Introduction
Glioblastoma is the deadliest primary central
nervous system tumor and has a prevalence of 3.19% in the United
States (1). In the past 15 years,
treatment of glioblastoma has included maximal safe surgical
resection combined with radiotherapy and temozolomide chemotherapy
(2). Despite this, the overall
five-year survival is still less than 5%, with an average survival
of just 14 months following initial diagnosis (3). Immunotherapy is an alternative approach
which has the potential to overcome the limitations of the current
standard therapies. In the past five years, chimeric antigen
receptor (CAR)-T immunotherapy has had substantial success in the
treatment of chronic lymphocytic leukemia and acute lymphoblastic
leukemia patients who have relapsed from chemotherapy treatment
(4–6).
The CAR-T immunotherapy method uses genetically
engineered T cells to express CAR. First generation CAR is composed
of an antigen recognition domain (scFv) and the essential
activating signal CD3ζ, while second generation CAR includes one
co-stimulatory molecule and the third generation includes two
co-stimulatory molecules (7). The
antigen recognition domain confers a specificity of CAR-T cells for
tumor-associated antigens. Thus, finding a suitable tumor antigen
is important for the use of CAR-T cells to treat glioblastoma.
Human epidermal growth factor receptor 2 (HER2), a member of the
EGFR family, encodes a 185-kDa transmembrane protein via tyrosine
protein kinase activity. The gene amplification and protein
overexpression of HER2 in human tumors have been associated with
more aggressive cancers (8,9). The HER2 antigen is overexpressed in
approximately 25–30% of patients living with breast cancer, 30–40%
of patients with primary renal cell carcinoma and 30–35% of
patients with lung adenocarcinoma (10–12). HER2
expression is absent in the adult central nervous system (13). CAR-T cells targeting HER2 have been
demonstrated to be safe and effective in clinical trials (14,15).
According to previous studies, HER2 is expressed in
as many as 80% of glioblastoma cases (16,17).
Furthermore, overexpression of HER2 has been revealed to be
correlated with the malignancy of glioma (18) and with high-grade glioma (19) as well as with early mortality
(9). These data indicate that HER2 is
an attractive therapeutic target for glioblastoma. In the
preclinical model of glioblastoma, CAR-T cells being studied for
glioma-targeted antigens IL-13Rα2, EphA2, HER2 (using a second
generation anti-HER2 CAR) and EGFRvIII (20) demonstrated strong anti-tumor activity.
In addition to careful selection of the tumor-associated antigen,
the optimal design of the CAR architecture (such as using a new
generation anti-HER2 CAR) is an important prerequisite for
achieving significant responses in CAR-mediated immunotherapy.
While CAR-T cells have exhibited significant
responses in intractable hematological malignancies, they have low
therapeutic effects for solid tumors. This may be due to the many
obstacles in the tumor microenvironment of solid tumors (21–26), such
as the intrinsic inhibition pathway mediated by T cell surface
inhibitory receptors binding to their tumor cell surface ligands
(26). One of the most frequently
studied T cell inhibitory receptors is programmed death-1
(PD1/CD279), which is upregulated following the engagement of T
cell receptor with its ligand. Currently, the known ligand of PD1
in various cancers is programmed cell death 1 ligand 1 (PDL1/CD274)
(27). However, PDL1 on the cell
surface of solid tumors is typically upregulated in response to
cytokines (such as IL-2 and IFN-γ) which have been secreted by T
cells, thus serving as tumor immune evasion (24).
In the present study, a third generation anti-HER2
CAR (anti-HER2 scFv-CD28-CD137-CD3ζ) was used, which we previously
designed and constructed, and which carried two co-stimulatory
molecules (CD28 and CD137). The third generation anti-HER2 CAR has
not been reported in clinical trials for treatment of malignant
glioblastoma. Lentivirus-mediated T cell transduction was utilized
to generate CAR-T cells. It was examined whether anti-HER2 CAR-T
cells alone have specific and efficient cytotoxicity, as well as
whether their combination with PD1 blockade enhances the
therapeutic activity against HER2+/PDL1+
glioblastoma cells in vitro. The use of anti-HER2 CAR-T
cells in combination with anti-PD1 antibody has not been reported
in clinical trials for malignant glioma.
Materials and methods
Cell lines and media
293T-17 cells [American Tissue Culture Collection
(ATCC)], human malignant glioblastoma cells U87 (glioblastoma of
unknown origin, HTB-14™) (ATCC) as well as U251 (China Center for
Type Culture Collection) were cultured in Dulbecco's Modified
Eagle's Medium (DMEM) (Gibco; Thermo Fisher Scientific, Inc.) and
supplemented with 10% fetal bovine serum (FBS) (both from Gibco;
Thermo Fisher Scientific, Inc.). The U251 cell line was
authenticated using STR analysis (Cell Bank, Type Culture
Collection, Chinese Academy of Sciences).
Construction of anti-HER2 CAR and
production of recombinant lentivirus
The third generation anti-HER2 CAR was developed by
our research group and synthesized by the Beijing Genomics
Institute in China. The recombinant lentivirus was produced using
293T-17 cells co-transfected with recombinant lentiviral vector
pLVX-EF1α-CAR-IRES-ZsGreen1 as well as packaging plasmids psPAX2
and pMD2.G. Packaging, concentration and purification of the
recombinant lentivirus were performed following protocols we had
optimized (28).
Preparation of human peripheral T
lymphocytes
Preparation of human peripheral T lymphocytes,
including isolation, activation, and purification, was performed
following the protocols we have previously described (28,29) but
with some further optimization. The present study was approved by
the Ethical Committee of Wenzhou Medical University (Wenzhou,
China). Peripheral blood materials used in this study were obtained
from healthy donors who provided informed consent. The total number
of healthy donors used was three (including one male and two
females), the age distribution was 24–28, collected from January
2017 to December 2018 at the First Affiliated Hospital of Wenzhou
Medical University. Peripheral blood mononuclear cells (PBMCs) were
isolated from heparinized peripheral blood by density-gradient
centrifugation (at 900 × g) utilizing a lymphocyte separation
medium (Sigma-Aldrich; Merck KGaA). PBMCs were activated
(stimulated) using anti-CD2-, anti-CD3- and anti-CD28-coated
microbeads (Miltenyi Biotec GmbH) at a 1:1 bead to cell ratio for
two days. Primary human peripheral T lymphocytes were cultured in
GT-T551 medium (Takara Bio, Inc.) which had been supplemented with
10% FBS and 300 IU/ml Interleukin-2 (IL-2) (PeproTech, Inc.).
Transduction and expansion of T
cells
The CD3+ human peripheral T cells were
transduced with recombinant lentivirus at a multiplicity of
infection (MOI) value of 20, as previously described (29). For transduction, the cell plate was
centrifuged at 1,200 × g for 2 h and cultured for 10–14 h, then
replaced with fresh medium and cultured for expansion. After
examining the proper GFP expression level (~40% GFP-positive cell
ratio) under fluorescence microscope (Nikon Eclipse Ti), the
transduced cells were subjected to flow cytometry and western
blotting to evaluate the GFP-positive cell percentage and CAR
expression level respectively.
