Introduction
Lung cancer has a high incidence and mortality rate,
and 70–80% of patients are diagnosed with advanced disease and are
unsuitable for surgery (1). Recently,
the diagnosis and treatment of lung cancer has entered the era of
individualized treatment (2).
Non-small cell lung cancer (NSCLC) is the major histological
subtype of lung cancer, and the molecular classification of NSCLC
is developing rapidly (3). In China,
the epidermal growth factor receptor (EGFR) molecular variant
subtypes account for approximately 20–30% of NSCLC, and tyrosine
kinase inhibitors of EGFR (EGFR-TKIs), such as gefitinib, have
achieved wide success in the treatment of NSCLC (4). EGFR is a transmembrane receptor tyrosine
kinase and plays an important role in cell growth, proliferation,
differentiation, and other physiological processes (5). In NSCLC, EGFR mutations, which result in
abnormal activation of EGFR, occur mainly in the intracellular
tyrosine kinase coding region, and gefitinib can bind this region
to inhibit the abnormal activation of EGFR (6). However, during the course of treatment
with gefitinib, many patients have been found to be resistant to
gefitinib, which eventually leads to tumor recurrence or
progression (7). It has been found
that approximately 50% of gefitinib resistance is associated with
resistant EGFR mutations (such as T790M) and 20% is associated with
amplification of the proto-oncogene MET; however, the molecular
mechanism of approximately 30% of gefitinib resistance remains
unclear (8). Therefore, the in-depth
study of gefitinib resistance mechanisms and the identification of
approaches to overcome gefitinib resistance are essential in
NSCLC.
miRNAs are endogenous non-coding small RNAs of
approximately 18–25 nucleotides in length that are highly conserved
in evolution and highly specific in tissues (9). miRNAs have post-transcriptional gene
regulatory functions, and can degrade mRNA or inhibit mRNA
translation by binding to the 3′UTR of the target gene mRNA. At
present, more than 1,000 miRNAs have been identified in humans, and
these miRNAs can regulate the expression of at least 30% of genes
that control various biological functions, such as cell
development, differentiation, proliferation, and apoptosis
(10). In recent years, studies have
found that many miRNAs exhibited aberrant expression in tumors and
played a key role in controlling the occurrence, development,
metastasis, and drug resistance of cancers, including NSCLC
(11,12).
In order to investigate the molecular mechanism of
gefitinib resistance in NSCLC, we induced PC9 cells (EGFR single
mutation) to form PC9/gefitinib-resistant (GR) cells by gradually
increasing the concentration of gefitinib. We found that the
expression of let-7 was downregulated and the expression of miR-17
was upregulated in PC9/GR cells compared with PC9 cells. In NSCLC,
it was found that the aberrant expression of let-7 and miR-17 was
associated with tumor progression and poor prognosis (13–15).
However, there were no available data at the time of this study on
the involvement of let-7 and miR-17 in EGFR-TKI resistance of
NSCLC. In the present study, it was revealed that let-7 and miR-17
were involved in the regulation of gefitinib resistance by
targeting MYC and CDKN1A, which promote self-renewal. In addition,
clinical analysis revealed that the expression levels of let-7 and
miR-17 in NSCLC tissues were associated with the response to
gefitinib. These findings indicated that let-7 and miR-17 were
involved in EGFR-TKI resistance by regulating self-renewal, and
that let-7 and miR-17 were potential new biomarkers for EGFR-TKI
resistance in NSCLC.
Materials and methods
Cell culture and cell
transfection
Human NSCLC cells PC9 (parental) cells, PC9/GR
(gefitinib-resistant) cells, and HCC827 cells were cultured in
RPMI-1640 medium containing 10% fetal bovine serum (FBS) at 37°C.
PC9/GR cells were induced using progressive concentrations of
gefitinib. Briefly, PC9 cells in logarithmic growth were treated
with 0.5 µmol/l of gefitinib. After 48 h, gefitinib was removed and
the cells were cultured without gefitinib until they recovered. The
same treatment was then performed again, and when the cells were
resistant to the current concentration, the gefitinib concentration
was gradually increased to 1, 2 µmol/l, and finally to 3 µmol/l.
When the induced cells survived 3 µmol/l of gefitinib for ~2 months
with normal activity, the cells were confirmed to be
gefitinib-resistant and named PC9/GR. The PC9/GR cells were
cultured with 1 µmol/l gefitinib.
Cells were transfected with miRNA mimics, miRNA
inhibitors, siRNAs, and plasmids using Lipofectamine Transfection
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). In the
present study, we used a mimic mixture of nine let-7 members or six
miR-17 members and inhibitors mixture of nine let-7 members or six
miR-17 members to enhance and inhibit the let-7 family or miR-17
family simultaneously (Qiagen GmbH). The sequences of miRNA mimics
and miRNA inhibitors are listed in Table
I.
 | Table I.Sequences of miRNA mimics and miRNA
inhibitors. |
Table I.
Sequences of miRNA mimics and miRNA
inhibitors.
| Primer | Sequence |
|---|
| let-7a mimics |
5′-UGAGGUAGUAGGUUGUAUAGUU-3′ |
| let-7a
inhibitors |
5′-ACTATACAACCTACTACCTC-3′ |
| let-7b mimics |
5′-UGAGGUAGUAGGUUGUGUGGUU-3′ |
| let-7b
inhibitors |
5′-ACCACACAACCTACTACCTC-3′ |
| let-7c mimics |
5′-UGAGGUAGUAGGUUGUAUGGUU-3′ |
| let-7c
inhibitors |
5′-ACCATACAACCTACTACCTC-3′ |
| let-7d mimics |
5′-AGAGGUAGUAGGUUGCAUAGUU-3′ |
| let-7d
inhibitors |
5′-ACTATGCAACCTACTACCTC-3′ |
| let-7e mimics |
5′-UGAGGUAGGAGGUUGUAUAGUU-3′ |
| let-7e
inhibitors |
5′-ACTATACAACCTCCTACCTC-3′ |
| let-7f mimics |
5′-UGAGGUAGUAGAUUGUAUAGUU-3′ |
| let-7f
inhibitors |
5′-ACTATACAATCTACTACCTCA-3′ |
| let-7g mimics |
5′-UGAGGUAGUAGUUUGUACAGUU-3′ |
| let-7g
inhibitors |
5′-ACTGTACAAACTACTACCTC-3′ |
| let-7i mimics |
5′-UGAGGUAGUAGUUUGUGCUGUU-3′ |
| let-7i
inhibitors |
5′-ACAGCACAAACTACTACCTC-3′ |
| miR-98 mimics |
5′-UGAGGUAGUAAGUUGUAUUGUU-3′ |
| miR-98
inhibitors |
5′-ACAATACAACTTACTACCTC-3′ |
| miR-17 mimics |
5′-CAAAGUGCUUACAGUGCAGGUAG-3′ |
| miR-17
inhibitors |
5′-CTACCTGCACTGTAAGCAC-3′ |
| miR-20a mimics |
5′-UAAAGUGCUUAUAGUGCAGGUAG-3′ |
| miR-20a
inhibitors |
5′-CTACCTGCACTATAAGCAC-3′ |
| miR-20b mimics |
5′-CAAAGUGCUCAUAGUGCAGGUAG-3′ |
| miR-20b
inhibitors |
5′-ACCTGCACTATGAGCACTTT-3′ |
| miR-93 mimics |
5′-CAAAGUGCUGUUCGUGCAGGUAG-3′ |
| miR-93
inhibitors |
5′-TACCTGCACGAACAGCACTTT-3′ |
| miR-106a
mimics |
5′-AAAAGUGCUUACAGUGCAGGUAG-3′ |
| miR-106a
inhibitors |
5′-TACCTGCACTGTAAGCACTTTT-3′ |
| miR-106b
mimics |
5′-UAAAGUGCUGACAGUGCAGAU-3′ |
| miR-106b
inhibitors |
5′-ATCTGCACTGTCAGCACTT-3′ |
| Control mimics |
5′-GAUGCUACGGUCAAUGUCUAAG-3′ |
| Control
inhibitors |
5′-TAACACGTCTATACGCCCA-3′ |
Collection of NSCLC samples
Fifty-six NSCLC patients (Table II) were recruited for this study.
