Introduction
PARP inhibitors are toxic to cells with defects in
homologous recombination (HR)-mediated DNA double-strand break
(DSB) repair, including cells with mutations in BRCA1 and
BRCA2, genes whose loss of function predisposes patients to
breast and ovarian cancer (1). BRCA1
and BRCA2 are the most important genetic factors in hereditary
breast cancer (2). PARP1/2 inhibitors
induce synthetic lethality in cancer cells defective in the HR
repair pathway including BRCA1/2 (3).
Previous research indicates that PARP1 [poly(ADP-ribose) polymerase
1] facilitates DNA repair by binding to DNA breaks and attracting
DNA repair proteins to the site of damage (4). Moreover, the local chromatin relaxation
at DNA damage sites is regulated by PARP1 enzymatic activity
(5). Many patients benefit from the
treatment of PARP inhibitors (6).
Furthermore, PARP inhibitor, niraparib, also showed significant
clinical benefit in patients without HR deficiencies (7). Given the expanding clinical use of PARP
inhibitors and the high likelihood of acquired resistance, there is
a significant need to identify and overcome the mechanisms of
resistance.
Amplified in liver cancer 1 (ALC1) [also
known as CHD1L (chromodomain-helicase-DNA-binding protein
1-like], a poly(ADP-ribose) and ATP-dependent remodeler is involved
in the chromatin-relaxation process (8,9). The
presence of ALC1 overexpression has been suggested to be associated
with aggressive tumor biology in breast cancer, multiple myeloma
and lung cancer (10–13). Additionally, ALC1 interacts with
PARP1/PARylation in base excision repair (14). Notably, this interaction is mediated
through the interplay of the ALC1 macro-domain and the PAR moiety
of PARylated-PARP1 (15), which
activates ALC1 at sites of DNA damage (16). CHFR (checkpoint with
forehead-associated and RING finger domains) is a nuclear protein
and functions as a tumor suppressor in the early mitotic checkpoint
by actively delaying passage into mitosis in response to mitotic
stress (17,18). Previous research has demonstrated that
CHFR functions as an E3 ubiquitin ligase, resulting in interaction
between CHFR and PARP1 induced by mitotic stress (19). Additionally, the interaction between
CHFR and PARP1 plays an important role in cell cycle regulation and
cancer therapy (20). Given the
evidence that primary and secondary resistance to PARP inhibition
have led to treatment failure (21),
the development of new biomarkers and the ability to identify
potential mechanisms of resistance are vital. However, the
associations among PARP, CHFR and ALC1 in regards to cancer
development and therapeutic response remain undefined.
Materials and methods
Breast cancer tissues
A cohort of 28 paired human breast and peripheral
non-tumor tissues extracted during surgical resection were
collected from breast cancer patients in Shanghai Changhai Hospital
from 2013 January to 2015 January. All cancer specimens were frozen
in liquid nitrogen and stored at −80°C after surgical resection.
This study was approved by the Institutional Review Board of the
Second Military Medical University (Shanghai, China). All
participants gave informed consent before they entered the study.
The patients included all women, with a median age of 54 years
(range 31–85 years).
Quantitative Real-time PCR
Total RNA was extracted from breast cancer tissues
or peripheral non-cancer tissues samples using the TRIzol Reagent
(Invitrogen) according to the manufacturer's instructions, and 1–2
µg of RNA was treated by RNase-free DNaseI (Takara) to remove
genomic DNA contamination. qRT-PCR analysis was conducted using a
SYBR Green Supermix kit (Toyobo, Osaka, Japan) with a Light Cycler
480 II (Roche, Basel, Switzerland). The cycle parameters were 95°C
for a 1 min hot start and 45 cycles of 95°C for 10 sec, 60°C for 10
sec and 72°C for 20 sec. The fold change in expression was
calculated using the ΔΔCt method with the B2M mRNA as an internal
control. Experiments for each sample were performed with two
duplicates.
Cell lines and cell culture
The human breast carcinoma cell line (MCF-7), and
293T cells (CRL-3216) were obtained from the Shanghai Cell Bank of
the Chinese Academy of Sciences (Shanghai, China). The cells were
maintained in Dulbecco's modified Eagle's medium (DMEM) (Thermo
Fisher Scientific, Inc.) and 1% penicillin/streptomycin (P/S)
(Sigma-Aldrich; Merck KGaA) supplemented with 10% (v/v) fetal
bovine serum (FBS) (Pan). All cells were grown at 37°C with 5%
CO2 in a humidified incubator.
Plasmids
Full length UB (ubiquitin) was cloned into a
modified pCDNA3 vector to generate encoding hemagglutinin
(HA)-tagged UB. Meanwhile, ALC1 was cloned into the pRIES2-EGFP
vector to construct S-FLAG-SBP (SFB)-tagged ALC1. The
pCMV-Myc/HA-CHFR recombinant plasmid was constructed. The deletion
mutants of CHFR were generated by using the QuikChange
Site-Directed Mutagenesis kit (Stratagene; Agilent Technologies,
Inc.) according to the manufacturer's protocol.
Antibodies, chemicals and
reagents
Antibodies against ALC1 (cat. no. ab51324; Abcam),
anti-HA (cat. no. ab18181; Abcam) and GAPDH (cat. no. ab8245;
Abcam) were purchased from Abcam. Anti-FLAG (cat. no. F3165;
Sigma), anti-β-actin (cat. no. A5441; Sigma), anti-Myc (cat. no.
M4439; Sigma) antibodies were purchased from Sigma-Aldrich/Merck
KGaA. Anti-CHFR antibody (cat. no. PA5-28079) was purchased from
Thermo Fisher Scientific, Inc. The working concentration of
antigens was 1:1,000 dilution.
MG132, dimethyl sulphoxide (DMSO) and cycloheximide
(CHX) were purchased from Sigma. MG132 and CHX were prepared in
DMSO to obtain 10 and 100 µM stock solutions, respectively.
Aliquots were stored at −20°C to avoid freeze-thaw cycles, and a
working solution was freshly prepared with culture medium
immediately prior to use.
The PARP inhibitors AZD2281 (olaparib), and PJ34
(iniparib) were purchased from Selleck Chemicals. AZD2281 and PJ34
were dissolved in DMSO, and stored as per the manufacturers'
recommendations. Cells were seeded at 1×106 cells in 10
ml of medium and 24 h after seeding, the cells were treated with 10
µM AZD2281 or 10 µM PJ34 for 24 h in fresh medium.