Flow cytometric analysis
The expression of HER2 in glioblastoma cells U87 and
U251 was determined using APC anti-human CD340 (erbB2/HER2)
antibody (cat. no. 324407), with APC mouse IgG1κ (both from
BioLegend, Inc.) used as an isotype control. The expression of PDL1
in glioblastoma cells U87 and U251 was evaluated using PE
anti-human CD274 (PDL1) antibody (BD Biosciences), while PE mouse
IgG2aκ (BD Biosciences) was used as an isotype control.
Transduction efficiency to T cells was measured by the percentage
of transduced T cells expressing GFP (that is, CAR). Flow
cytometric data were analyzed using FlowJo v10 software (FlowJo
LLC).
Western blot analysis
Transduced human peripheral T cells were detected
for the expression level of CAR by western blot analysis. T cells
were lysed in modified RIPA lysis buffer [1% Triton X-100, 50 mM
Tris-HCl, pH 7.4, 1 mM EDTA, 150 mM NaCl, 0.25% Na-deoxycholate,
0.05% SDS, 10 mM NaF, 1 mM sodium vanadate, 1 mM
phenylmethylsulfonyl fluoride and protease inhibitor cocktail
(Sigma-Aldrich; Merck KGaA)]. The BCA assay kit (Beyotime Institute
of Biotechnology) was used to determine the protein concentration.
Lysates (10 µg per lane) were resolved by 10% SDS-PAGE, and
transferred to polyvinylidene difluoride (PVDF) membranes (EMD
Millipore). The membranes were blocked in TBST containing 5%
non-fat dry milk, immunoblotted with anti-human CD3ζ antibody (cat.
no. ab226475; dilution 1:1,000; Abcam) and anti-rabbit
HRP-conjugated secondary antibody (cat. no. sc-2004; dilution
1:5,000; Santa Cruz Biotechnology, Inc.) and then developed
utilizing ECL reagent (Beyotime Institute of Biotechnology). Light
emission was detected using the ChemiDoc MP Imaging System (Bio-Rad
Laboratories, Inc.). Expression levels of CAR (containing exogenous
CD3ζ) were detected, using endogenous CD3ζ as a loading
control.
Examination of the targeting ability
of CAR-T cells
In order to examine the targeting of anti-HER2 CAR-T
cells on HER2-positive glioblastoma cells in vitro,
HER2-positive U251cells (or HER2-negative control U87cells) were
seeded in a 96-well plate at 1×104 cells/well for 6 h.
Anti-HER2 CAR-T cells were then added at an E:T (effector to
target) ratio of 4:1 to co-culture for 24 h. The binding ability of
transduced T cells to target cells as well as the status of target
cells were observed using a fluorescence microscope.
Detection of cytokine secretion of
CAR-T cells
Anti-HER2 CAR-T cells (effector cells) (or in
combination with 20 µg/ml anti-PD1 antibody) were co-cultured with
HER2+ glioblastoma cells U251 (target cells) at an E:T
ratio of 4:1, for 24, 48 or 72 h in a 96-well plate. Anti-PD1
antibody (clone RMP1-14) was obtained from Bio X Cell. ELISA kits
(R&D Systems, Inc.) for detecting IL-2 and IFN-γ were utilized
to analyze supernatants of cells according to the manufacturer's
instructions. Supernatants from co-culture of anti-HER2 CAR-T cells
with HER2− glioblastoma cells U87 as well as from
co-culture of (untransduced) blank T cells (or in combination with
anti-PD1 antibody) with HER2+ glioblastoma cells U251
were the negative controls.
Cytotoxicity assay
Anti-HER2 CAR-T cells (effector cells) (or in
combination with 20 µg/ml anti-PD1 antibody) were co-cultured with