Inclusion criteria were as follows: Patients with primary NSCLC;
with a histological diagnosis of NSCLC with at least one measurable
lesion; with a TNM clinical stage of IIIB to IV; who had undergone
molecular-targeted therapy with gefitinib. Tissue samples were
obtained and divided into two groups according to patient responses
assessed using Response Evaluation Criteria in Solid Tumors
(RECIST). Patients with a response or partial response to treatment
were considered to be gefitinib sensitive (GS), and patients with
stable or progressive disease were considered to be gefitinib
resistant (GR).
| Table II.Overall patient characteristics. |
Table II.
Overall patient characteristics.
| Clinicopathological
factors | Data |
|---|
| Total no. | 56 |
| Sex, n (%) |
|
|
Male | 33 (58.9%) |
|
Female | 23 (41.1%) |
| Age, years
(range) |
|
|
Mean | 59.3 (37–75) |
| TNM clinical stage,
n (%) |
|
|
III | 39 (69.6%) |
| IV | 17 (30.4%) |
All patients provided written informed consent.
NSCLC tissues were collected from the Cancer Center of Guangzhou
Medical University (Guangzhou, China) with permission from the
Institutional Review Board. The study protocol was approved by the
Ethics Committee of the Cancer Center of Guangzhou Medical
University [approval no. (2014) 66].
Microarray detection of miRNA
expression
Total RNA from PC9 and PC9/GR cells was isolated
using a Total RNA Purification kit (Qiagen GmbH). Microarray
hybridization assays were carried out in two experiments: PC9 cells
(Cy5-labeled) compared with PC9/GR cells (Cy3-labeled). Data were
analyzed using LOWESS filters and t-tests.
RNA extraction and quantitative
reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA in cells and tissue samples was extracted
by TRIzol™ (Invitrogen; Thermo fisher Scientific, Inc.). For the
detection of mRNA and miRNA, RT-PCR was performed using gene
specific primers or the miScript™ Primer assay kit (Qiagen GmbH)
after reverse transcription from RNA to cDNA. Relative expression
of mRNA and miRNA was normalized by GAPDH and U6, respectively.
RT-PCR primers were designed as follows: MYC
forward, 5′-CGTCCTCGGATTCTCTGCTC-3′ and reverse,
5′-GCTGGTGCATTTTCGGTTGT-3′; CDKN1A forward,
5′-TGCCGAAGTCAGTTCCTTGT-3′ and reverse, 5′-CATTAGCGCATCACAGTCGC-3′;
CD44 forward, 5′-TTACAGCCTCAGCAGAGCAC-3′ and reverse,
5′-TGACCTAAGACGGAGGGAGG-3′; ALDH1 forward,
5′-CTGTGTTCCAGGAGCCGAAT-3′ and reverse, 5′-AGCATCCATAGTACGCCACG-3′;
OCT4 forward, 5′-TGTCAGGGCTCTTTGTCCAC-3′ and reverse,
5′-TCTCCCCAGCTTGCTTTGAG-3′; GAPDH forward,
5′-TGACTTCAACAGCGACACCCA-3′ and reverse,
5′-CACCCTGTTGCTGTAGCCAAA-3′.
Dual-luciferase reporter assay
By using TargetScan prediction system (version 7.1;
http://www.targetscan.org/vert_71/),
we identified a number of potential miRNA targets. The wild-type
(WT) and mutant-type (MUT) 3′untranslated region (3′-UTR) of MYC
and CDKN1A were synthesized chemically and inserted into the
psiCHECK™-2 vector (Promega Corp., Madison, WI, USA) to obtain
psiCHECK™-2-MYC-3′-UTR (WT or MUT plasmids) and
psiCHECK™-2-CDKN1A-3′-UTR (WT or MUT plasmids). Cells were
transfected with WT plasmids or MUT plasmids in the presence of
miRNA mimics or a non-target control (NC). At 48 h after
transfection, the luciferase activity of cells was assessed
according to the Dual-Luciferase reporter assay system (Promega
Corp.).
Western blotting
Proteins were extracted from cells by RIPA buffer
(Thermo Fisher Scientific, Inc.) for 30 min at 4°C. A total of 50
µg proteins per lane were determined by BCA method, loaded into 15%
SDS-PAGE and blotted onto polyvinylidene fluoride (PVDF) membranes
for analysis. Blocking buffer (5% skim milk), was then added and
shaken gently for at least 1 h at 25°C. The primary antibody was
rabbit polyclonal anti-CD44 (cat. no. 37259), anti-ALDH1 (cat. no.
36671) and anti-OCT4 (cat. no. 2890) (1:1,000 dilution; shaken
gently overnight at 4°C; Cell Signaling Technology, Inc., Danvers,
MA, USA). The secondary antibody was anti-rabbit IgG, HRP-linked
antibody (cat. no. 7074) (1:1,000 dilution; shaken gently for at
least 1 h at 25°C; Cell Signaling Technology, Inc.). Proteins were
detected by SuperSignal chemiluminescence substrate (Pierce,
Rockford, IL, USA). Actin was used as an internal control.
Microsphere-forming assay
Cells were incubated in DMEM F12 serum-free medium
with 20 ng/ml of EGF, 20 ng/ml of bFGF, 2% B27 and
1%methylcellulose (Invitrogen; Thermo Fisher Scientific, Inc.).
After 4–7 days, microsphere-like structures were visible, and
images of the microspheres were captured using a light
microscope.
Cell cytotoxicity assays
The cell cytotoxicity assay was performed using a
CCK-8 kit (Beyotime Institute of Biotechnology, Haimen, China).
Cells were incubated with different concentrations of gefitinib.
After 48 h, the medium was removed, and 90 µl of medium and 10 µl
of CCK-8 solution were added to each well of the plate. The plate
was incubated for 3 h at 37°C. The absorbance at a wavelength of
450 nm was measured by an automated reader. Gefitinib-induced
cytotoxicity was represented as the IC50 value
(µmol/l).
Cell apoptosis assay
Cell apoptosis was determined using the Apoptosis
Analysis kit (Beyotime Institute of Biotechnology). Cells were
incubated with gefitinib for 48 h and then collected and fixed in
70% ethanol overnight at 4°C. The cells were labeled with Annexin
V-FITC and propidium iodide (PI) and analyzed using flow cytometry.
The cell apoptosis ratio was analyzed using FlowJo software (FlowJo
LLC, Ashland, OR, USA).
Cell cycle assay
The cell cycle assay was performed using the Cell
Cycle Analysis kit (Beyotime Institute of Biotechnology). Cells
were collected and fixed in 70% ethanol overnight at 4°C. The cells
were stained with PI and analyzed using flow cytometry. The cell
cycle was analyzed using FlowJo software (FlowJo LLC).