Protein affinity purification
The soluble fraction was incubated with 0.5 ml of
streptavidin-conjugated beads (Thermo Fisher Scientific, Inc.) at
4°C for 2 h. The beads were washed three times with NETN100 buffer.
Associated proteins were eluted with 2 mM biotin in 1X PBS and
incubated further with 50 µl of S beads (Novagen) at 4°C for an
additional 2 h, and then washed by NETN buffer five times. The
bound proteins were eluted with SDS sample buffer (4% SDS, 20%
glycerol, 10% 2-mercaptoethanol, 0.004% bromphenol blue, 0.125 M
Tris-HCl), analyzed by SDS-PAGE (polyacrylamide gel
electrophoresis) and silver staining. The objective band was
analyzed by mass spectrometry.
Plasmid transfection
The 293T cells were seeded in 6-well plates
(4×105 cells/well) one day before transfection. The next
day, transfection was performed using Lipofectamine 2000 reagent
(Thermo Fisher Scientific, Inc.) when the cells reached ~80–90%
confluence according to the manufacturer's protocol. Medium was
replaced after 4–6 h with complete medium with FBS and P/S. Cells
were harvested for use after 48 h of incubation.
Western blot analysis
The cells were harvested and washed with
phosphate-buffered saline (PBS). Next, the cells were lysed with 30
ml of ice-cold NETN100 buffer [150 mM NaCl, 1% Triton X-100, 1 mM
phenylmethyl-sulfonyl fluoride, and 25 mM Tris (pH 7.5)] containing
cocktail which was the protease inhibitor (Santa Cruz
Biotechnology, Inc.). The cell lysates were centrifuged at 12,000 ×
g at 4°C for 8 min. The clear supernatant extract was boiled in SDS
buffer (SDS, glycerol, bromic acid, 1 M Tris·HCl) for 8 min and
stored at −20°C. A total of 100 µl of the cell lysates were
subjected to electrophoresis on SDS-12.5% polyacrylamide gels.
Then, the separated proteins were blotted on a PVDF (polyvinylidene
fluoride) membrane (GE Healthcare) using a semi-dry transfer unit
(Bio-Rad). The membranes were incubated in TBS with 5% non-fat milk
and 0.1% Tween-20 for 1 h at room temperature (RT) and then
incubated with the primary antibodies overnight at 4°C. After
washing with Tris-buffered saline containing 0.1% Tween-20, the
membranes were incubated with a secondary antibody, either
HRP-conjugated anti-mouse IgG (Santa Cruz Biotechnology, Inc.;
sc-516176, dilution 1:2,000) or HRP-conjugated anti-rabbit IgG
(Santa Cruz Biotechnology, Inc.; cat. no. sc-516087; dilution
1:2,000) for 40 min at RT. The blots were visualized with a
chemiluminescent ECL kit (Santa Cruz Biotechnology, Inc.).
Co-immunoprecipitation and
immunoblotting
Cells were lysed with 1X cell lysis buffer (Cell
Signaling Technology) and rotated at 12,000 × g at 4°C for 8 min.
Cell debris was removed by centrifugation and the soluble fraction
was collected and precleared with protein A/G agarose beads for 2 h
at 4°C. The precleared cell lysate was incubated with the indicated
antibodies (anti-FLAG antibody) overnight followed by incubation
with protein A/G beads for at least 2 h at 4°C. Immunoprecipitates
were then washed 6 times with cell lysis buffer and boiled in 1X
SDS loading buffer. Samples were resolved on SDS-PAGE and
transferred to PVDF membranes and immunoblotting was carried out
with antibodies as indicated.
Statistical analysis
The statistical analyses were performed with the
ANOVA and a relevant post hoc test. A value of P<0.05 was
considered indicative of a statistically significant result. All
data were analyzed using either GraphPad Prism 6 software (GraphPad
Software, Inc.) or SPSS 20.0 (IBM). Values are shown as mean ± SEM
for each group.
Results
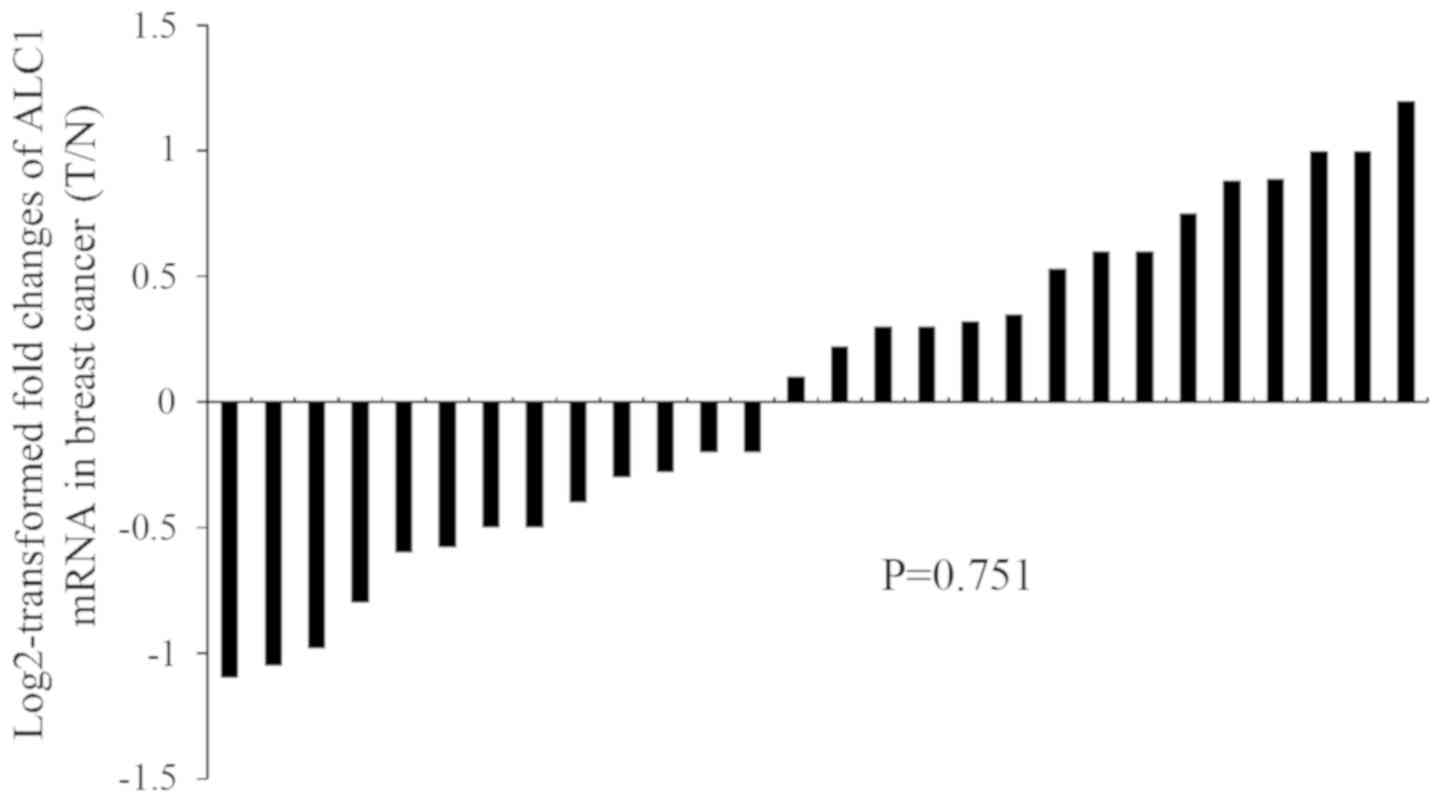
Transcription level of ALC1 is not
regulated in breast cancer tissues
Although the presence of ALC1 overexpression has
been observed in breast cancer tissues (10), little is known concerning the mRNA
level of ALC1 in breast cancer development. To address this issue,
we evaluated the transcriptional expression level of ALC1 in breast
cancer tissues and peripheral non-tumor tissues. There was no
difference in relative mRNA levels in 28 breast cancer samples
compared with the adjacent non-cancerous tissues as analyzed by
quantitative real-time PCR (qPCR) (Fig.