HER2+ glioblastoma cells U251 (target cells) at E:T
ratios of 2:1, 4:1, 8:1 and 16:1 for 18 h in a 96-well plate. The
co-cultures of anti-HER2 CAR-T cells with HER2−
glioblastoma cells U87 and the blank T cells (or in combination
with anti-PD1 antibody) with HER2+ glioblastoma cells
U251 were used as negative controls. Specific lactate dehydrogenase
(LDH) which had been released from target cells in cell-free
supernatant was detected using a cytotoxicity LDH detection kit
(Genmed) according to the manufacturer's instructions. The amount
of LDH released was used to assess the lysis of target cells, which
may be translated into the effectiveness of effector cells. Percent
cytotoxicity was calculated according to OD values utilizing the
following formula: Cytotoxicity (%)=(Experimental-Effector
spontaneous-Target spontaneous)/(Target maximum-Target spontaneous)
×100%.
Statistical analysis
Probability (P)-values were calculated with GraphPad
Prism 5.0 software (GraphPad Software). All experiments were
repeated at least three times. Group means were compared via
one-way analysis of variance/Newman-Keuls. Differences of P<0.05
were considered to indicate a statistically significant
difference.
Results
Expression of HER2 and PDL1 in
glioblastoma cells
In order to assess the efficacy of anti-HER2 CAR-T
cells in combination with anti-PD1 antibody against tumor cells, it
is crucial to determine the expression level of HER2 and PDL1 in
tumor cells. Flow cytometry was used to detect HER2 and PDL1
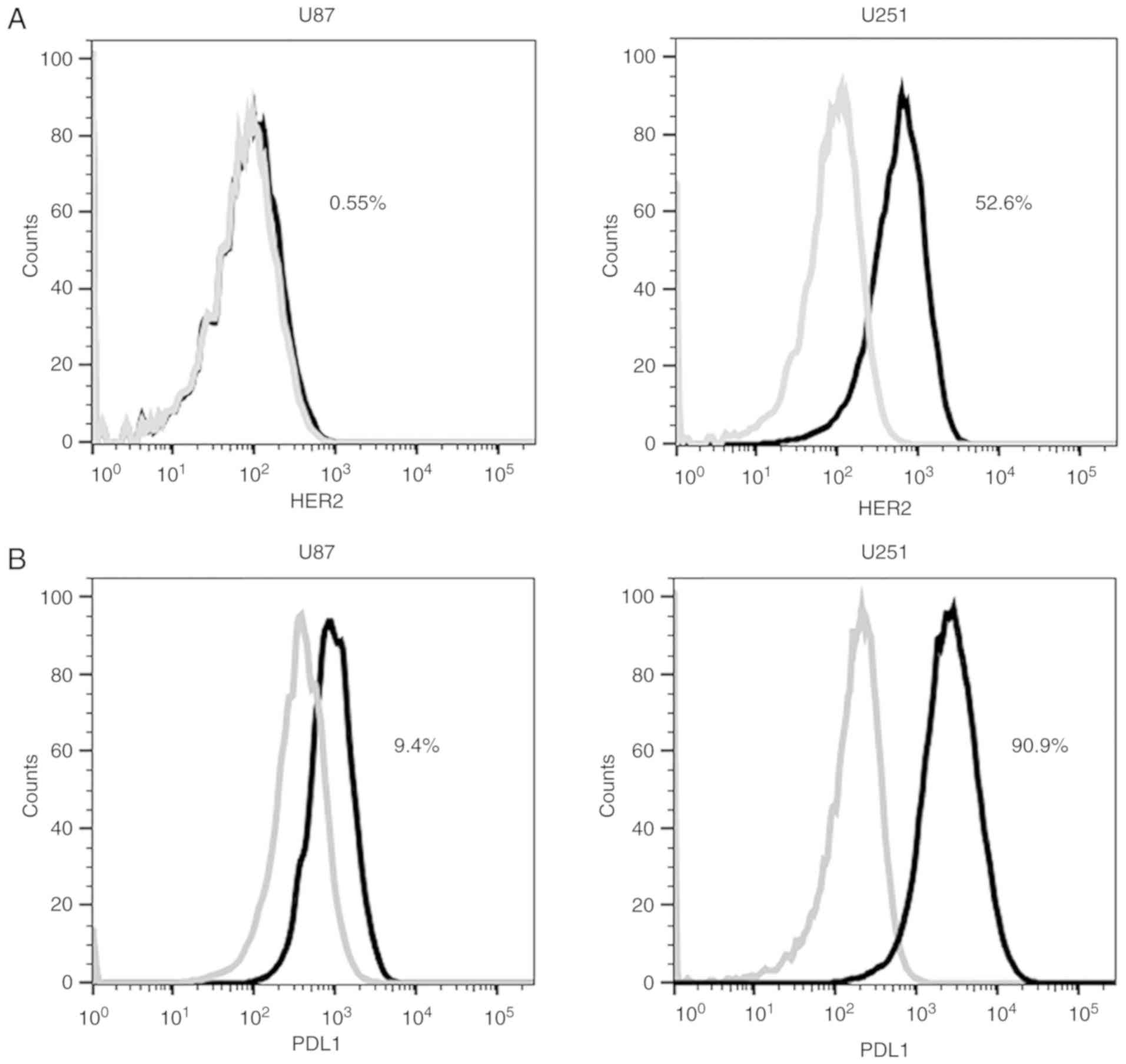
expression. The expression levels of HER2 in glioblastoma cells
U251 and U87 were 52.6 and 0.55% respectively (Fig. 1A). The expression levels of PDL1 in
glioblastoma cells U251 and U87 were 90.9 and 9.4% respectively
(Fig. 1B). Glioblastoma cell line
U251 was set as a HER2+/PDL1+ positive target
cell, while glioblastoma cell U87 was set as a
HER2−/PDL1− negative control cell.
Preparation of the third generation
anti-HER2 CAR-T cells
The structure of anti-HER2 CAR constructed by our
research group is presented in Fig.
2A. Human peripheral T cells were separated and activated. Once
human peripheral T cells had been transduced with recombinant
lentivirus at a multiplicity of infection (MOI) of 20 for five
days, they were observed under a fluorescence microscope in visible
light (Fig. 2B-a) and fluorescence
(Fig. 2B-b) to evaluate the
percentage of GFP-positive cells. Percentages of viable cells
(7-AAD negative cells) and GFP-positive cells (that is,
transduction efficiency) were analyzed using flow cytometry
(Fig. 2C). Transduction efficiency
was 38.2% (Fig. 2C). The expression
levels of CAR (containing exogenous CD3ζ) were detected utilizing
western blotting, using endogenous CD3ζ as a loading control. CAR-T
cells expressed not only endogenous CD3ζ (16 kDa) but also the
expected exogenous CD3ζ included in CAR (58 kDa) (Fig. 2D).
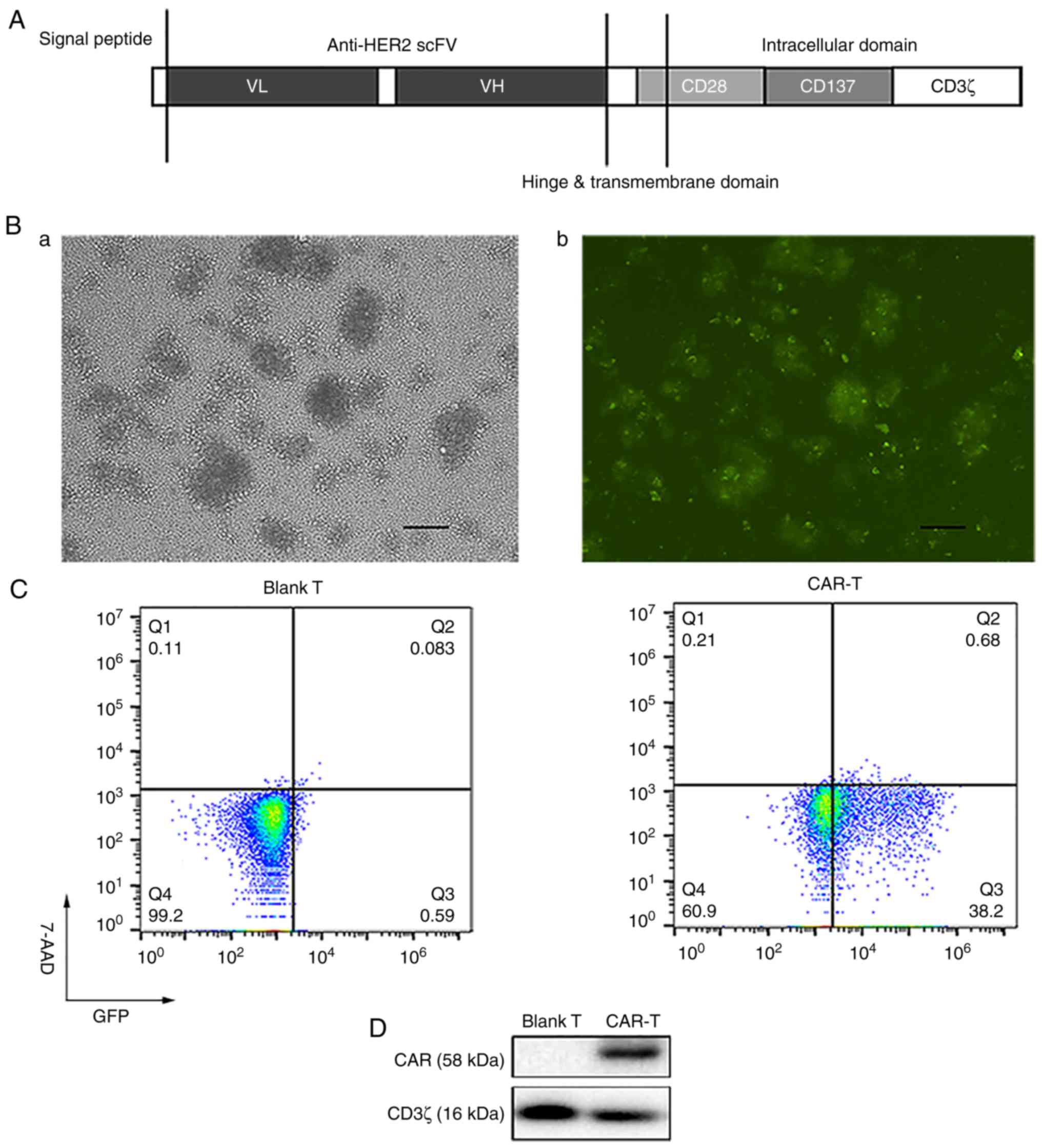 | Figure 2.Preparation of the third generation
anti-HER2 CAR-T cells. (A) Structure of anti-HER2 CAR. (B) After
human peripheral T cells were transduced with recombinant
lentivirus at MOI 20 for five days, T cells were observed under a