Statistical analyses
Values were presented as the mean ± standard
deviation (SD) of at least three separate experiments. The
IC50 values were assessed by linear regression analysis.
The Student's unpaired t-test, one-way analysis of variance (ANOVA)
and receiver operating characteristic (ROC) curves were performed
using SPSS, version 21.0 (IBM Corp., Armonk, New York, USA).
Multiple comparisons between the groups was performed using
Bonferroni method. P<0.05 (two-tailed) was considered to
indicate a statistically significant difference.
Results
Gefitinib-resistant NSCLC cells reduce
let-7 and induce miR-17
miRNA microarray chip analysis was used to detect
the miRNA expression profiles of gefitinib-resistant PC9/GR cells
and gefitinib-sensitive PC9 cells. The results of microarray
analyses and RT-PCR confirmed that in comparison with PC9 cells,
the expression levels of nine members of the let-7 family (let-7a,
let-7b, let-7c, let-7d, let-7e, let-7f, let-7g, let-7i and miR-98)
were downregulated in PC9/GR cells, and the expression levels of
six members of the miR-17 family (miR-17, miR-20a, miR-20b, miR-93,
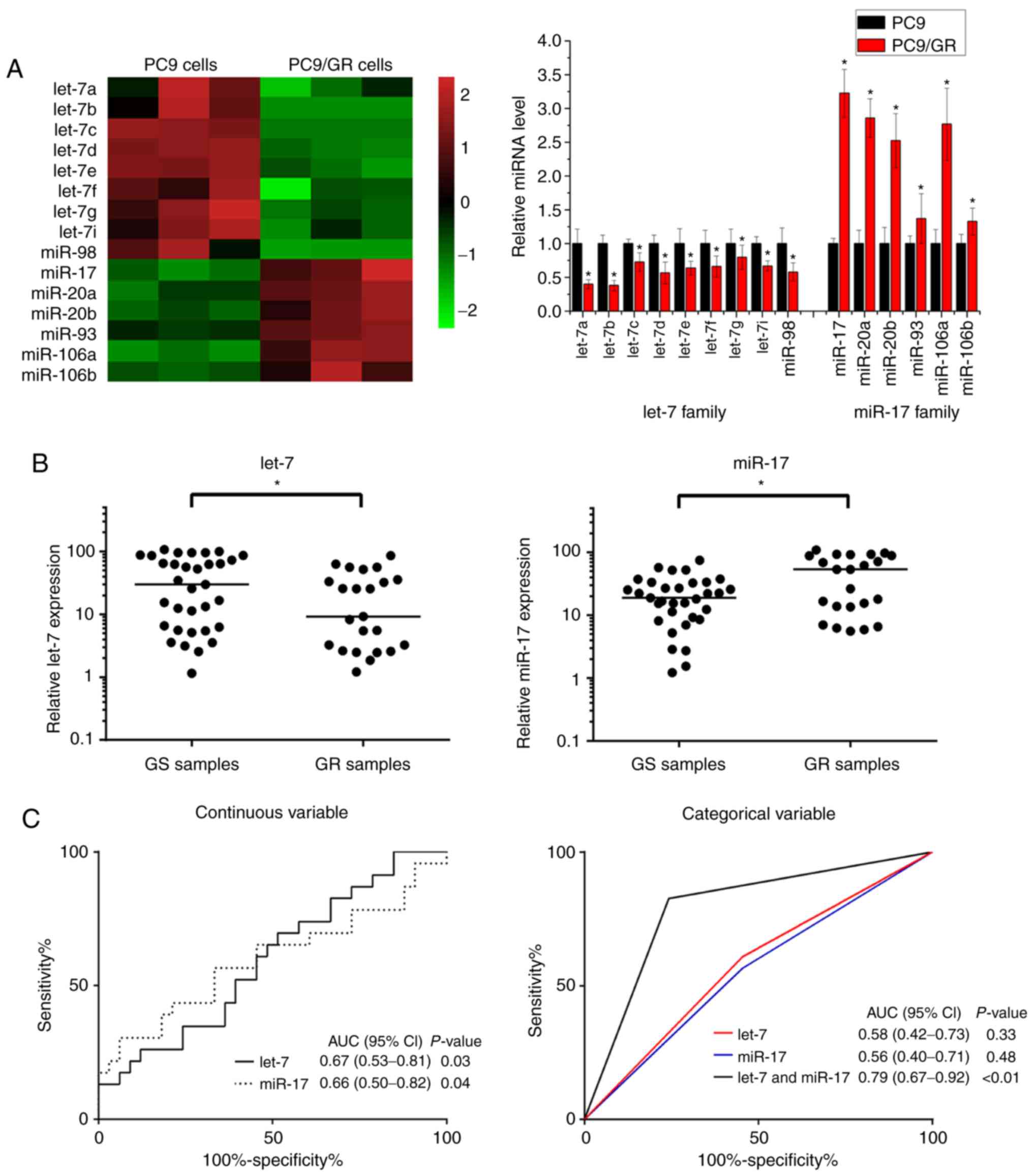
miR-106a and miR-106b) were upregulated in PC9/GR cells (Fig. 1A and B). Therefore, these results
indicated that the let-7 family and miR-17 family were involved in
NSCLC gefitinib resistance.
It was then determined whether let-7 and miR-17 were
associated with the outcome of gefitinib therapy in NSCLC patients.
Using RT-PCR, the expression levels of let-7 were found to be
significantly lower in gefitinib-resistant patients (GR samples)
compared with gefitinib-sensitive patients (GS samples) (Fig. 1B). In contrast to let-7, the
expression levels of miR-17 were significantly higher in GR samples
than in GS samples (Fig. 1B). These
results indicated that low expression levels of let-7 and high
expression levels of miR-17 were positively associated with a poor
response to gefitinib therapy in NSCLC patients. To determine the
potential of let-7 and miR-17 as biomarkers, the expression levels
of let-7 and miR-17 were analyzed as continuous or categorical
variables in ROC analyses. Compared with separate analyses of let-7
and miR-17, combined analyses of let-7 and miR-17 produced a higher
area under the curve (AUC) score (Fig.
1C). Moreover, according to the association analysis between
the expression levels of the let-7 and miR-17 and the
clinicopathological factors of NSCLC patients, the clinical outcome
of gefitinib therapy was associated with the expression levels of
the let-7 in NSCLC tissues (P=0.057, Table III) and significantly correlated
with the expression levels of the miR-17 in NSCLC tissues (P=0.015,
Table IV). These results indicate
that let-7 and miR-17 could be potential biomarkers for predicting
the clinical outcome of gefitinib therapy in NSCLC.
| Table III.Association between the expression of
the let-7 family and the clinicopathological features of NSCLC
patients. |
Table III.
Association between the expression of
the let-7 family and the clinicopathological features of NSCLC
patients.
|
| Expression level of
let-7a |
|
|
|---|
|
|
|
|
|
|---|
| Clinicopathological
factors | High (n=28) | Low (n=28) | χ2
value |
P-valueb |
|---|
| Sex |
|
| 0.074 | 0.786 |
|
Male | 16 | 17 |
|
|
|
Female | 12 | 11 |
|
|
| Age (years) |
|
| 0.650 | 0.420 |
|
<60 | 11 | 14 |
|
|
|
≥60 | 17 | 14 |
|
|
| TNM clinical
stage |
|
| 0.760 | 0.383 |
|
III | 21 | 18 |
|
|
| IV | 7 | 10 |
|
|
| Therapy
response |
|
| 3.615 | 0.057 |
|
CR+PR | 20 | 13 |
|
|
|
SD+PD | 8 | 15 |
|
|
| Table IV.Association between the expression of
the miR-17 family and the clinicopathological features of NSCLC
patients. |
Table IV.