1).
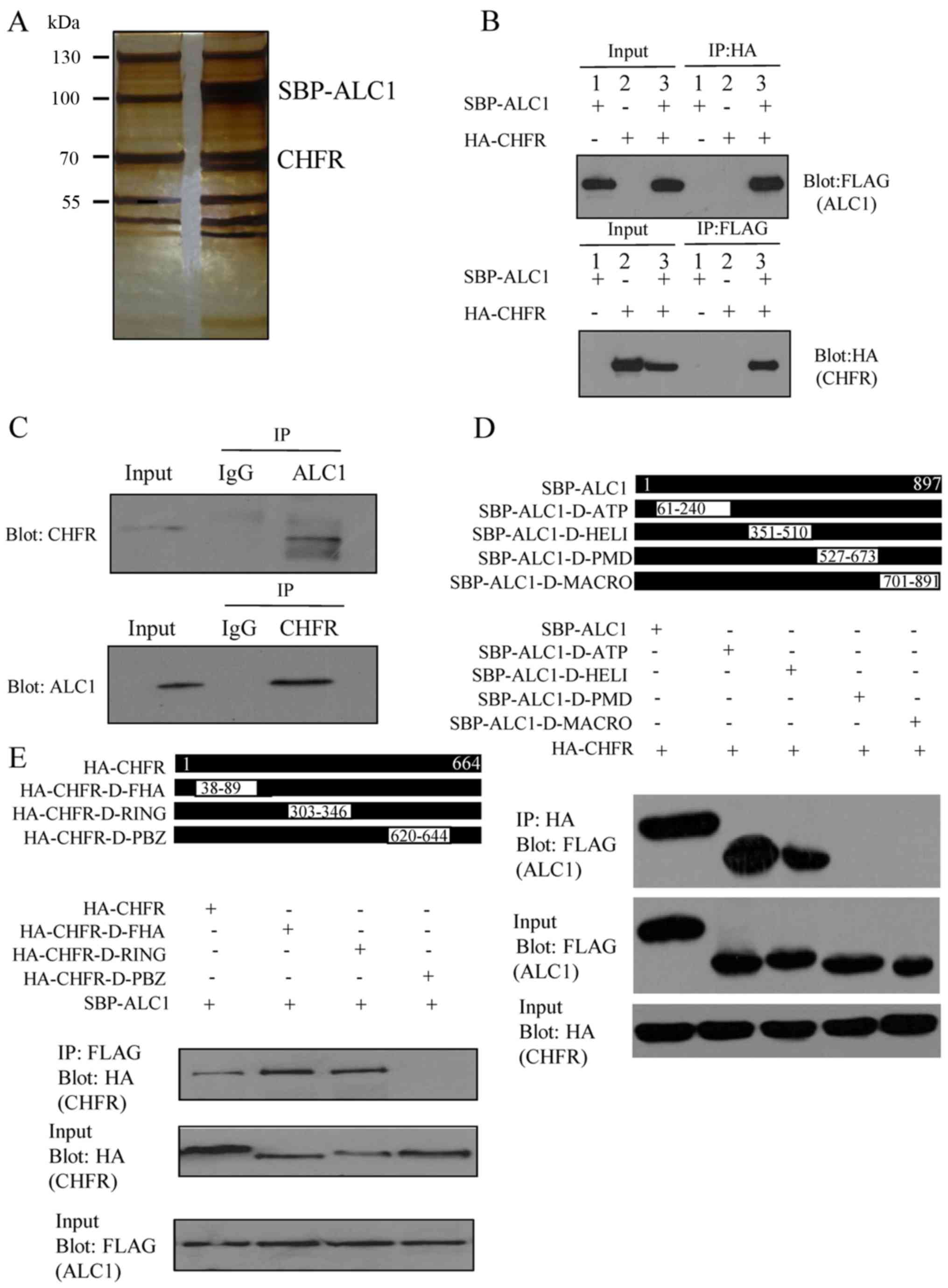
CHFR interacts with ALC1
In order to investigate the high level of ALC1 in
breast cancer, mass spectrometry analysis was used to analyze the
associated proteins. Stably expressing SBP-tagged ALC1 293T cells
were generated and used to identify the associated proteins of ALC1
by tandem affinity purification. Compared with the control group, a
component with a molecular weight of ~70 kDa was observed in the
purified complex by silver staining. Mass spectrometry analysis
demonstrated that the band represented CHFR (Fig. 2A).
