk Lymphoma.
56:2056–2060. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ni M, Rui Y, Chen Q, Wang Y and Li G:
Effect of growth differentiation factor 7 on tenogenic
differentiation of bone marrow mesenchymal stem cells of rat in
vitro. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 25:1103–1109.
2011.(In Chinese). PubMed/NCBI
|
|
23
|
Weich N, Ferri C, Moiraghi B, Bengió R,
Giere I, Pavlovsky C, Larripa IB and Fundia AF: GSTM1 and GSTP1,
but not GSTT1 genetic polymorphisms are associated with chronic
myeloid leukemia risk and treatment response. Cancer Epidemiol.
44:16–21. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Čokić VP, Mojsilović S, Jauković A,
Kraguljac-Kurtović N, Mojsilović S, Šefer D, Mitrović Ajtić O,
Milošević V, Bogdanović A, Đikić D, et al: Gene expression profile
of circulating CD34(+) cells and granulocytes in chronic myeloid
leukemia. Blood Cells Mol Dis. 55:373–381. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rahman MA, Amin AR, Wang D, Koenig L,
Nannapaneni S, Chen Z, Wang Z, Sica G, Deng X, Chen ZG and Shin DM:
RRM2 regulates Bcl-2 in head and neck and lung cancers: A potential
target for cancer therapy. Clin Cancer Res. 19:3416–3428. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jordheim LP, Sève P, Trédan O and Dumontet
C: The ribonucleotide reductase large subunit (RRM1) as a
predictive factor in patients with cancer. Lancet Oncol.
12:693–702. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Engström Y, Eriksson S, Jildevik I, Skog
S, Thelander L and Tribukait B: Cell cycle-dependent expression of
mammalian ribonucleotide reductase. Differential regulation of the
two subunits. J Biol Chem. 260:9114–9116. 1985.PubMed/NCBI
|
|
29
|
Liang WH, Li N, Yuan ZQ, Qian XL and Wang
ZH: DSCAM-AS1 promotes tumor growth of breast cancer by reducing
miR-204-5p and up-regulating RRM2. Mol Carcinog. 58:461–473. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kang W, Tong JH, Chan AW, Zhao J, Wang S,
Dong Y, Sin FM, Yeung S, Cheng AS, Yu J and To K: Targeting
ribonucleotide reductase M2 subunit by small interfering RNA exerts
anti-oncogenic effects in gastric adenocarcinoma. Oncol Rep.
31:2579–2586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang N, Zhan T, Ke T, Huang X, Ke D, Wang
Q and Li H: Increased expression of RRM2 by human papillomavirus E7
oncoprotein promotes angiogenesis in cervical cancer. Br J Cancer.
110:1034–1044. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li C, Zheng J, Chen S, Huang B, Li G, Feng
Z, Wang J and Xu S: RRM2 promotes the progression of human
glioblastoma. J Cell Physiol. 233:6759–6767. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rahman MA, Amin AR, Wang X, Zuckerman JE,
Choi CH, Zhou B, Wang D, Nannapaneni S, Koenig L, Chen Z, et al:
Systemic delivery of siRNA nanoparticles targeting RRM2 suppresses
head and neck tumor growth. J Control Release. 159:384–392. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Duxbury MS, Ito H, Zinner MJ, Ashley SW
and Whang EE: RNA interference targeting the M2 subunit of
ribonucleotide reductase enhances pancreatic adenocarcinoma
chemosensitivity to gemcitabine. Oncogene. 23:1539–1548. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang L, Zhao S, Frasor JM and Dai Y: An
integrated bioinformatics approach identifies elevated cyclin E2
expression and E2F activity as distinct features of tamoxifen
resistant breast tumors. PLoS One. 6:e222742011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chipuk JE, Moldoveanu T, Llambi F, Parsons
MJ and Green DR: The BCL-2 family reunion. Mol Cell. 37:299–310.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.PubMed/NCBI
|
|
38
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Singh N, Tripathi AK, Sahu DK, Mishra A,
Linan M, Argente B, Varkey J, Parida N, Chowdhry R, Shyam H, et al:
Differential genomics and transcriptomics between tyrosine kinase
inhibitor-sensitive and -resistant BCR-ABL-dependent chronic
myeloid leukemia. Oncotarget. 9:30385–30418. 2018. View Article : Google Scholar : PubMed/NCBI
|