Association between the expression of
the miR-17 family and the clinicopathological features of NSCLC
patients.
|
| Expression level of
miR-17a |
|
|
|---|
|
|
|
|
|
|---|
| Clinicopathological
factors | High (n=28) | Low (n=28) | χ2
value |
P-valueb |
|---|
| Sex |
|
| 0.664 | 0.415 |
|
Male | 15 | 18 |
|
|
|
Female | 13 | 10 |
|
|
| Age (years) |
|
| 0.072 | 0.788 |
|
<60 | 13 | 12 |
|
|
|
≥60 | 15 | 16 |
|
|
| TNM clinical
stage |
|
| 2.112 | 0.146 |
|
III | 17 | 22 |
|
|
| IV | 11 | 6 |
|
|
| Therapy
response |
|
| 5.976 | 0.015 |
|
CR+PR | 12 | 21 |
|
|
|
SD+PD | 16 | 7 |
|
|
let-7 and miR-17 are involved in the
regulation of gefitinib resistance in NSCLC
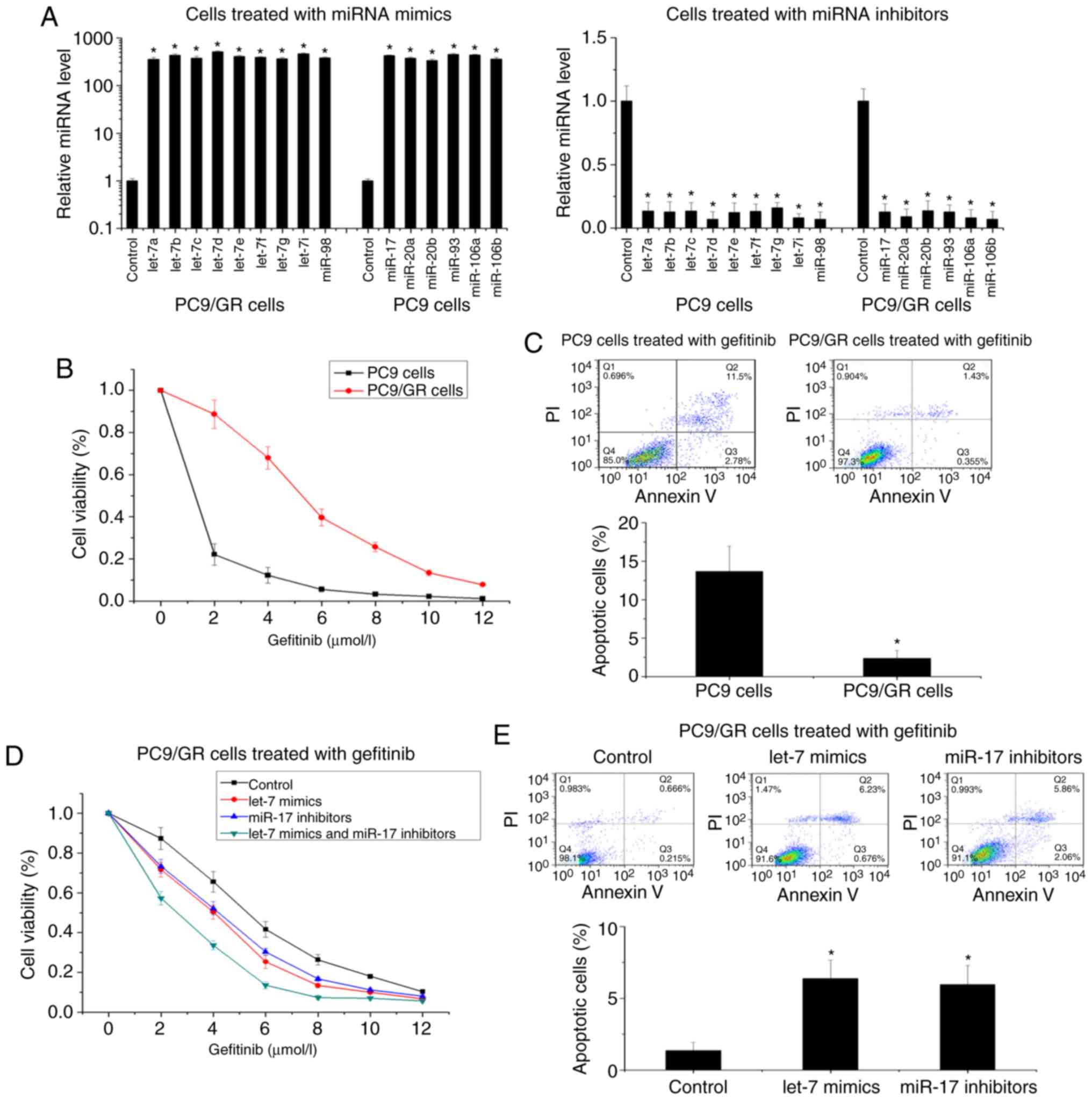
In order to investigate the regulatory functions of
let-7 and miR-17 in gefitinib resistance, we used a miRNA mimics
mixture and a miRNA inhibitors mixture of the let-7 family or the
miR-17 family to determine the influence of let-7 and miR-17 on
gefitinib resistance (Fig. 2A). Cell
cytotoxicity and apoptosis assays were used to compare the
gefitinib resistance of PC9/GR cells and PC9 cells. Compared with
PC9 cells, PC9/GR cells revealed increased resistance to gefitinib
(the IC50 increased from 0.49 to 5.66 µmol/l) and
decreased gefitinib-induced cellular apoptosis (Fig. 2B and C). In PC9/GR cells, upregulation
of let-7 or downregulation of miR-17 restored sensitivity to
gefitinib (the IC50 decreased from 5.37 to 3.92 µmol/l
and from 5.37 to 3.85 µmol/l, respectively) and increased
gefitinib-induced cellular apoptosis (Fig. 2D and E). Conversely, in PC9 cells,
downregulation of let-7 and upregulation of miR-17 protected PC9
cells from gefitinib (the IC50 increased from 0.51 to
1.13 µmol/l and from 0.51 to 0.96 µmol/l) and decreased
gefitinib-induced cellular apoptosis (Fig. 2F and G). Moreover, combined regulation
of let-7 and miR-17 influenced gefitinib resistance more
significantly compared with single regulation of let-7 or miR-17
(the IC50 decreased from 5.37 to 2.39 µmol/l in PC9/GR
cells, and the IC50 increased from 0.51 to 2.07 µmol/l
in PC9 cells) (Fig. 2D and F). Using
another NSCLC cell line, HCC827, the effects of let-7 and miR-17 on
gefitinib resistance were also observed (Fig. 2H and I). Therefore, these findings
indicated that both let-7 and miR-17 may play important roles in
the regulation of gefitinib resistance in NSCLC.