To confirm the interaction between CHFR and ALC1 in
the physiological condition, we performed reciprocal
immunoprecipitation of the exogenous proteins. SBP-ALC1 plasmids
were transfected into the first group and HA-CHFR plasmids were
transfected into the second group. SBP-ALC1 and HA-CHFR plasmids
were co-transfected into the third group. Cell lysates were
collected 30 h later. The samples were analyzed by western blot
analysis. The protein bands on the PVDF membranes were detected
with anti-FLAG antibody and anti-HA antibody. As a result, the
bands of the interacting proteins were detected in the third group,
and the interaction was not found in the first or second group. As
expected, exogenously expressed HA-Tagged CHFR interacted with
SBP-Tagged ALC1 in vitro (Fig.
2B). To explore whether endogenous ALC1 interacts with
CHFR-like exogenous proteins, endogenous immunoprecipitation
experiments were performed. MCF-7 cells were collected, some were
incubated with CHFR antibody with NETN lysate, and the others were
incubated with IgG as control. Then the proteins were detected by
western blot analysis. The PVDF membranes were incubated with the
ALC1 antibody. The ALC1 band was found in the panel incubated with
CHFR antibody previously, but no band was detected in the other
panel (Fig. 2C). To map the
ALC1-interacting region with CHFR, ATP (adenosine triphosphate)
(aa61-240) domain, Helicase (aa351-510) domain, PMD (PAR
modification domain) (aa527-673) domain, and MACRO (aa701-891)
domain deletion mutants of ALC1 were generated. Immunoprecipitation
experiments showed that both the MACRO domain (376-592aa) and PMD
(aa527-673) deletion mutants failed to associate with CHFR
(Fig. 2D). Meanwhile, to map the
CHFR-interacting region with ALC1, FHA (filamentous hemagglutinin)
(aa38-89) domain, RING (aa303-346) domain and PBZ
(poly(ADP-ribose)-binding zinc finger) (aa620-644) deletion mutants
of CHFR were generated and their abilities to interact with ALC1
were tested. Only the PBZ deletion mutant failed to interact with
ALC1 (Fig. 2E).
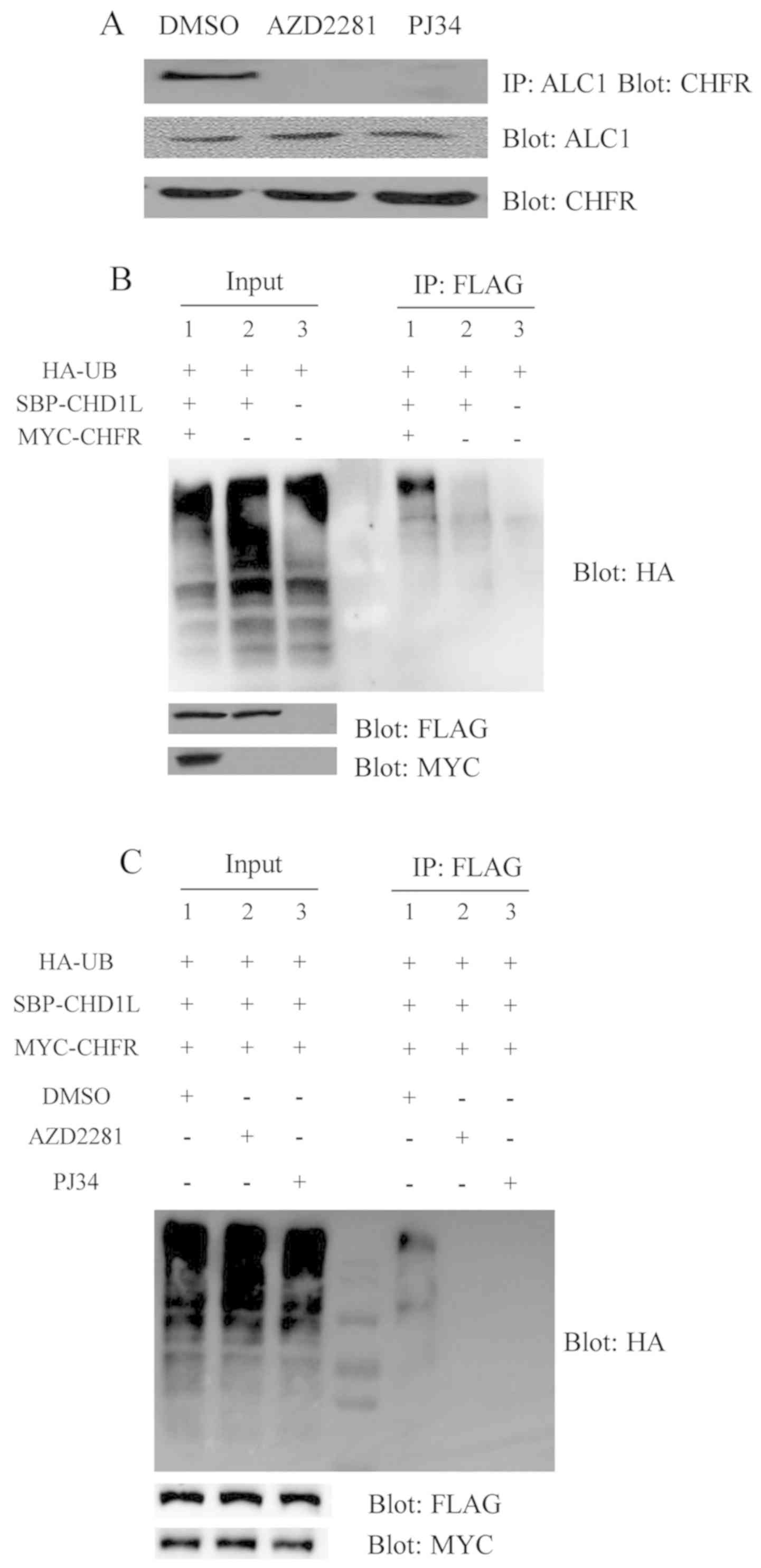
Ubiquitination of ALC1 by CHFR is
dependent on PARylation
We sought to determine whether PAR is crucial for
the interaction of ALC1 and CHFR. The results showed that the
endogenous ALC1 protein cannot bind to CHFR with PARP inhibitors
which disrupt the PAR interaction. However, the endogenous ALC1
proteins were bound to CHFR without PARP inhibitors, suggesting
that PARP1/2 inhibitors influence the interaction between ALC1 and
CHFR proteins (Fig. 3A). PARP
inhibitors were found to abolish the PAR-dependent recruitment of
CHFR and ALC1 to the DSB sites. These data indicated that PAR
deficiency could suppress the interaction between CHFR and ALC1.