fluorescence microscope in (a) visible light and (b) fluorescence
to evaluate the percentage of GFP-positive cells (scale bar, 100
µm). (C) Percentages of viable cells (7-AAD negative cells) and
GFP-positive cells (i.e., transduction efficiency) were analyzed
using flow cytometry after the human peripheral T cells were
transduced with recombinant lentivirus at MOI 20 for five days. (D)
Recombinant lentivirus was transduced into human peripheral T
cells. Expression levels of CAR (containing exogenous CD3ζ) were
detected by western blotting, using endogenous CD3ζ as a loading
control. HER2, human epidermal growth factor 2; CAR, chimeric
antigen receptor; MOI, multiplicity of infection; scFv, single
chain variable fragment; VL, light chain variable region; VH, heavy
chain variable region; 7-AAD, 7-aminoactinomycin D; Blank T, T
cells not transduced; CAR-T, T cells transduced with CAR. |
Targeting of anti-HER2 CAR-T cells to
HER2-positive glioblastoma cells
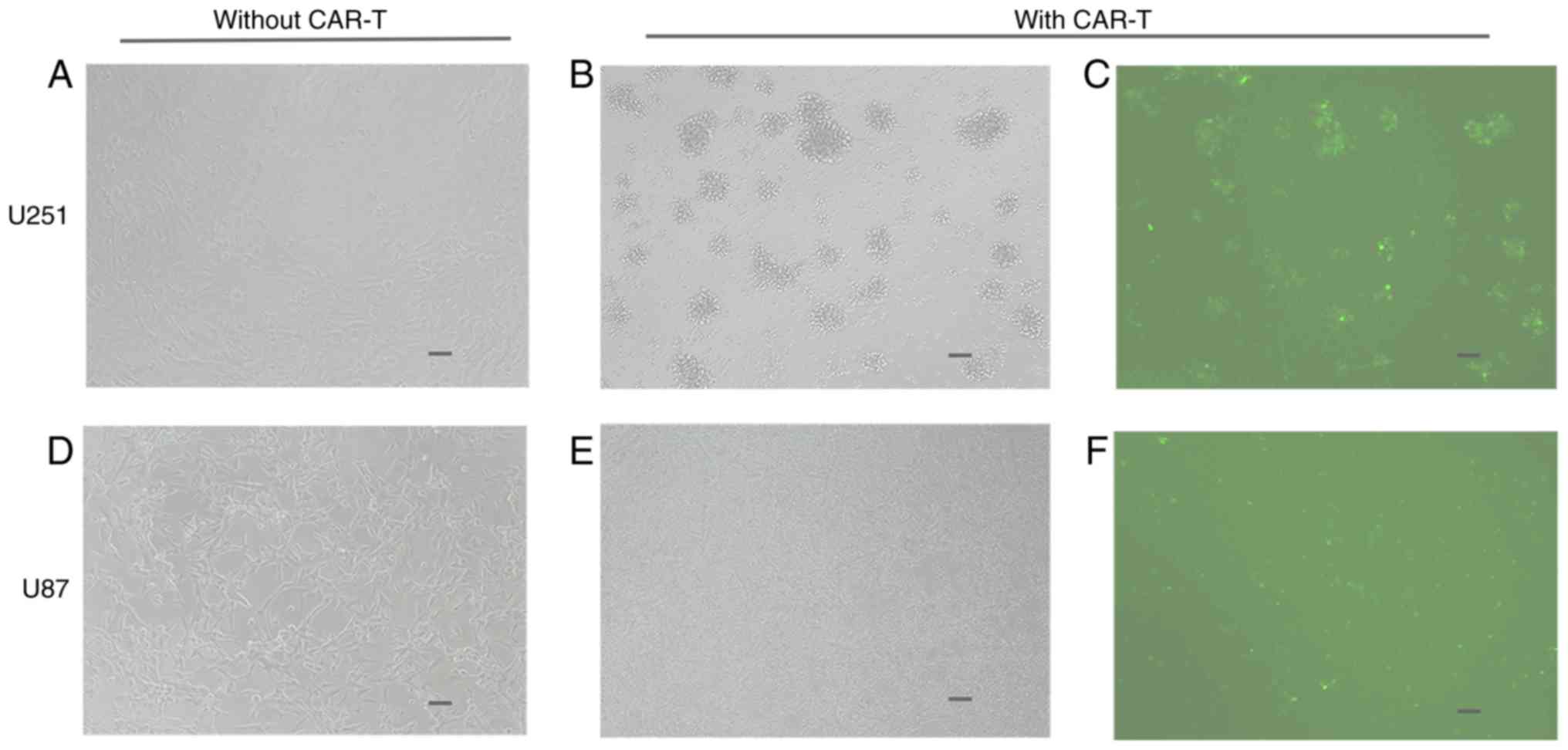
In order to examine the targeting of anti-HER2 scFv
on the anti-HER2 CAR-T cell surface, anti-HER2 CAR-T cells were
co-cultured with HER2-positive U251 cells for 24 h. The binding
ability of anti-HER2 CAR-T cells to U251 cells was examined under a
fluorescence microscope in both visible light and fluorescence.
CAR-T cells which had been co-cultured with U251 cells formed
clusters with U251 cells (Fig. 3C),
while the number of viable U251 cells decreased and seemingly
disappeared (Fig. 3B) when compared
to the number of viable U251 cells not co-cultured with CAR-T cells
(Fig. 3A). CAR-T cells did not
combine with U87 cells, while most U87 cells remained alive
(Fig. 3D-F).
Activation levels of anti-HER2 CAR-T
cells induced by HER2-positive glioblastoma cells
In order to determine whether anti-HER2 CAR-T cells
alone or in combination with anti-PD1 antibody were activated when
co-cultured with target cells, cytokine secretion of anti-HER2
CAR-T cells was detected. The results demonstrated that IL-2 or
IFN-γ secretion of the CAR-T group after being co-cultured with the
U251 cells for 24, 48 or 72 h significantly increased when compared
to the IL-2 or IFN-γ secretion of the blank T group which had been
co-cultured with U251 cells for the same time (P<0.01 or
P<0.001) (Fig. 4). Secreted IL-2
and IFN-γ from the CAR-T group, after being co-cultured with U251
cells for 24 h, increased 27-fold and 15-fold, respectively when
compared to those of the blank T group. Secreted IL-2 and IFN-γ
from the CAR-T group, after being co-cultured with U251 cells for
24 h, increased 5.4-fold and 12.5-fold respectively when compared
to those of the CAR-T group which had been co-cultured with U87
cells for 24 h. Notably, following the addition of anti-PD1
antibody, CAR-T cells could significantly increase production of
IL-2 and IFN-γ after being co-cultured with U251 cells for 72 h,
when compared to those of CAR-T cells alone (P<0.05 and
P<0.01) (Fig. 4). CAR-T cells in
combination with anti-PD1 antibody could significantly increase
production of IL-2 and IFN-γ after being co-cultured with U251
cells for 24, 48 or 72 h, when compared to those of blank T cells
in combination with anti-PD1 antibody (P<0.001) (Fig. 4).