let-7 and miR-17 regulates gefitinib
resistance by promoting self-renewal of NSCLC cells
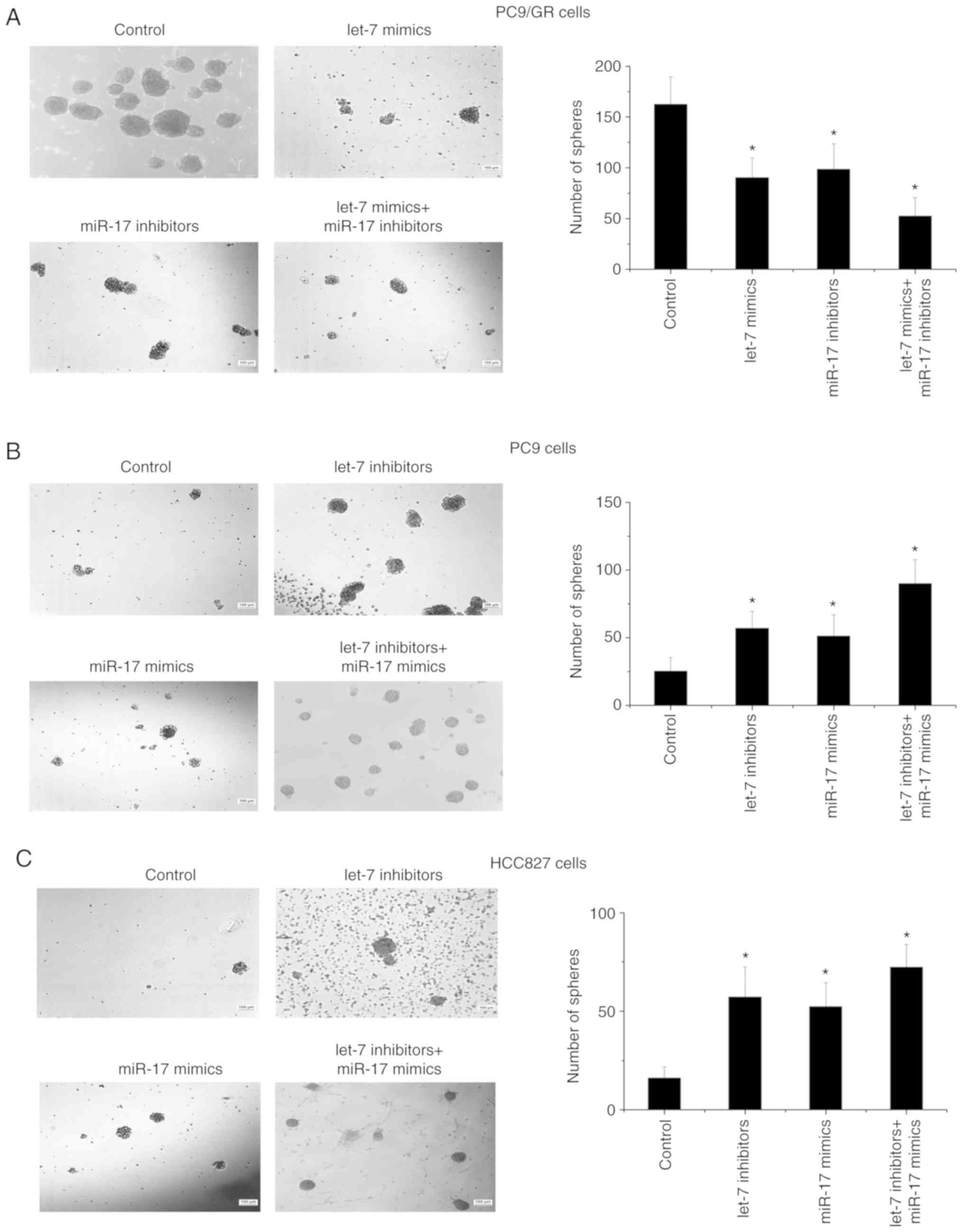
The self-renewal capacity of PC9/GR cells was
compared with that of PC9 cells using the microsphere formation
assay. It was found that the number and size of microspheres formed
by PC9/GR cells were greater than the microspheres formed by PC9
cells (Fig. 3A and B). The effects of
let-7 and miR-17 on the self-renewal capability were then
investigated. The results revealed that the number of microspheres
formed by PC9/GR cells decreased when let-7 was upregulated or
miR-17 was downregulated, and the number of microspheres formed by
PC9 cells increased when let-7 was downregulated or miR-17 was
upregulated (Fig. 3A and B).
Moreover, consistent with the results of the cytotoxicity analyses,
the combined regulation of let-7 and miR-17 expression increased
the number of microspheres more significantly than single
regulation of let-7 or miR-17 (Fig. 3A
and B). Using another NSCLC cell line, HCC827, the effects of
let-7 and miR-17 on self-renewal were also observed (Fig. 3C). Therefore, these data indicated
that let-7 and miR-17 influenced gefitinib resistance by regulating
the self-renewal capability of NSCLC cells.
let-7 regulates self-renewal by
targeting MYC essential for stemness maintenance
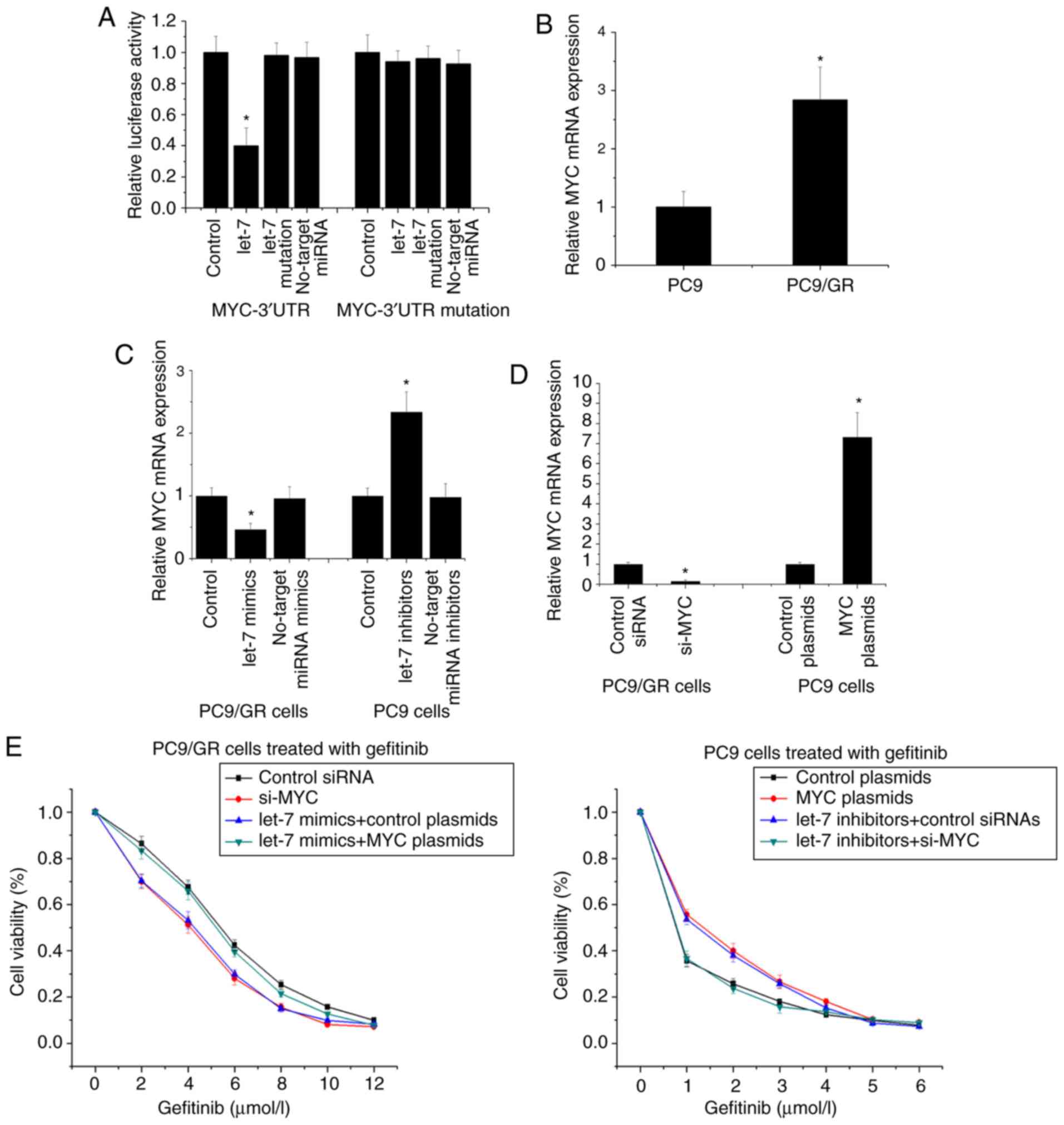
Since miRNAs function by silencing target genes, we
used the TargetScan prediction system and Dual-Luciferase reporter
assay to determine whether let-7 binds to the 3′-UTR of MYC
(Fig. 4A). RT-PCR revealed that MYC
was higher in PC9/GR cells than in PC9 cells, and upregulation of
let-7 decreased MYC expression in PC9/GR cells and downregulation
of let-7 increased MYC expression in PC9 cells (Fig. 4B and C). Furthermore, we used
overexpression plasmids and siRNA to determine the influence of MYC
on gefitinib resistance (Fig. 4D).