Yet, it was not ascertained whether CHFR regulates the level of
ALC1 protein. The CHFR contains a RING domain, which has the
function of UB ligase E3. Since the E3 enzymes play the most
important role in the ubiquitination reaction, we hypothesized that
UB may be able to bind to the ALC1 through CHFR. ALC1 is
ubiquitinated and degraded by proteasome through the
ubiquitin-proteasome pathway, thereby expression of ALC1 is
reduced. Based on this hypothesis, experiments were designed.
First, to verify whether there is ubiquitination, three groups were
used. HA-UB was transfected in the first group, HA-UB and SBP-ALC1
were transfected in the second group, HA-UB, SBP-ALC1 and MYC-CHFR
were transfected in the third group. The experiments were performed
using 293T cells, respectively. After 24 h of transfection, MG132
at a concentration of 10 µM was added to inhibit proteolysis and
the cells were harvested after 4 h. The cells were lysed with a
NETN lysate containing 10 µM MG132. The transferred PVDF membranes
were incubated with HA, FLAG and MYC antibodies to evaluate the
transfection effect. The results showed that UB bands were
significantly attenuated in the third panel compared with the other
groups in 293T cells. The results suggest that ALC1 can be
ubiquitinated mediated by CHFR (Fig.
3B). Downregulation of the level of ALC1 protein is related to
the ubiquitination of itself. It is not known whether the
regulation is affected by PARP inhibitors. To verify this issue,
the following experiment was performed. HA-UB, SBP-ALC1 and
MYC-CHFR plasmids were all transfected into the three groups of
293T cells at the same time. To one group AZD2281was added, PJ34
was added to another group, and DMSO was added to the control
group. After 24 h of transfection, MG132 was added at the
concentration of 10 µM to inhibit proteolysis, and the cells were
harvested after 4 h. UB bands were observed in the control group,
but not in the group with the addition of PARP1/2 inhibitors
(Fig. 3C). These results suggested
that PARP1/2 inhibitors can inhibit the ubiquitination of ALC1.
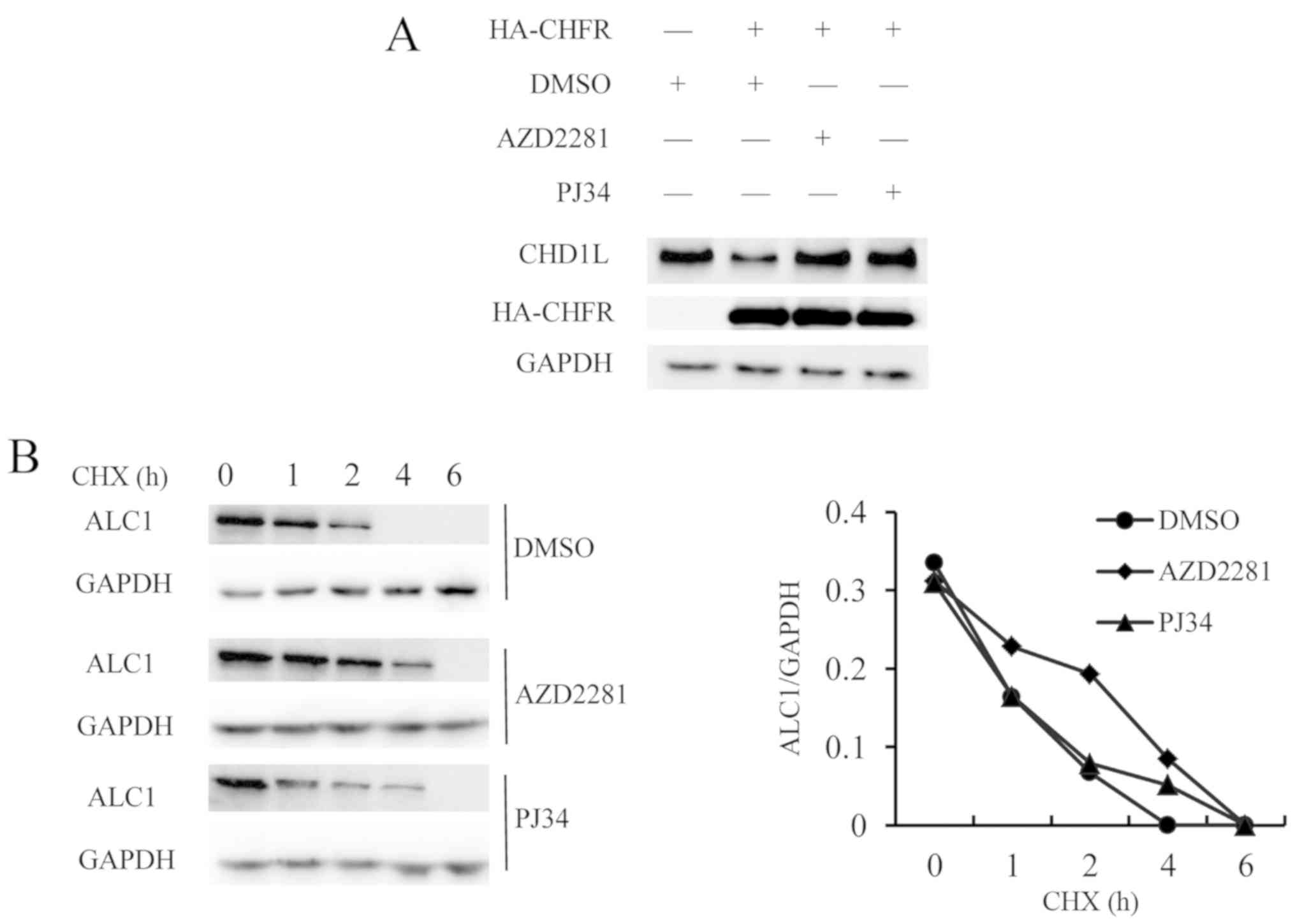
CHFR regulates the stability of
ALC1
As a result of ubiquitination of ALC1 mediated by
CHFR, the level of ALC1 protein may be decreased. The plasmids
HA-CHFR-WT, HA-CHFR-D-RING and HA-CHFR-D-PBZ were transfected into
293T cells to express CHFR proteins respectively. The expression of
CHFR was detected by HA antibody and the expression of ALC1 protein
was detected by ALC1 antibody, and GAPDH was used as internal
reference. The expression of the endogenous ALC1 protein was
decreased in cells with CHFR overexpression. To investigate the
effect of PARP inhibitors on ubiquitination of ALC1, AZD22811 and
PJ34 were added to the cells transfected with HA-CHFR. In addition,
untransfected cells were used as negative controls, and transfected
cells treated with DMSO were used as positive controls. The results
showed that the expression of endogenous ALC1 was not decreased
with the addition of PARP inhibitors, suggesting that PARP
inhibitor could inhibit the ubiquitination of ALC1 mediated by CHFR
(Fig. 4A).