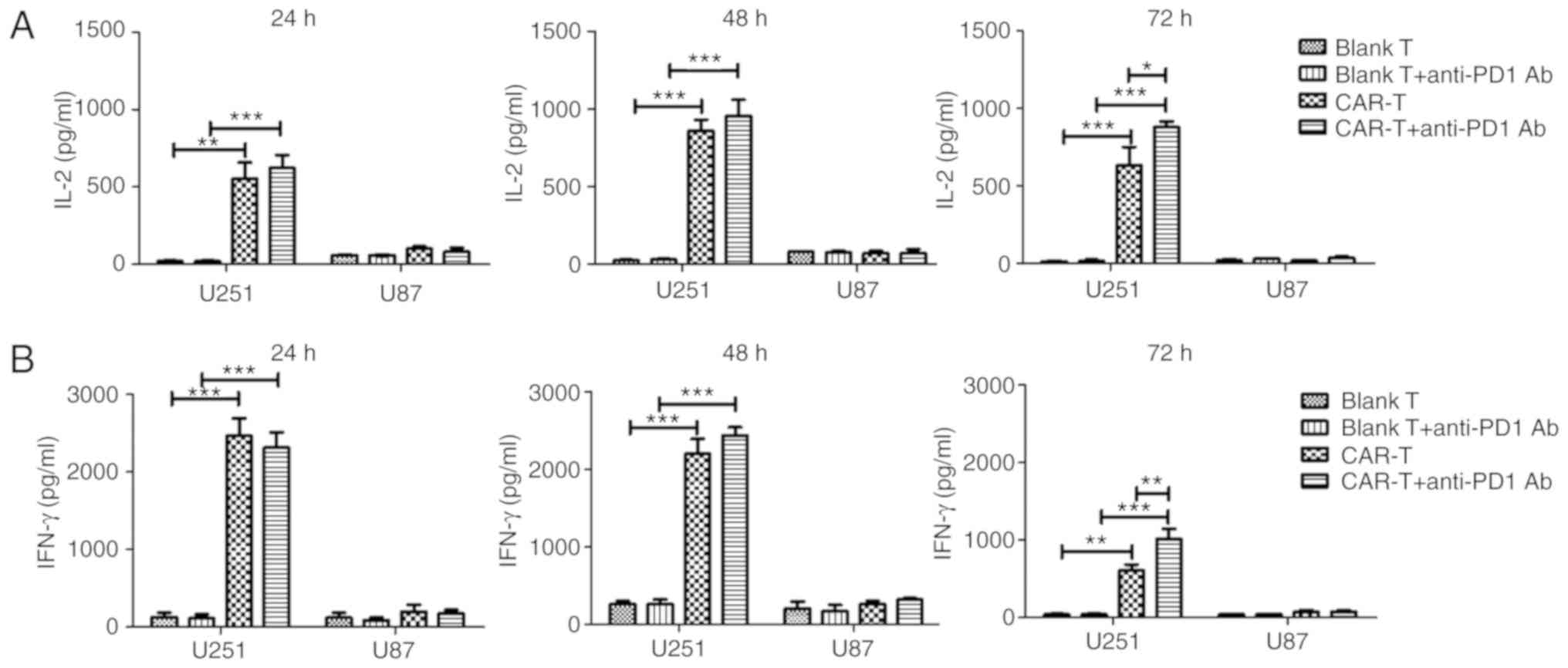 | Figure 4.Cytokine secretion of anti-HER2 CAR-T
cells. After anti-HER2 CAR-T cells (effector cells) (or in
combination with anti-PD1 antibody) were co-cultured with
HER2+ glioblastoma cells U251 (target cells) at an
effector to target ratio of 4:1 for 24, 48 or 72 h, supernatant was
collected. ELISA kits for detecting (A) IL-2 and (B) IFN-γ were
used to analyze supernatants. Supernatants from the co-culture of
anti-HER2 CAR-T cells with HER2- glioblastoma cells U87, as well as
from the co-culture of (untransduced) blank T cells (or in
combination with anti-PD1 antibody) with HER2+
glioblastoma cells U251 were used as negative controls. Three
determinations were performed and the mean values are presented.
For (A) IL-2 or (B) IFN-γ secretion, the CAR-T group was compared
with the blank T group, or the CAR-T in combination with anti-PD1
Ab group was compared with blank T in combination with anti-PD1 Ab
group, or the CAR-T in combination with anti-PD1 Ab group was
compared with the CAR-T group. *P<0.05; **P<0.01;
***P<0.001. HER2, human epidermal growth factor 2; CAR, chimeric
antigen receptor; CAR-T, T cells transduced with CAR; IL-2,
interleukin-2; IFN-γ, interferon-γ. |
Cytotoxicity of anti-HER2 CAR-T cells
alone or in combination with anti-PD1 antibody against
HER2-positive glioblastoma cells
In order to evaluate the efficacy of effector cells
alone or in combination with PD1 blockade on target cells in
vitro, anti-HER2 CAR-T cells alone or those in combination with
anti-PD1 antibody were co-cultured with HER2-positive glioblastoma
cells U251 at E:T ratios of 2:1, 4:1, 8:1 and 16:1 for 18 h. The
cytotoxicity (%) of CAR-T cells to lyse U251 cells was revealed to
be significantly higher than that of blank T cells at every E:T
ratio (P<0.05 and P<0.001) (Fig.
5A). Notably, the addition of anti-PD1 Ab significantly
increased cytotoxicity (%) of CAR-T cells against U251 cells at the
E:T ratios of 8:1 and 16:1 compared to that of CAR-T cells alone
(P<0.05 and P<0.01) (Fig. 5A).
CAR-T cells in combination with anti-PD1 antibody could
significantly increase cytotoxicity (%) against U251 cells at the
E:T ratios of 2:1, 4:1, 8:1 and 16:1, when compared to that of
blank T cells in combination with anti-PD1 antibody (P<0.05,
P<0.01 and P<0.001) (Fig. 5A).
As a negative control, anti-HER2 CAR-T cells were co-cultured with
HER2− glioblastoma cells U87 (Fig. 5B). At an E:T of 16:1, cytotoxicity (%)
of CAR-T cells against U251 cells reached 46.65%, while
cytotoxicity (%) against HER2-negtive U87 cells was much lower, at
just 15.74% (Fig. 5C) (P<0.01).
Furthermore, at the E:T ratio of 16:1, following the addition of
anti-PD1 Ab, cytotoxicity (%) of CAR-T cells against U251 cells
reached 63.08%, while cytotoxicity (%) against U87 cells remained
low at 17.76%, meaning the former was significantly higher
(Fig. 5C) (P<0.01).