The results revealed that in PC9/GR cells, transfection with si-MYC
decreased gefitinib resistance and microsphere formation capacity,
and transfection with MYC plasmid inhibited the reducing effect of
let-7 upregulation on gefitinib resistance and microsphere
formation capacity (Fig. 4E and F).
Transfection with MYC plasmid in PC9 cells increased gefitinib
resistance and microsphere formation capacity, and transfection
with si-MYC inhibited the promoting effect of let-7 downregulation
on gefitinib resistance and microsphere formation capacity
(Fig. 4E and F). These results
indicated that let-7 influenced gefitinib resistance and
self-renewal capacity by directly regulating MYC in NSCLC
cells.
MYC is an important transcription factor in stem
cells, and both RT-PCR and western blotting assays were used to
detect the effect of let-7 and MYC on the expression of stem cell
markers CD44, ALDH1 and OCT4. The expression of CD44, ALDH1, and
OCT4 in PC9/GR cells increased compared with PC9 cells (Fig. 4G). In PC9/GR cells, upregulation of
let-7 or transfection with si-MYC decreased CD44, ALDH1, and OCT4
expression, and transfection with MYC plasmid inhibited the
reducing effect of let-7 upregulation on CD44, ALDH1 and OCT4
expression (Fig. 4G). In PC9 cells,
downregulation of let-7 or transfection with MYC plasmid increased
CD44, ALDH1 and OCT4 expression, and transfection with si-MYC
inhibited the promoting effect of let-7 downregulation on CD44,
ALDH1 and OCT4 expression (Fig. 4G).
Using another NSCLC cell line, HCC827, the effects of let-7 on
stemness maintenance were also observed (Fig. 4H). Therefore, these findings indicated
that let-7 regulated self-renewal by targeting MYC essential for
stemness maintenance.
miR-17 regulates self-renewal by
targeting CDKN1A
The TargetScan prediction system and dual-luciferase
reporter assay were used determine whether miR-17 binds to the
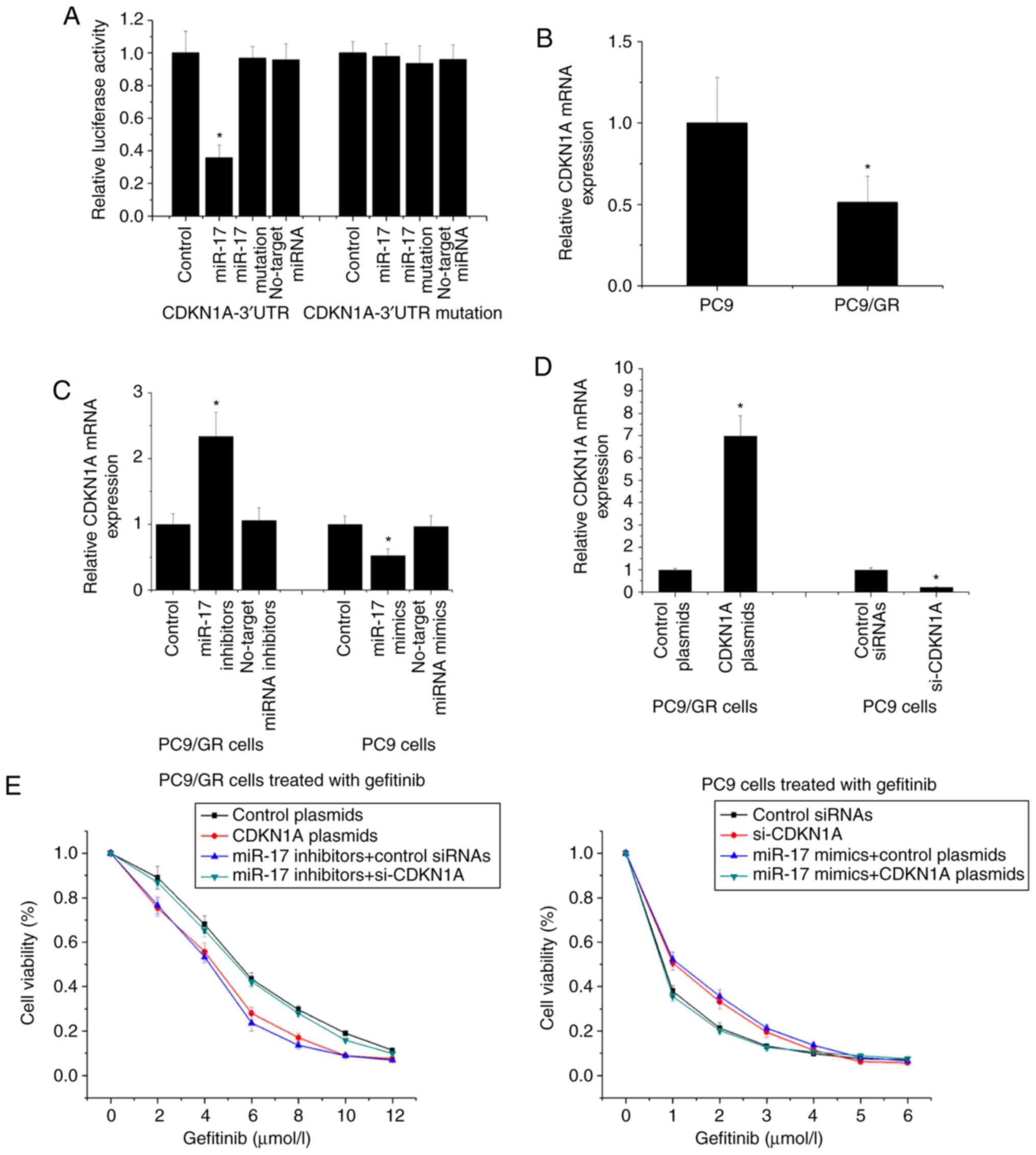
3′-UTR of CDKN1A (Fig. 5A). It was
revealed that CDKN1A was lower in PC9/GR cells compared with PC9
cells, and downregulation of miR-17 increased CDKN1A expression in
PC9/GR cells and upregulation of miR-17 decreased CDKN1A expression
in PC9 cells (Fig. 5B and C).