To compare the expression of ALC1 protein in the
presence and absence of PARP1/2 inhibitors, the effect of PARP1/2
inhibitors on ALC1 half-life was examined. The MCF-7 cells were
cultured for 24 h, and then the culture media were replaced with
the addition of PARP1/2 inhibitors, AZD2281, PJ34 or DMSO at a
concentration of 10 µM. After 10 h, CHX was added at a final
concentration of 100 µM. At 0, 1, 2, 4 and 6 h, the cells were
harvested. The results showed that the PARP1/2 inhibitors extended
the half-life of ALC1, indicating that PARP1/2 inhibitors increased
the expression of ALC1 (Fig. 4B).
Discussion
Local chromatin relaxation at DNA damage sites is
regulated by PARP1 enzymatic activity which is one of the earliest
cellular responses to DNA damage (22). PARP1/2 inhibitors lead to the
inhibition of PARylation with various oncogenic proteins to inhibit
the DNA repair pathway (23). Yet
PARP inhibitor resistance is also a growing concern in the clinical
setting. The most widely accepted mechanism of PARP1/2 inhibitor
resistance is the restoration of the HR pathway through secondary
reversion mutations (24). Moreover,
a previous study showed that acquisition of PARP1/2 inhibitors and
cisplatin resistance is associated with replication fork protection
in BRCA2-deficient tumor cells (25).
Our study revealed a new mechanism. ALC1, a poly(ADP-ribose)- and
ATP-dependent remodeler, is involved in the chromatin-relaxation
process regulated by PARP1 (8).
Moreover, ALC1 is an oncogene located at Chr1q21 and it is
amplified in many solid tumors (26).
ALC1 is highly expressed in breast cancer tissues, and high
expression of ALC1 protein suggests poor prognosis (13). The relationship among drug resistance,
high expression of ALC1 and PARP inhibition remains unclear.
Furthermore, the mRNA of ALC1 is not regulated in breast cancer
tissues. The results indicated that there was no significant
difference in the transcription level of ALC1 between breast cancer
tissues and adjacent non-cancerous tissues. It was speculated that
epigenetic modifiers regulate ALC1 expression. Then we further
analyzed the associated proteins interacting with ALC1. CHFR was
found to bind with ALC1 by mass spectrometry. CHFR functions as an
E3 Ub-ligase of associated proteins and is responsible for its
proteasome degradation (27). The
results indicate that CHFR may play a crucial role in the
regulation of ALC1.
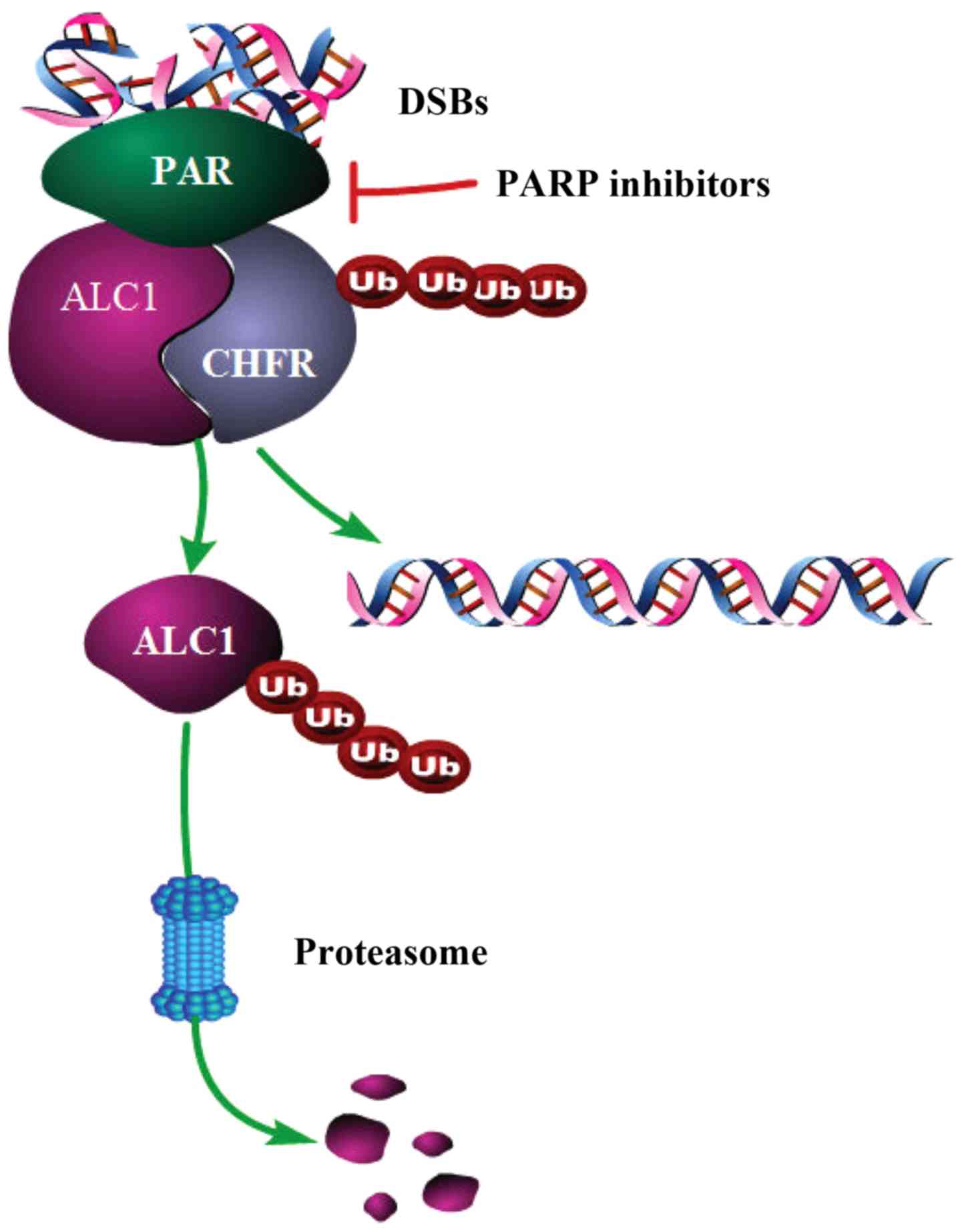
Based on the results, we speculated how the ALC1
protein interacts with the CHFR protein and a schematic
representation of the interaction is shown (Fig. 5). First, PARP1/2 binds rapidly to the
site of DNA double-strand breaks (DSBs) and undergoes PARylation
when the DNA is damaged. Next, ALC1 and CHFR are recruited to the
site of DNA damage. The macro domain of ALC1 binds to PAR, and then
CHFR can recognize ALC1 through the PBZ domain. It was found that
CHFR could mediate the ubiquitination of ALC1 and affect the
stability of ALC1 protein, which could lead to the degradation of
ALC1. The presence of CHFR can keep ALC1 from playing a role only
in the DNA damage response, but not in mediating malignant
biological behavior. In the treatment of breast cancer, the use of
PARP inhibitors may inhibit PARylation. Then ALC1 and CHFR would
not be recruited together at the sites of DSBs. Thus,
ubiquitination of ALC1 would be reduced and accumulation of ALC1
would be higher than before. Once ALC1 protein is highly expressed,
its potential malignant biological behavior may affect the
prognosis of the patient. For most BRCA1/2-deficient patients, high
expression of ALC1 has no impact on the therapeutic effect of
PARP1/2 inhibitors. However, for patients who exhibit resistance to
PARP1/2 inhibitors, high ALC1 should not be ignored. This study
also elucidated the reason why the expression of ALC1 protein is
high in breast cancer, but the mRNA level is normal. This is a type
of epigenetic modification. In brief, PARP1/2 inhibitors may turn
on another tumor proliferation pathway while shutting down DNA
damage repair, which may be one of the causes for the failure of
the efficacy of PARP inhibitors. ALC1 is recruited by PARP1/2 at