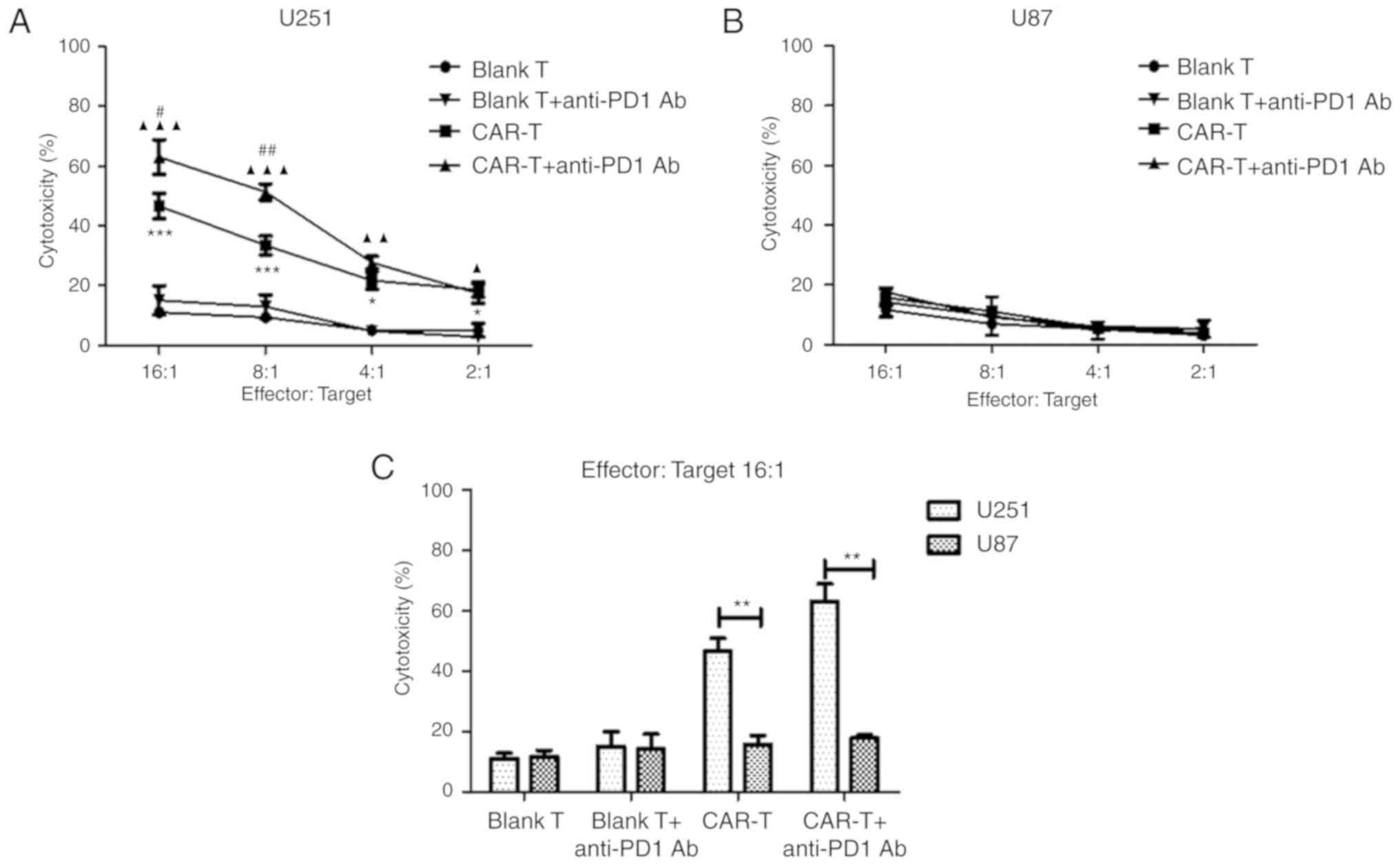 | Figure 5.The cytotoxicity of anti-HER2 CAR-T
cells (or in combination with anti-PD1 antibody) against
HER2-positive glioblastoma cells. (A) After anti-HER2 CAR-T cells
(effector cells) were co-cultured with HER2+
glioblastoma cells U251 (target cells) (or in combination with
anti-PD1 antibody) at different ratios of E:T for 18 h, supernatant
was detected using a cytotoxicity LDH detection kit for LDH
released from lysed target cells. Cytotoxicity (%) was calculated.
At E:T ratios of 2:1, 4:1, 8:1 or 16:1 respectively, the
cytotoxicity (%) of the CAR-T group was compared with the Blank T
group, *P<0.05, ***P<0.001; or the CAR-T in combination with
anti-PD1 Ab group was compared with blank T in combination with
anti-PD1 Ab group, ▲P<0.05, ▲▲P<0.01,
▲▲▲P<0.001; or the CAR-T in combination with anti-PD1
Ab group was compared with the CAR-T group, #P<0.05,
##P<0.01. (B) As a negative control, anti-HER2 CAR-T
cells were co-cultured with HER2- glioblastoma cells U87. (C) At
E:T ratio 16:1, the cytotoxicity (%) of the U251 group was compared
with the U87 group, **P<0.01. Three determinations were
performed and the mean values are presented. HER2, human epidermal
growth factor 2; CAR, chimeric antigen receptor; CAR-T, T cells
transduced with CAR; PD1, programmed death 1; E:T, effector to
target; LDH, lactate dehydrogenase. |
Discussion
The anti-HER2 CAR developed by our group contains
CD28-CD137-CD3ζ, which belongs to third-generation CAR (Fig. 2A). In a reported preclinical study for
glioblastoma, the anti-HER2 CAR utilized was second-generation
(20). Most CAR-T clinical trials
have used second-generation CAR for treating hematological
malignancies. To date, there has been no successful clinical trial
of anti-HER2 CAR-T cells which has utilized third-generation CAR
(https://www.clinicaltrials.gov). In the
present study, anti-HER2 CAR was transduced into T cells by
lentiviral vector. To date, there has also been no successful
clinical trial for anti-HER2 CAR-T cells which used lentiviral
vectors (https://www.clinicaltrials.gov).
The present study demonstrated that third generation
anti-HER2 CAR-T cells (with co-stimulatory molecules CD28 and
4-1BB/CD137) are able to eliminate HER2-positive malignant
glioblastoma cells both specifically and efficiently. The specialty
for binding of CAR to antigen can be affected by the affinity of
the selected scFv. The present study used the anti-HER2 scFv from
antibody 4D5 for CAR construction, and the CAR-T cells demonstrated
HER2-specific tumor cell lysis in vitro. A functionality
assay revealed that these redirected T cells could secret both
cytokine IL-2 and IFN-γ as well as exert efficient cytotoxic T cell
response in an HER2-specific manner. Major progress has been made
since the introduction of co-stimulatory signaling into the
architecture of CAR. Given the in-depth understanding of
co-stimulatory signaling in T cell immune response, several
co-stimulatory molecules (including CD28, 4-1BB/CD137, CD27, OX40)
were embedded in the CAR, following which their roles in
coordinating antitumor immunity were explored (30). When compared with other co-stimulatory
molecules, CD28 was more effective at enhancing IL-2 production,
improving clonal expansion and maintaining persistence of CAR-T
cells. CD28 used with 4-1BB/CD137 signaling is more effective than
CD28 alone in terms of cytotoxicity and IFN-γ production (31). The inclusion of the CD137
co-stimulatory molecule in CAR appears more key than CD28. Finney
et al demonstrated that CD137 in the CAR induced a maximal
effect on target cell lysis (32).