Moreover, we used overexpression plasmids and siRNA to determine
the influence of CDKN1A on gefitinib resistance (Fig. 5D). In PC9/GR cells, transfection with
CDKN1A plasmids decreased gefitinib resistance and microsphere
formation capacity, and transfection with si-CDKN1A inhibited the
reducing effect of miR-17 downregulation on gefitinib resistance
and microsphere formation capacity; in PC9 cells, transfection with
si-CDKN1A increased gefitinib resistance and microsphere formation
capacity, and transfection with CDKN1A plasmid inhibited the
promoting effect of miR-17 upregulation on gefitinib resistance and
microsphere formation capacity (Fig. 5E
and F). These results indicated that miR-17 influenced
gefitinib resistance and self-renewal capacity by directly
regulating CDKN1A in NSCLC cells.
CDKN1A is a cyclin-dependent kinase inhibitor, and
flow cytometric assays were used to determine the effect of miR-17
and CDKN1A on the cell cycle. It was found that the G2/M phase
increased and the G1 phase decreased in PC9/GR cells compared with
PC9 cells (Fig. 5G). In PC9/GR cells,
downregulation of miR-17 or transfection with CDKN1A plasmid
decreased the G2/M phase and increased the G1 phase, and
transfection with si-CDKN1A inhibited the reducing effect of miR-17
downregulation on the increased G2/M phase (Fig. 5G). In PC9 cells, upregulation of
miR-17 or transfection with si-CDKN1A increased the G2/M phase and
decreased the G1 phase, and transfection with CDKN1A plasmid
inhibited the promoting effect of upregulation of let-7 on the
decreased G2/M phase (Fig. 5G). Using
another NSCLC cell line, HCC827, the effects of miR-17 on the cell
cycle were also observed (Fig. 5H).
Therefore, these findings indicated that miR-17 regulated
self-renewal by targeting CDKN1A.
Discussion
Cancer stem cells are considered to have important
roles in many cancers including NSCLC (16–18).
Similar to normal stem cells, cancer stem cells have stem cell-like
characteristics which can promote cancer development and metastasis
(19–21). It has also been found that cancer stem
cells are drug resistant due to their capacity for self-renewal
which promotes cancer stem cell resistance to the cytotoxic and
killing effect induced by drugs (22,23). In
the present study, PC9 cells (EGFR single mutation) were induced to
form gefitinib-resistant PC9/GR cells by gradually increasing the
concentration of gefitinib, and found that PC9/GR cells had
increased microsphere formation capacity compared with PC9 cells,
indicating that PC9/GR cells exhibited self-renewal capacity.
Therefore, it is suggested that self-renewal was involved in the
regulation of EGFR-TKI resistance in NSCLC.
A miRNA is an important endogenous non-coding RNA,
and miRNA-mediated silencing of gene expression is important in
many physiological and pathological processes including
self-renewal (24–26). In this study, compared with PC9 cells,
the expression of the let-7 family was downregulated and the
expression of the miR-17 family was upregulated in PC9/GR cells. We
further examined the expression levels of let-7 and miR-17 in 56
NSCLC tissue samples, and found that low expression levels of let-7
and high expression levels of miR-17 had significant clinical
relevance in gefitinib resistance. Based on the ROC analysis, it is
worth noting that both let-7 and miR-17 combined had the potential
to be therapeutic response biomarkers of gefitinib treatment in
NSCLC. Moreover, cellular assays revealed that let-7 and miR-17
influenced gefitinib resistance and self-renewal capacity.
Therefore, let-7 and miR-17 were involved in the regulation of
EGFR-TKI resistance in NSCLC by regulating self-renewal.
let-7 was one of the first miRNAs to be discovered
and has been revealed to play an important role in the regulation
of nematode development (27). The
let-7 family is highly conserved among species including mammals
and is widely expressed in different tissue types (28). In humans, nine members of the let-7
family share the same core sequence, and the coding loci of these
nine members are distributed over eight chromosomal DNA sequences
(29). Recently, let-7 was revealed
to be an important tumor suppressor and play an important role in
tumor suppression in various cancers including NSCLC (30,31). Many
target genes of the let-7 family have been identified, such as MYC,
LIN28, HMGA2, IGF2BP, and IMP1, and these genes are mainly related
to the regulation of cell differentiation (32). Our research revealed that let-7
influenced the formation of microspheres and participated in the
maintenance of stem cell markers (CD44, ALDH1, and OCT4) by
regulating the expression of the target gene, MYC. MYC is a
proto-oncogene that has been implicated in the pathogenesis of many
types of human tumors and is associated with the formation of
cancer stem cells (33). Therefore,
let-7 affected the EGFR-TKI resistance of NSCLC and self-renewal by
regulating MYC.
The miR-17 family consists of six members that have
the same conserved nucleotide core sequence, and the coding sites
of these six members are located in the miR-17~92 cluster and its
two paralogs (miR-106b~25 clusters and miR-106a~363 clusters) on
the DNA sequence (34). It was found
that amplification and high expression of the miR-17 family were
involved in the occurrence and development of various cancers
including NSCLC (35–37). At present, the miR-17 family target
genes that have been discovered and confirmed in cancer research
mainly include CDKN1A, cyclin D1, E2F1, and TGFβR2, and these genes
are closely related to regulation of the cell cycle (38). Our research group found that miR-17
influenced the formation of microspheres and participated in
regulation of the cell cycle by regulating the expression of the
t3arget gene, CDKN1A. CDKN1A is a cyclin-dependent kinase inhibitor
that prevents cells from passing the G1 checkpoint, thereby
inhibiting cell proliferation (39).
Therefore, miR-17 affected the EGFR-TKI resistance of NSCLC and
self-renewal by regulating CDKN1A.
The present study indicated that let-7 and miR-17
influenced self-renewal and gefitinib resistance by regulating MYC
and CDKN1A, respectively. On the one hand, low let-7 levels
increased MYC expression to help maintain the undifferentiated
status. On the other hand, high miR-17 levels decreased CDKN1A
expression to help maintain the proliferative potential. Thus, both
low let-7 and high miR-17 combined could regulate self-renewal by
promoting cancer stem cell expansion, and protect cancer cells from
gefitinib-induced cytotoxicity resulting in gefitinib resistance.
Therefore, we propose that let-7 and miR-17, and target genes MYC
and CDKN1A form a joint regulatory network that promotes
self-renewal of NSCLC cells, thereby forming EGFR-TKI resistance.
This study provided novel molecular mechanisms of EGFR-TKI
resistance in NSCLC, and also provided new biomarkers and
strategies to predict and reverse EGFR-TKI resistance in NSCLC in
the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81501969 and
81572258), the Guangzhou Science and Technology Program (nos.
201607010031 and 201804010077), the Scientific Research Project of
Guangzhou Municipal University (nos. 1201410198 and 1201630087) and
the Guangzhou Key Medical Discipline Construction Project Fund.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JY, WH and JZ conceived and designed the
experiments. JY, WH and LP performed the experiments and assembled
the data. WF, LD and ZJ obtained the tumor and tissues with
clinical information where it pertained. JY and FZ performed the
statistical analysis. JY and JZ analyzed the data and drafted the
manuscript. LP, WF, LD, ZJ and FZ revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
NSCLC tissues were collected from the Cancer Center
of Guangzhou Medical University (Guangzhou, China) with written
informed consent and permission from the Institutional Review
Board. All patients provided written informed consent. The study
protocol was approved by the Ethics Committee of the Cancer Center
of Guangzhou Medical University [approval no. (2014) 66].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2016. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mitsudomi T, Suda K and Yatabe Y: Surgery
for NSCLC in the era of personalized medicine. Nat Rev Clin Oncol.