DNA damage sites, and degraded by CHFR. However, PARP1/2 inhibitors
can reverse the degradation of ALC1 by CHFR. High ALC1 accumulation
contributes to development of cancer. In other words, PARP1/2
inhibitors, DNA damage drugs combined with anti-ALC1 drugs may help
to improve the therapeutic effect for breast cancer patients. This
should be considered when choosing therapeutic strategies for
patients. By inhibiting ALC1 expression, PARP1 inhibitors can be
effective for more breast cancer patients that exhibit potential
drug resistance. However, one limitation of the study is that
PARP1/2 gene knockout cell lines were not constructed, which can
validate the expression of ALC1 without PARP1/2.
To conclude, in the present study, we demonstrated
that ALC1 can interact with CHFR depending on PAR. Ubiquitination
mediated by CHFR functioning as an E3 ubiquitin ligase results in
the degradation of ALC1. PARP1/2 inhibitors decrease the
ubiquitination of ALC1 and lead to the accumulation of ALC1, which
affects the therapeutic effects of DNA damage response drugs in
breast cancer treatment.
Acknowledgements
We thank Yongjun Dang of the School of Basic Medical
Science, Fudan University for the fruitful scientific discussion
and the sharing of various reagents.
Funding
This study was supported by the National Natural
Science Foundation of China (81572591).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YaW and JW conceived and designed the experiments.
YinW and YirW performed the experiments. YirW, YinW and FL
collected and analyzed the data. LY and CS contributed to
acquisition of the reagents/materials/analysis tools and
interpretation of data for the study. YirW and NW wrote the
manuscript and JW and NW revised the manuscript. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Board of the Second Military Medical University (Shanghai, China).
All participants gave informed consent before they entered the
study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PARP
|
poly(ADP-ribose) polymerase
|
|
CHFR
|
checkpoint with forehead-associated
and RING finger domains
|
|
ALC1
|
amplified in liver cancer protein
1
|
|
CHD1L
|
chromodomain-helicase-DNA-binding
protein 1-like
|
|
UB
|
ubiquitin
|
|
PAR
|
poly-ADP-ribose
|
|
CHX
|
cycloheximide
|
|
BRCA
|
breast cancer susceptibility gene
|
|
PBS
|
phosphate-buffered saline
|
|
PVDF
|
polyvinylidene fluoride
|
|
DMSO
|
dimethyl sulphoxide
|
|
ATP
|
adenosine triphosphate
|
|
PMD
|
PAR modification domain
|
|
FHA
|
filamentous hemagglutinin
|
|
PBZ
|
poly(ADP-ribose)-binding zinc
finger
|
|
DSB
|
double strand break
|
|
HR
|
homologous recombination
|
References
|
1
|
Kolb AL, Gunn AR and Lakin ND: Redundancy
between nucleases required for homologous recombination promotes
PARP inhibitor resistance in the eukaryotic model organism
Dictyostelium. Nucleic Acids Res. 45:10056–10067. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sie AS, Spruijt L, van Zelst-Stams WA,
Mensenkamp AR, Ligtenberg MJ, Brunner HG, Prins JB and Hoogerbrugge
N: High satisfaction and low distress in breast cancer patients one
year after BRCA-mutation testing without prior Face-to-Face genetic
counseling. J Genet Couns. 25:504–514. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Drew Y, Ledermann J, Hall G, Rea D,
Glasspool R, Highley M, Jayson G, Sludden J, Murray J, Jamieson D,
et al: Phase 2 multicentre trial investigating intermittent and
continuous dosing schedules of the poly(ADP-ribose) polymerase
inhibitor rucaparib in germline BRCA mutation carriers with
advanced ovarian and breast cancer. Br J Cancer. 114:723–730. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kulkarni A, Oza J, Yao M, Sohail H,
Ginjala V, Tomas-Loba A, Horejsi Z, Tan AR, Boulton SJ and Ganesan
S: Tripartite Motif-containing 33 (TRIM33) protein functions in the
poly(ADP-ribose) polymerase (PARP)-dependent DNA damage response
through interaction with amplified in liver cancer 1 (ALC1)
protein. J Biol Chem. 288:32357–32369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rugo HS, Olopade OI, DeMichele A, Yau C,
van't Veer LJ, Buxton MB, Hogarth M, Hylton NM, Paoloni M,
Perlmutter J, et al: Adaptive randomization of
veliparib-carboplatin treatment in breast cancer. N Engl J Med.