The present results revealed that the presence of
anti-PD1 antibody is critical for enhancing T cell response to
tumor cells. CAR-T cells, in combination with anti-PD1 antibody,
could release more IL-2 and IFN-γ after targeting tumor cells, and
were more efficient for eliminating tumor cells when compared with
CAR-T cells alone. The main function of PD1 is to reduce the
sensitivity of cancer patients to T cell-mediated anti-tumor immune
response. Blocking PD1 signaling can rescue exhausted T lymphocytes
and is an effective treatment for cancer. In recent years,
checkpoint inhibitors for the PD1/PDLl checkpoint pathway have
demonstrated significant antitumor effects in advanced melanoma,
classic Hodgkin's lymphoma, non-small cell lung cancer (NSCLC) and
other conditions (33–36). Research has suggested that CAR-T cell
therapy (including targeting CD19, PSMA) in combination with PD1
blockade is an ideal therapy combination for the treatment of solid
tumors (37,38).
Liu et al confirmed that treatment of mice
that have large, established solid tumors with PD1-CD28 CAR-T cells
led to a significant regression in tumor volume due to to enhanced
CAR tumor-infiltrating lymphocyte (TIL) infiltrate, decreased
susceptibility to tumor-induced hypofunction and attenuation of
inhibitory receptor expression when compared to treatments with
CAR-T cells alone or PD1 antibodies (39). Cherkassky et al have reported
that anti-MSLN CAR-T cells used in combination with PD1 blockade
could enhance the function of CAR-T cells in PDL1+ tumor
tissues, and may lead to a longer, tumor-free survival period for
cancer patients (40).
Future research will verify the in vivo
effect of third generation anti-HER2 CAR-T cells in combination
with anti-PD1 antibody via a glioblastoma animal model and thus
ascertain whether this therapy can prolong the survival of a
glioblastoma animal model or even achieve tumor-free survival.
In summary, the present study demonstrated that
third generation anti-HER2 CAR-T cells are able to eliminate
malignant glioblastoma cells specifically and efficiently. Blocking
PD1 immuno-suppression can increase activation of CAR-T cells after
activation by targeting antigens. This study revealed that, when
used in combination with anti-PD1 antibody, anti-HER2 CAR-T cells
have a greater therapeutic activity against
HER2+/PDL1+ malignant glioblastoma cells when
compared with anti-HER2 CAR-T cells alone.
Acknowledgments
Not applicable.
Funding
The present research was supported by grants from
the National Natural Science Foundation of China [grant no.
81572980] and the Wenzhou Science and Technology Bureau of China
[grant no. H20170001] awarded to HL, as well as from the National
Natural Science Foundation of China [grant no. 81772819] awarded to
HG.
Availability of data and materials
Datasets used and analyzed during the current study
are available from the corresponding authors on reasonable
request.
Authors' contributions
WY and LS undertook the research, analyzed the data
and wrote the paper. SB, PL and JC helped to perform the research
and experiments. HL and HG designed the research, wrote and revised
the paper. All of the authors read and approved the final
manuscript and agree to be for all aspects of the work in ensuring
that questions related to the accuracy or integrity of any part of
the work are appropriately investigated and resolved. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Wenzhou Medical University (Wenzhou, China).
Peripheral blood materials used in this study were obtained from
healthy donors who provided informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CAR
|
chimeric antigen receptor
|
|
PD1
|
programmed death-1
|
|
ATCC
|
American Tissue Culture Collection
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
PBMCs
|
peripheral blood mononuclear cells
|
|
IL-2
|
interleukin-2
|
|
IFN-γ
|
interferon-γ
|
|
MOI
|
multiplicity of infection
|
|
LDH
|
lactate dehydrogenase
|
|
7-AAD
|
7-aminoactinomycin D
|
|
TIL
|
tumor-infiltrating lymphocyte
|
References
|
1
|
Reardon DA and Mitchell DA: The
development of dendritic cell vaccine-based immunotherapies for
glioblastoma. Semin Immunopathol. 39:225–239. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohba S and Hirose Y: Current and Future
Drug Treatments for Glioblastomas. Curr Med Chem. 23:4309–4316.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Batash R, Asna N, Schaffer P, Francis N
and Schaffer M: Glioblastoma multiforme, diagnosis and treatment;
recent literature review. Curr Med Chem. 24:3002–3009. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hao L, Li T, Chang LJ and Chen X: Adoptive
immunotherapy for B-cell malignancies using CD19-targeted chimeric
antigen receptor T-cells: A systematic review of efficacy and
safety. Curr Med Chem. Aug 1–2017.(Epub ahead of print). doi:
10.2174/0929867324666170801101842. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jacoby E, Bielorai B, Avigdor A, Itzhaki
O, Hutt D, Nussboim V, Meir A, Kubi A, Levy M, Zikich D, et al:
Locally produced CD19 CAR T cells leading to clinical remissions in
medullary and extramedullary relapsed acute lymphoblastic leukemia.
Am J Hematol. 93:1485–1492. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang D, Shi R, Wang Q and Li J:
Extramedullary relapse of acute lymphoblastic leukemia after
allogeneic hematopoietic stem cell transplantation treated by CAR
T-cell therapy: A case report. Onco Targets Ther. 11:6327–6332.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jena B, Dotti G and Cooper LJ: Redirecting
T-cell specificity by introducing a tumor-specific chimeric antigen
receptor. Blood. 116:1035–1044. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Potti A, Forseen SE, Koka VK, Pervez H,
Koch M, Fraiman G, Mehdi SA and Levitt R: Determination of
HER-2/neu overexpression and clinical predictors of survival in a
cohort of 347 patients with primary malignant brain tumors. Cancer
Invest. 22:537–544. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Koka V, Potti A, Forseen SE, Pervez H,
Fraiman GN, Koch M and Levitt R: Role of Her-2/neu overexpression
and clinical determinants of early mortality in glioblastoma
multiforme. Am J Clin Oncol. 26:332–335. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seliger B, Rongcun Y, Atkins D, Hammers S,
Huber C, Storkel S and Kiessling R: HER-2/neu is expressed in human
renal cell carcinoma at heterogeneous levels independently of tumor
grading and staging and can be recognized by HLA-A2.1-restricted
cytotoxic T lymphocytes. Int J Cancer. 87:349–359. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang S, Saboorian MH, Frenkel E, Hynan L,
Gokaslan ST and Ashfaq R: Laboratory assessment of the status of
Her-2/neu protein and oncogene in breast cancer specimens:
Comparison of immunohistochemistry assay with fluorescence in situ
hybridisation assays. J Clin Pathol. 53:374–381. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li BT, Ross DS, Aisner DL, Chaft JE, Hsu
M, Kako SL, Kris MG, Varella-Garcia M and Arcila ME: HER2
amplification and HER2 mutation are distinct molecular targets in
lung cancers. J Thorac Oncol. 11:414–419. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Press MF, Cordon-Cardo C and Slamon DJ:
Expression of the HER-2/neu proto-oncogene in normal human adult
and fetal tissues. Oncogene. 5:953–962. 1990.PubMed/NCBI
|
|
14
|
Whilding LM and Maher J: ErbB-targeted CAR
T-cell immunotherapy of cancer. Immunotherapy. 7:229–241. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nowakowska P, Romanski A, Miller N,
Odendahl M, Bonig H, Zhang C, Seifried E, Wels WS and Tonn T:
Clinical grade manufacturing of genetically modified,
CAR-expressing NK-92 cells for the treatment of ErbB2-positive
malignancies. Cancer Immunol Immunother. 67:25–38. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu G, Ying H, Zeng G, Wheeler CJ, Black
KL and Yu JS: HER-2, gp100, and MAGE-1 are expressed in human
glioblastoma and recognized by cytotoxic T cells. Cancer Res.