10:235–244. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park SR, Davis M, Doroshow JH and Kummar
S: Safety and feasibility of targeted agent combinations in solid
tumours. Nat Rev Clin Oncol. 10:154–168. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tebbutt N, Pedersen MW and Johns TG:
Targeting the ERBB family in cancer: Couples therapy. Nat Rev
Cancer. 13:663–673. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Blakely CM, Watkins TBK, Wu W, Gini B,
Chabon JJ, McCoach CE, McGranahan N, Wilson GA, Birkbak NJ, Olivas
VR, et al: Evolution and clinical impact of co-occurring genetic
alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet.
49:1693–1704. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rotow J and Bivona TG: Understanding and
targeting resistance mechanisms in NSCLC. Nat Rev Cancer.
17:637–658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sequist LV, Bell DW, Lynch TJ and Haber
DA: Molecular predictors of response to epidermal growth factor
receptor antagonists in non-small-cell lung cancer. J Clin Oncol.
25:587–595. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ha M and Kim VN: Regulation of microRNA
biogenesis. Nat Rev Mol Cell Biol. 15:509–524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: Are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin S and Gregory RI: MicroRNA biogenesis
pathways in cancer. Nat Rev Cancer. 15:321–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee YS and Dutta A: MicroRNAs in cancer.
Annu Rev Pathol. 4:199–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shin JI and Brusselle GG: Mechanistic
links between COPD and lung cancer: A role of microRNA let-7? Nat
Rev Cancer. 14:702014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guinot A, Oeztuerk-Winder F and Ventura
JJ: miR-17-92/p38α dysregulation enhances Wnt signaling and selects
Lgr6+ cancer stem-like cells during lung adenocarcinoma
progression. Cancer Res. 76:4012–4022. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Osada H and Takahashi T: let-7 and
miR-17-92: Small-sized major players in lung cancer development.
Cancer Sci. 102:9–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takebe N, Miele L, Harris PJ, Jeong W,
Bando H, Kahn M, Yang SX and Ivy SP: Targeting notch, hedgehog, and
wnt pathways in cancer stem cells: Clinical update. Nat Rev Clin
Oncol. 12:445–464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
MacDonagh L, Gray SG, Breen E, Cuffe S,
Finn SP, O'Byrne KJ and Barr MP: Lung cancer stem cells: The root
of resistance. Cancer Lett. 372:147–156. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pattabiraman DR and Weinberg RA: Tackling
the cancer stem cells-What challenges do they pose? Nat Rev Drug
Discov. 13:497–512. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lopez-Ayllon BD, Moncho-Amor V, Abarrategi
A, Ibañez de Cáceres I, Castro-Carpeño J, Belda-Iniesta C, Perona R
and Sastre L: Cancer stem cells and cisplatin-resistant cells
isolated from non-small-lung cancer cell lines constitute related
cell populations. Cancer Med. 3:1099–1111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shien K, Toyooka S, Yamamoto H, Soh J,
Jida M, Thu KL, Hashida S, Maki Y, Ichihara E, Asano H, et al:
Acquired resistance to EGFR inhibitors is associated with a
manifestation of stem cell-like properties in cancer cells. Cancer
Res. 73:3051–3061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Flemming A: Cancer stem cells: Targeting
the root of cancer relapse. Nat Rev Drug Discov. 14:1652015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wicha MS: Targeting self-renewal, an
achilles' heel of cancer stem cells. Nat Med. 20:14–15. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ravasio R, Ceccacci E and Minucci S:
Self-renewal of tumor cells: Epigenetic determinants of the cancer
stem cell phenotype. Curr Opin Genet Dev. 36:92–99. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hwang WL, Jiang JK, Yang SH, Huang TS, Lan
HY, Teng HW, Yang CY, Tsai YP, Lin CH, Wang HW and Yang MH:
MicroRNA-146a directs the symmetric division of snail-dominant
colorectal cancer stem cells. Nat Cell Biol. 16:268–280. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lv C, Li F, Li X, Tian Y, Zhang Y, Sheng
X, Song Y, Meng Q, Yuan S, Luan L, et al: miR-31 promotes mammary
stem cell expansion and breast tumorigenesis by suppressing wnt
signaling antagonists. Nat Commun. 8:10362017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Deng L, Shang L, Bai S, Chen J, He X,
Martin-Trevino R, Chen S, Li XY, Meng X, Yu B, et al: MicroRNA100
inhibits self-renewal of breast cancer stem-like cells and breast
tumor development. Cancer Res. 74:6648–6660. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Reinhart BJ, Slack FJ, Basson M,
Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR and Ruvkun G:
The 21-nucleotide let-7 RNA regulates developmental timing in
caenorhabditis elegans. Nature. 403:901–906. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nimmo RA and Slack FJ: An elegant miRror:
MicroRNAs in stem cells, developmental timing and cancer.
Chromosoma. 118:405–418. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hertel J, Bartschat S, Wintsche A and Otto
C: Students of the bioinformatics computer lab and stadler PF:
Evolution of the let-7 microRNA family. RNA Biol. 9:231–241. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boyerinas B, Park SM, Hau A, Murmann AE
and Peter ME: The role of let-7 in cell differentiation and cancer.
Endocr Relat Cancer. 17:F19–F36. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takamizawa J, Konishi H, Yanagisawa K,
Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y,
et al: Reduced expression of the let-7 microRNAs in human lung
cancers in association with shortened postoperative survival.
Cancer Res. 64:3753–3756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong
C, Huang Y, Hu X, Su F, Lieberman J and Song E: let-7 regulates
self renewal and tumorigenicity of breast cancer cells. Cell.
131:1109–1123. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gabay M, Li Y and Felsher DW: MYC
activation is a hallmark of cancer initiation and maintenance. Cold
Spring Harb Perspect Med. 4:a0142412014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mendell JT: MiRiad roles for the miR-17-92
cluster in development and disease. Cell. 133:217–222. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jin HY, Oda H, Lai M, Skalsky RL, Bethel
K, Shepherd J, Kang SG, Liu WH, Sabouri-Ghomi M, Cullen BR, et al:
MicroRNA-17~92 plays a causative role in lymphomagenesis by
coordinating multiple oncogenic pathways. EMBO J. 32:2377–2391.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hayashita Y, Osada H, Tatematsu Y, Yamada
H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y and
Takahashi T: A polycistronic microRNA cluster, miR-17-92, is
overexpressed in human lung cancers and enhances cell
proliferation. Cancer Res. 65:9628–9632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Matsubara H, Takeuchi T, Nishikawa E,
Yanagisawa K, Hayashita Y, Ebi H, Yamada H, Suzuki M, Nagino M,
Nimura Y, et al: Apoptosis induction by antisense oligonucleotides
against miR-17-5p and miR-20a in lung cancers overexpressing
miR-17-92. Oncogene. 26:6099–6105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Murphy BL, Obad S, Bihannic L, Ayrault O,
Zindy F, Kauppinen S and Roussel MF: Silencing of the miR-17~92
cluster family inhibits medulloblastoma progression. Cancer Res.
73:7068–7078. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Abbas T and Dutta A: P21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|