375:23–34. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mirza MR, Monk BJ, Herrstedt J, Oza AM,
Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I,
et al: Niraparib maintenance therapy in Platinum-sensitive,
recurrent ovarian cancer. N Engl J Med. 375:2154–2164. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sellou H, Lebeaupin T, Chapuis C, Smith R,
Hegele A, Singh HR, Kozlowski M, Bultmann S, Ladurner AG, Timinszky
G, et al: The poly(ADP-ribose)-dependent chromatin remodeler Alc1
induces local chromatin relaxation upon DNA damage. Mol Biol Cell.
27:3791–3799. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ahel D, Horejsi Z, Wiechens N, Polo SE,
Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, et
al: Poly(ADP-ribose)-dependent regulation of DNA repair by the
chromatin remodeling enzyme ALC1. Science. 325:1240–1243. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu J, Zong Y, Fei X, Chen X, Huang O, He
J, Chen W, Li Y, Shen K and Zhu L: Presence of CHD1L
over-expression is associated with aggressive tumor biology and is
a novel prognostic biomarker for patient survival in human breast
cancer. PLoS One. 9:e986732014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu X, He Y, Miao X, Wu Y, Han J, Wang Q,
Liu J, Zhong F, Ou Y, Wang Y and He S: Cell adhesion induces
overexpression of chromodomain helicase/ATPase DNA binding protein
1-like gene (CHD1L) and contributes to cell adhesion-mediated drug
resistance (CAM-DR) in multiple myeloma cells. Leuk Res. 47:54–62.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He LR, Ma NF, Chen JW, Li BK, Guan XY, Liu
MZ and Xie D: Overexpression of CHD1L is positively associated with
metastasis of lung adenocarcinoma and predicts patients poor
survival. Oncotarget. 6:31181–31190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mu QJ, Li HL, Yao Y, Liu SC, Yin CG and Ma
XZ: Chromodomain Helicase/ATPase DNA-binding protein 1-Like Gene
(CHD1L) expression and implications for invasion and metastasis of
breast cancer. PLoS One. 10:e01430302015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsuda M, Cho K, Ooka M, Shimizu N,
Watanabe R, Yasui A, Nakazawa Y, Ogi T, Harada H, Agama K, et al:
ALC1/CHD1L, a chromatin-remodeling enzyme, is required for
efficient base excision repair. PLoS One. 12:e01883202017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang BH, Chen WY, Li HY, Chien Y, Chang
WC, Hsieh PC, Wu P, Chen CY, Song HY, Chien CS, et al: CHD1L
regulated PARP1-driven pluripotency and chromatin remodeling during
the early-stage cell reprogramming. Stem Cells. 33:2961–2972. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lehmann LC, Hewitt G, Aibara S, Leitner A,
Marklund E, Maslen SL, Maturi V, Chen Y, van der Spoel D, Skehel
JM, et al: Mechanistic insights into autoinhibition of the
oncogenic chromatin remodeler ALC1. Mol Cell. 68:847–859.e7. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun Z, Liu J, Jing H, Dong SX and Wu J:
The diagnostic and prognostic value of CHFR hypermethylation in
colorectal cancer, a meta-analysis and literature review.
Oncotarget. 8:89142–89148. 2017.PubMed/NCBI
|
|
18
|
Privette LM, Gonzalez ME, Ding L, Kleer CG
and Petty EM: Altered expression of the early mitotic checkpoint
protein, CHFR, in breast cancers: Implications for tumor
suppression. Cancer Res. 67:6064–6074. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kashima L, Idogawa M, Mita H, Shitashige
M, Yamada T, Ogi K, Suzuki H, Toyota M, Ariga H, Sasaki Y and
Tokino T: CHFR protein regulates mitotic checkpoint by targeting
PARP-1 protein for ubiquitination and degradation. J Biol Chem.
287:12975–12984. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brodie SA, Li G, Donald H, Khuri FR,
Vertino PM and Brandes JC: Small molecule inhibition of the
CHFR-PARP1 interaction as novel approach to overcome intrinsic
taxane resistance in cancer. Oncotarget. 6:30773–30786. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lim JS and Tan DS: Understanding
resistance mechanisms and expanding the therapeutic utility of PARP
inhibitors. Cancers (Basel). 9:E1092017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Luijsterburg MS, de Krijger I, Wiegant WW,
Shah RG, Smeenk G, de Groot AJ, Pines A, Vertegaal AC, Jacobs JJ,
Shah GM and van Attikum H: PARP1 Links CHD2-mediated chromatin
expansion and H3.3 deposition to DNA Repair by Non-homologous
End-Joining. Molecular Cell. 61:547–562. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rajawat J, Shukla N and Mishra DP:
Therapeutic targeting of Poly(ADP-Ribose) Polymerase-1 (PARP1) in
cancer: Current developments, therapeutic strategies, and future
opportunities. Med Res Rev. 37:1461–1491. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barber LJ, Sandhu S, Chen L, Campbell J,
Kozarewa I, Fenwick K, Assiotis I, Rodrigues DN, Reis Filho JS,
Moreno V, et al: Secondary mutations in BRCA2 associated with
clinical resistance to a PARP inhibitor. J Pathol. 229:422–429.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang YT, Yuan B, Chen HD, Xu L, Tian YN,
Zhang A, He JX and Miao ZH: Acquired resistance of PTEN-deficient
cells to PARP inhibitor and Ara-C mediated by 53BP1 loss and SAMHD1
overexpression. Cancer Sci. 109:821–831. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Singh HR, Nardozza AP, Möller IR, Knobloch
G, Kistemaker HA, Hassler M, Harrer N, Blessing C, Eustermann S,
Kotthoff C, et al: A Poly-ADP-Ribose trigger releases the
Auto-inhibition of a chromatin remodeling oncogene. Mol Cell.
68:860–871.e7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim M, Kwon YE, Song JO, Bae SJ and Seol
JH: CHFR negatively regulates SIRT1 activity upon oxidative stress.
Sci Rep. 6:375782016. View Article : Google Scholar : PubMed/NCBI
|