64:4980–4986. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ahmed N, Salsman VS, Kew Y, Shaffer D,
Powell S, Zhang YJ, Grossman RG, Heslop HE and Gottschalk S:
HER2-specific T cells target primary glioblastoma stem cells and
induce regression of autologous experimental tumors. Clin Cancer
Res. 16:474–485. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thomas CY, Chouinard M, Cox M, Parsons S,
Stallings-Mann M, Garcia R, Jove R and Wharen R: Spontaneous
activation and signaling by overexpressed epidermal growth factor
receptors in glioblastoma cells. Int J Cancer. 104:19–27. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Andersson U, Guo D, Malmer B, Bergenheim
AT, Brannstrom T, Hedman H and Henriksson R: Epidermal growth
factor receptor family (EGFR, ErbB2-4) in gliomas and meningiomas.
Acta Neuropathol. 108:135–142. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuramitsu S, Yamamichi A, Ohka F, Motomura
K, Hara M and Natsume A: Adoptive immunotherapy for the treatment
of glioblastoma: Progress and possibilities. Immunotherapy.
8:1393–1404. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tlsty TD and Coussens LM: Tumor stroma and
regulation of cancer development. Annu Rev Pathol. 1:119–150. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Denko NC: Hypoxia, HIF1 and glucose
metabolism in the solid tumour. Nat Rev Cancer. 8:705–713. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zheng Y, Zha Y and Gajewski TF: Molecular
regulation of T-cell anergy. EMBO Rep. 9:50–55. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gajewski TF, Schreiber H and Fu YX: Innate
and adaptive immune cells in the tumor microenvironment. Nat
Immunol. 14:1014–1022. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Al-Zoughbi W, Huang J, Paramasivan GS,
Till H, Pichler M, Guertl-Lackner B and Hoefler G: Tumor
macroenvironment and metabolism. Semin Oncol. 41:281–295. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zou W and Chen L: Inhibitory B7-family
molecules in the tumour microenvironment. Nat Rev Immunol.
8:467–477. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Keir ME, Butte MJ, Freeman GJ and Sharpe
AH: PD-1 and its ligands in tolerance and immunity. Annu Rev
Immunol. 26:677–704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yuan W, Chen J, Cao Y, Yang L, Shen L,
Bian Q, Bin S, Li P, Cao J, Fang H, et al: Comparative analysis and
optimization of protocols for producing recombinant lentivirus
carrying the anti-Her2 chimeric antigen receptor gene. J Gene Med.
20:e30272018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang C, Hu W, Shen L, Dou R, Zhao S, Shan
D, Yu K, Huang R and Li H: Adoptive antitumor immunotherapy in
vitro and in vivo using genetically activated erbB2-specific T
cells. J Immunother. 37:351–359. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhong XS, Matsushita M, Plotkin J, Riviere
I and Sadelain M: Chimeric antigen receptors combining 4-1BB and
CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and
CD8+ T cell-mediated tumor eradication. Mol Ther.
18:413–420. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brentjens RJ, Santos E, Nikhamin Y, Yeh R,
Matsushita M, La Perle K, Quintas-Cardama A, Larson SM and Sadelain
M: Genetically targeted T cells eradicate systemic acute
lymphoblastic leukemia xenografts. Clin Cancer Res. 13:5426–5435.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Finney HM, Akbar AN and Lawson AD:
Activation of resting human primary T cells with chimeric
receptors: Costimulation from CD28, inducible costimulator, CD134,
and CD137 in series with signals from the TCR zeta chain. J
Immunol. 172:104–113. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Roger A, Finet A, Boru B, Beauchet A,
Mazeron JJ, Otzmeguine Y, Blom A, Longvert C, de Maleissye MF,
Funck-Brentano E and Saiag P: Efficacy of combined
hypo-fractionated radiotherapy and anti-PD-1 monotherapy in
difficult-to-treat advanced melanoma patients. Oncoimmunology.
7:e14421662018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Taquin H, Fontas E, Massol O, Chevallier
P, Balloti R, Beranger G, Lacour JP, Passeron T and Montaudie H:
Efficacy and safety data for checkpoint inhibitors in advanced
melanoma under real-life conditions: A monocentric study conducted
in Nice from 2010 to 2016. Ann Dermatol Venereol. 145:649–658.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Y, Zhang X, Yang L, Xue J and Hu G:
Blockade of CCL2 enhances immunotherapeutic effect of anti-PD1 in
lung cancer. J Bone Oncol. 11:27–32. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zibetti Dal Molin G, Abrahao CM, Coleman
RL and Maluf FC: Response to pembrolizumab in a heavily treated
patient with metastatic ovarian carcinosarcoma. Gynecol Oncol Res
Pract. 5:62018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li S, Siriwon N, Zhang X, Yang S, Jin T,
He F, Kim YJ, Mac J, Lu Z, Wang S, et al: Enhanced cancer
immunotherapy by chimeric antigen receptor-modified t cells
engineered to secrete checkpoint inhibitors. Clin Cancer Res.
23:6982–6992. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Serganova I, Moroz E, Cohen I, Moroz M,
Mane M, Zurita J, Shenker L, Ponomarev V and Blasberg R:
Enhancement of PSMA-directed CAR adoptive immunotherapy by
PD-1/PD-L1 blockade. Mol Ther Oncolytics. 4:41–54. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu X, Ranganathan R, Jiang S, Fang C, Sun
J, Kim S, Newick K, Lo A, June CH, Zhao Y and Moon EK: A chimeric
switch-receptor targeting PD1 augments the efficacy of
second-generation car t cells in advanced solid tumors. Cancer Res.
76:1578–1590. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cherkassky L, Morello A, Villena-Vargas J,
Feng Y, Dimitrov DS, Jones DR, Sadelain M and Adusumilli PS: Human
CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist
tumor-mediated inhibition. J Clin Invest. 126:3130–3144. 2016.
View Article : Google Scholar : PubMed/NCBI
|